

Novel Variant in CEP250 Causes Protein Mislocalization and Leads to Nonsyndromic Autosomal Recessive Type of Progressive Hearing Loss

 , , and
, , and
Abstract
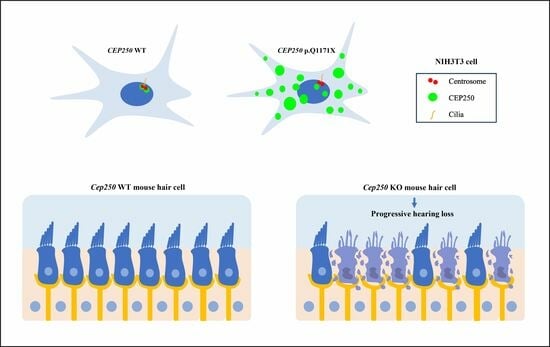
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
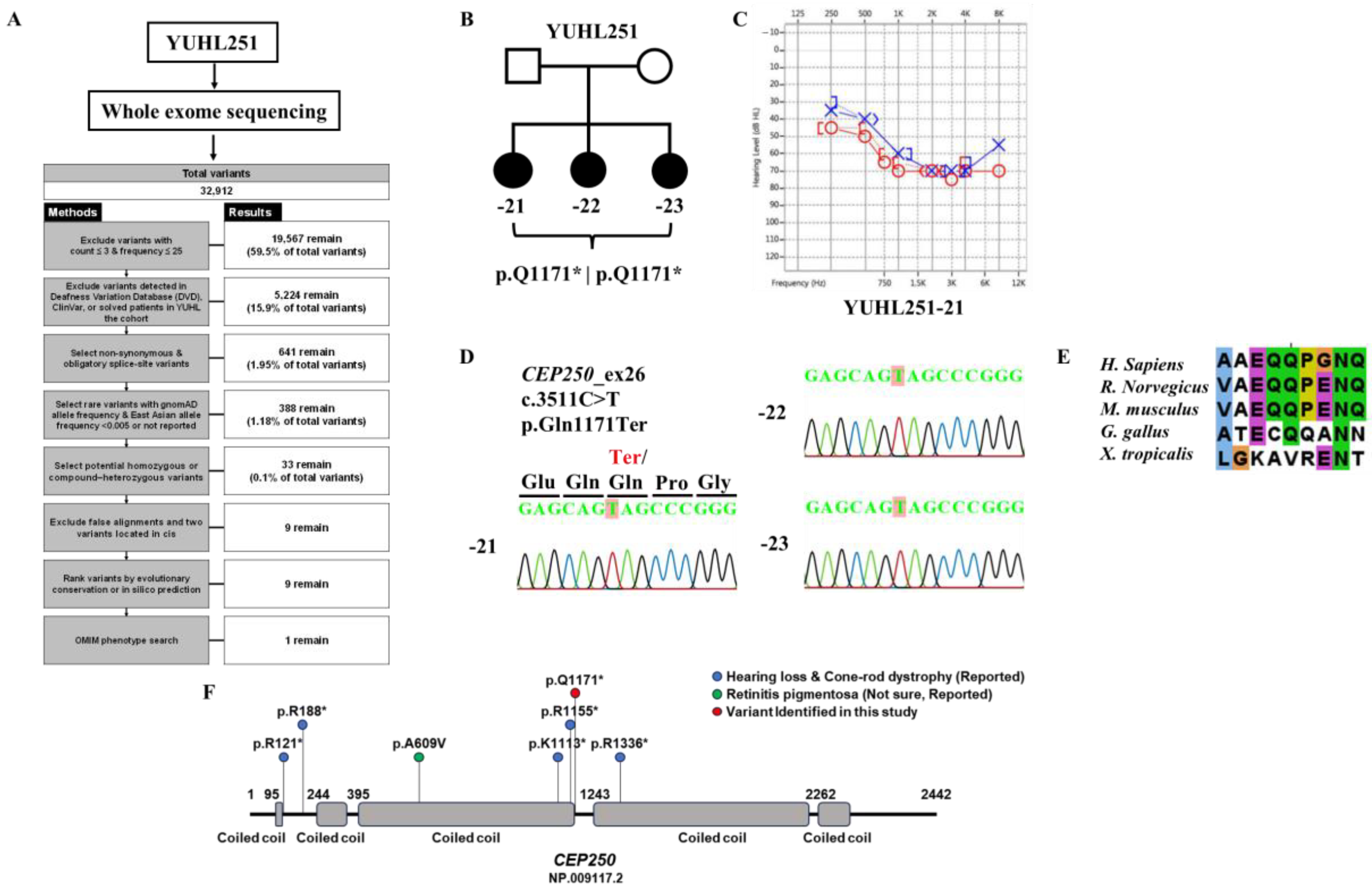
2.1. Patients and Diagnosis of Genetic Hearing Loss
2.2. WES and Analyses
2.3. pRK5-Myc-CEP250 Recombinant Vector Construction and Mutagenesis
2.4. Cell Culture and Transfection
2.5. Immunoblotting
2.6. Immunocytochemistry
2.7. Cep250 Knockout Mouse
2.8. Inner Ear Immunoblotting
2.9. Inner Ear Immunohistochemistry
2.10. Auditory Brainstem Response Test
2.11. Statistical Analysis
3. Results
3.1. Identification of CEP250 as a Candidate Deafness Gene for High-Frequency Sensorineural Hearing Loss
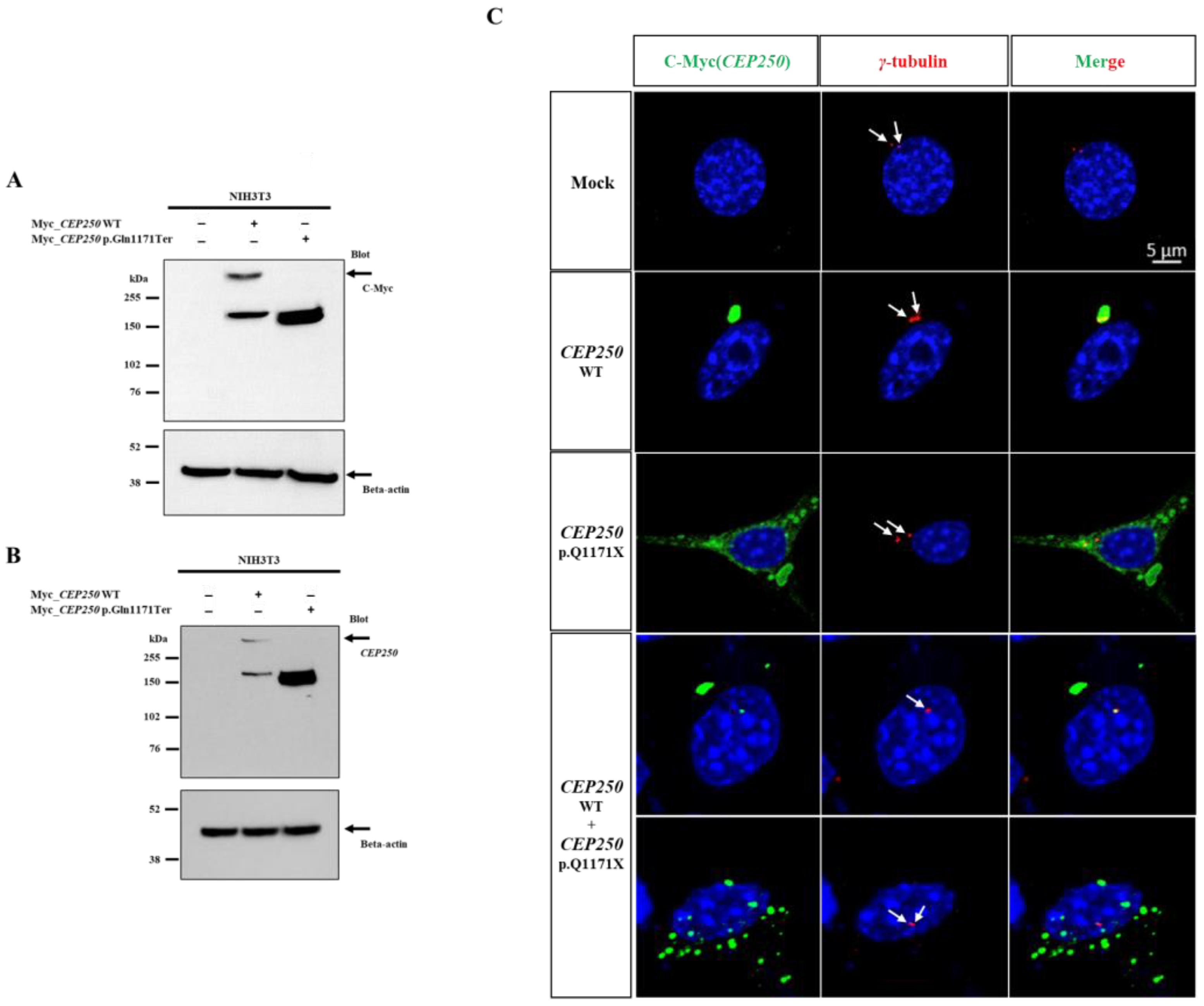
3.2. Expression of CEP250 WT and p.Gln1171Ter Variant in NIH3T3 Cells
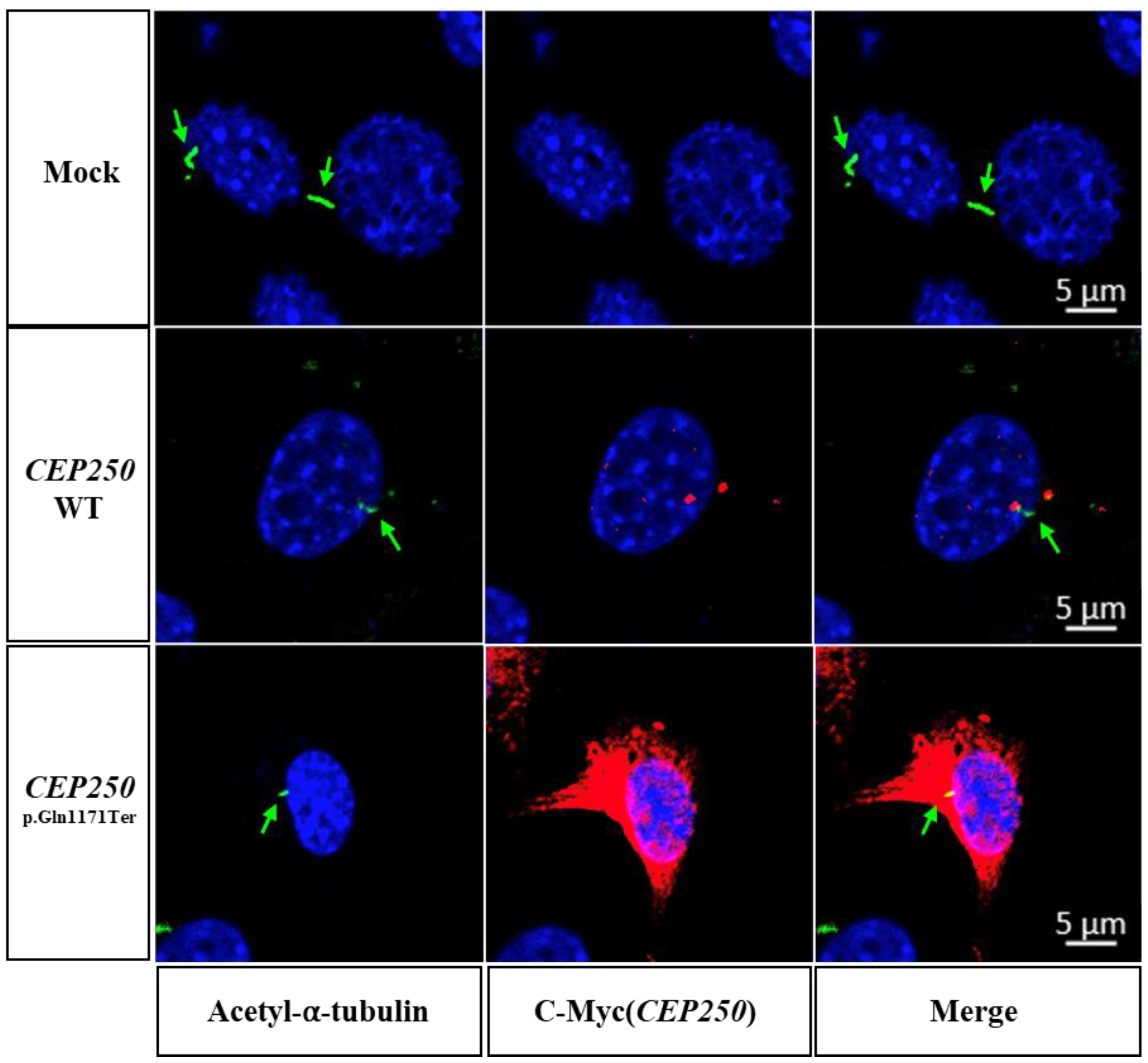
3.3. Effect of CEP250 p.Gln1171Ter Variant on the Cilia Development in NIH3T3 Cells
3.4. Cep250 Is Expressed in the Cochlear Hair Cells and Spiral Ganglion
3.5. Hearing Loss in the Cep250 Knockout Mice
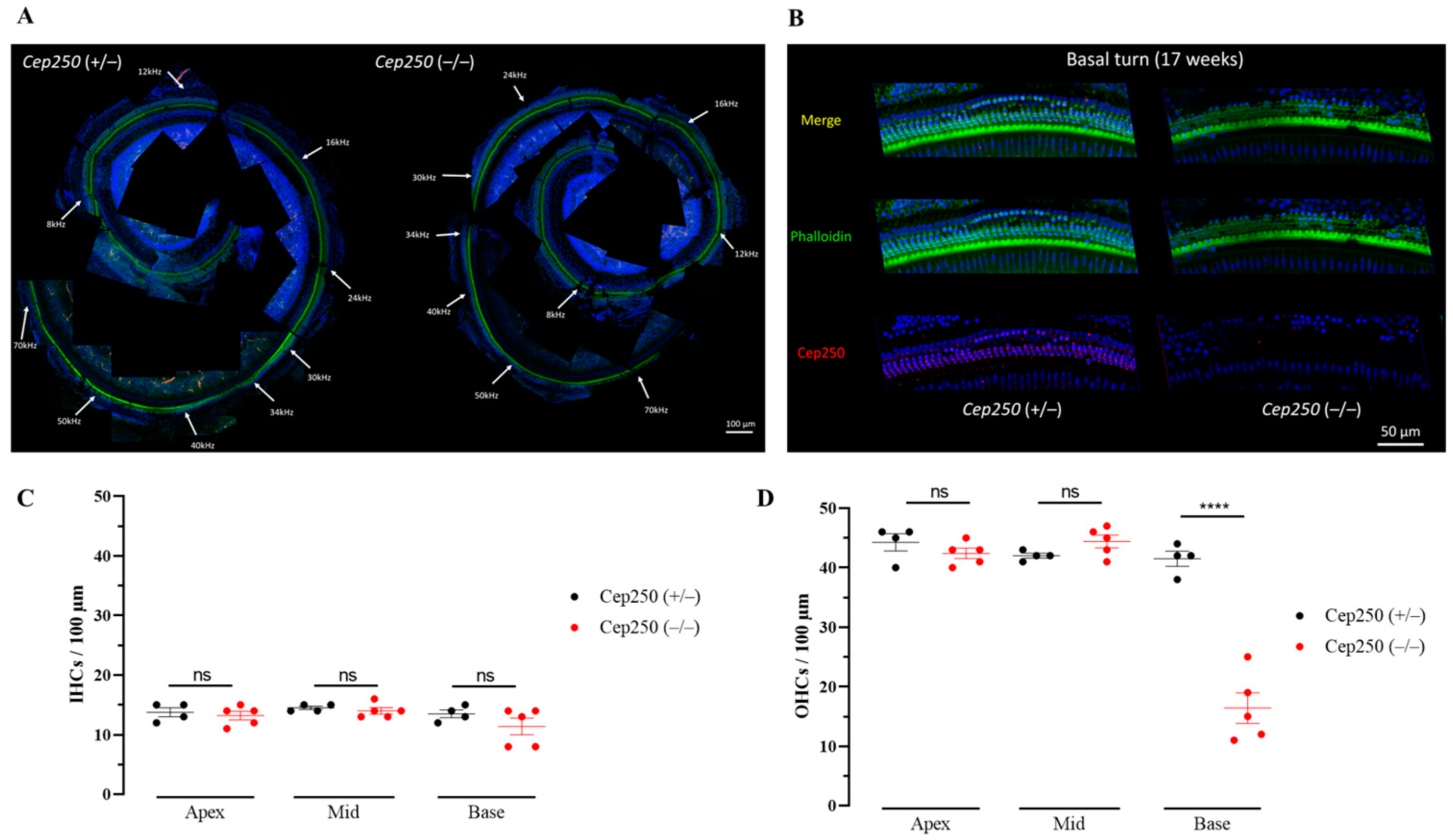
3.6. Hair Cell Degeneration in the Cep250 Knockout Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fortnum, H.M.; Summerfield, A.Q.; Marshall, D.H.; Davis, A.C.; Bamford, J.M. Prevalence of permanent childhood hearing impairment in the United Kingdom