
Insight and Recommendations for Fragile X-Premutation-Associated Conditions from the Fifth International Conference on FMR1 Premutation

 , , ,
, , ,  , , , , , ,
, , , , , ,  , ,
, ,  , , , , , , , , ,
, , , , , , , , ,  and add
Show full author list
and add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
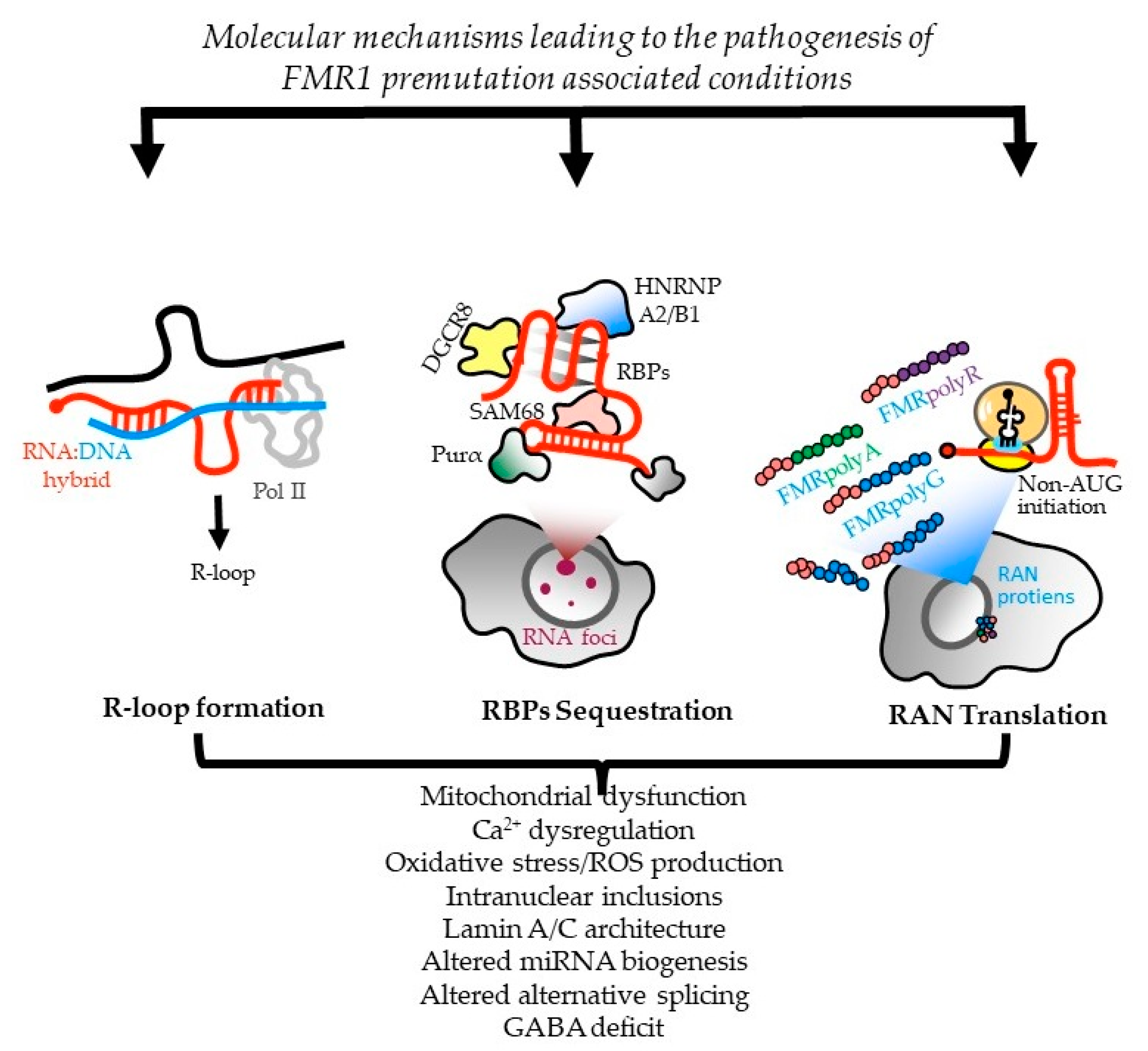
2. The Molecular Basis of FXPAC
2.1. Molecular Basis of the FMR1 Locus
2.2. Molecular Mechanisms Leading to FXTAS Pathology—RNA Toxicity and RAN Translation at CGG Repeats: Mechanistic Insights and Their Contribution to Disease Pathology
2.3. Therapeutic Perspectives to FXTAS from a RAN-Translation Perspective
2.4. Genetic Modifiers in Fragile X-Associated Tremor Ataxia Syndrome (FXTAS)
2.5. The Use of Human Pluripotent-Stem-Cell-Based Neurodevelopmental Models for FXTAS
2.6. Shared Molecular Mechanism with Other Repeat Expansion Disorders
2.7. Mitochondrial Dysfunction in PM Carriers
2.8. Omics Studies (Metabolomics and Proteomics) in PM Carriers
2.9. CGG Short Tandem Repeat (STR) Expansions
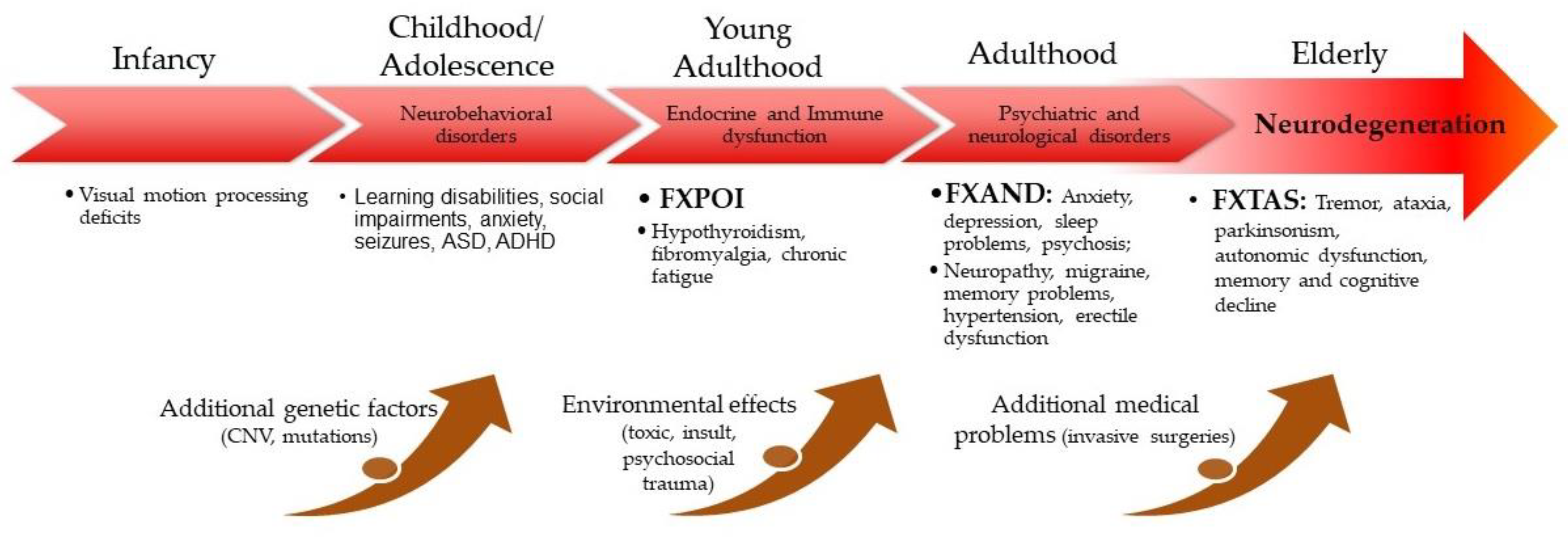
3. Clinical Involvement in Children Who Have a PM
- Increasing efforts to prepare support organizations, genetic counselors, and healthcare practitioners to be able to respond to and treat children who have a PM and who are symptomatic;
- Detailed characterization of the pediatric phenotype—both at clinically actionable and subthreshold levels;
- Efforts to study outcomes at a population scale through newborn screening that may provide an evidence-base around developmental trajectories and risks;
- Clarified testing indications and, potentially, modified diagnostic testing workflows to ensure that symptomatic children with PMs do not miss out on comprehensive genetic testing with microarrays and potentially other methodologies (WES or WGS).
4. FXPAC and Relationships with Genetic Markers
4.1. FXTAS: Neurological/Cognitive Phenotypes
4.2. FXTAS Spectrum: Nonsyndromic Neurological, Cognitive, and Psychiatric Involvements
4.3. Do PM Cognitive and Motor Deficits Represent a Distinct Form of Neural Involvement, or Are They Prodromal to FXTAS?
4.4. Major Psychiatric Issues (FXAND)
4.4.1. Anxiety
4.4.2. Depression
4.4.3. Substance Abuse
4.4.4. Autism Spectrum Disorder (ASD) and the Broad Autism Phenotype (BAP)
4.5. Other FXPAC-Related Symptoms and Conditions
4.5.1. Hypertension
4.5.2. Metabolic Syndrome
4.5.3. Chronic Fatigue
4.5.4. Chronic Pain and Fibromyalgia
4.5.5. Sleep Problems
5. FXTAS Clinical and Protective Mechanisms
6. Reproductive and Health Implications in Women Who Carry the PM
6.1. Fragile X-Associated Primary Ovarian Insufficiency (FXPOI)
6.2. Medications to Treat FXAND in FXPOI
6.3. Psychotherapy to Treat FXAND in FXPOI
6.4. Early Diagnosis and Carrier Screening
6.5. Future Directions
7. Neuroimaging Findings in FXTAS
7.1. Structural Brain Differences Associated with FXTAS
7.2. Functional Brain Differences Associated with FXTAS
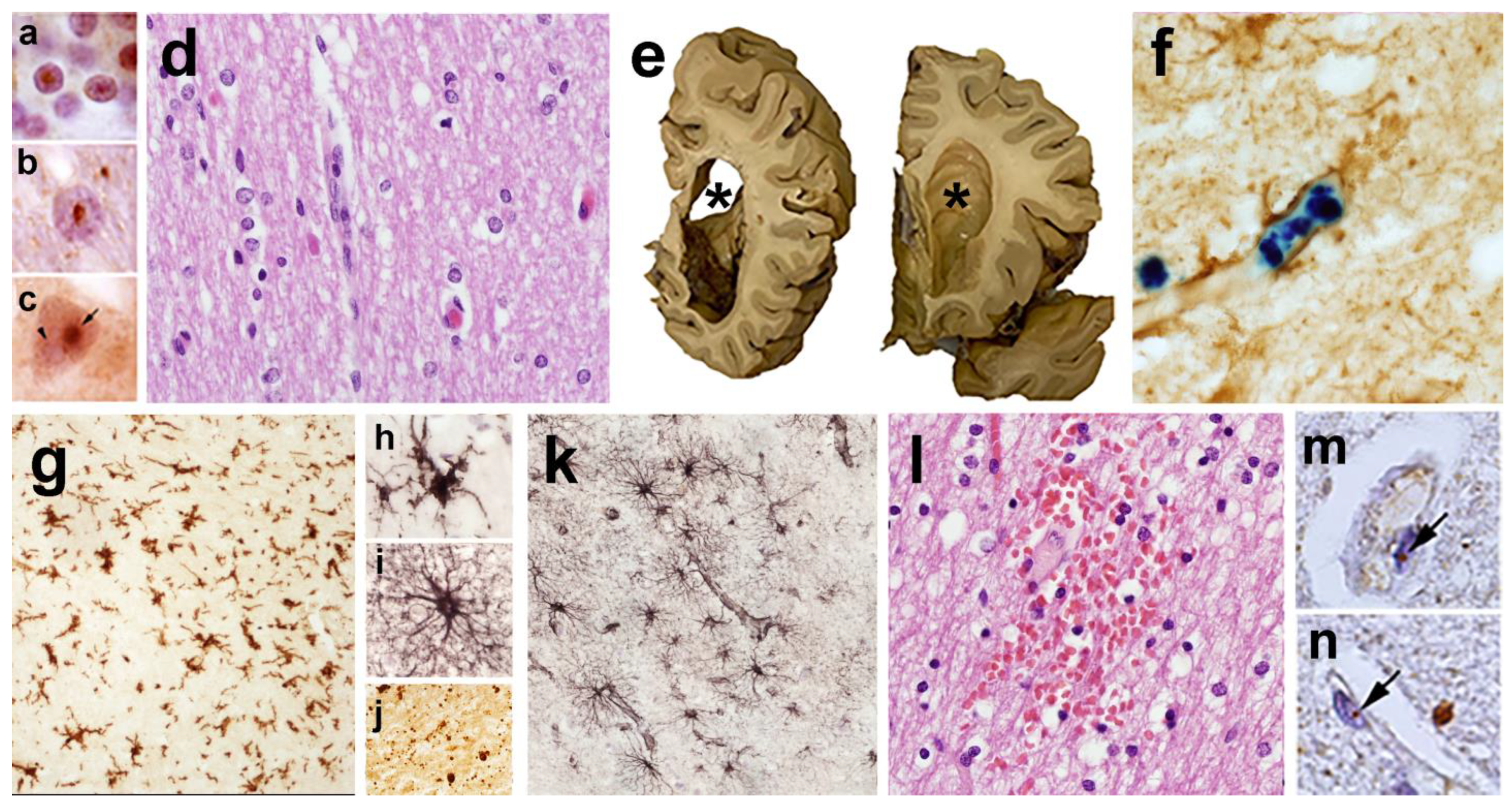
8. The Neuropathology of FXTAS
9. FXTAS Treatment
9.1. Treatment Trials Specific to FXTAS
9.2. Management of Neurologic Symptoms in FXTAS
9.3. Lifestyle Changes in FXTAS
9.4. Future Directions to Advance Treatment in FXTAS
10. Screening for Fragile X and FXPAC
10.1. Diagnosis via Cascade Testing
10.2. Newborn Screening
10.3. Carrier and Prenatal Screening
10.4. Genetic Testing Pathways
11. Shining a Light on the FMR1 PM: What We Know, What We Think We Know, and What We Need to Know
- Fertility-related issues—the need for increased knowledge and better pathways for fertility-related issues associated with the PM gene, particularly for younger women;
- The CGG repeat number is recognized as only part of the evolving picture—research indicating AR, FMR1 mRNA, FMRP levels, AGG interruptions, and allelic instability as also important factors to consider;
- Lifestyle measures—multiple presenters mentioned the importance of healthy lifestyle as a protective measure against risk factors associated with the PM, including an emphasis on limiting alcohol, not smoking, the importance of exercise and a good diet, and avoiding excess environmental toxins and high stress;
- It was noted that many PMs have high levels of functioning and achievement;
- Many PMs also face the challenges of children with developmental issues and FXS.
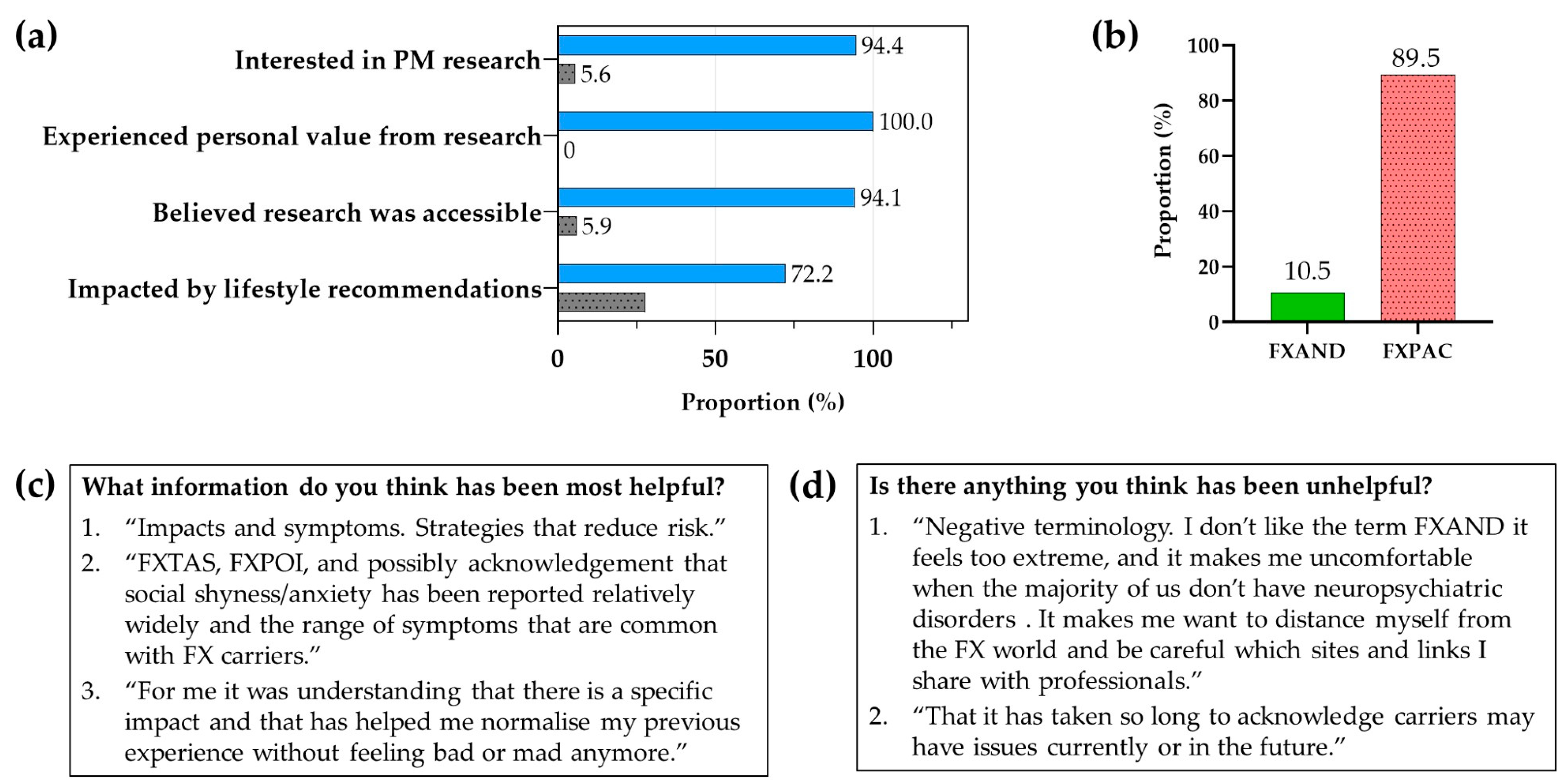
12. NZ Fragile X Community Response to PM Research (Fragile X New Zealand)
13. What Is in a Name? (National Fragile X Foundation (NFXF) and Fragile X Association of Australia (FXAA))
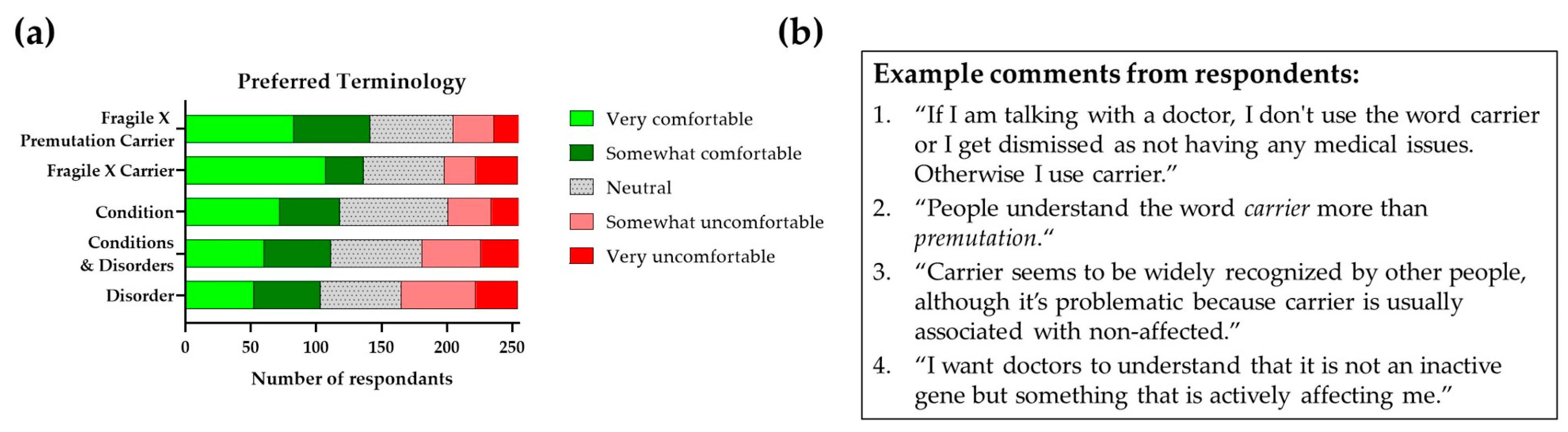
13.1. Terminology
13.2. The Importance of Appropriate Terminology
14. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ADHD | attention deficit hyperactivity disorder |
| AR | activation ratio |
| ASD | autism spectrum disorder |
| ASFMR1 | antisense FMR1 |
| ASOs | antisense oligonucleotides |
| BAP | broad autism phenotype |
| BDNF | brain-derived nerve growth factor |
| BRIEF | The Behavior Rating Inventory of Executive Function Questionnaire |
| CANTAB | Cambridge Automated Neuropsychological Test Battery |
| CBT | cognitive-behavioral therapy |
| CRHR1 | corticotropin releasing hormone type 1 receptor |
| CSF | cerebrospinal fluid |
| DBS | deep brain stimulation |
| DGCR8 | DiGeorge syndrome critical region 8 |
| DM1 | myotonic dystrophy type 1 |
| EBV | Epstein–Barr virus |
| EEG | electroencephalography |
| EGFP | green florescent protein |
| ERP | event-related potentials |
| FDA | Food and Drug Administration Agency |
| FECD | Fuchs endothelial corneal dystrophy |
| FMR1 | fragile X messenger ribonucleoprotein 1 gene |
| fMRI | functional magnetic resonance imaging |
| FMRP | FMR1 protein |
| FRAXI | Fragile X International |
| FXAA | Fragile X Association of Australia |
| FXAND | fragile X-associated neuropsychiatric disorders |
| FXPAC | fragile X-premutation-associated conditions |
| FXPOI | fragile X-associated primary ovarian insufficiency |
| FXR | farnesoid X receptor |
| FXTAS | fragile X-associated tremor/ataxia syndrome |
| HNRNP A2/B1 | heterogeneous nuclear ribonucleoprotein A1 |
| hESC | human embryonic stem cell |
| hPSCs | human pluripotent stem cells |
| ID | intellectual disabilities |
| iPSCs | induced pluripotent stem cells |
| LFB-PAS | luxol fast blue/periodic acid-Schiff stain |
| LXR/RXR | liver X receptor/retinoid X receptor |
| mTOR | the mechanistic target of rapamycin |
| MBNL1 | muscleblind-like protein 1 |
| MCP sign | MRI findings of white-matter disease usually in the middle cerebellar peduncles |
| MDD | major depressive disorder |
| MOCA | Montreal cognitive assessment |
| MRI | magnetic resonance imaging |
| NFXF | National Fragile X Foundation |
| NIID | neuronal intranuclear inclusion disease |
| NZ | New Zealand |
| OPDM | oculopharyngodistal myopathy |
| OPML | oculopharyngeal myopathy with leukoencephalopathy |
| OT | occupational therapy |
| p62/SQSTM1 | p62/sequestosome-1 |
| PBMCs | peripheral blood mononuclear cells |
| PCOS | polycystic ovary syndrome |
| PD | Parkinson’s disease |
| PGD | preimplantation genetic diagnosis |
| PKR | protein kinase R |
| PM | premutation |
| PMDD | premenstrual dysphoric disorder |
| POI | primary ovarian insufficiency |
| PSAT1 | phosphoserine aminotransferase 1 |
| PSCs | pluripotent stem cells |
| PSMB5 | proteasome subunit beta type-5 |
| RAN | repeat-associated non-AUG |
| RBP | RNA-binding proteins |
| RCT | randomized placebo-controlled trial |
| Rm 62 | ATP-dependent RNA helicase p62 |
| RNAi | RNA interference |
| ROS | reactive oxygen species |
| SAM68 | Src-associated substrate during mitosis of 68-kDa |
| SCAD | sudden coronary artery dissection |
| Sk2 | sphingosine kinase |
| SNRIs | serotonin and norepinephrine reuptake inhibitors |
| SSRIs | selective serotonin reuptake inhibitors |
| SOD | superoxide dismutase 1 |
| SUMO | small ubiquitin-like modifier |
| TDP-43 | TAR DNA-binding protein 43 |
| TNC | tenascin-C |
| WES | whole-exome sequencing |
| WGS | whole-genome sequencing |
| WMH | white-matter hyperintensities |
References
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Cronister, A.; Schreiner, R.; Wittenberger, M.; Amiri, K.; Harris, K.; Hagerman, R.J. Heterozygous fragile X female: Historical, physical, cognitive, and cytogenetic features. Am. J. Med. Genet. 1991, 38, 269–274. [Google Scholar] [CrossRef]
- Sherman, S.L. Premature ovarian failure in the fragile X syndrome. Am. J. Med. Genet. 2000, 97, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Mailick, M.R.; Hong, J.; Greenberg, J.; Smith, L.; Sherman, S. Curvilinear association of CGG repeats and age at menopause in women with FMR1 premutation expansions. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.K.; Marcus, M.; Epstein, M.P.; Allen, E.G.; Anido, A.E.; Paquin, J.J.; Yadav-Shah, M.; Sherman, S.L. Association of FMR1 repeat size with ovarian dysfunction. Hum. Reprod. 2005, 20, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hagerman, R.J.; Taylor, A.K.; Gane, L.W.; Godfrey, T.E.; Hagerman, P.J. Elevated levels of FMR1 mRNA in carrier males: A new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000, 66, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.; Grigsby, J.; Zhang, L.; Brunberg, J.A.; Greco, C.; Des Portes, V.; Jardini, T.; Levine, R.; et al. Fragile X premutation tremor/ataxia syndrome: Molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet. 2003, 72, 869–878. [Google Scholar] [CrossRef]
- Jacquemont, S.; Hagerman, R.J.; Leehey, M.A.; Hall, D.A.; Levine, R.A.; Brunberg, J.A.; Zhang, L.; Jardini, T.; Gane, L.W.; Harris, S.W.; et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004, 291, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Birch, R.C.; Anheim, M.; Jønch, A.E.; Pintado, E.; O’Keefe, J.; Trollor, J.N.; Stebbins, G.T.; Hagerman, R.J.; Fahn, S.; et al. Emerging topics in FXTAS. J. Neurodev. Disord. 2014, 6, 31. [Google Scholar] [CrossRef]
- Greco, C.M.; Berman, R.F.; Martin, R.M.; Tassone, F.; Schwartz, P.H.; Chang, A.; Trapp, B.D.; Iwahashi, C.; Brunberg, J.; Grigsby, J.; et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006, 129 Pt 1, 243–255. [Google Scholar] [CrossRef]
- Cabal-Herrera, A.M.; Tassanakijpanich, N.; Salcedo-Arellano, M.J.; Hagerman, R.J. Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS): Pathophysiology and Clinical Implications. Int. J. Mol. Sci. 2020, 21, 4391. [Google Scholar] [CrossRef] [PubMed]
- Aydin, E.Y.; Schneider, A.; Protic, D.; Wang, J.Y.; Martínez-Cerdeño, V.; Tassone, F.; Tang, H.T.; Perlman, S.; Hagerman, R.J. Rapidly Progressing Neurocognitive Disorder in a Male with FXTAS and Alzheimer’s Disease. Clin. Intervig. Aging 2020, 15, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Cerdeño, V.; Wang, J.Y.; Grigsby, J.; Hall, D.; Hagerman, R.J. FXTAS New Advances and Treatments. In Fragile X Syndrome and Premutation Disorders; Hagerman, R.J., Hagerman, P.J., Eds.; Mac Keith Press: London, UK, 2020; pp. 83–96. [Google Scholar]
- Famula, J.; Ferrer, E.; Hagerman, R.J.; Tassone, F.; Schneider, A.; Rivera, S.M.; Hessl, D. Neuropsychological changes in FMR1 premutation carriers and onset of fragile X-associated tremor/ataxia syndrome. J. Neurodev. Disord. 2022, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F.; Hall, D.A. FXTAS, FXPOI, and Other Premutation Disorders; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Wang, J.; Napoli, E.; Kim, K.; McLennan, Y.A.; Hagerman, R.J.; Giulivi, C. Brain Atrophy and White Matter Damage Linked to Peripheral Bioenergetic Deficits in the Neurodegenerative Disease FXTAS. Int. J. Mol. Sci. 2021, 22, 9171. [Google Scholar] [CrossRef] [PubMed]
- Greco, C.M.; Hagerman, R.J.; Tassone, F.; Chudley, A.E.; Del Bigio, M.R.; Jacquemont, S.; Leehey, M.; Hagerman, P.J. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, 125 Pt 8, 1760–1771. [Google Scholar] [CrossRef]
- Ariza, J.; Steward, C.; Rueckert, F.; Widdison, M.; Coffman, R.; Afjei, A.; Noctor, S.C.; Hagerman, R.; Hagerman, P.; Martínez-Cerdeño, V. Dysregulated iron metabolism in the choroid plexus in fragile X-associated tremor/ataxia syndrome. Brain Res. 2015, 1598, 88–96. [Google Scholar] [CrossRef]
- Salcedo-Arellano, M.J.; Wang, J.Y.; McLennan, Y.A.; Doan, M.; Cabal-Herrera, A.M.; Jimenez, S.; Wolf-Ochoa, M.W.; Sanchez, D.; Juarez, P.; Tassone, F.; et al. Cerebral Microbleeds in Fragile X-Associated Tremor/Ataxia Syndrome. Mov. Disord. 2021, 36, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Salcedo-Arellano, M.J.; Wolf-Ochoa, M.W.; Hong, T.; Amina, S.; Tassone, F.; Lechpammer, M.; Hagerman, R.; Martínez-Cerdeño, V. Parkinsonism Versus Concomitant Parkinson’s Disease in Fragile X-Associated Tremor/Ataxia Syndrome. Mov. Disord. Clin. Pract. 2020, 7, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Seritan, A.L.; Kim, K.; Benjamin, I.; Seritan, I.; Hagerman, R.J. Risk Factors for Cognitive Impairment in Fragile X-Associated Tremor/Ataxia Syndrome. J. Geriatr. Psychiatry Neurol. 2016, 29, 328–337. [Google Scholar] [CrossRef]
- Schneider, A.; Summers, S.; Tassone, F.; Seritan, A.; Hessl, D.; Hagerman, P.; Hagerman, R. Women with Fragile X-Associated Tremor/Ataxia Syndrome. Mov. Disord. Clin. Pract. 2020, 7, 910–919. [Google Scholar] [CrossRef]
- Sellier, C.; Freyermuth, F.; Tabet, R.; Tran, T.; He, F.; Ruffenach, F.; Alunni, V.; Moine, H.; Thibault, C.; Page, A.; et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013, 3, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Duan, R.; Qurashi, A.; Qin, Y.; Tian, D.; Rosser, T.C.; Liu, H.; Feng, Y.; Warren, S.T. Pur alpha binds to rCGG repeats and modulates repeat-mediated neurodegeneration in a Drosophila model of fragile X tremor/ataxia syndrome. Neuron 2007, 55, 556–564. [Google Scholar] [CrossRef]
- Sofola, O.A.; Jin, P.; Qin, Y.; Duan, R.; Liu, H.; de Haro, M.; Nelson, D.L.; Botas, J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron 2007, 55, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Holm, K.N.; Herren, A.W.; Taylor, S.L.; Randol, J.L.; Kim, K.; Espinal, G.; Martiínez-Cerdeño, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Human Cerebral Cortex Proteome of Fragile X-Associated Tremor/Ataxia Syndrome. Front. Mol. Biosci. 2020, 7, 600840. [Google Scholar]
