Identification of BRCC3 and BRCA1 as Regulators of TAZ Stability and Activity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Transfection
2.3. Viral Production and Cells’ Transduction
2.4. Cloning
2.5. Immunoblot Analysis
2.6. TAZ Immunoprecipitation
2.7. RNA Extraction and RT–qPCR
2.8. Proximity Ligation Assay and Immunofluorescence Analyses
2.9. siRNA Screening
2.10. Statistical Analyses
3. Results
3.1. A Genetic Screen to Identify Regulators of TAZ Activity
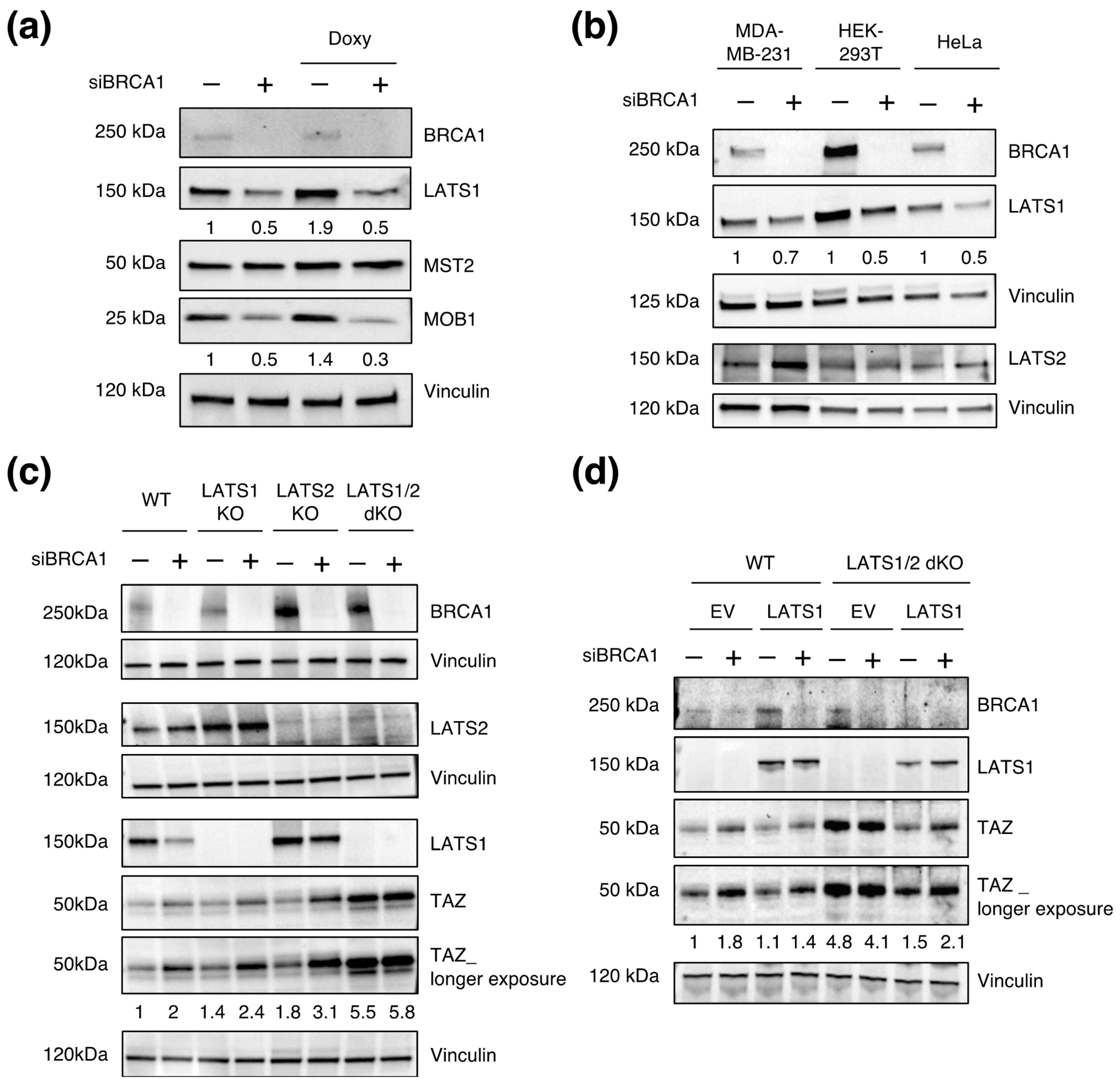
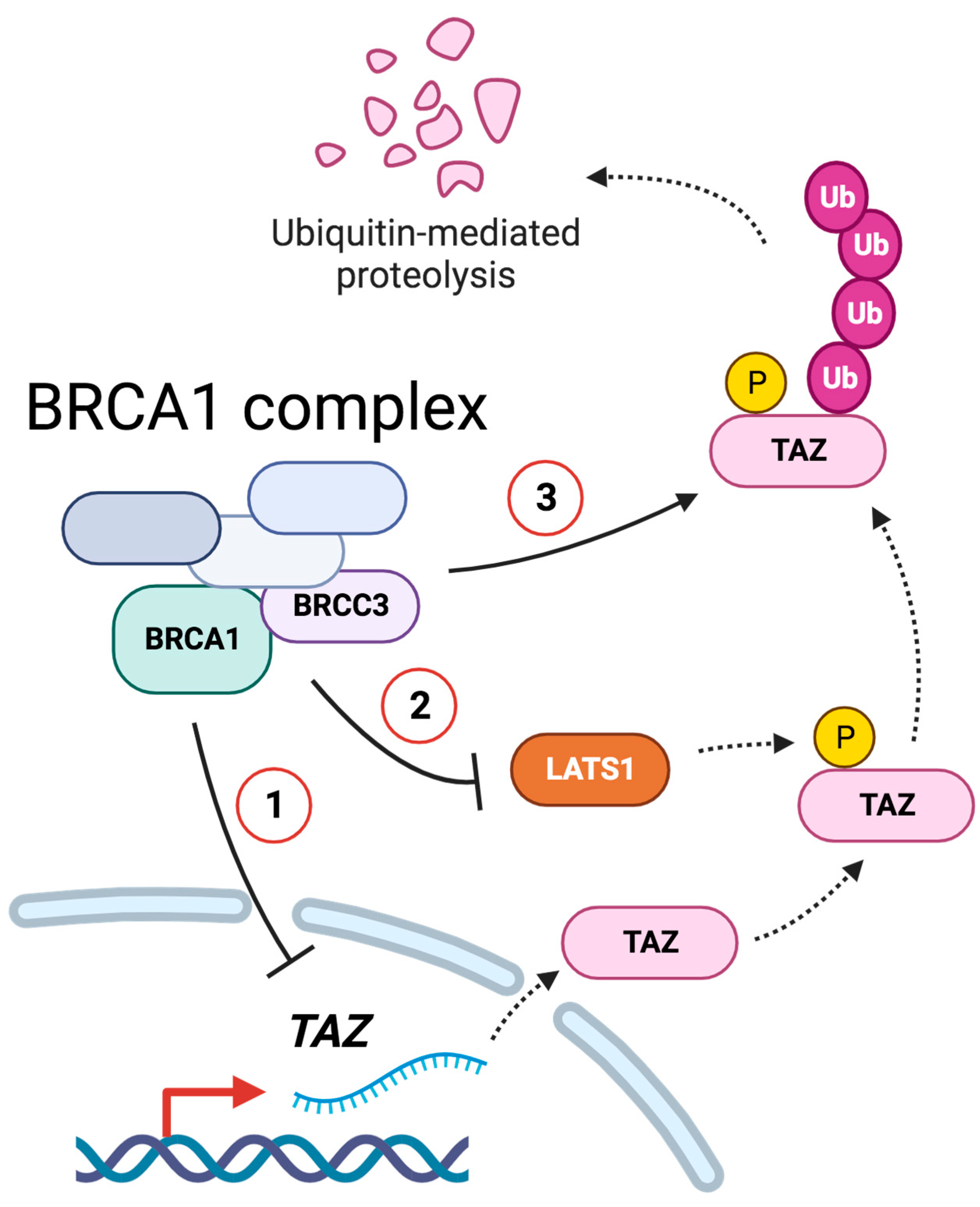
3.2. BRCC3 and the BRCA1–A Complex Regulates TAZ
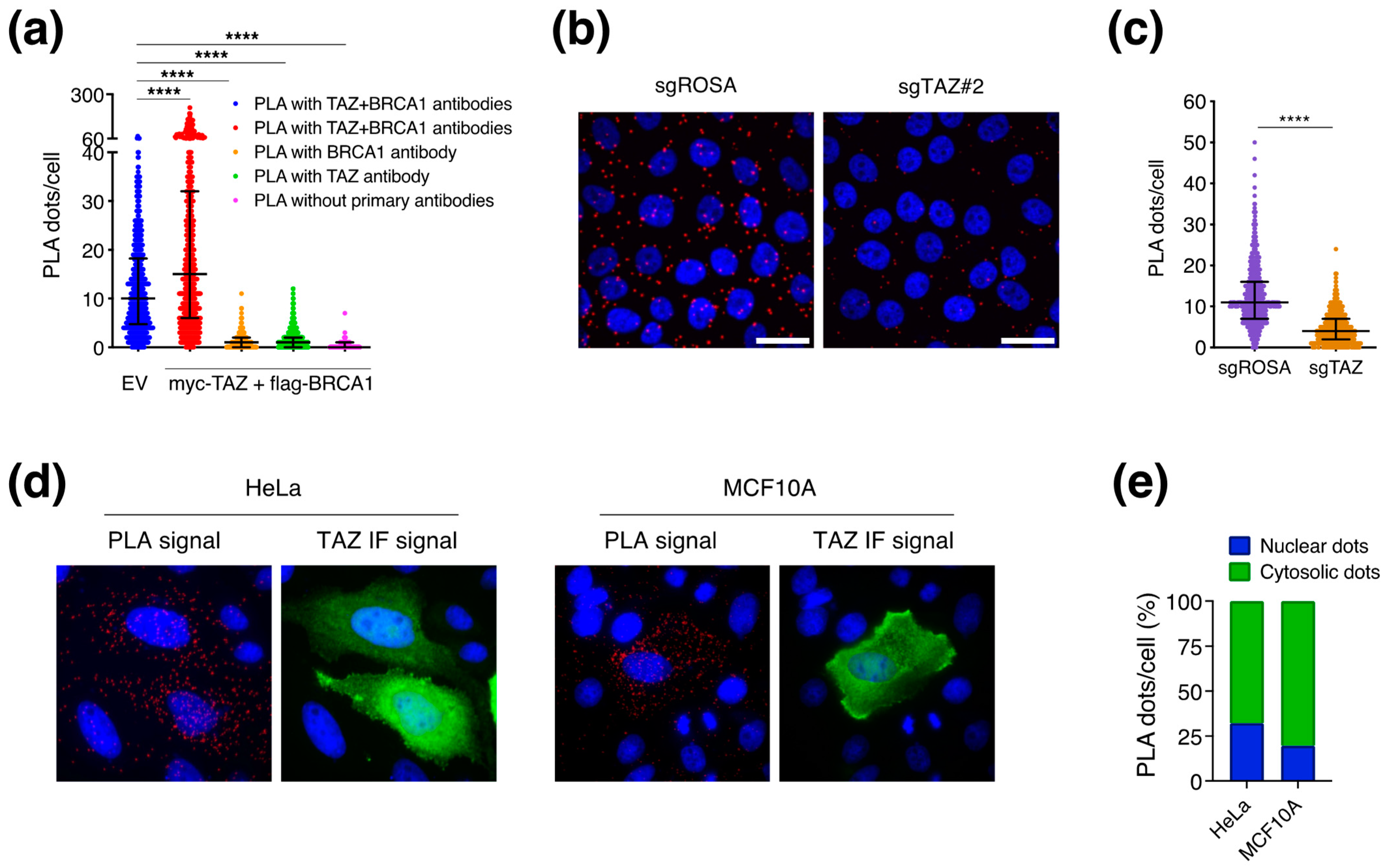
3.3. BRCA1 Does Not Control TAZ Cellular Localization
3.4. BRCA1 May Regulate TAZ in the Cytoplasm
3.5. LATS1/2 Are Epistatic over BRCA1 in Regulating TAZ Levels
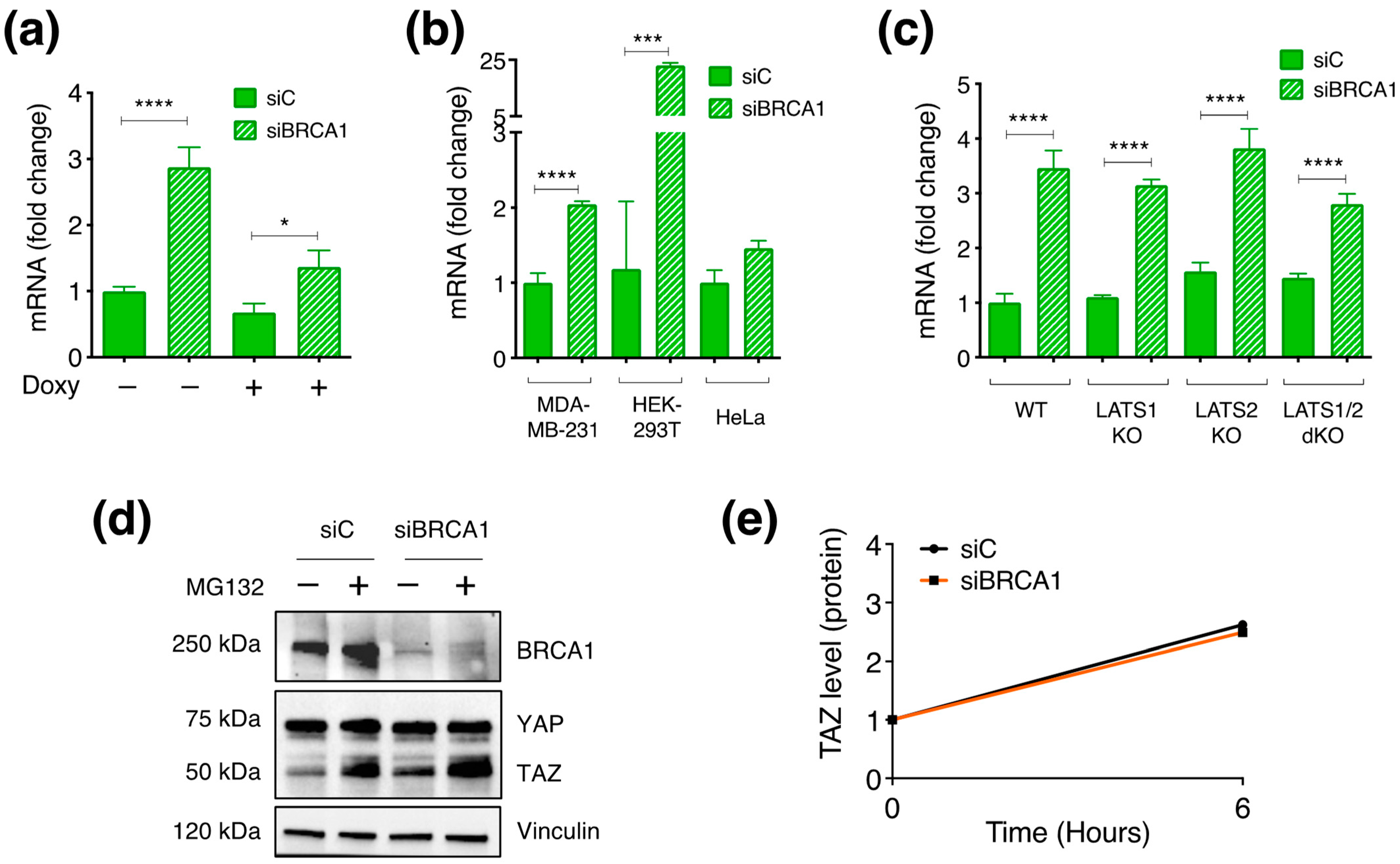
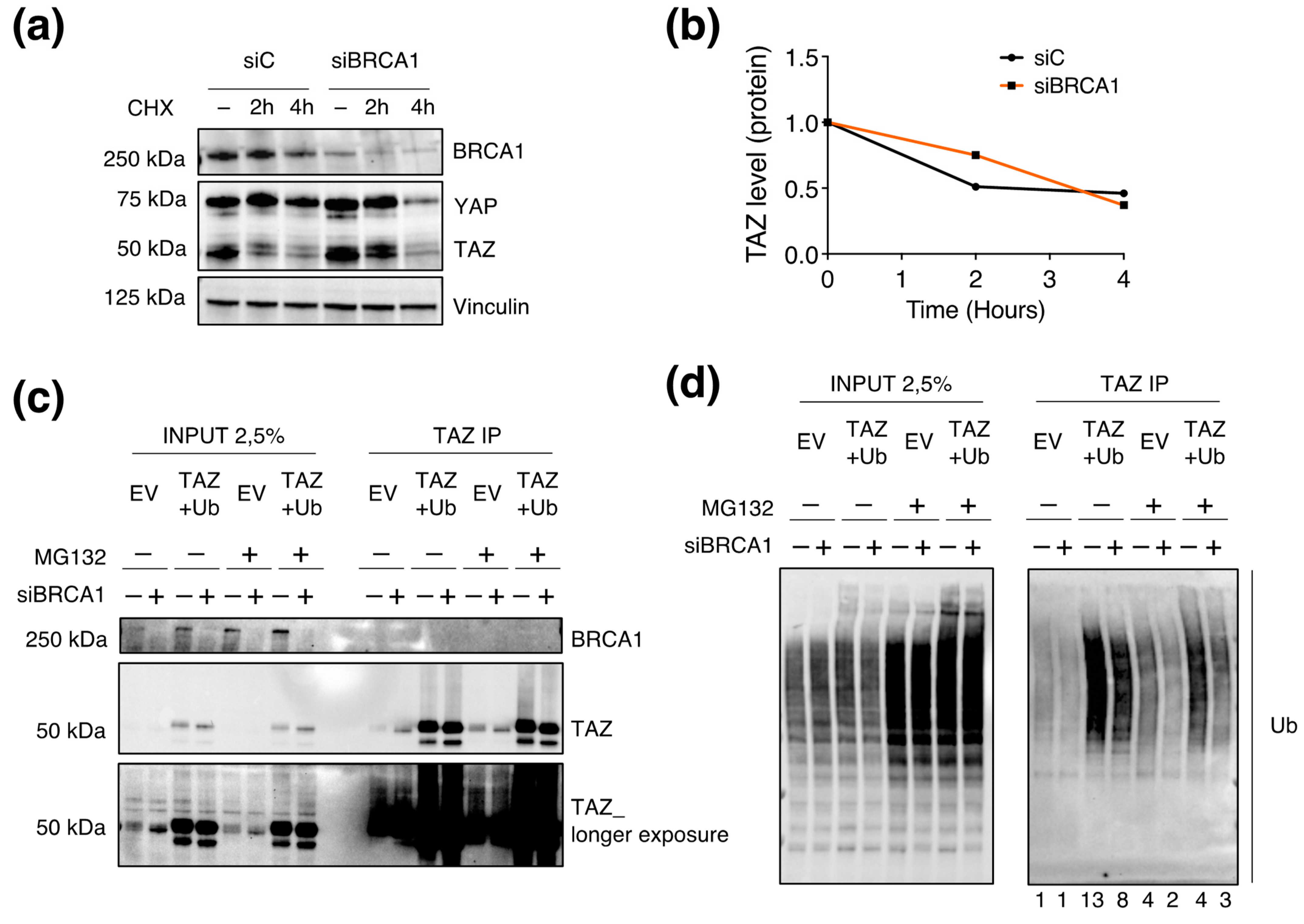
3.6. BRCA1 Controls TAZ Expression Mainly by Post-Translational Mechanisms
3.7. BRCA1 Regulates TAZ Poly-Ubiquitination
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, J.; Feldmann, G.; Huang, J.; Wu, S.; Zhang, N.; Comerford, S.A.; Gayyed, M.F.; Anders, R.A.; Maitra, A.; Pan, D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007, 130, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Zha, Z.Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCFbeta-TrCP E3 ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef]
- Zhao, B.; Li, L.; Tumaneng, K.; Wang, C.Y.; Guan, K.L. A coordinated phosphorylation by Lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes. Dev. 2010, 24, 72–85. [Google Scholar] [CrossRef]