and implications for universal neonatal hearing screening: Questionnaire based ascertainment study. BMJ 2001, 323, 536–540. [Google Scholar] [CrossRef]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Function and expression pattern of nonsyndromic deafness genes. Curr. Mol. Med. 2009, 9, 546–564. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Horta, O.; Duman, D.; Foster, J., 2nd; Sırmacı, A.; Gonzalez, M.; Mahdieh, N.; Fotouhi, N.; Bonyadi, M.; Cengiz, F.B.; Menendez, I.; et al. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS ONE 2012, 7, e50628. [Google Scholar] [CrossRef]
- Kim, N.K.; Kim, A.R.; Park, K.T.; Kim, S.Y.; Kim, M.Y.; Nam, J.Y.; Woo, S.J.; Oh, S.H.; Park, W.Y.; Choi, B.Y. Whole-exome sequencing reveals diverse modes of inheritance in sporadic mild to moderate sensorineural hearing loss in a pediatric population. Genet. Med. 2015, 17, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.J.; Botuyan, M.V.; Wu, Y.; Ward, C.J.; Nicholson, G.A.; Hammans, S.; Hojo, K.; Yamanishi, H.; Karpf, A.R.; Wallace, D.C.; et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat. Genet. 2011, 43, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Shahin, H.; Elkan-Miller, T.; Lee, M.K.; Thornton, A.M.; Roeb, W.; Abu Rayyan, A.; Loulus, S.; Avraham, K.B.; King, M.C.; et al. Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am. J. Hum. Genet. 2010, 87, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. CEP proteins: The knights of centrosome dynasty. Protoplasma 2013, 250, 965–983. [Google Scholar] [CrossRef]