- Todd, P.K.; Oh, S.Y.; Krans, A.; He, F.; Sellier, C.; Frazer, M.; Renoux, A.J.; Chen, K.C.; Scaglione, K.M.; Basrur, V.; et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 2013, 78, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Rosario, R.; Stewart, H.L.; Choudhury, N.R.; Michlewski, G.; Charlet-Berguerand, N.; Anderson, R.A. Evidence for a fragile X messenger ribonucleoprotein 1 (FMR1) mRNA gain-of-function toxicity mechanism contributing to the pathogenesis of fragile X-associated premature ovarian insufficiency. Faseb J. 2022, 36, e22612. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Ross-Inta, C.; Wong, S.; Omanska-Klusek, A.; Barrow, C.; Iwahashi, C.; Garcia-Arocena, D.; Sakaguchi, D.; Berry-Kravis, E.; Hagerman, R.; et al. Altered zinc transport disrupts mitochondrial protein processing/import in fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2011, 20, 3079–3092. [Google Scholar] [CrossRef]
- Napoli, E.; Song, G.; Wong, S.; Hagerman, R.; Giulivi, C. Altered Bioenergetics in Primary Dermal Fibroblasts from Adult Carriers of the FMR1 Premutation before the Onset of the Neurodegenerative Disease Fragile X-Associated Tremor/Ataxia Syndrome. Cerebellum 2016, 15, 552–564. [Google Scholar] [CrossRef]
- Giulivi, C.; Napoli, E.; Tassone, F.; Halmai, J.; Hagerman, R. Plasma metabolic profile delineates roles for neurodegeneration, pro-inflammatory damage and mitochondrial dysfunction in the FMR1 premutation. Biochem. J. 2016, 473, 3871–3888. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Duffy, D.L.; Martin, N.G.; Tassone, F.; Atkinson, A.; Storey, E. ‘Essential Tremor’ Phenotype in FMR1 Premutation/Gray Zone Sibling Series: Exploring Possible Genetic Modifiers. Twin Res. Hum. Genet. 2021, 24, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.; Santos, E.; Kim, K.; Ponzini, M.D.; McLennan, Y.A.; Schneider, A.; Tassone, F.; Hagerman, R.J. Increased Pain Symptomatology among Females vs. Males with Fragile X-Associated Tremor/Ataxia Syndrome. Front. Psychiatry 2021, 12, 762915. [Google Scholar] [CrossRef] [PubMed]
- Coffey, S.M.; Cook, K.; Tartaglia, N.; Tassone, F.; Nguyen, D.V.; Pan, R.; Bronsky, H.E.; Yuhas, J.; Borodyanskaya, M.; Grigsby, J.; et al. Expanded clinical phenotype of women with the FMR1 premutation. Am. J. Med. Genet. A 2008, 146, 1009–1016. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, L.; Abucayan, F.; Hagerman, R.; Tassone, F.; Hessl, D. Anxiety disorders in fragile X premutation carriers: Preliminary characterization of probands and non-probands. Intractable Rare Dis. Res. 2015, 4, 123–130. [Google Scholar] [CrossRef]
- Farzin, F.; Perry, H.; Hessl, D.; Loesch, D.; Cohen, J.; Bacalman, S.; Gane, L.; Tassone, F.; Hagerman, P.; Hagerman, R. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J. Dev. Behav. Pediatr. 2006, 27 (Suppl. S2), S137–S144. [Google Scholar] [CrossRef]
- Hunter, J.E.; Leslie, M.; Novak, G.; Hamilton, D.; Shubeck, L.; Charen, K.; Abramowitz, A.; Epstein, M.P.; Lori, A.; Binder, E.; et al. Depression and anxiety symptoms among women who carry the FMR1 premutation: Impact of raising a child with fragile X syndrome is moderated by CRHR1 polymorphisms. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 549–559. [Google Scholar] [CrossRef]
- Aishworiya, R.; Protic, D.; Tang, S.J.; Schneider, A.; Tassone, F.; Hagerman, R. Fragile X-Associated Neuropsychiatric Disorders (FXAND) in Young Fragile X Premutation Carriers. Genes 2022, 13, 2399. [Google Scholar] [CrossRef]
- Clifford, S.; Dissanayake, C.; Bui, Q.M.; Huggins, R.; Taylor, A.K.; Loesch, D.Z. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J. Autism Dev. Disord. 2007, 37, 738–747. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Raspa, M.; Olmsted, M.; Holiday, D.B. Co-occurring conditions associated with FMR1 gene variations: Findings from a national parent survey. Am. J. Med. Genet. A 2008, 146, 2060–2069. [Google Scholar] [CrossRef]
- Aziz, M.; Stathopulu, E.; Callias, M.; Taylor, C.; Turk, J.; Oostra, B.; Willemsen, R.; Patton, M. Clinical features of boys with fragile X premutations and intermediate alleles. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2003, 121, 119–127. [Google Scholar] [CrossRef]
- Chonchaiya, W.; Au, J.; Schneider, A.; Hessl, D.; Harris, S.W.; Laird, M.; Mu, Y.; Tassone, F.; Nguyen, D.V.; Hagerman, R.J. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum. Genet. 2012, 131, 581–589. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lozano, R.; Hagerman, R.J.; Duyzend, M.; Budimirovic, D.B.; Eichler, E.E.; Tassone, F. Genomic studies in fragile X premutation carriers. J. Neurodev. Disord. 2014, 6, 27. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial dynamics—Fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Song, G.; Napoli, E.; Wong, S.; Hagerman, R.; Liu, S.; Tassone, F.; Giulivi, C. Altered redox mitochondrial biology in the neurodegenerative disorder fragile X-tremor/ataxia syndrome: Use of antioxidants in precision medicine. Mol. Med. 2016, 22, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Ligsay, A.; El-Deeb, M.; Salcedo-Arellano, M.J.; Schloemerkemper, N.; Grayson, J.S.; Hagerman, R. General Anesthetic Use in Fragile X Spectrum Disorders. J. Neurosurg. Anesth. 2019, 31, 285–290. [Google Scholar] [CrossRef]
- Muzar, Z.; Adams, P.E.; Schneider, A.; Hagerman, R.J.; Lozano, R. Addictive substances may induce a rapid neurological deterioration in fragile X-associated tremor ataxia syndrome: A report of two cases. Intractable Rare Dis. Res. 2014, 3, 162–165. [Google Scholar] [CrossRef]
- Muzar, Z.; Lozano, R.; Schneider, A.; Adams, P.E.; Faradz, S.M.; Tassone, F.; Hagerman, R.J. Methadone use in a male with the FMRI premutation and FXTAS. Am. J. Med. Genet. A 2015, 167, 1354–1359. [Google Scholar] [CrossRef]
- Keil Stietz, K.P.; Sethi, S.; Klocke, C.R.; de Ruyter, T.E.; Wilson, M.D.; Pessah, I.N.; Lein, P.J. Sex and Genotype Modulate the Dendritic Effects of Developmental Exposure to a Human-Relevant Polychlorinated Biphenyls Mixture in the Juvenile Mouse. Front. Neurosci. 2021, 15, 766802. [Google Scholar] [CrossRef]
- Saldarriaga, W.; Salcedo-Arellano, M.J.; Rodriguez-Guerrero, T.; Ríos, M.; Fandiño-Losada, A.; Ramirez-Cheyne, J.; Lein, P.J.; Tassone, F.; Hagerman, R.J. Increased severity of fragile X spectrum disorders in the agricultural community of Ricaurte, Colombia. Int. J. Dev. Neurosci. 2019, 72, 1–5. [Google Scholar] [CrossRef]
- Saldarriaga, W.; Lein, P.; González Teshima, L.Y.; Isaza, C.; Rosa, L.; Polyak, A.; Hagerman, R.; Girirajan, S.; Silva, M.; Tassone, F. Phenobarbital use and neurological problems in FMR1 premutation carriers. Neurotoxicology 2016, 53, 141–147. [Google Scholar] [CrossRef]
- Sodhi, D.K.; Hagerman, R. Fragile X Premutation: Medications, Therapy and Lifestyle Advice. Pharmgenom. Pers. Med. 2021, 14, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, E.S.; Cao, Z.; Hulsizer, S.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Pessah, I.N. Early mitochondrial abnormalities in hippocampal neurons cultured from FMR1 pre-mutation mouse model. J. Neurochem. 2012, 123, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Hulsizer, S.; Tassone, F.; Tang, H.T.; Hagerman, R.J.; Rogawski, M.A.; Hagerman, P.J.; Pessah, I.N. Clustered burst firing in FMR1 premutation hippocampal neurons: Amelioration with allopregnanolone. Hum. Mol. Genet. 2012, 21, 2923–2935. [Google Scholar] [CrossRef]
- Aishworiya, R.; Protic, D.; Hagerman, R. Autism spectrum disorder in the fragile X premutation state: Possible mechanisms and implications. J. Neurol. 2022, 269, 4676–4683. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.M.; Cogswell, J.; Goodrich, J.E.; Mu, Y.; Nguyen, D.V.; Brass, S.D.; Hagerman, R.J. Fatigue and body mass index in the Fragile X premutation carrier. Fatigue Biomed. Health Behav. 2014, 2, 64–72. [Google Scholar] [CrossRef]
- Summers, S.M.; Cogswell, J.; Goodrich, J.E.; Mu, Y.; Nguyen, D.V.; Brass, S.D.; Hagerman, R.J. Prevalence of restless legs syndrome and sleep quality in carriers of the fragile X premutation. Clin. Genet. 2014, 86, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Wheeler, A.C.; Allen, E.G.; Wald, K.; Rajkovic, A.; Hagerman, R.J.; Sherman, S.L. Fragile X Syndrome and Premutation Disorders: New Developments and Treatments; Hagerman, R.J., Hagerman, P.J., Eds.; Mac Keith Press: London, UK, 2020; pp. 75–82. [Google Scholar]
- Hagerman, R.J.; Protic, D.; Rajaratnam, A.; Salcedo-Arellano, M.J.; Aydin, E.Y.; Schneider, A. Fragile X-Associated Neuropsychiatric Disorders (FXAND). Front. Psychiatry 2018, 9, 564. [Google Scholar] [CrossRef]
- Winarni, T.I.; Chonchaiya, W.; Sumekar, T.A.; Ashwood, P.; Morales, G.M.; Tassone, F.; Nguyen, D.V.; Faradz, S.M.; Van de Water, J.; Cook, K.; et al. Immune-mediated disorders among women carriers of fragile X premutation alleles. Am. J. Med. Genet. A 2012, 158, 2473–2481. [Google Scholar] [CrossRef]
- Hamlin, A.A.; Sukharev, D.; Campos, L.; Mu, Y.; Tassone, F.; Hessl, D.; Nguyen, D.V.; Loesch, D.; Hagerman, R.J. Hypertension in FMR1 premutation males with and without fragile X-associated tremor/ataxia syndrome (FXTAS). Am. J. Med. Genet. A 2012, 158, 1304–1309. [Google Scholar] [CrossRef]
- Au, J.; Akins, R.S.; Berkowitz-Sutherland, L.; Tang, H.T.; Chen, Y.; Boyd, A.; Tassone, F.; Nguyen, D.V.; Hagerman, R. Prevalence and risk of migraine headaches in adult fragile X premutation carriers. Clin. Genet. 2013, 84, 546–551. [Google Scholar] [CrossRef]
- Tassanakijpanich, N.; McKenzie, F.J.; McLennan, Y.A.; Makhoul, E.; Tassone, F.; Jasoliya, M.J.; Romney, C.; Petrasic, I.C.; Napalinga, K.; Buchanan, C.B.; et al. Hypermobile Ehlers-Danlos syndrome (hEDS) phenotype in fragile X premutation carriers: Case series. J. Med. Genet. 2022, 59, 687–690. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, F.J.; Tassankijpanich, N.; Epps, K.C.; March, S.K.; Hagerman, R.J. Spontaneous Coronary Artery Dissection in Females with the Fragile X FMR1 Premutation. JACC Case Rep. 2020, 2, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Hunsaker, M.R.; Greco, C.M.; Spath, M.A.; Smits, A.P.; Navarro, C.S.; Tassone, F.; Kros, J.M.; Severijnen, L.A.; Berry-Kravis, E.M.; Berman, R.F.; et al. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol. 2011, 122, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, J.A.; Coffey, S.M.; Rivera, S.M.; Hessl, D.; Gane, L.W.; Tassone, F.; Greco, C.; Finucane, B.; Nelson, L.; Berry-Kravis, E.; et al. A review of fragile X premutation disorders: Expanding the psychiatric perspective. J. Clin. Psychiatry 2009, 70, 852–862. [Google Scholar] [CrossRef]
- Bourgeois, J.A.; Seritan, A.L.; Casillas, E.M.; Hessl, D.; Schneider, A.; Yang, Y.; Kaur, I.; Cogswell, J.B.; Nguyen, D.V.; Hagerman, R.J. Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J. Clin. Psychiatry 2011, 72, 175–182. [Google Scholar] [CrossRef]
- Losh, M.; Klusek, J.; Martin, G.E.; Sideris, J.; Parlier, M.; Piven, J. Defining genetically meaningful language and personality traits in relatives of individuals with fragile X syndrome and relatives of individuals with autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 660–668. [Google Scholar] [CrossRef]
- Roberts, J.E.; Tonnsen, B.L.; McCary, L.M.; Ford, A.L.; Golden, R.N.; Bailey, D.B., Jr. Trajectory and Predictors of Depression and Anxiety Disorders in Mothers with the FMR1 Premutation. Biol. Psychiatry 2016, 79, 850–857. [Google Scholar] [CrossRef]
- Gossett, A.; Sansone, S.; Schneider, A.; Johnston, C.; Hagerman, R.; Tassone, F.; Rivera, S.M.; Seritan, A.L.; Hessl, D. Psychiatric disorders among women with the fragile X premutation without children affected by fragile X syndrome. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 1139–1147. [Google Scholar] [CrossRef]
- Kraan, C.M.; Hocking, D.R.; Georgiou-Karistianis, N.; Metcalfe, S.A.; Archibald, A.D.; Fielding, J.; Trollor, J.; Bradshaw, J.L.; Cohen, J.; Cornish, K.M. Impaired response inhibition is associated with self-reported symptoms of depression, anxiety, and ADHD in female FMR1 premutation carriers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2014, 165, 41–51. [Google Scholar] [CrossRef]
- Movaghar, A.; Page, D.; Brilliant, M.; Baker, M.W.; Greenberg, J.; Hong, J.; DaWalt, L.S.; Saha, K.; Kuusisto, F.; Stewart, R.; et al. Data-driven phenotype discovery of FMR1 premutation carriers in a population-based sample. Sci. Adv. 2019, 5, eaaw7195. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Bui, M.Q.; Hammersley, E.; Schneider, A.; Storey, E.; Stimpson, P.; Burgess, T.; Francis, D.; Slater, H.; Tassone, F.; et al. Psychological status in female carriers of premutation FMR1 allele showing a complex relationship with the size of CGG expansion. Clin. Genet. 2015, 87, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Herring, J.; Richstein, J. Fragile X Premutation Associated Conditions (FXPAC). Front. Pediatr. 2020, 8, 266. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Zhang, F.; Hagedorn, C.H.; Warren, S.T. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum. Mol. Genet. 2001, 10, 1449–1454. [Google Scholar] [CrossRef]
- Allen, E.G.; He, W.; Yadav-Shah, M.; Sherman, S.L. A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum. Genet. 2004, 114, 439–447. [Google Scholar] [PubMed]
- Tassone, F.; Beilina, A.; Carosi, C.; Albertosi, S.; Bagni, C.; Li, L.; Glover, K.; Bentley, D.; Hagerman, P.J. Elevated FMR1 mRNA in premutation carriers is due to increased transcription. Rna 2007, 13, 555–562. [Google Scholar] [CrossRef]
- Primerano, B.; Tassone, F.; Hagerman, R.J.; Hagerman, P.; Amaldi, F.; Bagni, C. Reduced FMR1 mRNA translation efficiency in fragile X patients with premutations. Rna 2002, 8, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Peprah, E.; He, W.; Allen, E.; Oliver, T.; Boyne, A.; Sherman, S.L. Examination of FMR1 transcript and protein levels among 74 premutation carriers. J. Hum. Genet. 2010, 55, 66–68. [Google Scholar] [CrossRef]
- Yrigollen, C.M.; Martorell, L.; Durbin-Johnson, B.; Naudo, M.; Genoves, J.; Murgia, A.; Polli, R.; Zhou, L.; Barbouth, D.; Rupchock, A.; et al. AGG interruptions and maternal age affect FMR1 CGG repeat allele stability during transmission. J. Neurodev. Disord. 2014, 6, 24. [Google Scholar] [CrossRef]
- Nolin, S.L.; Glicksman, A.; Ersalesi, N.; Dobkin, C.; Brown, W.T.; Cao, R.; Blatt, E.; Sah, S.; Latham, G.J.; Hadd, A.G. Fragile X full mutation expansions are inhibited by one or more AGG interruptions in premutation carriers. Genet. Med. 2015, 17, 358–364. [Google Scholar] [CrossRef]
- Yrigollen, C.M.; Tassone, F.; Durbin-Johnson, B.; Tassone, F. The role of AGG interruptions in the transcription of FMR1 premutation alleles. PLoS ONE 2011, 6, e21728. [Google Scholar] [CrossRef]