- Heng, B.C.; Zhang, X.; Aubel, D.; Bai, Y.; Li, X.; Wei, Y.; Fussenegger, M.; Deng, X. An overview of signaling pathways regulating YAP/TAZ activity. Cell Mol. Life Sci. 2021, 78, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Martin-Belmonte, F.; Perez-Moreno, M. Epithelial cell polarity, stem cells and cancer. Nat. Rev. Cancer 2011, 12, 23–38. [Google Scholar] [CrossRef]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Dobrokhotov, O.; Samsonov, M.; Sokabe, M.; Hirata, H. Mechanoregulation and pathology of YAP/TAZ via Hippo and non-Hippo mechanisms. Clin. Transl. Med. 2018, 7, 23. [Google Scholar] [CrossRef]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef]
- DeRan, M.; Yang, J.; Shen, C.H.; Peters, E.C.; Fitamant, J.; Chan, P.; Hsieh, M.; Zhu, S.; Asara, J.M.; Zheng, B.; et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014, 9, 495–503. [Google Scholar] [CrossRef]
- Biagioni, F.; Croci, O.; Sberna, S.; Donato, E.; Sabo, A.; Bisso, A.; Curti, L.; Chiesa, A.; Campaner, S. Decoding YAP dependent transcription in the liver. Nucleic Acids Res. 2022, 50, 7959–7971. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Forcato, M.; Battilana, G.; Azzolin, L.; Quaranta, E.; Bodega, B.; Rosato, A.; Bicciato, S.; Cordenonsi, M.; Piccolo, S. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol. 2015, 17, 1218–1227. [Google Scholar] [CrossRef]