- Badano, J.L.; Teslovich, T.M.; Katsanis, N. The centrosome in human genetic disease. Nat. Rev. Genet. 2005, 6, 194–205. [Google Scholar] [CrossRef]
- Fry, A.M.; Mayor, T.; Meraldi, P.; Stierhof, Y.D.; Tanaka, K.; Nigg, E.A. C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J. Cell Biol. 1998, 141, 1563–1574. [Google Scholar] [CrossRef]
- de Castro-Miró, M.; Tonda, R.; Escudero-Ferruz, P.; Andrés, R.; Mayor-Lorenzo, A.; Castro, J.; Ciccioli, M.; Hidalgo, D.A.; Rodríguez-Ezcurra, J.J.; Farrando, J.; et al. Novel Candidate Genes and a Wide Spectrum of Structural and Point Mutations Responsible for Inherited Retinal Dystrophies Revealed by Exome Sequencing. PLoS ONE 2016, 11, e0168966. [Google Scholar] [CrossRef]
- Khateb, S.; Zelinger, L.; Mizrahi-Meissonnier, L.; Ayuso, C.; Koenekoop, R.K.; Laxer, U.; Gross, M.; Banin, E.; Sharon, D. A homozygous nonsense CEP250 mutation combined with a heterozygous nonsense C2orf71 mutation is associated with atypical Usher syndrome. J. Med. Genet. 2014, 51, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Fuster-Garcia, C.; Garcia-Garcia, G.; Jaijo, T.; Fornes, N.; Ayuso, C.; Fernandez-Burriel, M.; Sanchez-De la Morena, A.; Aller, E.; Millan, J.M. High-throughput sequencing for the molecular diagnosis of Usher syndrome reveals 42 novel mutations and consolidates CEP250 as Usher-like disease causative. Sci. Rep. 2018, 8, 17113. [Google Scholar] [CrossRef] [PubMed]