- Ludwig, A.L.; Raske, C.; Tassone, F.; Garcia-Arocena, D.; Hershey, J.W.; Hagerman, P.J. Translation of the FMR1 mRNA is not influenced by AGG interruptions. Nucleic Acids Res. 2009, 37, 6896–6904. [Google Scholar] [CrossRef] [PubMed]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef]
- Hwang, Y.H.; Hayward, B.E.; Zafarullah, M.; Kumar, J.; Durbin Johnson, B.; Holmans, P.; Usdin, K.; Tassone, F. Both cis and trans-acting genetic factors drive somatic instability in female carriers of the FMR1 premutation. Sci. Rep. 2022, 12, 10419. [Google Scholar] [CrossRef] [PubMed]
- Zafarullah, M.; Li, J.; Tseng, E.; Tassone, F. Structure and Alternative Splicing of the Antisense FMR1 (ASFMR1) Gene. Mol. Neurobiol. 2023, 60, 2051–2061. [Google Scholar] [CrossRef] [PubMed]
- Zafarullah, M.; Palczewski, G.; Rivera, S.M.; Hessl, D.R.; Tassone, F. Metabolic profiling reveals dysregulated lipid metabolism and potential biomarkers associated with the development and progression of Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). Faseb J. 2020, 34, 16676–16692. [Google Scholar] [CrossRef]
- Vittal, P.; Pandya, S.; Sharp, K.; Berry-Kravis, E.; Zhou, L.; Ouyang, B.; Jackson, J.; Hall, D.A. ASFMR1 splice variant: A predictor of fragile X-associated tremor/ataxia syndrome. Neurol. Genet. 2018, 4, e246. [Google Scholar] [CrossRef]
- Pretto, D.I.; Mendoza-Morales, G.; Lo, J.; Cao, R.; Hadd, A.; Latham, G.J.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet. 2014, 51, 309–318. [Google Scholar] [CrossRef]
- Aishworiya, R.; Hwang, Y.H.; Santos, E.; Hayward, B.; Usdin, K.; Durbin-Johnson, B.; Hagerman, R.; Tassone, F. Clinical implications of somatic allele expansion in female FMR1 premutation carriers. Sci. Rep. 2023, 13, 7050. [Google Scholar] [CrossRef]
- Dobkin, C.S.; Nolin, S.L.; Cohen, I.; Sudhalter, V.; Bialer, M.G.; Ding, X.H.; Jenkins, E.C.; Zhong, N.; Brown, W.T. Tissue differences in fragile X mosaics: Mosaicism in blood cells may differ greatly from skin. Am. J. Med. Genet. 1996, 64, 296–301. [Google Scholar] [CrossRef]
- Maddalena, A.; Yadvish, K.N.; Spence, W.C.; Howard-Peebles, P.N. A fragile X mosaic male with a cryptic full mutation detected in epithelium but not in blood. Am. J. Med. Genet. 1996, 64, 309–312. [Google Scholar] [CrossRef]
- Taylor, A.K.; Tassone, F.; Dyer, P.N.; Hersch, S.M.; Harris, J.B.; Greenough, W.T.; Hagerman, R.J. Tissue heterogeneity of the FMR1 mutation in a high-functioning male with fragile X syndrome. Am. J. Med. Genet. 1999, 84, 233–239. [Google Scholar] [CrossRef]
- Fernández, E.; Gennaro, E.; Pirozzi, F.; Baldo, C.; Forzano, F.; Turolla, L.; Faravelli, F.; Gastaldo, D.; Coviello, D.; Grasso, M.; et al. FXS-Like Phenotype in Two Unrelated Patients Carrying a Methylated Premutation of the FMR1 Gene. Front. Genet. 2018, 9, 442. [Google Scholar] [CrossRef] [PubMed]
- Jiraanont, P.; Sweha, S.R.; AlOlaby, R.R.; Silva, M.; Tang, H.T.; Durbin-Johnson, B.; Schneider, A.; Espinal, G.M.; Hagerman, P.J.; Rivera, S.M.; et al. Clinical and molecular correlates in fragile X premutation females. eNeurologicalSci 2017, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Del Hoyo Soriano, L.; Thurman, A.J.; Harvey, D.J.; Ted Brown, W.; Abbeduto, L. Genetic and maternal predictors of cognitive and behavioral trajectories in females with fragile X syndrome. J. Neurodev. Disord. 2018, 10, 22. [Google Scholar] [CrossRef]
- Lyon, M.F. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Sun, Z.; Fan, J.; Wang, Y. X-Chromosome Inactivation and Related Diseases. Genet. Res. 2022, 2022, 1391807. [Google Scholar] [CrossRef]
- Devys, D.; Lutz, Y.; Rouyer, N.; Bellocq, J.P.; Mandel, J.L. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 1993, 4, 335–340. [Google Scholar] [CrossRef]
- Franke, P.; Leboyer, M.; Hardt, J.; Sohne, E.; Weiffenbach, O.; Biancalana, V.; Cornillet-Lefebre, P.; Delobel, B.; Froster, U.; Schwab, S.G.; et al. Neuropsychological profiles of FMR-1 premutation and full-mutation carrier females. Psychiatry Res. 1999, 87, 223–231. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Huggins, R.M.; Hagerman, R.J. Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 31–41. [Google Scholar] [CrossRef]
- Godler, D.E.; Slater, H.R.; Bui, Q.M.; Ono, M.; Gehling, F.; Francis, D.; Amor, D.J.; Hopper, J.L.; Hagerman, R.; Loesch, D.Z. FMR1 intron 1 methylation predicts FMRP expression in blood of female carriers of expanded FMR1 alleles. J. Mol. Diagn. 2011, 13, 528–536. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Potanos, K.; Weinberg, D.; Zhou, L.; Goetz, C.G. Fragile X-associated tremor/ataxia syndrome in sisters related to X-inactivation. Ann. Neurol. 2005, 57, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Robertson-Dick, E.E.; O’Keefe, J.A.; Hadd, A.G.; Zhou, L.; Berry-Kravis, E. X-inactivation in the clinical phenotype of fragile X premutation carrier sisters. Neurol. Genet. 2016, 2, e45. [Google Scholar] [CrossRef] [PubMed]
- Abrams, M.T.; Reiss, A.L.; Freund, L.S.; Baumgardner, T.L.; Chase, G.A.; Denckla, M.B. Molecular-neurobehavioral associations in females with the fragile X full mutation. Am. J. Med. Genet. 1994, 51, 317–327. [Google Scholar] [CrossRef]
- Hessl, D.; Dyer-Friedman, J.; Glaser, B.; Wisbeck, J.; Barajas, R.G.; Taylor, A.; Reiss, A.L. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics 2001, 108, E88. [Google Scholar] [CrossRef] [PubMed]
- Heine-Suñer, D.; Torres-Juan, L.; Morlà, M.; Busquets, X.; Barceló, F.; Picó, G.; Bonilla, L.; Govea, N.; Bernués, M.; Rosell, J. Fragile-X syndrome and skewed X-chromosome inactivation within a family: A female member with complete inactivation of the functional X chromosome. Am. J. Med. Genet. Part. A 2003, 122, 108–114. [Google Scholar] [CrossRef]
- Talebizadeh, Z.; Bittel, D.C.; Veatch, O.J.; Kibiryeva, N.; Butler, M.G. Brief report: Non-random X chromosome inactivation in females with autism. J. Autism Dev. Disord. 2005, 35, 675–681. [Google Scholar] [CrossRef]
- Stembalska, A.; Łaczmańska, I.; Gil, J.; Pesz, K.A. Fragile X syndrome in females—A familial case report and review of the literature. Dev. Period. Med. 2016, 20, 99–104. [Google Scholar]
- Sobesky, W.E.; Taylor, A.K.; Pennington, B.F.; Bennetto, L.; Porter, D.; Riddle, J.; Hagerman, R.J. Molecular/clinical correlations in females with fragile X. Am. J. Med. Genet. 1996, 64, 340–345. [Google Scholar] [CrossRef]
- Tassone, F.; Pan, R.; Amiri, K.; Taylor, A.K.; Hagerman, P.J. A rapid polymerase chain reaction-based screening method for identification of all expanded alleles of the fragile X (FMR1) gene in newborn and high-risk populations. J. Mol. Diagn. 2008, 10, 43–49. [Google Scholar] [CrossRef]
- Godler, D.E.; Tassone, F.; Loesch, D.Z.; Taylor, A.K.; Gehling, F.; Hagerman, R.J.; Burgess, T.; Ganesamoorthy, D.; Hennerich, D.; Gordon, L.; et al. Methylation of novel markers of fragile X alleles is inversely correlated with FMRP expression and FMR1 activation ratio. Hum. Mol. Genet. 2010, 19, 1618–1632. [Google Scholar] [CrossRef]
- Hadd, A.G.; Filipovic-Sadic, S.; Zhou, L.; Williams, A.; Latham, G.J.; Berry-Kravis, E.; Hall, D.A. A methylation PCR method determines FMR1 activation ratios and differentiates premutation allele mosaicism in carrier siblings. Clin. Epigenetics 2016, 8, 130. [Google Scholar] [CrossRef] [PubMed]
- Protic, D.; Polli, R.; Hwang, Y.H.; Mendoza, G.; Hagerman, R.; Durbin-Johnson, B.; Hayward, B.E.; Usdin, K.; Murgia, A.; Tassone, F. Activation Ratio Correlates with IQ in Female Carriers of the FMR1 Premutation. Cells 2023, 12, 1711. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Hagerman, P. Fragile X-associated tremor/ataxia syndrome—Features, mechanisms and management. Nat. Rev. Neurol. 2016, 12, 403–412. [Google Scholar] [CrossRef]
- Loomis, E.W.; Sanz, L.A.; Chédin, F.; Hagerman, P.J. Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Author Correction: Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 644. [Google Scholar] [CrossRef] [PubMed]
- Iwahashi, C.K.; Yasui, D.H.; An, H.J.; Greco, C.M.; Tassone, F.; Nannen, K.; Babineau, B.; Lebrilla, C.B.; Hagerman, R.J.; Hagerman, P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain 2006, 129 Pt 1, 256–271. [Google Scholar] [CrossRef]
- Sellier, C.; Rau, F.; Liu, Y.; Tassone, F.; Hukema, R.K.; Gattoni, R.; Schneider, A.; Richard, S.; Willemsen, R.; Elliott, D.J.; et al. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. Embo J. 2010, 29, 1248–1261. [Google Scholar] [CrossRef]
- Qurashi, A.; Liu, H.; Ray, L.; Nelson, D.L.; Duan, R.; Jin, P. Chemical screen reveals small molecules suppressing fragile X premutation rCGG repeat-mediated neurodegeneration in Drosophila. Hum. Mol. Genet. 2012, 21, 2068–2075. [Google Scholar] [CrossRef]
- Tan, H.; Poidevin, M.; Li, H.; Chen, D.; Jin, P. MicroRNA-277 modulates the neurodegeneration caused by Fragile X premutation rCGG repeats. PLoS Genet. 2012, 8, e1002681. [Google Scholar] [CrossRef]
- Khalili, K.; Del Valle, L.; Muralidharan, V.; Gault, W.J.; Darbinian, N.; Otte, J.; Meier, E.; Johnson, E.M.; Daniel, D.C.; Kinoshita, Y.; et al. Puralpha is essential for postnatal brain development and developmentally coupled cellular proliferation as revealed by genetic inactivation in the mouse. Mol. Cell Biol. 2003, 23, 6857–6875. [Google Scholar] [CrossRef]
- Hokkanen, S.; Feldmann, H.M.; Ding, H.; Jung, C.K.; Bojarski, L.; Renner-Müller, I.; Schüller, U.; Kretzschmar, H.; Wolf, E.; Herms, J. Lack of Pur-alpha alters postnatal brain development and causes megalencephaly. Hum. Mol. Genet. 2012, 21, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.N.; Shaw, C.; Yu, P.; Parghi, D.; Poidevin, M.; Jin, P.; Nelson, D.L. CGG repeats in RNA modulate expression of TDP-43 in mouse and fly models of fragile X tremor ataxia syndrome. Hum. Mol. Genet. 2014, 23, 5906–5915. [Google Scholar] [CrossRef] [PubMed][Green Version]
- He, F.; Krans, A.; Freibaum, B.D.; Taylor, J.P.; Todd, P.K. TDP-43 suppresses CGG repeat-induced neurotoxicity through interactions with HnRNP A2/B1. Hum. Mol. Genet. 2014, 23, 5036–5051. [Google Scholar] [CrossRef] [PubMed]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef]
- Banez-Coronel, M.; Ranum, L.P.W. Repeat-associated non-AUG (RAN) translation: Insights from pathology. Lab. Investig. 2019, 99, 929–942. [Google Scholar] [CrossRef]
- Kearse, M.G.; Green, K.M.; Krans, A.; Rodriguez, C.M.; Linsalata, A.E.; Goldstrohm, A.C.; Todd, P.K. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol. Cell 2016, 62, 314–322. [Google Scholar] [CrossRef]
- Krans, A.; Skariah, G.; Zhang, Y.; Bayly, B.; Todd, P.K. Neuropathology of RAN translation proteins in fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2019, 7, 152. [Google Scholar] [CrossRef]
- Wright, S.E.; Rodriguez, C.M.; Monroe, J.; Xing, J.; Krans, A.; Flores, B.N.; Barsur, V.; Ivanova, M.I.; Koutmou, K.S.; Barmada, S.J.; et al. CGG repeats trigger translational frameshifts that generate aggregation-prone chimeric proteins. Nucleic Acids Res. 2022, 50, 8674–8689. [Google Scholar] [CrossRef]
- Sellier, C.; Buijsen, R.A.M.; He, F.; Natla, S.; Jung, L.; Tropel, P.; Gaucherot, A.; Jacobs, H.; Meziane, H.; Vincent, A.; et al. Translation of Expanded CGG Repeats into FMRpolyG Is Pathogenic and May Contribute to Fragile X Tremor Ataxia Syndrome. Neuron 2017, 93, 331–347. [Google Scholar] [CrossRef]
- Buijsen, R.A.; Visser, J.A.; Kramer, P.; Severijnen, E.A.; Gearing, M.; Charlet-Berguerand, N.; Sherman, S.L.; Berman, R.F.; Willemsen, R.; Hukema, R.K. Presence of inclusions positive for polyglycine containing protein, FMRpolyG, indicates that repeat-associated non-AUG translation plays a role in fragile X-associated primary ovarian insufficiency. Hum. Reprod. 2016, 31, 158–168. [Google Scholar] [CrossRef]
- Ma, L.; Herren, A.W.; Espinal, G.; Randol, J.; McLaughlin, B.; Martinez-Cerdeño, V.; Pessah, I.N.; Hagerman, R.J.; Hagerman, P.J. Composition of the Intranuclear Inclusions of Fragile X-associated Tremor/Ataxia Syndrome. Acta Neuropathol. Commun. 2019, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Glineburg, M.R.; Basrur, V.; Conlon, K.; Wright, S.E.; Krans, A.; Hall, D.A.; Todd, P.K. Mechanistic convergence across initiation sites for RAN translation in fragile X associated tremor ataxia syndrome. Hum. Mol. Genet. 2022, 31, 2317–2332. [Google Scholar] [CrossRef] [PubMed]
- Asamitsu, S.; Yabuki, Y.; Ikenoshita, S.; Kawakubo, K.; Kawasaki, M.; Usuki, S.; Nakayama, Y.; Adachi, K.; Kugoh, H.; Ishii, K.; et al. CGG repeat RNA G-quadruplexes interact with FMRpolyG to cause neuronal dysfunction in fragile X-related tremor/ataxia syndrome. Sci. Adv. 2021, 7, eabd9440. [Google Scholar] [CrossRef] [PubMed]
- Linsalata, A.E.; He, F.; Malik, A.M.; Glineburg, M.R.; Green, K.M.; Natla, S.; Flores, B.N.; Krans, A.; Archbold, H.C.; Fedak, S.J.; et al. DDX3X and specific initiation factors modulate FMR1 repeat-associated non-AUG-initiated translation. EMBO Rep. 2019, 20, e47498. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.M.; Wright, S.E.; Kearse, M.G.; Haenfler, J.M.; Flores, B.N.; Liu, Y.; Ifrim, M.F.; Glineburg, M.R.; Krans, A.; Jafar-Nejad, P.; et al. A native function for RAN translation and CGG repeats in regulating fragile X protein synthesis. Nat. Neurosci. 2020, 23, 386–397. [Google Scholar] [CrossRef] [PubMed]
- Haify, S.N.; Mankoe, R.S.D.; Boumeester, V.; van der Toorn, E.C.; Verhagen, R.F.M.; Willemsen, R.; Hukema, R.K.; Bosman, L.W.J. Lack of a Clear Behavioral Phenotype in an Inducible FXTAS Mouse Model Despite the Presence of Neuronal FMRpolyG-Positive Aggregates. Front. Mol. Biosci. 2020, 7, 599101. [Google Scholar] [CrossRef]
- Tseng, Y.J.; Sandwith, S.N.; Green, K.M.; Chambers, A.E.; Krans, A.; Raimer, H.M.; Sharlow, M.E.; Reisinger, M.A.; Richardson, A.E.; Routh, E.D.; et al. The RNA helicase DHX36-G4R1 modulates C9orf72 GGGGCC hexanucleotide repeat-associated translation. J. Biol. Chem. 2021, 297, 100914. [Google Scholar] [CrossRef]
- Green, K.M.; Glineburg, M.R.; Kearse, M.G.; Flores, B.N.; Linsalata, A.E.; Fedak, S.J.; Goldstrohm, A.C.; Barmada, S.J.; Todd, P.K. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nat. Commun. 2017, 8, 2005. [Google Scholar] [CrossRef]
- Zafarullah, M.; Durbin-Johnson, B.; Fourie, E.S.; Hessl, D.R.; Rivera, S.M.; Tassone, F. Metabolomic Biomarkers Are Associated with Area of the Pons in Fragile X Premutation Carriers at Risk for Developing FXTAS. Front. Psychiatry 2021, 12, 691717. [Google Scholar] [CrossRef]
- In Proceedings of the 5th International Conference on FMR1 Premutation: Molecular Mechanism, Clinical Involvements and Target. Waitangi, New Zealand, 27 February–3 March 2023.