- Galli, G.G.; Carrara, M.; Yuan, W.C.; Valdes-Quezada, C.; Gurung, B.; Pepe-Mooney, B.; Zhang, T.; Geeven, G.; Gray, N.S.; de Laat, W.; et al. YAP Drives Growth by Controlling Transcriptional Pause Release from Dynamic Enhancers. Mol. Cell 2015, 60, 328–337. [Google Scholar] [CrossRef]
- Stein, C.; Bardet, A.F.; Roma, G.; Bergling, S.; Clay, I.; Ruchti, A.; Agarinis, C.; Schmelzle, T.; Bouwmeester, T.; Schubeler, D.; et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet. 2015, 11, e1005465. [Google Scholar] [CrossRef]
- Lopez-Hernandez, A.; Sberna, S.; Campaner, S. Emerging Principles in the Transcriptional Control by YAP and TAZ. Cancers 2021, 13, 4242. [Google Scholar] [CrossRef]
- Croci, O.; De Fazio, S.; Biagioni, F.; Donato, E.; Caganova, M.; Curti, L.; Doni, M.; Sberna, S.; Aldeghi, D.; Biancotto, C.; et al. Transcriptional integration of mitogenic and mechanical signals by Myc and YAP. Genes. Dev. 2017, 31, 2017–2022. [Google Scholar] [CrossRef]
- Luo, M.; Meng, Z.; Moroishi, T.; Lin, K.C.; Shen, G.; Mo, F.; Shao, B.; Wei, X.; Zhang, P.; Wei, Y.; et al. Heat stress activates YAP/TAZ to induce the heat shock transcriptome. Nat. Cell Biol. 2020, 22, 1447–1459. [Google Scholar] [CrossRef]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Bartucci, M.; Dattilo, R.; Moriconi, C.; Pagliuca, A.; Mottolese, M.; Federici, G.; Benedetto, A.D.; Todaro, M.; Stassi, G.; Sperati, F.; et al. TAZ is required for metastatic activity and chemoresistance of breast cancer stem cells. Oncogene 2015, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yu, X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013, 23, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Coleman, K.A.; Greenberg, R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011, 286, 13669–13680. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.J.; et al. Isomerization of BRCA1-BARD1 promotes replication fork protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef]
- Starita, L.M.; Horwitz, A.A.; Keogh, M.C.; Ishioka, C.; Parvin, J.D.; Chiba, N. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J. Biol. Chem. 2005, 280, 24498–24505. [Google Scholar] [CrossRef]
- Plouffe, S.W.; Meng, Z.; Lin, K.C.; Lin, B.; Hong, A.W.; Chun, J.V.; Guan, K.L. Characterization of Hippo Pathway Components by Gene Inactivation. Mol. Cell 2016, 64, 993–1008. [Google Scholar] [CrossRef]
- Zhang, X.D. Illustration of SSMD, z score, SSMD*, z* score, and t statistic for hit selection in RNAi high-throughput screens. J. Biomol. Screen. 2011, 16, 775–785. [Google Scholar] [CrossRef]
- Gill, M.K.; Christova, T.; Zhang, Y.Y.; Gregorieff, A.; Zhang, L.; Narimatsu, M.; Song, S.; Xiong, S.; Couzens, A.L.; Tong, J.; et al. A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat. Commun. 2018, 9, 3510. [Google Scholar] [CrossRef]
- Shen, S.; Guo, X.; Yan, H.; Lu, Y.; Ji, X.; Li, L.; Liang, T.; Zhou, D.; Feng, X.H.; Zhao, J.C.; et al. A miR-130a-YAP positive feedback loop promotes organ size and tumorigenesis. Cell Res. 2015, 25, 997–1012. [Google Scholar] [CrossRef]