- Igelman, A.D.; Ku, C.; da Palma, M.M.; Georgiou, M.; Schiff, E.R.; Lam, B.L.; Sankila, E.M.; Ahn, J.; Pyers, L.; Vincent, A.; et al. Expanding the clinical phenotype in patients with disease causing variants associated with atypical Usher syndrome. Ophthalmic Genet. 2021, 42, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Kubota, D.; Gocho, K.; Kikuchi, S.; Akeo, K.; Miura, M.; Yamaki, K.; Takahashi, H.; Kameya, S. CEP250 mutations associated with mild cone-rod dystrophy and sensorineural hearing loss in a Japanese family. Ophthalmic Genet. 2018, 39, 500–507. [Google Scholar] [CrossRef]
- Song, M.H.; Jung, J.; Rim, J.H.; Choi, H.J.; Lee, H.J.; Noh, B.; Lee, J.S.; Gee, H.Y.; Choi, J.Y. Genetic Inheritance of Late-Onset, Down-Sloping Hearing Loss and Its Implications for Auditory Rehabilitation. Ear. Hear. 2020, 41, 114–124. [Google Scholar] [CrossRef]
- Oh, K.S.; Walls, D.; Joo, S.Y.; Kim, J.A.; Yoo, J.E.; Koh, Y.I.; Kim, D.H.; Rim, J.H.; Choi, H.J.; Kim, H.Y.; et al. COCH-related autosomal dominant nonsyndromic hearing loss: A phenotype-genotype study. Hum. Genet. 2022, 141, 889–901. [Google Scholar] [CrossRef]
- Jung, J.; Choi, H.B.; Koh, Y.I.; Rim, J.H.; Choi, H.J.; Kim, S.H.; Lee, J.H.; An, J.; Kim, A.; Lee, J.S.; et al. Whole-exome sequencing identifies two novel mutations in KCNQ4 in individuals with nonsyndromic hearing loss. Sci. Rep. 2018, 8, 16659. [Google Scholar] [CrossRef]