- Zafarullah, M.; Li, J.; Salemi, M.; Phinney, B.; Durbin-Johnson, B.P.; Hagerman, R.; Hessl, D.; Rivera, S.M.; Tassone, F. Blood proteome profiling reveals biomarkers and pathways alterations in Fragile X premutation carriers at risk for developing FXTAS. Int. J. Mol. Biol. 2023, 24, 13477. [Google Scholar]
- Derbis, M.; Kul, E.; Niewiadomska, D.; Sekrecki, M.; Piasecka, A.; Taylor, K.; Hukema, R.K.; Stork, O.; Sobczak, K. Short antisense oligonucleotides alleviate the pleiotropic toxicity of RNA harboring expanded CGG repeats. Nat. Commun. 2021, 12, 1265. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.E.; Lim, J.; Linsalata, A.; Kang, Y.; Malik, I.; Allen, E.G.; Cao, Y.; Shubeck, L.; Johnston, R.; Huang, Y.; et al. Identification of PSMB5 as a genetic modifier of fragile X-associated tremor/ataxia syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2118124119. [Google Scholar] [CrossRef] [PubMed]
- Konieczny, P.; Mukherjee, S.; Stepniak-Konieczna, E.; Taylor, K.; Niewiadomska, D.; Piasecka, A.; Walczak, A.; Baud, A.; Dohno, C.; Nakatani, K.; et al. Cyclic mismatch binding ligands interact with disease-associated CGG trinucleotide repeats in RNA and suppress their translation. Nucleic Acids Res. 2021, 49, 9479–9495. [Google Scholar] [CrossRef] [PubMed]
- Filley, C.M.; Brown, M.S.; Onderko, K.; Ray, M.; Bennett, R.E.; Berry-Kravis, E.; Grigsby, J. White matter disease and cognitive impairment in FMR1 premutation carriers. Neurology 2015, 84, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.E.; Lim, J.; Zhang, F.; Huang, L.; Gu, Y.; Nelson, D.L.; Allen, E.G.; Jin, P. Metabolic pathways modulate the neuronal toxicity associated with fragile X-associated tremor/ataxia syndrome. Hum. Mol. Genet. 2019, 28, 980–991. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Tao, W.; Li, X.; Li, H.; Zhang, J.; Wei, D.; Chen, Y.; Zhang, Z. The Contribution of Genetic Factors to Cognitive Impairment and Dementia: Apolipoprotein E Gene, Gene Interactions, and Polygenic Risk. Int. J. Mol. Sci. 2019, 20, 1177. [Google Scholar] [CrossRef]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Blanchard, J.W.; Akay, L.A.; Davila-Velderrain, J.; von Maydell, D.; Mathys, H.; Davidson, S.M.; Effenberger, A.; Chen, C.Y.; Maner-Smith, K.; Hajjar, I.; et al. APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 2022, 611, 769–779. [Google Scholar] [CrossRef]
- Tassone, F.; Greco, C.M.; Hunsaker, M.R.; Seritan, A.L.; Berman, R.F.; Gane, L.W.; Jacquemont, S.; Basuta, K.; Jin, L.W.; Hagerman, P.J.; et al. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes. Brain Behav. 2012, 11, 577–585. [Google Scholar] [CrossRef]
- Silva, F.; Rodriguez-Revenga, L.; Madrigal, I.; Alvarez-Mora, M.I.; Oliva, R.; Milà, M. High apolipoprotein E4 allele frequency in FXTAS patients. Genet. Med. 2013, 15, 639–642. [Google Scholar] [CrossRef] [PubMed]
- Avitzour, M.; Mor-Shaked, H.; Yanovsky-Dagan, S.; Aharoni, S.; Altarescu, G.; Renbaum, P.; Eldar-Geva, T.; Schonberger, O.; Levy-Lahad, E.; Epsztejn-Litman, S.; et al. FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Rep. 2014, 3, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Koscielska, K.A.; Cao, Z.; Hulsizer, S.; Grace, N.; Mitchell, G.; Nacey, C.; Githinji, J.; McGee, J.; Garcia-Arocena, D.; et al. Signaling defects in iPSC-derived fragile X premutation neurons. Hum. Mol. Genet. 2012, 21, 3795–3805. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, J.; Zaninovic, N.; Zhan, Q.; Madireddy, A.; Nolin, S.L.; Ersalesi, N.; Yan, Z.; Rosenwaks, Z.; Schildkraut, C.L. Cis-acting DNA sequence at a replication origin promotes repeat expansion to fragile X full mutation. J. Cell Biol. 2014, 206, 599–607. [Google Scholar] [CrossRef]
- Gerhardt, J.; Tomishima, M.J.; Zaninovic, N.; Colak, D.; Yan, Z.; Zhan, Q.; Rosenwaks, Z.; Jaffrey, S.R.; Schildkraut, C.L. The DNA replication program is altered at the FMR1 locus in fragile X embryonic stem cells. Mol. Cell 2014, 53, 19–31. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a dish: Modeling development and disease using organoid technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Kraff, J.; Tang, H.T.; Cilia, R.; Canesi, M.; Pezzoli, G.; Goldwurm, S.; Hagerman, P.J.; Tassone, F. Screen for excess FMR1 premutation alleles among males with parkinsonism. Arch. Neurol. 2007, 64, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Wenzel, H.J.; Hunsaker, M.R.; Greco, C.M.; Willemsen, R.; Berman, R.F. Ubiquitin-positive intranuclear inclusions in neuronal and glial cells in a mouse model of the fragile X premutation. Brain Res. 2010, 1318, 155–166. [Google Scholar] [CrossRef]
- Vardinon, N.; Spirer, Z.; Goldhar, J.; Kacevman, B.; Eylan, E. Human milk anti-E. coli antibodies: Relationship to maternal parity. Eur. J. Pediatr. 1979, 130, 173–180. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Gutekunst, C.A.; Li, S.H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.S.L.; Xu, Z.; Chen, Z.; Tan, Y.J.; Lim, W.K.; Ting, S.K.S.; Yu, W.Y.; Cheng, Q.H.; Foo, J.N.; Tan, E.K.; et al. NOTCH2NLC-linked neuronal intranuclear inclusion body disease and fragile X-associated tremor/ataxia syndrome. Brain 2020, 143, e69. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 589–607. [Google Scholar] [CrossRef]
- Todd, P.K.; Paulson, H.L. RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol. 2010, 67, 291–300. [Google Scholar] [CrossRef]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 69, 385. [Google Scholar] [CrossRef]
- Mahadevan, M.S.; Yadava, R.S.; Yu, Q.; Balijepalli, S.; Frenzel-McCardell, C.D.; Bourne, T.D.; Phillips, L.H. Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat. Genet. 2006, 38, 1066–1070. [Google Scholar] [CrossRef]
- Oh, S.Y.; He, F.; Krans, A.; Frazer, M.; Taylor, J.P.; Paulson, H.L.; Todd, P.K. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Hum. Mol. Genet. 2015, 24, 4317–4326. [Google Scholar] [CrossRef]
- Koehorst, E.; Núñez-Manchón, J.; Ballester-López, A.; Almendrote, M.; Lucente, G.; Arbex, A.; Chojnacki, J.; Vázquez-Manrique, R.P.; Gómez-Escribano, A.P.; Pintos-Morell, G.; et al. Characterization of RAN Translation and Antisense Transcription in Primary Cell Cultures of Patients with Myotonic Dystrophy Type 1. J. Clin. Med. 2021, 10, 5520. [Google Scholar] [CrossRef]
- Furling, D.; LaM, L.T.; Agbulut, O.; Butler-Browne, G.S.; Morris, G.E. Changes in myotonic dystrophy protein kinase levels and muscle development in congenital myotonic dystrophy. Am. J. Pathol. 2003, 162, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, G.; Pizza, F.; Scaglione, C.; Tonon, C.; Lodi, R.; Barbiroli, B.; Ambrosetto, P.; Martinelli, P. A case of fragile X premutation tremor/ataxia syndrome with evidence of mitochondrial dysfunction. Mov. Disord. 2006, 21, 1541–1542. [Google Scholar] [CrossRef] [PubMed]
- Ross-Inta, C.; Omanska-Klusek, A.; Wong, S.; Barrow, C.; Garcia-Arocena, D.; Iwahashi, C.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Giulivi, C. Evidence of mitochondrial dysfunction in fragile X-associated tremor/ataxia syndrome. Biochem. J. 2010, 429, 545–552. [Google Scholar] [CrossRef]
- Chen, Y.; Tassone, F.; Berman, R.F.; Hagerman, P.J.; Hagerman, R.J.; Willemsen, R.; Pessah, I.N. Murine hippocampal neurons expressing FMR1 gene premutations show early developmental deficits and late degeneration. Hum. Mol. Genet. 2010, 19, 196–208. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Mora, M.I.; Podlesniy, P.; Gelpi, E.; Hukema, R.; Madrigal, I.; Pagonabarraga, J.; Trullas, R.; Mila, M.; Rodriguez-Revenga, L. Fragile X-associated tremor/ataxia syndrome: Regional decrease of mitochondrial DNA copy number relates to clinical manifestations. Genes. Brain Behav. 2019, 18, e12565. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Annesley, S.J.; Trost, N.; Bui, M.Q.; Lay, S.T.; Storey, E.; De Piazza, S.W.; Sanislav, O.; Francione, L.M.; Hammersley, E.M.; et al. Novel Blood Biomarkers Are Associated with White Matter Lesions in Fragile X-Associated Tremor/Ataxia Syndrome. Neurodegener. Dis. 2017, 17, 22–30. [Google Scholar] [CrossRef]
- Fisher, P.R.; Allan, C.Y.; Sanislav, O.; Atkinson, A.; Ngoei, K.R.W.; Kemp, B.E.; Storey, E.; Loesch, D.Z.; Annesley, S.J. Relationships between Mitochondrial Function, AMPK, and TORC1 Signaling in Lymphoblasts with Premutation Alleles of the FMR1 Gene. Int. J. Mol. Sci. 2021, 22, 10393. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Kemp, B.E.; Bui, M.Q.; Fisher, P.R.; Allan, C.Y.; Sanislav, O.; Ngoei, K.R.W.; Atkinson, A.; Tassone, F.; Annesley, S.J.; et al. Cellular Bioenergetics and AMPK and TORC1 Signalling in Blood Lymphoblasts Are Biomarkers of Clinical Status in FMR1 Premutation Carriers. Front. Psychiatry 2021, 12, 747268. [Google Scholar] [CrossRef]
- Cid-Samper, F.; Gelabert-Baldrich, M.; Lang, B.; Lorenzo-Gotor, N.; Ponti, R.D.; Severijnen, L.; Bolognesi, B.; Gelpi, E.; Hukema, R.K.; Botta-Orfila, T.; et al. An Integrative Study of Protein-RNA Condensates Identifies Scaffolding RNAs and Reveals Players in Fragile X-Associated Tremor/Ataxia Syndrome. Cell Rep. 2018, 25, 3422–3434.e7. [Google Scholar] [CrossRef]
- Hu, Y.; Deng, H.; Xu, S.; Zhang, J. MicroRNAs Regulate Mitochondrial Function in Cerebral Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2015, 16, 24895–24917. [Google Scholar] [CrossRef]
- Han, J.; Lee, Y.; Yeom, K.H.; Kim, Y.K.; Jin, H.; Kim, V.N. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004, 18, 3016–3027. [Google Scholar] [CrossRef] [PubMed]
- Tafuri, F.; Ronchi, D.; Magri, F.; Comi, G.P.; Corti, S. SOD1 misplacing and mitochondrial dysfunction in amyotrophic lateral sclerosis pathogenesis. Front. Cell Neurosci. 2015, 9, 336. [Google Scholar] [CrossRef] [PubMed]
- Gohel, D.; Sripada, L.; Prajapati, P.; Singh, K.; Roy, M.; Kotadia, D.; Tassone, F.; Charlet-Berguerand, N.; Singh, R. FMRpolyG alters mitochondrial transcripts level and respiratory chain complex assembly in Fragile X associated tremor/ataxia syndrome [FXTAS]. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Jové, M.; Portero-Otín, M.; Naudí, A.; Ferrer, I.; Pamplona, R. Metabolomics of human brain aging and age-related neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2014, 73, 640–657. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Li, H.; Peng, X.X. Functional metabolomics: From biomarker discovery to metabolome reprogramming. Protein Cell 2015, 6, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Song, G.; Schneider, A.; Hagerman, R.; Eldeeb, M.A.; Azarang, A.; Tassone, F.; Giulivi, C. Warburg effect linked to cognitive-executive deficits in FMR1 premutation. Faseb J. 2016, 30, 3334–3351. [Google Scholar] [CrossRef]
- Napoli, E.; Schneider, A.; Wang, J.Y.; Trivedi, A.; Carrillo, N.R.; Tassone, F.; Rogawski, M.; Hagerman, R.J.; Giulivi, C. Allopregnanolone Treatment Improves Plasma Metabolomic Profile Associated with GABA Metabolism in Fragile X-Associated Tremor/Ataxia Syndrome: A Pilot Study. Mol. Neurobiol. 2019, 56, 3702–3713. [Google Scholar] [CrossRef]
- Abbasi, D.A.; Nguyen, T.T.A.; Hall, D.A.; Robertson-Dick, E.; Berry-Kravis, E.; Cologna, S.M. Characterization of the Cerebrospinal Fluid Proteome in Patients with Fragile X-Associated Tremor/Ataxia Syndrome. Cerebellum 2022, 21, 86–98. [Google Scholar] [CrossRef]
- Deng, J.; Yu, J.; Li, P.; Luan, X.; Cao, L.; Zhao, J.; Yu, M.; Zhang, W.; Lv, H.; Xie, Z.; et al. Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am. J. Hum. Genet. 2020, 106, 793–804. [Google Scholar] [CrossRef]
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019, 51, 1222–1232. [Google Scholar] [CrossRef]
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019, 51, 1215–1221. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Wang, Y. Tonotopic differentiation of presynaptic neurotransmitter-releasing machinery in the auditory brainstem during the prehearing period and its selective deficits in FMR1 knockout mice. J. Comp. Neurol. 2022, 530, 3248–3269. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.H.; Yang, K.; Du, G.Q.; Chen, Y.K.; Cao, C.Y.; Qiu, Y.S.; He, J.; Lv, H.D.; Qu, Q.Q.; Chen, J.N.; et al. GGC Repeat Expansion of RILPL1 is Associated with Oculopharyngodistal Myopathy. Ann. Neurol. 2022, 92, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Annear, D.J.; Vandeweyer, G.; Elinck, E.; Sanchis-Juan, A.; French, C.E.; Raymond, L.; Kooy, R.F. Abundancy of polymorphic CGG repeats in the human genome suggest a broad involvement in neurological disease. Sci. Rep. 2021, 11, 2515. [Google Scholar] [CrossRef]
- Pearson, C.E.; Nichol Edamura, K.; Cleary, J.D. Repeat instability: Mechanisms of dynamic mutations. Nat. Rev. Genet. 2005, 6, 729–742. [Google Scholar] [CrossRef]
- Essop, F.B.; Krause, A. Diagnostic, carrier and prenatal genetic testing for fragile X syndrome and other FMR-1-related disorders in Johannesburg, South Africa: A 20-year review. S. Afr. Med. J. 2013, 103 (Suppl. S1), 994–998. [Google Scholar] [CrossRef]
- Kraan, C.M.; Bui, Q.M.; Field, M.; Archibald, A.D.; Metcalfe, S.A.; Christie, L.M.; Bennetts, B.H.; Oertel, R.; Smith, M.J.; du Sart, D.; et al. FMR1 allele size distribution in 35,000 males and females: A comparison of developmental delay and general population cohorts. Genet. Med. 2018, 20, 1627–1634. [Google Scholar] [CrossRef]
- Madrigal, I.; Xunclà, M.; Tejada, M.I.; Martínez, F.; Fernández-Carvajal, I.; Pérez-Jurado, L.A.; Rodriguez-Revenga, L.; Milà, M. Intermediate FMR1 alleles and cognitive and/or behavioural phenotypes. Eur. J. Hum. Genet. 2011, 19, 921–923. [Google Scholar] [CrossRef][Green Version]
- Tassone, F.; Choudhary, N.S.; Tassone, F.; Durbin-Johnson, B.; Hansen, R.; Hertz-Picciotto, I.; Pessah, I. Identification of expanded alleles of the FMR1 Gene in the CHildhood Autism Risks from Genes and Environment (CHARGE) study. J. Autism Dev. Disord. 2013, 43, 530–539. [Google Scholar] [CrossRef]
- Raspa, M.; Wylie, A.; Wheeler, A.C.; Kolacz, J.; Edwards, A.; Heilman, K.; Porges, S.W. Sensory Difficulties in Children with an FMR1 Premutation. Front. Genet. 2018, 9, 351. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Sideris, J.; Roberts, J.; Hatton, D. Child and genetic variables associated with maternal adaptation to fragile X syndrome: A multidimensional analysis. Am. J. Med. Genet. A 2008, 146, 720–729. [Google Scholar] [CrossRef]
- Kraan, C.M.; Hocking, D.R.; Bradshaw, J.L.; Fielding, J.; Cohen, J.; Georgiou-Karistianis, N.; Cornish, K.M. Neurobehavioural evidence for the involvement of the FMR1 gene in female carriers of fragile X syndrome. Neurosci. Biobehav. Rev. 2013, 37, 522–547. [Google Scholar] [CrossRef] [PubMed]
- Lachiewicz, A.M.; Dawson, D.V.; Spiridigliozzi, G.A.; McConkie-Rosell, A. Arithmetic difficulties in females with the fragile X premutation. Am. J. Med. Genet. A 2006, 140, 665–672. [Google Scholar] [CrossRef]
- Wheeler, A.C.; Bailey, D.B., Jr.; Berry-Kravis, E.; Greenberg, J.; Losh, M.; Mailick, M.; Milà, M.; Olichney, J.M.; Rodriguez-Revenga, L.; Sherman, S.; et al. Associated features in females with an FMR1 premutation. J. Neurodev. Disord. 2014, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Cornish, K.M.; Kraan, C.M.; Bui, Q.M.; Bellgrove, M.A.; Metcalfe, S.A.; Trollor, J.N.; Hocking, D.R.; Slater, H.R.; Inaba, Y.; Li, X.; et al. Novel methylation markers of the dysexecutive-psychiatric phenotype in FMR1 premutation women. Neurology 2015, 84, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Brooker, R.J.; Buss, K.A.; Lemery-Chalfant, K.; Aksan, N.; Davidson, R.J.; Goldsmith, H.H. The development of stranger fear in infancy and toddlerhood: Normative development, individual differences, antecedents, and outcomes. Dev. Sci. 2013, 16, 864–878. [Google Scholar] [CrossRef]
- Brooker, R.J.; Kiel, E.J.; Buss, K.A. Early social fear predicts kindergarteners’ socially anxious behaviors: Direct associations, moderation by inhibitory control, and differences from nonsocial fear. Emotion 2016, 16, 997–1010. [Google Scholar] [CrossRef]
- Klusek, J.; Thurman, A.J.; Abbeduto, L. Maternal Pragmatic Language Difficulties in the FMR1 Premutation and the Broad Autism Phenotype: Associations with Individual and Family Outcomes. J. Autism Dev. Disord. 2022, 52, 835–851. [Google Scholar] [CrossRef]
- Maltman, N.; Guilfoyle, J.; Nayar, K.; Martin, G.E.; Winston, M.; Lau, J.C.Y.; Bush, L.; Patel, S.; Lee, M.; Sideris, J.; et al. The Phenotypic Profile Associated with the FMR1 Premutation in Women: An Investigation of Clinical-Behavioral, Social-Cognitive, and Executive Abilities. Front. Psychiatry 2021, 12, 718485. [Google Scholar] [CrossRef]
- Strawn, J.R.; Lu, L.; Peris, T.S.; Levine, A.; Walkup, J.T. Research Review: Pediatric anxiety disorders—What have we learnt in the last 10 years? J. Child. Psychol. Psychiatry 2021, 62, 114–139. [Google Scholar] [CrossRef]
- Tolan, P.H.; Dodge, K.A. Children’s mental health as a primary care and concern: A system for comprehensive support and service. Am. Psychol. 2005, 60, 601–614. [Google Scholar] [CrossRef]
- Walter, H.J.; Bukstein, O.G.; Abright, A.R.; Keable, H.; Ramtekkar, U.; Ripperger-Suhler, J.; Rockhill, C. Clinical Practice Guideline for the Assessment and Treatment of Children and Adolescents with Anxiety Disorders. J. Am. Acad. Child. Adolesc. Psychiatry 2020, 59, 1107–1124. [Google Scholar] [CrossRef]