- Gu, Y.; Wang, Y.; Sha, Z.; He, C.; Zhu, Y.; Li, J.; Yu, A.; Zhong, Z.; Wang, X.; Sun, Y.; et al. Transmembrane protein KIRREL1 regulates Hippo signaling via a feedback loop and represents a therapeutic target in YAP/TAZ-active cancers. Cell Rep. 2022, 40, 111296. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Park, H.W.; Qin, B.; Chen, Q.; Meng, Z.; Plouffe, S.W.; Taniguchi, K.; Yu, F.X.; Karin, M.; Pan, D.; et al. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes. Dev. 2015, 29, 1271–1284. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.V.; Irvine, K.D. Regulation of Hippo signaling by EGFR-MAPK signaling through Ajuba family proteins. Dev. Cell 2013, 24, 459–471. [Google Scholar] [CrossRef]
- Zanconato, F.; Battilana, G.; Forcato, M.; Filippi, L.; Azzolin, L.; Manfrin, A.; Quaranta, E.; Di Biagio, D.; Sigismondo, G.; Guzzardo, V.; et al. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat. Med. 2018, 24, 1599–1610. [Google Scholar] [CrossRef]
- Zheng, H.; Gupta, V.; Patterson-Fortin, J.; Bhattacharya, S.; Katlinski, K.; Wu, J.; Varghese, B.; Carbone, C.J.; Aressy, B.; Fuchs, S.Y.; et al. A BRISC-SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Rep. 2013, 5, 180–193. [Google Scholar] [CrossRef]
- Walden, M.; Tian, L.; Ross, R.L.; Sykora, U.M.; Byrne, D.P.; Hesketh, E.L.; Masandi, S.K.; Cassel, J.; George, R.; Ault, J.R.; et al. Metabolic control of BRISC-SHMT2 assembly regulates immune signalling. Nature 2019, 570, 194–199. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38, e100376. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef]
- Rabl, J. BRCA1-A and BRISC: Multifunctional Molecular Machines for Ubiquitin Signaling. Biomolecules 2020, 10, 1503. [Google Scholar] [CrossRef]