- Oza, A.M.; DiStefano, M.T.; Hemphill, S.E.; Cushman, B.J.; Grant, A.R.; Siegert, R.K.; Shen, J.; Chapin, A.; Boczek, N.J.; Schimmenti, L.A. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 2018, 39, 1593–1613. [Google Scholar] [CrossRef]
- Jung, J.; Kim, J.; Roh, S.H.; Jun, I.; Sampson, R.D.; Gee, H.Y.; Choi, J.Y.; Lee, M.G. The HSP70 co-chaperone DNAJC14 targets misfolded pendrin for unconventional protein secretion. Nat. Commun. 2016, 7, 11386. [Google Scholar] [CrossRef]
- Jung, J.; Yoo, J.E.; Choe, Y.H.; Park, S.C.; Lee, H.J.; Lee, H.J.; Noh, B.; Kim, S.H.; Kang, G.Y.; Lee, K.M.; et al. Cleaved Cochlin Sequesters Pseudomonas aeruginosa and Activates Innate Immunity in the Inner Ear. Cell Host. Microbe. 2019, 25, 513–525.e516. [Google Scholar] [CrossRef]
- Koh, Y.I.; Oh, K.S.; Kim, J.A.; Noh, B.; Choi, H.J.; Joo, S.Y.; Rim, J.H.; Kim, H.Y.; Kim, D.Y.; Yu, S.; et al. OSBPL2 mutations impair autophagy and lead to hearing loss, potentially remedied by rapamycin. Autophagy 2022, 18, 2593–2614. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.M.; Cai, M.; Hoyer-Fender, S. Heterogeneity of the NIH3T3 Fibroblast Cell Line. Cells 2022, 11, 2677. [Google Scholar] [CrossRef] [PubMed]
- Orvis, J.; Gottfried, B.; Kancherla, J.; Adkins, R.S.; Song, Y.; Dror, A.A.; Olley, D.; Rose, K.; Chrysostomou, E.; Kelly, M.C. gEAR: Gene Expression Analysis Resource portal for community-driven, multi-omic data exploration. Nat. Methods 2021, 18, 843–844. [Google Scholar] [CrossRef]