- Leehey, M.A.; Berry-Kravis, E.; Min, S.J.; Hall, D.A.; Rice, C.D.; Zhang, L.; Grigsby, J.; Greco, C.M.; Reynolds, A.; Lara, R.; et al. Progression of tremor and ataxia in male carriers of the FMR1 premutation. Mov. Disord. 2007, 22, 203–206. [Google Scholar] [CrossRef]
- O’Keefe, J.A.; Robertson-Dick, E.; Dunn, E.J.; Li, Y.; Deng, Y.; Fiutko, A.N.; Berry-Kravis, E.; Hall, D.A. Characterization and Early Detection of Balance Deficits in Fragile X Premutation Carriers with and without Fragile X-Associated Tremor/Ataxia Syndrome (FXTAS). Cerebellum 2015, 14, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Fraint, A.; Vittal, P.; Szewka, A.; Bernard, B.; Berry-Kravis, E.; Hall, D.A. New observations in the fragile X-associated tremor/ataxia syndrome (FXTAS) phenotype. Front. Genet. 2014, 5, 365. [Google Scholar] [CrossRef]
- Hall, D.A.; Leehey, M.A.; Hagerman, R.J.; Pelak, V.S. Eye Movements in Fragile X-Associated Tremor/Ataxia Syndrome. J. Neuroophthalmol. 2021, 41, e661–e664. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.M.; Goodrich-Hunsaker, N.J.; McLennan, Y.; Tassone, F.; Zhang, M.; Rivera, S.M.; Simon, T.J. Eye movements reveal impaired inhibitory control in adult male fragile X premutation carriers asymptomatic for FXTAS. Neuropsychology 2014, 28, 571–584. [Google Scholar] [CrossRef] [PubMed]
- Moser, C.; Schmitt, L.; Schmidt, J.; Fairchild, A.; Klusek, J. Response Inhibition Deficits in Women with the FMR1 Premutation are Associated with Age and Fall Risk. Brain Cogn. 2021, 148, 105675. [Google Scholar] [CrossRef]
- Grigsby, J.; Brega, A.G.; Jacquemont, S.; Loesch, D.Z.; Leehey, M.A.; Goodrich, G.K.; Hagerman, R.J.; Epstein, J.; Wilson, R.; Cogswell, J.B.; et al. Impairment in the cognitive functioning of men with fragile X-associated tremor/ataxia syndrome (FXTAS). J. Neurol. Sci. 2006, 248, 227–233. [Google Scholar] [CrossRef]
- Grigsby, J.; Brega, A.G.; Engle, K.; Leehey, M.A.; Hagerman, R.J.; Tassone, F.; Hessl, D.; Hagerman, P.J.; Cogswell, J.B.; Bennett, R.E.; et al. Cognitive profile of fragile X premutation carriers with and without fragile X-associated tremor/ataxia syndrome. Neuropsychology 2008, 22, 48–60. [Google Scholar] [CrossRef]
- Grigsby, J.; Brega, A.G.; Leehey, M.A.; Goodrich, G.K.; Jacquemont, S.; Loesch, D.Z.; Cogswell, J.B.; Epstein, J.; Wilson, R.; Jardini, T.; et al. Impairment of executive cognitive functioning in males with fragile X-associated tremor/ataxia syndrome. Mov. Disord. 2007, 22, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D.; Sherman, J.C. The cerebellar cognitive affective syndrome. Brain 1998, 121 Pt 4, 561–579. [Google Scholar] [CrossRef]
- Hocking, D.R.; Loesch, D.Z.; Stimpson, P.; Tassone, F.; Atkinson, A.; Storey, E. Relationships of Motor Changes with Cognitive and Neuropsychiatric Features in FMR1 Male Carriers Affected with Fragile X-Associated Tremor/Ataxia Syndrome. Brain Sci. 2022, 12, 1549. [Google Scholar] [CrossRef] [PubMed]
- Storey, E.; Bui, M.Q.; Stimpson, P.; Tassone, F.; Atkinson, A.; Loesch, D.Z. Relationships between motor scores and cognitive functioning in FMR1 female premutation X carriers indicate early involvement of cerebello-cerebral pathways. Cerebellum Ataxias 2021, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Hocking, D.R.; Loesch, D.Z.; Stimpson, P.; Tassone, F.; Atkinson, A.; Storey, E. Delineating the Relationships between Motor, Cognitive-Executive and Psychiatric Symptoms in Female FMR1 Premutation Carriers. Front. Psychiatry 2021, 12, 742929. [Google Scholar] [CrossRef] [PubMed]
- Fay-Karmon, T.; Hassin-Baer, S. The spectrum of tremor among carriers of the FMR1 premutation with or without the fragile X-associated tremor/ataxia syndrome (FXTAS). Park. Relat. Disord. 2019, 65, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Apartis, E.; Blancher, A.; Meissner, W.G.; Guyant-Maréchal, L.; Maltête, D.; De Broucker, T.; Legrand, A.P.; Bouzenada, H.; Thanh, H.T.; Sallansonnet-Froment, M.; et al. FXTAS: New insights and the need for revised diagnostic criteria. Neurology 2012, 79, 1898–1907. [Google Scholar] [CrossRef]
- Juncos, J.L.; Lazarus, J.T.; Graves-Allen, E.; Shubeck, L.; Rusin, M.; Novak, G.; Hamilton, D.; Rohr, J.; Sherman, S.L. New clinical findings in the fragile X-associated tremor ataxia syndrome (FXTAS). Neurogenetics 2011, 12, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Jennings, D.; Seibyl, J.; Tassone, F.; Marek, K. FMR1 gene expansion and scans without evidence of dopaminergic deficits in parkinsonism patients. Park. Relat. Disord. 2010, 16, 608–611. [Google Scholar] [CrossRef]
- Ceravolo, R.; Antonini, A.; Volterrani, D.; Rossi, C.; Goldwurm, S.; Di Maria, E.; Kiferle, L.; Bonuccelli, U.; Murri, L. Dopamine transporter imaging study in parkinsonism occurring in fragile X premutation carriers. Neurology 2005, 65, 1971–1973. [Google Scholar] [CrossRef]
- Wojtala, J.; Heber, I.A.; Neuser, P.; Heller, J.; Kalbe, E.; Rehberg, S.P.; Storch, A.; Linse, K.; Schneider, C.; Gräber, S.; et al. Cognitive decline in Parkinson’s disease: The impact of the motor phenotype on cognition. J. Neurol. Neurosurg. Psychiatry 2019, 90, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Jacquemont, S.; Leehey, M.A.; Hagerman, R.J.; Beckett, L.A.; Hagerman, P.J. Size bias of fragile X premutation alleles in late-onset movement disorders. J. Med. Genet. 2006, 43, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Leehey, M.A.; Berry-Kravis, E.; Goetz, C.G.; Zhang, L.; Hall, D.A.; Li, L.; Rice, C.D.; Lara, R.; Cogswell, J.; Reynolds, A.; et al. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology 2008, 70 Pt 2, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Tassone, F.; Atkinson, A.; Stimpson, P.; Trost, N.; Pountney, D.L.; Storey, E. Differential Progression of Motor Dysfunction between Male and Female Fragile X Premutation Carriers Reveals Novel Aspects of Sex-Specific Neural Involvement. Front. Mol. Biosci. 2020, 7, 577246. [Google Scholar] [CrossRef]
- Cornish, K.M.; Li, L.; Kogan, C.S.; Jacquemont, S.; Turk, J.; Dalton, A.; Hagerman, R.J.; Hagerman, P.J. Age-dependent cognitive changes in carriers of the fragile X syndrome. Cortex 2008, 44, 628–636. [Google Scholar] [CrossRef]
- Cornish, K.M.; Hocking, D.R.; Moss, S.A.; Kogan, C.S. Selective executive markers of at-risk profiles associated with the fragile X premutation. Neurology 2011, 77, 618–622. [Google Scholar] [CrossRef]
- Kogan, C.S.; Cornish, K.M. Mapping self-reports of working memory deficits to executive dysfunction in Fragile X Mental Retardation 1 (FMR1) gene premutation carriers asymptomatic for FXTAS. Brain Cogn. 2010, 73, 236–243. [Google Scholar] [CrossRef]
- Kogan, C.S.; Turk, J.; Hagerman, R.J.; Cornish, K.M. Impact of the Fragile X mental retardation 1 (FMR1) gene premutation on neuropsychiatric functioning in adult males without fragile X-associated Tremor/Ataxia syndrome: A controlled study. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147, 859–872. [Google Scholar] [CrossRef]
- Brown, S.S.G.; Whalley, H.C.; Kind, P.C.; Stanfield, A.C. Decreased functional brain response to emotional arousal and increased psychiatric symptomology in FMR1 premutation carriers. Psychiatry Res. Neuroimaging 2019, 285, 9–17. [Google Scholar] [CrossRef]
- Hashimoto, R.; Backer, K.C.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. An fMRI study of the prefrontal activity during the performance of a working memory task in premutation carriers of the fragile X mental retardation 1 gene with and without fragile X-associated tremor/ataxia syndrome (FXTAS). J. Psychiatr. Res. 2011, 45, 36–43. [Google Scholar] [CrossRef]
- Kim, S.Y.; Hashimoto, R.; Tassone, F.; Simon, T.J.; Rivera, S.M. Altered neural activity of magnitude estimation processing in adults with the fragile X premutation. J. Psychiatr. Res. 2013, 47, 1909–1916. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koldewyn, K.; Hessl, D.; Adams, J.; Tassone, F.; Hagerman, P.J.; Hagerman, R.J.; Rivera, S.M. Reduced Hippocampal Activation During Recall is Associated with Elevated FMR1 mRNA and Psychiatric Symptoms in Men with the Fragile X Premutation. Brain Imaging Behav. 2008, 2, 105–116. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gabis, L.V.; Shaham, M.; Attia, O.L.; Kowal, T.; David, S.; Banet-Levi, Y.; Shefer, S.; Gabis, D.; Mula-Topf, D.; Avrech Bar, M.; et al. An escalating continuum of learning and attention difficulties from premutation to full mutation in female carriers of FMR1 expansion. Front. Neurol. 2023, 14, 1135630. [Google Scholar] [CrossRef] [PubMed]
- Shelton, A.L.; Cornish, K.; Fielding, J. Long term verbal memory recall deficits in fragile X premutation females. Neurobiol. Learn. Mem. 2017, 144, 131–135. [Google Scholar] [CrossRef]
- Shelton, A.L.; Cornish, K.; Kraan, C.; Georgiou-Karistianis, N.; Metcalfe, S.A.; Bradshaw, J.L.; Hocking, D.R.; Archibald, A.D.; Cohen, J.; Trollor, J.N.; et al. Exploring inhibitory deficits in female premutation carriers of fragile X syndrome: Through eye movements. Brain Cogn. 2014, 85, 201–208. [Google Scholar] [CrossRef]
- Shelton, A.L.; Cornish, K.M.; Godler, D.E.; Clough, M.; Kraan, C.; Bui, M.; Fielding, J. Delineation of the working memory profile in female FMR1 premutation carriers: The effect of cognitive load on ocular motor responses. Behav. Brain Res. 2015, 282, 194–200. [Google Scholar] [CrossRef]
- Shelton, A.L.; Cornish, K.M.; Kraan, C.M.; Lozano, R.; Bui, M.; Fielding, J. Executive Dysfunction in Female FMR1 Premutation Carriers. Cerebellum 2016, 15, 565–569. [Google Scholar] [CrossRef]
- Sterling, A.M.; Mailick, M.; Greenberg, J.; Warren, S.F.; Brady, N. Language dysfluencies in females with the FMR1 premutation. Brain Cogn. 2013, 82, 84–89. [Google Scholar] [CrossRef][Green Version]
- Yang, J.C.; Simon, C.; Niu, Y.Q.; Bogost, M.; Schneider, A.; Tassone, F.; Seritan, A.; Grigsby, J.; Hagerman, P.J.; Hagerman, R.J.; et al. Phenotypes of hypofrontality in older female fragile X premutation carriers. Ann. Neurol. 2013, 74, 275–283. [Google Scholar] [CrossRef]
- Klusek, J.; Hong, J.; Sterling, A.; Berry-Kravis, E.; Mailick, M.R. Inhibition deficits are modulated by age and CGG repeat length in carriers of the FMR1 premutation allele who are mothers of children with fragile X syndrome. Brain Cogn. 2020, 139, 105511. [Google Scholar] [CrossRef]
- Klusek, J.; Porter, A.; Abbeduto, L.; Adayev, T.; Tassone, F.; Mailick, M.R.; Glicksman, A.; Tonnsen, B.L.; Roberts, J.E. Curvilinear Association Between Language Disfluency and FMR1 CGG Repeat Size Across the Normal, Intermediate, and Premutation Range. Front. Genet. 2018, 9, 344. [Google Scholar] [CrossRef] [PubMed]
- Maltman, N.; DaWalt, L.S.; Hong, J.; Baker, M.W.; Berry-Kravis, E.M.; Brilliant, M.H.; Mailick, M. FMR1 CGG Repeats and Stress Influence Self-Reported Cognitive Functioning in Mothers. Am. J. Intellect. Dev. Disabil. 2023, 128, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Goodrich-Hunsaker, N.J.; Wong, L.M.; McLennan, Y.; Tassone, F.; Harvey, D.; Rivera, S.M.; Simon, T.J. Adult Female Fragile X Premutation Carriers Exhibit Age- and CGG Repeat Length-Related Impairments on an Attentionally Based Enumeration Task. Front. Hum. Neurosci. 2011, 5, 63. [Google Scholar] [CrossRef]
- Goodrich-Hunsaker, N.J.; Wong, L.M.; McLennan, Y.; Srivastava, S.; Tassone, F.; Harvey, D.; Rivera, S.M.; Simon, T.J. Young adult female fragile X premutation carriers show age- and genetically-modulated cognitive impairments. Brain Cogn. 2011, 75, 255–260. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bredin-Oja, S.L.; Warren, S.F.; Swinburne Romine, R.E.; Fleming, K.K.; Brady, N.; Berry-Kravis, E. Word retrieval difficulty in adult females with the FMR1 premutation: Changes over time and across contexts. Brain Cogn. 2021, 148, 105694. [Google Scholar] [CrossRef] [PubMed]
- Klusek, J.; Fairchild, A.; Moser, C.; Mailick, M.R.; Thurman, A.J.; Abbeduto, L. Family history of FXTAS is associated with age-related cognitive-linguistic decline among mothers with the FMR1 premutation. J. Neurodev. Disord. 2022, 14, 7. [Google Scholar] [CrossRef]
- Maltman, N.; Klusek, J.; DaWalt, L.; Hong, J.; Sterling, A.; Berry-Kravis, E.; Mailick, M.R. Verbal inhibition declines among older women with high FMR1 premutation expansions: A prospective study. Brain Cogn. 2022, 159, 105851. [Google Scholar] [CrossRef]
- Segal, O.; Kowal, T.; Banet-Levi, Y.; Gabis, L.V. Executive Function and Working Memory Deficits in Females with Fragile X Premutation. Life 2023, 13, 813. [Google Scholar] [CrossRef]
- Grigsby, J.; Brega, A.G.; Bennett, R.E.; Bourgeois, J.A.; Seritan, A.L.; Goodrich, G.K.; Hagerman, R.J. Clinically significant psychiatric symptoms among male carriers of the fragile X premutation, with and without FXTAS, and the mediating influence of executive functioning. Clin. Neuropsychol. 2016, 30, 944–959. [Google Scholar] [CrossRef]
- Hippolyte, L.; Battistella, G.; Perrin, A.G.; Fornari, E.; Cornish, K.M.; Beckmann, J.S.; Niederhauser, J.; Vingerhoets, F.J.; Draganski, B.; Maeder, P.; et al. Investigation of memory, executive functions, and anatomic correlates in asymptomatic FMR1 premutation carriers. Neurobiol. Aging 2014, 35, 1939–1946. [Google Scholar] [CrossRef]
- Birch, R.C.; Hocking, D.R.; Cornish, K.M.; Menant, J.C.; Georgiou-Karistianis, N.; Godler, D.E.; Wen, W.; Hackett, A.; Rogers, C.; Trollor, J.N. Preliminary evidence of an effect of cerebellar volume on postural sway in FMR1 premutation males. Genes. Brain Behav. 2015, 14, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Epstein, M.P.; Tinker, S.W.; Abramowitz, A.; Sherman, S.L. The FMR1 premutation and attention-deficit hyperactivity disorder (ADHD): Evidence for a complex inheritance. Behav. Genet. 2012, 42, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Klusek, J.; Schmidt, J.; Fairchild, A.J.; Porter, A.; Roberts, J.E. Altered sensitivity to social gaze in the FMR1 premutation and pragmatic language competence. J. Neurodev. Disord. 2017, 9, 31. [Google Scholar] [CrossRef]
- Dembo, R.S.; Hong, J.; DaWalt, L.S.; Berry-Kravis, E.M.; Mailick, M.R. Health Effects of Sleep Quality in Premutation Carrier Mothers of Individuals with Fragile X Syndrome. Am. J. Intellect. Dev. Disabil. 2023, 128, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Kraan, C.M.; Hocking, D.R.; Georgiou-Karistianis, N.; Metcalfe, S.A.; Archibald, A.D.; Fielding, J.; Trollor, J.; Bradshaw, J.L.; Cohen, J.; Cornish, K.M. Age and CGG-repeat length are associated with neuromotor impairments in at-risk females with the FMR1 premutation. Neurobiol. Aging 2014, 35, 2179.e7–2179.e13. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hessl, D.; Hagerman, R.J.; Simon, T.J.; Tassone, F.; Ferrer, E.; Rivera, S.M. Abnormal trajectories in cerebellum and brainstem volumes in carriers of the fragile X premutation. Neurobiol. Aging 2017, 55, 11–19. [Google Scholar] [CrossRef]
- Hashimoto, R.; Srivastava, S.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. Diffusion tensor imaging in male premutation carriers of the fragile X mental retardation gene. Mov. Disord. 2011, 26, 1329–1336. [Google Scholar] [CrossRef]
- Mailick, M.; Hong, J.; Greenberg, J.; Dawalt, L.S.; Baker, M.W.; Rathouz, P.J. FMR1 genotype interacts with parenting stress to shape health and functional abilities in older age. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 399–412. [Google Scholar] [CrossRef]
- Cornish, K.M.; Kogan, C.S.; Li, L.; Turk, J.; Jacquemont, S.; Hagerman, R.J. Lifespan changes in working memory in fragile X premutation males. Brain Cogn. 2009, 69, 551–558. [Google Scholar] [CrossRef]
- Sévin, M.; Kutalik, Z.; Bergman, S.; Vercelletto, M.; Renou, P.; Lamy, E.; Vingerhoets, F.J.; Di Virgilio, G.; Boisseau, P.; Bezieau, S.; et al. Penetrance of marked cognitive impairment in older male carriers of the FMR1 gene premutation. J. Med. Genet. 2009, 46, 818–824. [Google Scholar] [CrossRef]
- O’Keefe, J.A.; Robertson, E.E.; Ouyang, B.; Carns, D.; McAsey, A.; Liu, Y.; Swanson, M.; Bernard, B.; Berry-Kravis, E.; Hall, D.A. Cognitive function impacts gait, functional mobility and falls in fragile X-associated tremor/ataxia syndrome. Gait Posture 2018, 66, 288–293. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, J.A.; Guan, J.; Robertson, E.; Biskis, A.; Joyce, J.; Ouyang, B.; Liu, Y.; Carnes, D.; Purcell, N.; Berry-Kravis, E.; et al. The Effects of Dual Task Cognitive Interference and Fast-Paced Walking on Gait, Turns, and Falls in Men and Women with FXTAS. Cerebellum 2021, 20, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Koldewyn, K.; Hashimoto, R.; Schneider, A.; Le, L.; Tassone, F.; Cheung, K.; Hagerman, P.; Hessl, D.; Rivera, S.M. Male carriers of the FMR1 premutation show altered hippocampal-prefrontal function during memory encoding. Front. Hum. Neurosci. 2012, 6, 297. [Google Scholar] [CrossRef] [PubMed]
- Cvejic, R.C.; Hocking, D.R.; Wen, W.; Georgiou-Karistianis, N.; Cornish, K.M.; Godler, D.E.; Rogers, C.; Trollor, J.N. Reduced caudate volume and cognitive slowing in men at risk of fragile X-associated tremor ataxia syndrome. Brain Imaging Behav. 2019, 13, 1128–1134. [Google Scholar] [CrossRef]