- Kofler, M.; Speight, P.; Little, D.; Di Ciano-Oliveira, C.; Szaszi, K.; Kapus, A. Mediated nuclear import and export of TAZ and the underlying molecular requirements. Nat. Commun. 2018, 9, 4966. [Google Scholar] [CrossRef]
- Savage, K.I.; Harkin, D.P. BRCA1, a ‘complex’ protein involved in the maintenance of genomic stability. FEBS J. 2015, 282, 630–646. [Google Scholar] [CrossRef]
- Shao, G.; Lilli, D.R.; Patterson-Fortin, J.; Coleman, K.A.; Morrissey, D.E.; Greenberg, R.A. The Rap80-BRCC36 de-ubiquitinating enzyme complex antagonizes RNF8-Ubc13-dependent ubiquitination events at DNA double strand breaks. Proc. Natl. Acad. Sci. USA 2009, 106, 3166–3171. [Google Scholar] [CrossRef]
- Chen, X.; Arciero, C.A.; Wang, C.; Broccoli, D.; Godwin, A.K. BRCC36 is essential for ionizing radiation-induced BRCA1 phosphorylation and nuclear foci formation. Cancer Res. 2006, 66, 5039–5046. [Google Scholar] [CrossRef]
- Verma, S.; Yeddula, N.; Soda, Y.; Zhu, Q.; Pao, G.; Moresco, J.; Diedrich, J.K.; Hong, A.; Plouffe, S.; Moroishi, T.; et al. BRCA1/BARD1-dependent ubiquitination of NF2 regulates Hippo-YAP1 signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 7363–7370. [Google Scholar] [CrossRef]
- Xiang, T.; Ohashi, A.; Huang, Y.; Pandita, T.K.; Ludwig, T.; Powell, S.N.; Yang, Q. Negative Regulation of AKT Activation by BRCA1. Cancer Res. 2008, 68, 10040–10044. [Google Scholar] [CrossRef]
- Kim, M.; Jho, E.H. Cross-talk between Wnt/beta-catenin and Hippo signaling pathways: A brief review. BMB Rep. 2014, 47, 540–545. [Google Scholar] [CrossRef]