- Kolla, L.; Kelly, M.C.; Mann, Z.F.; Anaya-Rocha, A.; Ellis, K.; Lemons, A.; Palermo, A.T.; So, K.S.; Mays, J.C.; Orvis, J. Characterization of the development of the mouse cochlear epithelium at the single cell level. Nat. Commun. 2020, 11, 2389. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, L.; Giffen, K.P.; Stringham, S.T.; Li, Y.; Judge, P.D.; Beisel, K.W.; He, D.Z. Cell-specific transcriptome analysis shows that adult pillar and Deiters’ cells express genes encoding machinery for specializations of cochlear hair cells. Front. Mol. Neurosci. 2018, 11, 356. [Google Scholar] [CrossRef]
- Li, C.; Li, X.; Bi, Z.; Sugino, K.; Wang, G.; Zhu, T.; Liu, Z. Comprehensive transcriptome analysis of cochlear spiral ganglion neurons at multiple ages. eLife 2020, 9, e50491. [Google Scholar] [CrossRef] [PubMed]
- Delmaghani, S.; El-Amraoui, A. The genetic and phenotypic landscapes of Usher syndrome: From disease mechanisms to a new classification. Hum. Genet. 2022, 141, 709–735. [Google Scholar] [CrossRef]
- Hartong, D.T.; Berson, E.L.; Dryja, T.P. Retinitis pigmentosa. Lancet 2006, 368, 1795–1809. [Google Scholar] [CrossRef]
- Rosenberg, T.; Haim, M.; Hauch, A.M.; Parving, A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin. Genet. 1997, 51, 314–321. [Google Scholar] [CrossRef]