- Filley, C.M. Fragile X tremor ataxia syndrome and white matter dementia. Clin. Neuropsychol. 2016, 30, 901–912. [Google Scholar] [CrossRef]
- Shelton, A.L.; Cornish, K.M.; Godler, D.; Bui, Q.M.; Kolbe, S.; Fielding, J. White matter microstructure, cognition, and molecular markers in fragile X premutation females. Neurology 2017, 88, 2080–2088. [Google Scholar] [CrossRef]
- Wang, J.Y.; Grigsby, J.; Placido, D.; Wei, H.; Tassone, F.; Kim, K.; Hessl, D.; Rivera, S.M.; Hagerman, R.J. Clinical and Molecular Correlates of Abnormal Changes in the Cerebellum and Globus Pallidus in Fragile X Premutation. Front. Neurol. 2022, 13, 797649. [Google Scholar] [CrossRef]
- Yang, J.C.; Chan, S.H.; Khan, S.; Schneider, A.; Nanakul, R.; Teichholtz, S.; Niu, Y.Q.; Seritan, A.; Tassone, F.; Grigsby, J.; et al. Neural substrates of executive dysfunction in fragile X-associated tremor/ataxia syndrome (FXTAS): A brain potential study. Cereb. Cortex 2013, 23, 2657–2666. [Google Scholar] [CrossRef]
- Flavell, J.; Franklin, C.; Nestor, P.J. A Systematic Review of Fragile X-Associated Neuropsychiatric Disorders. J. Neuropsychiatry Clin. Neurosci. 2023, 35, 110–120. [Google Scholar] [CrossRef]
- Association, A. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; The Fragile X Associated Tremor Ataxia Syndrome; American Psychiatric Association Publishing: Washington, DC, USA, 2013. [Google Scholar]
- Baxter, A.J.; Scott, K.M.; Vos, T.; Whiteford, H.A. Global prevalence of anxiety disorders: A systematic review and meta-regression. Psychol. Med. 2013, 43, 897–910. [Google Scholar] [CrossRef]
- Franke, P.; Leboyer, M.; Gänsicke, M.; Weiffenbach, O.; Biancalana, V.; Cornillet-Lefebre, P.; Croquette, M.F.; Froster, U.; Schwab, S.G.; Poustka, F.; et al. Genotype-phenotype relationship in female carriers of the premutation and full mutation of FMR-1. Psychiatry Res. 1998, 80, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.; Eliez, S.; Dyer-Friedman, J.; Hessl, D.; Glaser, B.; Blasey, C.; Taylor, A.; Reiss, A. Neurobehavioral phenotype in carriers of the fragile X premutation. Am. J. Med. Genet. 2001, 103, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Hessl, D.; Tassone, F.; Loesch, D.Z.; Berry-Kravis, E.; Leehey, M.A.; Gane, L.W.; Barbato, I.; Rice, C.; Gould, E.; Hall, D.A.; et al. Abnormal elevation of FMR1 mRNA is associated with psychological symptoms in individuals with the fragile X premutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2005, 139, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.E.; Rohr, J.K.; Sherman, S.L. Co-occurring diagnoses among FMR1 premutation allele carriers. Clin. Genet. 2010, 77, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Johnston, C.; Tassone, F.; Sansone, S.; Hagerman, R.J.; Ferrer, E.; Rivera, S.M.; Hessl, D. Broad autism spectrum and obsessive-compulsive symptoms in adults with the fragile X premutation. Clin. Neuropsychol. 2016, 30, 929–943. [Google Scholar] [CrossRef]
- Kenna, H.A.; Tartter, M.; Hall, S.S.; Lightbody, A.A.; Nguyen, Q.; de los Angeles, C.P.; Reiss, A.L.; Rasgon, N.L. High rates of comorbid depressive and anxiety disorders among women with premutation of the FMR1 gene. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2013, 162, 872–878. [Google Scholar] [CrossRef]
- Santos, E.; Emeka-Nwonovo, C.; Wang, J.Y.; Schneider, A.; Tassone, F.; Hagerman, P.; Hagerman, R. Developmental aspects of FXAND in a man with the FMR1 premutation. Mol. Genet. Genom. Med. 2020, 8, e1050. [Google Scholar] [CrossRef]
- Polussa, J.; Schneider, A.; Hagerman, R. Molecular Advances Leading to Treatment Implications for Fragile X Premutation Carriers. Brain Disord. Ther. 2014, 3, 1000119. [Google Scholar]
- Zhang, A.; Borhneimer, L.A.; Weaver, A.; Franklin, C.; Hai, A.H.; Guz, S.; Shen, L. Cognitive behavioral therapy for primary care depression and anxiety: A secondary meta-analytic review using robust variance estimation in meta-regression. J. Behav. Med. 2019, 42, 1117–1141. [Google Scholar] [CrossRef]
- Wheeler, A.; Raspa, M.; Hagerman, R.; Mailick, M.; Riley, C. Implications of the FMR1 Premutation for Children, Adolescents, Adults, and Their Families. Pediatrics 2017, 139 (Suppl. S3), S172–S182. [Google Scholar] [CrossRef]
- Hunter, J.E.; Allen, E.G.; Abramowitz, A.; Rusin, M.; Leslie, M.; Novak, G.; Hamilton, D.; Shubeck, L.; Charen, K.; Sherman, S.L. Investigation of phenotypes associated with mood and anxiety among male and female fragile X premutation carriers. Behav. Genet. 2008, 38, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.E.; Bailey, D.B., Jr.; Mankowski, J.; Ford, A.; Sideris, J.; Weisenfeld, L.A.; Heath, T.M.; Golden, R.N. Mood and anxiety disorders in females with the FMR1 premutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009, 150, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Seritan, A.L.; Bourgeois, J.A.; Schneider, A.; Mu, Y.; Hagerman, R.J.; Nguyen, D.V. Ages of Onset of Mood and Anxiety Disorders in Fragile X Premutation Carriers. Curr. Psychiatry Rev. 2013, 9, 65–71. [Google Scholar]
- Dorn, M.B.; Mazzocco, M.M.; Hagerman, R.J. Behavioral and psychiatric disorders in adult male carriers of fragile X. J. Am. Acad. Child. Adolesc. Psychiatry 1994, 33, 256–264. [Google Scholar] [CrossRef]
- Bush, L.; Scott, M.N. Neuropsychological and ASD phenotypes in rare genetic syndromes: A critical review of the literature. Clin. Neuropsychol. 2022, 36, 993–1027. [Google Scholar] [CrossRef] [PubMed]
- Klusek, J.; Martin, G.E.; Losh, M. Consistency between research and clinical diagnoses of autism among boys and girls with fragile X syndrome. J. Intellect. Disabil. Res. 2014, 58, 940–952. [Google Scholar] [CrossRef]
- Lee, M.; Martin, G.E.; Berry-Kravis, E.; Losh, M. A developmental, longitudinal investigation of autism phenotypic profiles in fragile X syndrome. J. Neurodev. Disord. 2016, 8, 47. [Google Scholar] [CrossRef]
- Martin, G.E.; Bush, L.; Klusek, J.; Patel, S.; Losh, M. A Multimethod Analysis of Pragmatic Skills in Children and Adolescents with Fragile X Syndrome, Autism Spectrum Disorder, and Down Syndrome. J. Speech Lang. Hear. Res. 2018, 61, 3023–3037. [Google Scholar] [CrossRef]
- Bolton, P.; Macdonald, H.; Pickles, A.; Rios, P.; Goode, S.; Crowson, M.; Bailey, A.; Rutter, M. A case-control family history study of autism. J. Child. Psychol. Psychiatry 1994, 35, 877–900. [Google Scholar] [CrossRef]
- Piven, J.; Palmer, P.; Jacobi, D.; Childress, D.; Arndt, S. Broader autism phenotype: Evidence from a family history study of multiple-incidence autism families. Am. J. Psychiatry 1997, 154, 185–190. [Google Scholar]
- Cornish, K.; Burack, J.A.; Rahman, A.; Munir, F.; Russo, N.; Grant, C. Theory of mind deficits in children with fragile X syndrome. J. Intellect. Disabil. Res. 2005, 49 Pt 5, 372–378. [Google Scholar] [CrossRef] [PubMed]
- White, S.J.; Gerber, D.; Sanchez Hernandez, R.D.; Efiannayi, A.; Chowdhury, I.; Partington, H.; Moss, J.F. Autistic traits and mental health in women with the fragile-X premutation: Maternal status versus genetic risk. Br. J. Psychiatry 2021, 218, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.C.; Van Driesche, S.J.; Zhang, C.; Hung, K.Y.; Mele, A.; Fraser, C.E.; Stone, E.F.; Chen, C.; Fak, J.J.; Chi, S.W.; et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 2011, 146, 247–261. [Google Scholar] [CrossRef] [PubMed]
- Klusek, J.; Fairchild, A.J.; Roberts, J.E. Vagal Tone as a Putative Mechanism for Pragmatic Competence: An Investigation of Carriers of the FMR1 Premutation. J. Autism Dev. Disord. 2019, 49, 197–208. [Google Scholar] [CrossRef]
- Winston, M.; Nayar, K.; Hogan, A.L.; Barstein, J.; La Valle, C.; Sharp, K.; Berry-Kravis, E.; Losh, M. Physiological regulation and social-emotional processing in female carriers of the FMR1 premutation. Physiol. Behav. 2020, 214, 112746. [Google Scholar] [CrossRef]
- Hessl, D.; Wang, J.M.; Schneider, A.; Koldewyn, K.; Le, L.; Iwahashi, C.; Cheung, K.; Tassone, F.; Hagerman, P.J.; Rivera, S.M. Decreased fragile X mental retardation protein expression underlies amygdala dysfunction in carriers of the fragile X premutation. Biol. Psychiatry 2011, 70, 859–865. [Google Scholar] [CrossRef]
- Klusek, J.; McGrath, S.E.; Abbeduto, L.; Roberts, J.E. Pragmatic Language Features of Mothers with the FMR1 Premutation Are Associated with the Language Outcomes of Adolescents and Young Adults with Fragile X Syndrome. J. Speech Lang. Hear. Res. 2016, 59, 49–61. [Google Scholar] [CrossRef]
- Coplan, R.J.; Weeks, M. Shy and soft-spoken: Shyness, pragmatic language, and socio-emotional adjustment in early childhood. Infant. Child. Dev. 2009, 18, 238–254. [Google Scholar] [CrossRef]
- Rodas, N.V.; Eisenhower, A.; Blacher, J. Structural and Pragmatic Language in Children with ASD: Longitudinal Impact on Anxiety and Externalizing Behaviors. J. Autism Dev. Disord. 2017, 47, 3479–3488. [Google Scholar] [CrossRef]
- Ostchega, Y.; Fryar, C.D.; Nwankwo, T.; Nguyen, D.T. Hypertension Prevalence Among Adults Aged 18 and Over: United States, 2017–2018. NCHS Data Brief 2020, 1–8. [Google Scholar]
- Tassanakijpanich, N.; Cohen, J.; Cohen, R.; Srivatsa, U.N.; Hagerman, R.J. Cardiovascular Problems in the Fragile X Premutation. Front. Genet. 2020, 11, 586910. [Google Scholar] [CrossRef] [PubMed]
- Gruber, N.; Haham, L.M.; Raanani, H.; Cohen, Y.; Gabis, L.; Berkenstadt, M.; Ries-Levavi, L.; Elizur, S.; Pinhas-Hamiel, O. Female fragile X premutation carriers are at increased risk for metabolic syndrome from early adulthood. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 1010–1018. [Google Scholar] [CrossRef] [PubMed]
- Griffith, J.; Zarrouf, F.A. A systematic review of chronic fatigue syndrome: Don’t assume it’s depression. Prim. Care Companion J. Clin. Psychiatry 2008, 10, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Hamlin, A.; Liu, Y.; Nguyen, D.V.; Tassone, F.; Zhang, L.; Hagerman, R.J. Sleep apnea in fragile X premutation carriers with and without FXTAS. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2011, 156, 923–928. [Google Scholar] [CrossRef]
- El-Deeb, M.; Adams, P.; Schneider, A.; Salcedo-Arellano, M.J.; Tassone, F.; Hagerman, R. Fentanyl overdose in a female with the FMR1 premutation and FXTAS. J. Mol. Genet. 2018, 1, 101. [Google Scholar]
- Leehey, M.A.; Legg, W.; Tassone, F.; Hagerman, R. Fibromyalgia in fragile X mental retardation 1 gene premutation carriers. Rheumatology 2011, 50, 2233–2236. [Google Scholar] [CrossRef]
- Hall, D.A.; Hermanson, M.; Dunn, E.; Stebbins, G.; Merkitch, D.; Ouyang, B.; Berry-Kravis, E.; Jhaveri, M. The Corpus Callosum Splenium Sign in Fragile X-Associated Tremor Ataxia Syndrome. Mov. Disord. Clin. Pr. 2017, 4, 383–388. [Google Scholar] [CrossRef]
- Yunus, M.B. Central sensitivity syndromes: A new paradigm and group nosology for fibromyalgia and overlapping conditions, and the related issue of disease versus illness. Semin. Arthritis Rheum. 2008, 37, 339–352. [Google Scholar] [CrossRef]
- Martorell, L.; Tondo, M.; Garcia-Fructuoso, F.; Naudo, M.; Alegre, C.; Gamez, J.; Genovés, J.; Poo, P. Screening for the presence of FMR1 premutation alleles in a Spanish population with fibromyalgia. Clin. Rheumatol. 2012, 31, 1611–1615. [Google Scholar] [CrossRef]
- Rodriguez-Revenga, L.; Madrigal, I.; Blanch-Rubió, J.; Elurbe, D.M.; Docampo, E.; Collado, A.; Vidal, J.; Carbonell, J.; Estivill, X.; Mila, M. Screening for the presence of FMR1 premutation alleles in women with fibromyalgia. Gene 2013, 512, 305–308. [Google Scholar] [CrossRef]
- Chonchaiya, W.; Nguyen, D.V.; Au, J.; Campos, L.; Berry-Kravis, E.M.; Lohse, K.; Mu, Y.; Utari, A.; Hervey, C.; Wang, L.; et al. Clinical involvement in daughters of men with fragile X-associated tremor ataxia syndrome. Clin. Genet. 2010, 78, 38–46. [Google Scholar] [CrossRef]
- Tentindo, G.S.; Fishman, S.M.; Li, C.S.; Wang, Q.; Brass, S.D. The prevalence and awareness of sleep apnea in patients suffering chronic pain: An assessment using the STOP-Bang sleep apnea questionnaire. Nat. Sci. Sleep. 2018, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Seltzer, M.M.; Barker, E.T.; Greenberg, J.S.; Hong, J.; Coe, C.; Almeida, D. Differential sensitivity to life stress in FMR1 premutation carrier mothers of children with fragile X syndrome. Health Psychol. 2012, 31, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Napoli, E.; Flores, A.; Mansuri, Y.; Hagerman, R.J.; Giulivi, C. Sulforaphane improves mitochondrial metabolism in fibroblasts from patients with fragile X-associated tremor and ataxia syndrome. Neurobiol. Dis. 2021, 157, 105427. [Google Scholar] [CrossRef] [PubMed]
- Muzar, Z.; Lozano, R. Current research, diagnosis, and treatment of fragile X-associated tremor/ataxia syndrome. Intractable Rare Dis. Res. 2014, 3, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Liddelow, S.A.; Temple, S.; Benowitz, L.I.; Di Polo, A.; Wellington, C.; Goldberg, J.L.; He, Z.; Duan, X.; Bu, G.; et al. Solving neurodegeneration: Common mechanisms and strategies for new treatments. Mol. Neurodegener. 2022, 17, 23. [Google Scholar] [CrossRef]
- Aiello Bowles, E.J.; Crane, P.K.; Walker, R.L.; Chubak, J.; LaCroix, A.Z.; Anderson, M.L.; Rosenberg, D.; Keene, C.D.; Larson, E.B. Cognitive Resilience to Alzheimer’s Disease Pathology in the Human Brain. J. Alzheimers Dis. 2019, 68, 1071–1083. [Google Scholar] [CrossRef]
- Kotagal, V.; Bohnen, N.I.; Müller, M.L.; Koeppe, R.A.; Frey, K.A.; Langa, K.M.; Albin, R.L. Educational attainment and motor burden in Parkinson’s disease. Mov. Disord. 2015, 30, 1143–1147. [Google Scholar] [CrossRef]
- Hartley, S.L.; DaWalt, L.S.; Hong, J.; Greenberg, J.S.; Mailick, M.R. Positive Emotional Support in Premutation Carrier Mothers of Adolescents and Adults With Fragile X Syndrome: Gene by Environment Interactions. Am. J. Intellect. Dev. Disabil. 2019, 124, 411–426. [Google Scholar] [CrossRef]
- Brega, A.G.; Reynolds, A.; Bennett, R.E.; Leehey, M.A.; Bounds, L.S.; Cogswell, J.B.; Hagerman, R.J.; Hagerman, P.J.; Grigsby, J. Functional status of men with the fragile X premutation, with and without the tremor/ataxia syndrome (FXTAS). Int. J. Geriatr. Psychiatry 2009, 24, 1101–1109. [Google Scholar] [CrossRef]
- Lozano, R.; Saito, N.; Reed, D.; Eldeeb, M.; Schneider, A.; Hessl, D.; Tassone, F.; Beckett, L.; Hagerman, R. Aging in Fragile X Premutation Carriers. Cerebellum 2016, 15, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Dembo, R.S.; DaWalt, L.S.; Brilliant, M.; Berry-Kravis, E.M.; Mailick, M. The effect of college degree attainment on neurodegenerative symptoms in genetically at-risk women. SSM Popul. Health 2022, 19, 101262. [Google Scholar] [CrossRef] [PubMed]
- Kraan, C.; Bui, M.; Archibald, A.; Davison, S.; Cvejic, R.C.; Metcalfe, S.; Amor, D.J.; Trollor, J.; Cohen, J.; Cornish, K. Social and physical predictors of mental health impact in adult women who have an FMR1 premutation. Genet. Med. 2023, in press. [CrossRef]
- Nelson, L.M. Primary ovarian insufficiency. N. Engl. J. Med. 2009, 360, 606–614. [Google Scholar] [CrossRef]
- Hipp, H.S.; Charen, K.H.; Spencer, J.B.; Allen, E.G.; Sherman, S.L. Reproductive and gynecologic care of women with fragile X primary ovarian insufficiency (FXPOI). Menopause 2016, 23, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Pan, M.L.; Chen, L.R.; Tsao, H.M.; Chen, K.H. Polycystic ovarian syndrome and the risk of subsequent primary ovarian insufficiency: A nationwide population-based study. Menopause 2017, 24, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Shuster, L.T.; Rhodes, D.J.; Gostout, B.S.; Grossardt, B.R.; Rocca, W.A. Premature menopause or early menopause: Long-term health consequences. Maturitas 2010, 65, 161–166. [Google Scholar] [CrossRef]
- Cedars, M.I. Biomarkers of ovarian reserve—Do they predict somatic aging? Semin. Reprod. Med. 2013, 31, 443–451. [Google Scholar] [CrossRef]
- Hundscheid, R.D.; Smits, A.P.; Thomas, C.M.; Kiemeney, L.A.; Braat, D.D. Female carriers of fragile X premutations have no increased risk for additional diseases other than premature ovarian failure. Am. J. Med. Genet. A 2003, 117, 6–9. [Google Scholar] [CrossRef]
- Allen, E.G.; Sullivan, A.K.; Marcus, M.; Small, C.; Dominguez, C.; Epstein, M.P.; Charen, K.; He, W.; Taylor, K.C.; Sherman, S.L. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum. Reprod. 2007, 22, 2142–2152. [Google Scholar] [CrossRef]
- Roeters van Lennep, J.E.; Heida, K.Y.; Bots, M.L.; Hoek, A. Cardiovascular disease risk in women with premature ovarian insufficiency: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2016, 23, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Reiss, S.; Zalles, L.; Gbekie, C.; Lozano, R. Identity and Reproductive Aspects in Females with Fragile X Syndrome. Women’s Health Rep. 2021, 2, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.G.; Charen, K.; Hipp, H.S.; Shubeck, L.; Amin, A.; He, W.; Hunter, J.E.; Sherman, S.L. Clustering of comorbid conditions among women who carry an FMR1 premutation. Genet. Med. 2020, 22, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Besterman, A.D.; Wilke, S.A.; Mulligan, T.E.; Allison, S.C.; Hagerman, R.; Seritan, A.L.; Bourgeois, J.A. Towards an Understanding of Neuropsychiatric Manifestations in Fragile X Premutation Carriers. Future Neurol. 2014, 9, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Protic, D.; Berry-Kravis, E.M. Medical, Psychopharmacological, and Targeted Treatment for FXS; Hagerman, R.J., Hagerman, P.J., Eds.; Mac Keith Press: London, UK, 2020; pp. 41–59. [Google Scholar]