- Lei, Q.Y.; Zhang, H.; Zhao, B.; Zha, Z.Y.; Bai, F.; Pei, X.H.; Zhao, S.; Xiong, Y.; Guan, K.L. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell Biol. 2008, 28, 2426–2436. [Google Scholar] [CrossRef]
- Chan, S.W.; Lim, C.J.; Guo, K.; Ng, C.P.; Lee, I.; Hunziker, W.; Zeng, Q.; Hong, W. A role for TAZ in migration, invasion, and tumorigenesis of breast cancer cells. Cancer Res. 2008, 68, 2592–2598. [Google Scholar] [CrossRef]
- Diaz-Martin, J.; Lopez-Garcia, M.A.; Romero-Perez, L.; Atienza-Amores, M.R.; Pecero, M.L.; Castilla, M.A.; Biscuola, M.; Santon, A.; Palacios, J. Nuclear TAZ expression associates with the triple-negative phenotype in breast cancer. Endocr. Relat. Cancer 2015, 22, 443–454. [Google Scholar] [CrossRef]
- Bai, F.; Wang, C.; Liu, X.; Hollern, D.; Liu, S.; Fan, C.; Liu, C.; Ren, S.; Herschkowitz, J.I.; Zhu, W.G.; et al. Loss of function of BRCA1 promotes EMT in mammary tumors through activation of TGFbetaR2 signaling pathway. Cell Death Dis. 2022, 13, 195. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Zhang, L.H.; Liu, X.; Wang, C.; Zheng, C.; Sun, J.; Li, M.; Zhu, W.G.; Pei, X.H. GATA3 functions downstream of BRCA1 to suppress EMT in breast cancer. Theranostics 2021, 11, 8218–8233. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Chan, H.L.; Scott, A.; Smith, M.D.; Fan, C.; Herschkowitz, J.I.; Perou, C.M.; Livingstone, A.S.; Robbins, D.J.; Capobianco, A.J.; et al. BRCA1 suppresses epithelial-to-mesenchymal transition and stem cell dedifferentiation during mammary and tumor development. Cancer Res. 2014, 74, 6161–6172. [Google Scholar] [CrossRef]