- Keats, B.J.; Savas, S. Genetic heterogeneity in Usher syndrome. Am. J. Med. Genet. A 2004, 130, 13–16. [Google Scholar] [CrossRef]
- Kimberling, W.J.; Orten, D.; Pieke-Dahl, S. Genetic heterogeneity of Usher syndrome. Adv. Otorhinolaryngol. 2000, 56, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Nolen, R.M.; Hufnagel, R.B.; Friedman, T.B.; Turriff, A.E.; Brewer, C.C.; Zalewski, C.K.; King, K.A.; Wafa, T.T.; Griffith, A.J.; Brooks, B.P.; et al. Atypical and ultra-rare Usher syndrome: A review. Ophthalmic Genet. 2020, 41, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Abu-Diab, A.; Gopalakrishnan, P.; Matsevich, C.; de Jong, M.; Obolensky, A.; Khalaileh, A.; Salameh, M.; Ejzenberg, A.; Gross, M.; Banin, E. Homozygous Knockout of Cep250 Leads to a Relatively Late-Onset Retinal Degeneration and Sensorineural Hearing Loss in Mice. Transl. Vis. Sci. Technol. 2023, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Groza, T.; Gomez, F.L.; Mashhadi, H.H.; Muñoz-Fuentes, V.; Gunes, O.; Wilson, R.; Cacheiro, P.; Frost, A.; Keskivali-Bond, P.; Vardal, B. The International Mouse Phenotyping Consortium: Comprehensive knockout phenotyping underpinning the study of human disease. Nucleic Acids Res. 2023, 51, D1038–D1045. [Google Scholar] [CrossRef]