- Kirwin, J.L.; Gören, J.L. Duloxetine: A dual serotonin-norepinephrine reuptake inhibitor for treatment of major depressive disorder. Pharmacotherapy 2005, 25, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Buoli, M.; Serati, M.; Cahn, W. Alternative pharmacological strategies for adult ADHD treatment: A systematic review. Expert. Rev. Neurother. 2016, 16, 131–144. [Google Scholar] [CrossRef]
- Mazza, M.; Harnic, D.; Catalano, V.; Janiri, L.; Bria, P. Duloxetine for premenstrual dysphoric disorder: A pilot study. Expert. Opin. Pharmacother. 2008, 9, 517–521. [Google Scholar] [CrossRef]
- Ramos, M.G.; Hara, C.; Rocha, F.L. Duloxetine treatment for women with premenstrual dysphoric disorder: A single-blind trial. Int. J. Neuropsychopharmacol. 2009, 12, 1081–1088. [Google Scholar] [CrossRef][Green Version]
- Shah, N.R.; Jones, J.B.; Aperi, J.; Shemtov, R.; Karne, A.; Borenstein, J. Selective serotonin reuptake inhibitors for premenstrual syndrome and premenstrual dysphoric disorder: A meta-analysis. Obs. Gynecol. 2008, 111, 1175–1182. [Google Scholar] [CrossRef]
- Hofmann, S.G.; Asnaani, A.; Vonk, I.J.; Sawyer, A.T.; Fang, A. The Efficacy of Cognitive Behavioral Therapy: A Review of Meta-analyses. Cogn. Ther. Res. 2012, 36, 427–440. [Google Scholar] [CrossRef]
- Twohig, M.; Levin, M.E. Acceptance and Commitment Therapy as a Treatment for Anxiety and Depression: A Review. Psychiatr. Clin. N. Am. 2017, 40, 751–770. [Google Scholar] [CrossRef] [PubMed]
- Otto, M.W.; Smits, J.A.; Reese, H.E. Combined Psychotherapy and Pharmacotherapy for Mood and Anxiety Disorders in Adults: Review and Analysis. FOCUS 2006, 4, 204–214. [Google Scholar] [CrossRef]
- Maina, G.; Mauri, M.; Rossi, A. Anxiety and depression. Med. Clin. N. Am. 2016, 72, 745–977. [Google Scholar]
- Curtiss, J.E.; Levine, D.S.; Ander, I.; Baker, A.W. Cognitive-Behavioral Treatments for Anxiety and Stress-Related Disorders. Focus 2021, 19, 184–189. [Google Scholar] [CrossRef]
- Sockol, L.E.; Epperson, C.N.; Barber, J. A meta-analysis of treatments for perinatal depression. Clin. Psychol. Rev. 2011, 31, 839–849. [Google Scholar] [CrossRef]
- Busse, J.W.; Montori, V.M.; Krasnik, C.; Patelis-Siotis, I.; Guyatt, G.H. Psychological intervention for premenstrual syndrome: A meta-analysis of randomized controlled trials. Psychother. Psychosom. 2009, 78, 6–15. [Google Scholar] [CrossRef]
- Pan, J.X.; Xia, J.J.; Deng, F.L.; Liang, W.W.; Wu, J.; Yin, B.M.; Dong, M.X.; Chen, J.J.; Ye, F.; Wang, H.Y.; et al. Diagnosis of major depressive disorder based on changes in multiple plasma neurotransmitters: A targeted metabolomics study. Transl. Psychiatry 2018, 8, 130. [Google Scholar] [CrossRef]
- Thoma, N.; Pilecki, B.; McKay, D. Contemporary Cognitive Behavior Therapy: A Review of Theory, History, and Evidence. Psychodyn. Psychiatry 2015, 43, 423–461. [Google Scholar] [CrossRef]
- AlHadi, A.N.; AlGhofili, H.H.; Almujaiwel, N.A.; Alsweirky, H.M.; Albeshr, M.F.; Almogbel, G.T. Perception and barriers to the use of cognitive-behavioral therapy in the treatment of depression in primary healthcare centers and family medicine clinics in Saudi Arabia. J. Fam. Community Med. 2021, 28, 77–84. [Google Scholar] [CrossRef]
- Steel, Z.; McDonald, R.; Silove, D.; Bauman, A.; Sandford, P.; Herron, J.; Minas, I.H. Pathways to the first contact with specialist mental health care. Aust. N. Z. J. Psychiatry 2006, 40, 347–354. [Google Scholar] [CrossRef]
- Juang, B.-T.; Ludwig, A.L.; Benedetti, K.L.; Gu, C.; Collins, K.; Morales, C.; Asundi, A.; Wittmann, T.; L’Etoile, N.; Hagerman, P.J. Expression of an expanded CGG-repeat RNA in a single pair of primary sensory neurons impairs olfactory adaptation in Caenorhabditis elegans. Hum. Mol. Genet. 2014, 23, 4945–4959. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Martinsen, E.W. Physical activity in the prevention and treatment of anxiety and depression. Nord. J. Psychiatry 2008, 62 (Suppl. S47), 25–29. [Google Scholar] [CrossRef] [PubMed]
- Shelly, K.E.; Candelaria, N.R.; Li, Z.; Allen, E.G.; Jin, P.; Nelson, D.L. Ectopic expression of CGG-repeats alters ovarian response to gonadotropins and leads to infertility in a murine FMR1 premutation model. Hum. Mol. Genet. 2021, 30, 923–938. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.E.; Le, W.W.; Entezam, A.; Otsuka, N.; Tong, Z.B.; Nelson, L.; Flaws, J.A.; McDonald, J.H.; Jafar, S.; Usdin, K. Ovarian abnormalities in a mouse model of fragile X primary ovarian insufficiency. J. Histochem. Cytochem. 2012, 60, 439–456. [Google Scholar] [CrossRef] [PubMed]
- Ennis, S.; Ward, D.; Murray, A. Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Eur. J. Hum. Genet. 2006, 14, 253–255. [Google Scholar] [CrossRef]
- Tejada, M.-I.; García-Alegría, E.; Bilbao, A.; Martínez-Bouzas, C.; Beristain, E.; Poch, M.; Ramos-Arroyo, M.A.; López, B.; Fernandez Carvajal, I.; Ribate, M.-P.; et al. Analysis of the molecular parameters that could predict the risk of manifesting premature ovarian failure in female premutation carriers of fragile X syndrome. Menopause 2008, 15, 945–949. [Google Scholar] [CrossRef]
- Spath, M.A.; Feuth, T.B.; Smits, A.P.; Yntema, H.G.; Braat, D.D.; Thomas, C.M.; van Kessel, A.G.; Sherman, S.L.; Allen, E.G. Predictors and risk model development for menopausal age in fragile X premutation carriers. Genet. Med. 2011, 13, 643–650. [Google Scholar] [CrossRef]
- Allen, E.G.; Charen, K.; Hipp, H.S.; Shubeck, L.; Amin, A.; He, W.; Nolin, S.L.; Glicksman, A.; Tortora, N.; McKinnon, B.; et al. Refining the risk for fragile X-associated primary ovarian insufficiency (FXPOI) by FMR1 CGG repeat size. Genet. Med. 2021, 23, 1648–1655. [Google Scholar] [CrossRef]
- Brunberg, J.A.; Jacquemont, S.; Hagerman, R.J.; Berry-Kravis, E.M.; Grigsby, J.; Leehey, M.A.; Tassone, F.; Brown, W.T.; Greco, C.M.; Hagerman, P.J. Fragile X premutation carriers: Characteristic MR imaging findings of adult male patients with progressive cerebellar and cognitive dysfunction. AJNR Am. J. Neuroradiol. 2002, 23, 1757–1766. [Google Scholar]
- Cohen, S.; Masyn, K.; Adams, J.; Hessl, D.; Rivera, S.; Tassone, F.; Brunberg, J.; DeCarli, C.; Zhang, L.; Cogswell, J.; et al. Molecular and imaging correlates of the fragile X-associated tremor/ataxia syndrome. Neurology 2006, 67, 1426–1431. [Google Scholar] [CrossRef]
- Hashimoto, R.; Javan, A.K.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. A voxel-based morphometry study of grey matter loss in fragile X-associated tremor/ataxia syndrome. Brain 2011, 134 Pt 3, 863–878. [Google Scholar] [CrossRef]
- Battistella, G.; Niederhauser, J.; Fornari, E.; Hippolyte, L.; Gronchi Perrin, A.; Lesca, G.; Forzano, F.; Hagmann, P.; Vingerhoets, F.J.; Draganski, B.; et al. Brain structure in asymptomatic FMR1 premutation carriers at risk for fragile X-associated tremor/ataxia syndrome. Neurobiol. Aging 2013, 34, 1700–1707. [Google Scholar] [CrossRef]
- Shelton, A.L.; Wang, J.Y.; Fourie, E.; Tassone, F.; Chen, A.; Frizzi, L.; Hagerman, R.J.; Ferrer, E.; Hessl, D.; Rivera, S.M. Middle Cerebellar Peduncle Width—A Novel MRI Biomarker for FXTAS? Front. Neurosci. 2018, 12, 379. [Google Scholar] [CrossRef]
- Birch, R.C.; Hocking, D.R.; Cornish, K.M.; Menant, J.C.; Lord, S.R.; Georgiou-Karistianis, N.; Godler, D.E.; Wen, W.; Rogers, C.; Trollor, J.N. Selective subcortical contributions to gait impairments in males with the FMR1 premutation. J. Neurol. Neurosurg. Psychiatry 2017, 88, 188–190. [Google Scholar] [CrossRef] [PubMed]
- Hocking, D.R.; Birch, R.C.; Bui, Q.M.; Menant, J.C.; Lord, S.R.; Georgiou-Karistianis, N.; Godler, D.E.; Wen, W.; Hackett, A.; Rogers, C.; et al. Cerebellar volume mediates the relationship between FMR1 mRNA levels and voluntary step initiation in males with the premutation. Neurobiol. Aging 2017, 50, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Hagerman, R.J.; Rivera, S.M. A multimodal imaging analysis of subcortical gray matter in fragile X premutation carriers. Mov. Disord. 2013, 28, 1278–1284. [Google Scholar] [CrossRef]
- Bostan, A.C.; Strick, P.L. The basal ganglia and the cerebellum: Nodes in an integrated network. Nat. Rev. Neurosci. 2018, 19, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Schmahmann, J.D.; Pandya, D.N. Disconnection syndromes of basal ganglia, thalamus, and cerebrocerebellar systems. Cortex 2008, 44, 1037–1066. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.S.; Adams, P.E.; Nguyen, D.; Brunberg, J.A.; Tassone, F.; Zhang, W.; Koldewyn, K.; Rivera, S.M.; Grigsby, J.; Zhang, L.; et al. Volumetric brain changes in females with fragile X-associated tremor/ataxia syndrome (FXTAS). Neurology 2007, 69, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Renaud, M.; Perriard, J.; Coudray, S.; Sévin-Allouet, M.; Marcel, C.; Meissner, W.G.; Chanson, J.B.; Collongues, N.; Philippi, N.; Gebus, O.; et al. Relevance of corpus callosum splenium versus middle cerebellar peduncle hyperintensity for FXTAS diagnosis in clinical practice. J. Neurol. 2015, 262, 435–442. [Google Scholar] [CrossRef]
- Wang, J.Y.; Hessl, D.H.; Hagerman, R.J.; Tassone, F.; Rivera, S.M. Age-dependent structural connectivity effects in fragile x premutation. Arch. Neurol. 2012, 69, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.E.; Adams, J.S.; Nguyen, D.V.; Hessl, D.; Brunberg, J.A.; Tassone, F.; Zhang, W.; Koldewyn, K.; Rivera, S.M.; Grigsby, J.; et al. Psychological symptoms correlate with reduced hippocampal volume in fragile X premutation carriers. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 775–785. [Google Scholar] [CrossRef]
- Jäkälä, P.; Hänninen, T.; Ryynänen, M.; Laakso, M.; Partanen, K.; Mannermaa, A.; Soininen, H. Fragile-X: Neuropsychological test performance, CGG triplet repeat lengths, and hippocampal volumes. J. Clin. Investig. 1997, 100, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Hessl, D.; Schneider, A.; Tassone, F.; Hagerman, R.J.; Rivera, S.M. Fragile X-associated tremor/ataxia syndrome: Influence of the FMR1 gene on motor fiber tracts in males with normal and premutation alleles. JAMA Neurol. 2013, 70, 1022–1029. [Google Scholar] [CrossRef]
- Hocking, D.R.; Loesch, D.Z.; Trost, N.; Bui, M.Q.; Hammersley, E.; Francis, D.; Tassone, F.; Storey, E. Total and Regional White Matter Lesions Are Correlated with Motor and Cognitive Impairments in Carriers of the FMR1 Premutation. Front. Neurol. 2019, 10, 832. [Google Scholar] [CrossRef] [PubMed]
- Loesch, D.Z.; Trost, N.; Bui, M.Q.; Hammersley, E.; Lay, S.T.; Annesley, S.J.; Sanislav, O.; Allan, C.Y.; Tassone, F.; Chen, Z.P.; et al. The Spectrum of Neurological and White Matter Changes and Premutation Status Categories of Older Male Carriers of the FMR1 Alleles Are Linked to Genetic (CGG and FMR1 mRNA) and Cellular Stress (AMPK) Markers. Front. Genet. 2018, 9, 531. [Google Scholar] [CrossRef] [PubMed]
- Giulivi, C.; Wang, J.Y.; Hagerman, R.J. Artificial neural network applied to fragile X-associated tremor/ataxia syndrome stage diagnosis based on peripheral mitochondrial bioenergetics and brain imaging outcomes. Sci. Rep. 2022, 12, 21382. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Hessl, D.; Tassone, F.; Kim, K.; Hagerman, R.J.; Rivera, S.M. Interaction between ventricular expansion and structural changes in the corpus callosum and putamen in males with FMR1 normal and premutation alleles. Neurobiol. Aging 2020, 86, 27–38. [Google Scholar] [CrossRef]
- Arpin, D.J.; Mitchell, T.; Archer, D.B.; Burciu, R.G.; Chu, W.T.; Gao, H.; Guttuso, T.; Hess, C.W.; Lai, S.; Malaty, I.A.; et al. Diffusion Magnetic Resonance Imaging Detects Progression in Parkinson’s Disease: A Placebo-Controlled Trial of Rasagiline. Mov. Disord. 2022, 37, 325–333. [Google Scholar] [CrossRef]
- Burciu, R.G.; Chung, J.W.; Shukla, P.; Ofori, E.; Li, H.; McFarland, N.R.; Okun, M.S.; Vaillancourt, D.E. Functional MRI of disease progression in Parkinson disease and atypical parkinsonian syndromes. Neurology 2016, 87, 709–717. [Google Scholar] [CrossRef]
- Yang, J.C.; Rodriguez, A.; Royston, A.; Niu, Y.Q.; Avar, M.; Brill, R.; Simon, C.; Grigsby, J.; Hagerman, R.J.; Olichney, J.M. Memantine Improves Attentional Processes in Fragile X-Associated Tremor/Ataxia Syndrome: Electrophysiological Evidence from a Randomized Controlled Trial. Sci. Rep. 2016, 6, 21719. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Chi, L.; Teichholtz, S.; Schneider, A.; Nanakul, R.; Nowacki, R.; Seritan, A.; Reed, B.; DeCarli, C.; Iragui, V.J.; et al. ERP abnormalities elicited by word repetition in fragile X-associated tremor/ataxia syndrome (FXTAS) and amnestic MCI. Neuropsychologia 2014, 63, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Niu, Y.Q.; Simon, C.; Seritan, A.L.; Chen, L.; Schneider, A.; Moghaddam, S.T.; Hagerman, P.J.; Hagerman, R.J.; Olichney, J.M. Memantine effects on verbal memory in fragile X-associated tremor/ataxia syndrome (FXTAS): A double-blind brain potential study. Neuropsychopharmacology 2014, 39, 2760–2768. [Google Scholar] [CrossRef] [PubMed]
- Seritan, A.L.; Nguyen, D.V.; Mu, Y.; Tassone, F.; Bourgeois, J.A.; Schneider, A.; Cogswell, J.B.; Cook, K.R.; Leehey, M.A.; Grigsby, J.; et al. Memantine for fragile X-associated tremor/ataxia syndrome: A randomized, double-blind, placebo-controlled trial. J. Clin. Psychiatry 2014, 75, 264–271. [Google Scholar] [CrossRef]
- McCormick, E.M.; Arnemann, K.L.; Ito, T.; Hanson, S.J.; Cole, M.W. Latent functional connectivity underlying multiple brain states. Netw. Neurosci. 2022, 6, 570–590. [Google Scholar] [CrossRef]
- Brown, S.S.G.; Basu, S.; Whalley, H.C.; Kind, P.C.; Stanfield, A.C. Age-related functional brain changes in FMR1 premutation carriers. Neuroimage Clin. 2018, 17, 761–767. [Google Scholar] [CrossRef]
- McKinney, W.S.; Bartolotti, J.; Khemani, P.; Wang, J.Y.; Hagerman, R.J.; Mosconi, M.W. Cerebellar-cortical function and connectivity during sensorimotor behavior in aging FMR1 gene premutation carriers. Neuroimage Clin. 2020, 27, 102332. [Google Scholar] [CrossRef]
- McKinney, W.S.; Wang, Z.; Kelly, S.; Khemani, P.; Lui, S.; White, S.P.; Mosconi, M.W. Precision Sensorimotor Control in Aging FMR1 Gene Premutation Carriers. Front. Integr. Neurosci. 2019, 13, 56. [Google Scholar] [CrossRef]
- Park, S.H.; Wang, Z.; McKinney, W.; Khemani, P.; Lui, S.; Christou, E.A.; Mosconi, M.W. Functional motor control deficits in older FMR1 premutation carriers. Exp. Brain Res. 2019, 237, 2269–2278. [Google Scholar] [CrossRef]
- Hessl, D.; Rivera, S.; Koldewyn, K.; Cordeiro, L.; Adams, J.; Tassone, F.; Hagerman, P.J.; Hagerman, R.J. Amygdala dysfunction in men with the fragile X premutation. Brain 2007, 130 Pt 2, 404–416. [Google Scholar] [CrossRef]
- Tassone, F.; Hagerman, R.J.; Garcia-Arocena, D.; Khandjian, E.W.; Greco, C.M.; Hagerman, P.J. Intranuclear inclusions in neural cells with premutation alleles in fragile X associated tremor/ataxia syndrome. J. Med. Genet. 2004, 41, e43. [Google Scholar] [CrossRef] [PubMed]
- Ariza, J.; Rogers, H.; Monterrubio, A.; Reyes-Miranda, A.; Hagerman, P.J.; Martínez-Cerdeño, V. A Majority of FXTAS Cases Present with Intranuclear Inclusions within Purkinje Cells. Cerebellum 2016, 15, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Buijsen, R.A.; Sellier, C.; Severijnen, L.A.; Oulad-Abdelghani, M.; Verhagen, R.F.; Berman, R.F.; Charlet-Berguerand, N.; Willemsen, R.; Hukema, R.K. FMRpolyG-positive inclusions in CNS and non-CNS organs of a fragile X premutation carrier with fragile X-associated tremor/ataxia syndrome. Acta Neuropathol. Commun. 2014, 2, 162. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.; Ariza, J.; Monterrubio, A.; Hagerman, P.; Martínez-Cerdeño, V. Cerebellar Mild Iron Accumulation in a Subset of FMR1 Premutation Carriers with FXTAS. Cerebellum 2016, 15, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Ariza, J.; Rogers, H.; Hartvigsen, A.; Snell, M.; Dill, M.; Judd, D.; Hagerman, P.; Martínez-Cerdeño, V. Iron accumulation and dysregulation in the putamen in fragile X-associated tremor/ataxia syndrome. Mov. Disord. 2017, 32, 585–591. [Google Scholar] [CrossRef]
- Wang, J.Y.; Sonico, G.J.; Salcedo-Arellano, M.J.; Hagerman, R.J.; Martínez-Cerdeño, V. A postmortem MRI study of cerebrovascular disease and iron content at end-stage of fragile X-associated tremor/ataxia syndrome. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Dufour, B.D.; Amina, S.; Martinez-Cerdeno, V. FXTAS presents with upregulation of the cytokines IL12 and TNFα. Park. Relat. Disord. 2021, 82, 117–120. [Google Scholar] [CrossRef]
- Niu, Y.Q.; Yang, J.C.; Hall, D.A.; Leehey, M.A.; Tassone, F.; Olichney, J.M.; Hagerman, R.J.; Zhang, L. Parkinsonism in fragile X-associated tremor/ataxia syndrome (FXTAS): Revisited. Park. Relat. Disord. 2014, 20, 456–459. [Google Scholar] [CrossRef]