- Zhang, Y.; Donaher, J.L.; Das, S.; Li, X.; Reinhardt, F.; Krall, J.A.; Lambert, A.W.; Thiru, P.; Keys, H.R.; Khan, M.; et al. Genome-wide CRISPR screen identifies PRC2 and KMT2D-COMPASS as regulators of distinct EMT trajectories that contribute differentially to metastasis. Nat. Cell Biol. 2022, 24, 554–564. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sberna, S.; Lopez-Hernandez, A.; Biancotto, C.; Motta, L.; Andronache, A.; Verhoef, L.G.G.C.; Caganova, M.; Campaner, S. Identification of BRCC3 and BRCA1 as Regulators of TAZ Stability and Activity. Cells 2023, 12, 2431. https://doi.org/10.3390/cells12202431
Sberna S, Lopez-Hernandez A, Biancotto C, Motta L, Andronache A, Verhoef LGGC, Caganova M, Campaner S. Identification of BRCC3 and BRCA1 as Regulators of TAZ Stability and Activity. Cells. 2023; 12(20):2431. https://doi.org/10.3390/cells12202431
Chicago/Turabian StyleSberna, Silvia, Alejandro Lopez-Hernandez, Chiara Biancotto, Luca Motta, Adrian Andronache, Lisette G. G. C. Verhoef, Marieta Caganova, and Stefano Campaner. 2023. "Identification of BRCC3 and BRCA1 as Regulators of TAZ Stability and Activity" Cells 12, no. 20: 2431. https://doi.org/10.3390/cells12202431
APA StyleSberna, S., Lopez-Hernandez, A., Biancotto, C., Motta, L., Andronache, A., Verhoef, L. G. G. C., Caganova, M., & Campaner, S. (2023). Identification of BRCC3 and BRCA1 as Regulators of TAZ Stability and Activity. Cells, 12(20), 2431. https://doi.org/10.3390/cells12202431