- Floriot, S.; Vesque, C.; Rodriguez, S.; Bourgain-Guglielmetti, F.; Karaiskou, A.; Gautier, M.; Duchesne, A.; Barbey, S.; Fritz, S.; Vasilescu, A.; et al. C-Nap1 mutation affects centriole cohesion and is associated with a Seckel-like syndrome in cattle. Nat. Commun. 2015, 6, 6894. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, M.; Kim, J.A.; Song, M.H.; Joo, S.Y.; Kim, S.J.; Jang, S.H.; Lee, H.; Seong, J.K.; Choi, J.Y.; Gee, H.Y.; et al. Novel Variant in CEP250 Causes Protein Mislocalization and Leads to Nonsyndromic Autosomal Recessive Type of Progressive Hearing Loss. Cells 2023, 12, 2328. https://doi.org/10.3390/cells12182328
Kang M, Kim JA, Song MH, Joo SY, Kim SJ, Jang SH, Lee H, Seong JK, Choi JY, Gee HY, et al. Novel Variant in CEP250 Causes Protein Mislocalization and Leads to Nonsyndromic Autosomal Recessive Type of Progressive Hearing Loss. Cells. 2023; 12(18):2328. https://doi.org/10.3390/cells12182328
Chicago/Turabian StyleKang, Minjin, Jung Ah Kim, Mee Hyun Song, Sun Young Joo, Se Jin Kim, Seung Hyun Jang, Ho Lee, Je Kyung Seong, Jae Young Choi, Heon Yung Gee, and et al. 2023. "Novel Variant in CEP250 Causes Protein Mislocalization and Leads to Nonsyndromic Autosomal Recessive Type of Progressive Hearing Loss" Cells 12, no. 18: 2328. https://doi.org/10.3390/cells12182328
APA StyleKang, M., Kim, J. A., Song, M. H., Joo, S. Y., Kim, S. J., Jang, S. H., Lee, H., Seong, J. K., Choi, J. Y., Gee, H. Y., & Jung, J. (2023). Novel Variant in CEP250 Causes Protein Mislocalization and Leads to Nonsyndromic Autosomal Recessive Type of Progressive Hearing Loss. Cells, 12(18), 2328. https://doi.org/10.3390/cells12182328