- Martínez-Cerdeño, V.; Lechpammer, M.; Hagerman, P.J.; Hagerman, R. Two FMR1 premutation cases without nuclear inclusions. Mov. Disord. 2017, 32, 1328–1329. [Google Scholar] [CrossRef]
- Paucar, M.; Nennesmo, I. Svenningsson, Pathological Study of a FMR1 Premutation Carrier with Progressive Supranuclear Palsy. Front. Genet. 2018, 9, 317. [Google Scholar] [CrossRef]
- Sacino, A.N.; Prokop, S.; Walsh, M.A.; Adamson, J.; Subramony, S.H.; Krans, A.; Todd, P.K.; Giasson, B.I.; Yachnis, A.T. Fragile X-associated tremor ataxia syndrome with co-occurrent progressive supranuclear palsy-like neuropathology. Acta Neuropathol. Commun. 2019, 7, 158. [Google Scholar] [CrossRef] [PubMed]
- Salcedo-Arellano, M.J.; Hagerman, R.J. Recent research in fragile X-associated tremor/ataxia syndrome. Curr. Opin. Neurobiol. 2022, 72, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.; Hagerman, P. Fragile X-associated tremor/ataxia syndrome: Pathophysiology and management. Curr. Opin. Neurol. 2021, 34, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; Berry-Kravis, E.; Hagerman, R.J.; Hagerman, P.J.; Rice, C.D.; Leehey, M.A. Symptomatic treatment in the fragile X-associated tremor/ataxia syndrome. Mov. Disord. 2006, 21, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Hall, D.A.; O’Keefe, J.A. Fragile x-associated tremor ataxia syndrome: The expanding clinical picture, pathophysiology, epidemiology, and update on treatment. Tremor Other Hyperkinetic Mov. 2012, 2, tre-02-56-352-1. [Google Scholar] [CrossRef]
- Hall, D.; Todorova-Koteva, K.; Pandya, S.; Bernard, B.; Ouyang, B.; Walsh, M.; Pounardjian, T.; Deburghraeve, C.; Zhou, L.; Losh, M.; et al. Neurological and endocrine phenotypes of fragile X carrier women. Clin. Genet. 2016, 89, 60–67. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Hall, D.A.; Coffey, S.; Leehey, M.; Bourgeois, J.; Gould, J.; Zhang, L.; Seritan, A.; Berry-Kravis, E.; Olichney, J.; et al. Treatment of fragile X-associated tremor ataxia syndrome (FXTAS) and related neurological problems. Clin. Interv. Aging 2008, 3, 251–262. [Google Scholar] [CrossRef]
- Wang, J.Y.; Trivedi, A.M.; Carrillo, N.R.; Yang, J.; Schneider, A.; Giulivi, C.; Adams, P.; Tassone, F.; Kim, K.; Rivera, S.M.; et al. Open-Label Allopregnanolone Treatment of Men with Fragile X-Associated Tremor/Ataxia Syndrome. Neurotherapeutics 2017, 14, 1073–1083. [Google Scholar] [CrossRef]
- Budimirovic, D.B. Can a Neurosteroid Ameliorate Fragile X-Associated Tremor/Ataxia Syndrome? Neurotherapeutics 2017, 14, 1070–1072. [Google Scholar] [CrossRef]
- Hall, D.A.; Robertson, E.E.; Leehey, M.; McAsey, A.; Ouyang, B.; Berry-Kravis, E.; O’Keefe, J.A. Open-label pilot clinical trial of citicoline for fragile X-associated tremor/ataxia syndrome (FXTAS). PLoS ONE 2020, 15, e0225191. [Google Scholar] [CrossRef]
- Santos, E.; Clark, K.; Maridith, B.; Biag, H.; Tang, S.J.; Kim, K.; Ponzini, M.; Schneider, A.; Montanaro, F.; Gipe, J.T.E.; et al. Open label trial of Sulforaphane in FMR1 Premutation Carriers with FXTAS. Cells, 2023; under review. [Google Scholar]
- Hall, D.A.; Berry-Kravis, E. Fragile X syndrome and fragile X-associated tremor ataxia syndrome. Handb. Clin. Neurol. 2018, 147, 377–391. [Google Scholar] [PubMed]
- Artusi, C.A.; Farooqi, A.; Romagnolo, A.; Marsili, L.; Balestrino, R.; Sokol, L.L.; Wang, L.L.; Zibetti, M.; Duker, A.P.; Mandybur, G.T.; et al. Deep brain stimulation in uncommon tremor disorders: Indications, targets, and programming. J. Neurol. 2018, 265, 2473–2493. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Wilmot, G.; Kuo, S.H.; Perlman, S.; Greenstein, P.E.; Ying, S.H.; Ashizawa, T.; Subramony, S.H.; Schmahmann, J.D.; Figueroa, K.P.; et al. Comprehensive Systematic Review Summary: Treatment of Cerebellar Motor Dysfunction and Ataxia: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 2018, 90, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Reisberg, B.; Doody, R.; Stöffler, A.; Schmitt, F.; Ferris, S.; Möbius, H.J. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 2003, 348, 1333–1341. [Google Scholar] [CrossRef]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Stamatakis, E.; Lee, I.M.; Bennie, J.; Freeston, J.; Hamer, M.; O’Donovan, G.; Ding, D.; Bauman, A.; Mavros, Y. Does Strength-Promoting Exercise Confer Unique Health Benefits? A Pooled Analysis of Data on 11 Population Cohorts with All-Cause, Cancer, and Cardiovascular Mortality Endpoints. Am. J. Epidemiol. 2018, 187, 1102–1112. [Google Scholar] [CrossRef]
- Northey, J.M.; Cherbuin, N.; Pumpa, K.L.; Smee, D.J.; Rattray, B. Exercise interventions for cognitive function in adults older than 50: A systematic review with meta-analysis. Br. J. Sports Med. 2018, 52, 154–160. [Google Scholar] [CrossRef]
- Rasmussen, P.; Brassard, P.; Adser, H.; Pedersen, M.V.; Leick, L.; Hart, E.; Secher, N.H.; Pedersen, B.K.; Pilegaard, H. Evidence for a release of brain-derived neurotrophic factor from the brain during exercise. Exp. Physiol. 2009, 94, 1062–1069. [Google Scholar] [CrossRef]
- Herold, F.; Törpel, A.; Schega, L.; Müller, N.G. Functional and/or structural brain changes in response to resistance exercises and resistance training lead to cognitive improvements—A systematic review. Eur. Rev. Aging Phys. Act. 2019, 16, 10. [Google Scholar] [CrossRef]
- Wang, P.; Song, M.; Eliassen, A.H.; Wang, M.; Fung, T.T.; Clinton, S.K.; Rimm, E.B.; Hu, F.B.; Willett, W.C.; Tabung, F.K.; et al. Optimal dietary patterns for prevention of chronic disease. Nat. Med. 2023, 29, 719–728. [Google Scholar] [CrossRef]
- Snetselaar, L.G.; de Jesus, J.M.; DeSilva, D.M.; Stoody, E.E. Dietary Guidelines for Americans, 2020–2025: Understanding the Scientific Process, Guidelines, and Key Recommendations. Nutr. Today 2021, 56, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Rapamycin for longevity: Opinion article. Aging 2019, 11, 8048–8067. [Google Scholar] [CrossRef] [PubMed]
- Mannick, J.B.; Lamming, D.W. Targeting the biology of aging with mTOR inhibitors. Nat. Aging 2023, 3, 642–660. [Google Scholar] [CrossRef]
- Moses, J.W.; Leon, M.B.; Popma, J.J.; Fitzgerald, P.J.; Holmes, D.R.; O’Shaughnessy, C.; Caputo, R.P.; Kereiakes, D.J.; Williams, D.O.; Teirstein, P.S.; et al. Sirolimus-eluting stents versus standard stents in patients with stenosis in a native coronary artery. N. Engl. J. Med. 2003, 349, 1315–1323. [Google Scholar] [CrossRef] [PubMed]
- Sasongko, T.H.; Ismail, N.F.; Zabidi-Hussin, Z. Rapamycin and rapalogs for tuberous sclerosis complex. Cochrane Database Syst. Rev. 2016, 7, Cd011272. [Google Scholar] [CrossRef]
- Arriola Apelo, S.I.; Neuman, J.C.; Baar, E.L.; Syed, F.A.; Cummings, N.E.; Brar, H.K.; Pumper, C.P.; Kimple, M.E.; Lamming, D.W. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 2016, 15, 28–38. [Google Scholar] [CrossRef]
- Lin, Y.; Tang, C.; He, H.; Duan, R. Activation of mTOR ameliorates fragile X premutation rCGG repeat-mediated neurodegeneration. PLoS ONE 2013, 8, e62572. [Google Scholar] [CrossRef]
- Lamming, D.W.; Ye, L.; Sabatini, D.M.; Baur, J.A. Rapalogs and mTOR inhibitors as anti-aging therapeutics. J. Clin. Investig. 2013, 123, 980–989. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef]
- Xu, K.; Li, Y.; Allen, E.G.; Jin, P. Therapeutic Development for CGG Repeat Expansion-Associated Neurodegeneration. Front. Cell Neurosci. 2021, 15, 655568. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Zarnescu, D.C.; Zhang, F.; Pearson, C.E.; Lucchesi, J.C.; Moses, K.; Warren, S.T. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron 2003, 39, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Alvarado, C.X.; Makarious, M.B.; Vitale, D.; Koretsky, M.J.; Bandres-Ciga, S.; Iwaki, H.; Levine, K.; Singleton, A.; Faghri, F.; Nalls, M.A.; et al. omicSynth: An Open Multi-omic Community Resource for Identifying Druggable Targets across Neurodegenerative Diseases. medRxiv 2023. medRxiv:06.23288266. [Google Scholar]
- Gabis, L.V.; Hochberg, O.; Leon Attia, O.; Banet-Levi, Y.; Topf, D.; Shefer, S. Prolonged Time Lag to Final Diagnosis of Fragile X Syndrome. J. Pediatr. 2018, 193, 217–221.e1. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Raspa, M.; Bishop, E.; Holiday, D. No change in the age of diagnosis for fragile x syndrome: Findings from a national parent survey. Pediatrics 2009, 124, 527–533. [Google Scholar] [CrossRef]
- McConkie-Rosell, A.; Abrams, L.; Finucane, B.; Cronister, A.; Gane, L.W.; Coffey, S.M.; Sherman, S.; Nelson, L.M.; Berry-Kravis, E.; Hessl, D.; et al. Recommendations from multi-disciplinary focus groups on cascade testing and genetic counseling for fragile X-associated disorders. J. Genet. Couns. 2007, 16, 593–606. [Google Scholar] [CrossRef]
- Raspa, M.; Edwards, A.; Wheeler, A.C.; Bishop, E.; Bailey, D.B., Jr. Family Communication and Cascade Testing for Fragile X Syndrome. J. Genet. Couns. 2016, 25, 1075–1084. [Google Scholar] [CrossRef]
- Berry-Kravis, E.; Abrams, L.; Coffey, S.M.; Hall, D.A.; Greco, C.; Gane, L.W.; Grigsby, J.; Bourgeois, J.A.; Finucane, B.; Jacquemont, S.; et al. Fragile X-associated tremor/ataxia syndrome: Clinical features, genetics, and testing guidelines. Mov. Disord. 2007, 22, 2018–2030, quiz 2140. [Google Scholar] [CrossRef]
- Acharya, K.; Schindler, A. Developmental and behavioral pediatricians’ attitudes toward screening for fragile X. Am. J. Intellect. Dev. Disabil. 2013, 118, 284–293. [Google Scholar] [CrossRef]
- Andermann, A.; Blancquaert, I.; Beauchamp, S.; Déry, V. Revisiting Wilson and Jungner in the genomic age: A review of screening criteria over the past 40 years. Bull. World Health Organ. 2008, 86, 317–319. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Berry-Kravis, E.; Gane, L.W.; Guarda, S.; Hagerman, R.; Powell, C.M.; Tassone, F.; Wheeler, A. Fragile X Newborn Screening: Lessons Learned from a Multisite Screening Study. Pediatrics 2017, 139 (Suppl. S3), S216–S225. [Google Scholar] [CrossRef] [PubMed]
- Christie, L.; Wotton, T.; Bennetts, B.; Wiley, V.; Wilcken, B.; Rogers, C.; Boyle, J.; Turner, C.; Hansen, J.; Hunter, M.; et al. Maternal attitudes to newborn screening for fragile X syndrome. Am. J. Med. Genet. A 2013, 161, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, A.C.; Gwaltney, A.; Raspa, M.; Okoniewski, K.C.; Berry-Kravis, E.; Botteron, K.N.; Budimirovic, D.; Hazlett, H.C.; Hessl, D.; Losh, M.; et al. Emergence of Developmental Delay in Infants and Toddlers with an FMR1 Mutation. Pediatrics 2021, 147, e2020011528. [Google Scholar] [CrossRef] [PubMed]
- Tassone, F. Newborn screening for fragile X syndrome. JAMA Neurol. 2014, 71, 355–359. [Google Scholar] [CrossRef]
- Lee, S.; Taylor, J.L.; Redmond, C.; Hadd, A.G.; Kemppainen, J.A.; Haynes, B.C.; Shone, S.; Bailey, D.B., Jr.; Latham, G.J. Validation of Fragile X Screening in the Newborn Population Using a Fit-for-Purpose FMR1 PCR Assay System. J. Mol. Diagn. 2020, 22, 346–354. [Google Scholar] [CrossRef]
- Tassone, F. Advanced technologies for the molecular diagnosis of fragile X syndrome. Expert. Rev. Mol. Diagn. 2015, 15, 1465–1473. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Wheeler, A.; Berry-Kravis, E.; Hagerman, R.; Tassone, F.; Powell, C.M.; Roche, M.; Gane, L.W.; Sideris, J. Maternal Consequences of the Detection of Fragile X Carriers in Newborn Screening. Pediatrics 2015, 136, e433–e440. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Gehtland, L.M.; Lewis, M.A.; Peay, H.; Raspa, M.; Shone, S.M.; Taylor, J.L.; Wheeler, A.C.; Cotten, M.; King, N.M.P.; et al. Early Check: Translational science at the intersection of public health and newborn screening. BMC Pediatr. 2019, 19, 238. [Google Scholar] [CrossRef]
- Gallego, P.K.; Burris, J.L.; Rivera, S.M. Visual motion processing deficits in infants with the fragile X premutation. J. Neurodev. Disord. 2014, 6, 29. [Google Scholar] [CrossRef]
- Wheeler, A.C.; Sideris, J.; Hagerman, R.; Berry-Kravis, E.; Tassone, F.; Bailey, D.B., Jr. Developmental profiles of infants with an FMR1 premutation. J. Neurodev. Disord. 2016, 8, 40. [Google Scholar] [CrossRef]
- Bailey, D.B., Jr.; Bishop, E.; Raspa, M.; Skinner, D. Caregiver opinions about fragile X population screening. Genet. Med. 2012, 14, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Archibald, A.D.; Hickerton, C.L.; Wake, S.A.; Jaques, A.M.; Cohen, J.; Metcalfe, S.A. “It gives them more options”: Preferences for preconception genetic carrier screening for fragile X syndrome in primary healthcare. J. Community Genet. 2016, 7, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, S.A.; Martyn, M.; Ames, A.; Anderson, V.; Archibald, A.D.; Couns, G.D.G.; Carter, R.; Cohen, J.; Cotter, M.; GenCouns, M.; et al. Informed decision making and psychosocial outcomes in pregnant and nonpregnant women offered population fragile X carrier screening. Genet. Med. 2017, 19, 1346–1355. [Google Scholar] [CrossRef] [PubMed]
- Xi, H.; Xie, W.; Chen, J.; Tang, W.; Deng, X.; Li, H.; Peng, Y.; Wang, D.; Yang, S.; Zhang, Y.; et al. Implementation of fragile X syndrome carrier screening during prenatal diagnosis: A pilot study at a single center. Mol. Genet. Genom. Med. 2021, 9, e1711. [Google Scholar] [CrossRef]
- Finucane, B.; Abrams, L.; Cronister, A.; Archibald, A.D.; Bennett, R.L.; McConkie-Rosell, A. Genetic counseling and testing for FMR1 gene mutations: Practice guidelines of the national society of genetic counselors. J. Genet. Couns. 2012, 21, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Martyn, M.; Anderson, V.; Archibald, A.; Carter, R.; Cohen, J.; Delatycki, M.; Donath, S.; Emery, J.; Halliday, J.; Hill, M.; et al. Offering fragile X syndrome carrier screening: A prospective mixed-methods observational study comparing carrier screening of pregnant and non-pregnant women in the general population. BMJ Open 2013, 3, e003660. [Google Scholar] [CrossRef]
- Miller, D.T.; Adam, M.P.; Aradhya, S.; Biesecker, L.G.; Brothman, A.R.; Carter, N.P.; Church, D.M.; Crolla, J.A.; Eichler, E.E.; Epstein, C.J.; et al. Consensus statement: Chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am. J. Hum. Genet. 2010, 86, 749–764. [Google Scholar] [CrossRef]
- Spector, E.; Behlmann, A.; Kronquist, K.; Rose, N.C.; Lyon, E.; Reddi, H.V. Laboratory testing for fragile X, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 799–812. [Google Scholar] [CrossRef]
- Biancalana, V.; Glaeser, D.; McQuaid, S.; Steinbach, P. EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders. Eur. J. Hum. Genet. 2015, 23, 417–425. [Google Scholar] [CrossRef]
- Hayward, B.; Loutaev, I.; Ding, X.; Nolin, S.L.; Thurm, A.; Usdin, K.; Smith, C.B. Fragile X syndrome in a male with methylated premutation alleles and no detectable methylated full mutation alleles. Am. J. Med. Genet. A 2019, 179, 2132–2137. [Google Scholar] [CrossRef]
- Tabolacci, E.; Pomponi, M.G.; Remondini, L.; Pietrobono, R.; Nobile, V.; Pennacchio, G.; Gurrieri, F.; Neri, G.; Genuardi, M.; Chiurazzi, P. Methylated premutation of the FMR1 gene in three sisters: Correlating CGG expansion and epigenetic inactivation. Eur. J. Hum. Genet. 2020, 28, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Jiraanont, P.; Kumar, M.; Tang, H.T.; Espinal, G.; Hagerman, P.J.; Hagerman, R.J.; Chutabhakdikul, N.; Tassone, F. Size and methylation mosaicism in males with Fragile X syndrome. Expert. Rev. Mol. Diagn. 2017, 17, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Archibald, A.D.; Smith, M.J.; Burgess, T.; Scarff, K.L.; Elliott, J.; Hunt, C.E.; McDonald, Z.; Barns-Jenkins, C.; Holt, C.; Sandoval, K.; et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: Outcomes of 12,000 tests. Genet. Med. 2018, 20, 513–523. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tassone, F.; Protic, D.; Allen, E.G.; Archibald, A.D.; Baud, A.; Brown, T.W.; Budimirovic, D.B.; Cohen, J.; Dufour, B.; Eiges, R.; et al. Insight and Recommendations for Fragile X-Premutation-Associated Conditions from the Fifth International Conference on FMR1 Premutation. Cells 2023, 12, 2330. https://doi.org/10.3390/cells12182330
Tassone F, Protic D, Allen EG, Archibald AD, Baud A, Brown TW, Budimirovic DB, Cohen J, Dufour B, Eiges R, et al. Insight and Recommendations for Fragile X-Premutation-Associated Conditions from the Fifth International Conference on FMR1 Premutation. Cells. 2023; 12(18):2330. https://doi.org/10.3390/cells12182330
Chicago/Turabian StyleTassone, Flora, Dragana Protic, Emily Graves Allen, Alison D. Archibald, Anna Baud, Ted W. Brown, Dejan B. Budimirovic, Jonathan Cohen, Brett Dufour, Rachel Eiges, and et al. 2023. "Insight and Recommendations for Fragile X-Premutation-Associated Conditions from the Fifth International Conference on FMR1 Premutation" Cells 12, no. 18: 2330. https://doi.org/10.3390/cells12182330
APA StyleTassone, F., Protic, D., Allen, E. G., Archibald, A. D., Baud, A., Brown, T. W., Budimirovic, D. B., Cohen, J., Dufour, B., Eiges, R., Elvassore, N., Gabis, L. V., Grudzien, S. J., Hall, D. A., Hessl, D., Hogan, A., Hunter, J. E., Jin, P., Jiraanont, P., ... Hagerman, R. J. (2023). Insight and Recommendations for Fragile X-Premutation-Associated Conditions from the Fifth International Conference on FMR1 Premutation. Cells, 12(18), 2330. https://doi.org/10.3390/cells12182330