

A Molecular Troika of Angiogenesis, Coagulopathy and Endothelial Dysfunction in the Pathology of Avascular Necrosis of Femoral Head: A Comprehensive Review

 and
and
Abstract
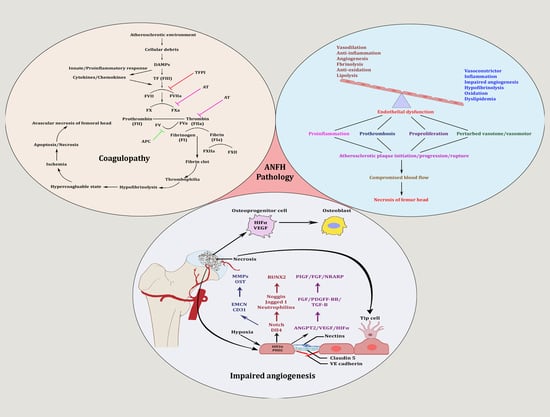
:
1. Introduction
2. Angiogenesis: Sprouting, Splitting and Stabilization
3. Angiogenesis: A Predominant Pacifier in Avascular Necrosis
4. Coagulopathy: A Culprit Alliance of Thrombophilia and Hypofibrinolysis
5. Endothelial Dysfunction: Holding Hands with Inflammation
6. Therapies Used in Other Diseases: A Possible Avenue for ANFH Management
7. Clinical Implications: A Call of a Crackling Tone of the Collapsing Bone
8. Conclusions
9. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Narayanan, A.; Khanchandani, P.; Borkar, R.M.; Ambati, C.R.; Roy, A.; Han, X.; Bhoskar, R.N.; Ragampeta, S.; Gannon, F.; Mysorekar, V.; et al. Avascular Necrosis of Femoral Head: A Metabolomic, Biophysical, Biochemical, Electron Microscopic and Histopathological Characterization. Sci. Rep. 2017, 7, 10721. [Google Scholar] [CrossRef] [PubMed]
- George, G.; Lane, J.M. Osteonecrosis of the Femoral Head. J. Am. Acad. Orthop. Surg. Glob. Res. Rev. 2022, 6, e21.00176. [Google Scholar] [CrossRef] [PubMed]
- Petek, D.; Hannouche, D.; Suva, D. Osteonecrosis of the Femoral Head: Pathophysiology and Current Concepts of Treatment. EFORT Open Rev. 2019, 4, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kerachian, M.A.; Harvey, E.J.; Cournoyer, D.; Chow, T.Y.K.; Séguin, C. Avascular Necrosis of the Femoral Head: Vascular Hypotheses. Endothelium 2006, 13, 237–244. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, C.; Yu, T.; Zhang, H.; Huang, J.; Chen, L.; Tong, P.; Zhen, G. The Therapeutic Effect of Adipose-Derived Lipoaspirate Cells in Femoral Head Necrosis by Improving Angiogenesis. Front. Cell Dev. Biol. 2022, 10, 1014789. [Google Scholar] [CrossRef]
- Kim, H.K.W.; Bian, H.; Randall, T.; Garces, A.; Gerstenfeld, L.C.; Einhorn, T.A. Increased VEGF Expression in the Epiphyseal Cartilage after Ischemic Necrosis of the Capital Femoral Epiphysis. J. Bone Miner. Res. 2004, 19, 2041–2048. [Google Scholar] [CrossRef]
- Palta, S.; Saroa, R.; Palta, A. Overview of the Coagulation System. Indian J. Anaesth. 2014, 58, 515–523. [Google Scholar] [CrossRef]
- Li, Y.; Liu, F.-X.; Yuan, C.; Meng, L. Association between Plasminogen Activator Inhibitor Gene Polymorphisms and Osteonecrosis of the Femoral Head Susceptibility: A Case-Control Study. Medicine 2017, 96, e7047. [Google Scholar] [CrossRef]
- Glueck, C.J.; Freiberg, R.A.; Wang, P. Heritable Thrombophilia-Hypofibrinolysis and Osteonecrosis of the Femoral Head. Clin. Orthop. Relat. Res. 2008, 466, 1034–1040. [Google Scholar] [CrossRef]
- Sandoo, A.; van Zanten, J.J.C.S.V.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The Endothelium and Its Role in Regulating Vascular Tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef]
- Poredos, P.; Poredos, A.V.; Gregoric, I. Endothelial Dysfunction and Its Clinical Implications. Angiology 2021, 72, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, D.; Ganz, P. Endothelial Function. From Vascular Biology to Clinical Applications. Am. J. Cardiol. 2002, 90, 40L–48L. [Google Scholar] [CrossRef] [PubMed]
- Kakar, P.; Lip, G.Y.H. Hypertension: Endothelial Dysfunction, the Prothrombotic State and Antithrombotic Therapy. Expert Rev. Cardiovasc. Ther. 2007, 5, 441–450. [Google Scholar] [CrossRef]
- Pouya, F.; Kerachian, M.A. Avascular Necrosis of the Femoral Head: Are Any Genes Involved? Arch. Bone Jt. Surg. 2015, 3, 149–155. [Google Scholar]
- Kretschmer, M.; Rüdiger, D.; Zahler, S. Mechanical Aspects of Angiogenesis. Cancers 2021, 13, 4987. [Google Scholar] [CrossRef]
- Semenza, G.L. Vasculogenesis, Angiogenesis, and Arteriogenesis: Mechanisms of Blood Vessel Formation and Remodeling. J. Cell. Biochem. 2007, 102, 840–847. [Google Scholar] [CrossRef]
- Ashraf, J.V.; Al Haj Zen, A. Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis. Int. J. Mol. Sci. 2021, 22, 10585. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L. Vascular Permeability--the Essentials. Upsala J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Stratman, A.N.; Davis, G.E. Endothelial Cell-Pericyte Interactions Stimulate Basement Membrane Matrix Assembly: Influence on Vascular Tube Remodeling, Maturation, and Stabilization. Microsc. Microanal. 2012, 18, 68–80. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Kwon, S.M. Angiogenesis and Its Therapeutic Opportunities. Mediat. Inflamm. 2013, 2013, 127170. [Google Scholar] [CrossRef]
- Smith, D.W. Is Avascular Necrosis of the Femoral Head the Result of Inhibition of Angiogenesis? Med. Hypotheses 1997, 49, 497–500. [Google Scholar] [CrossRef]
- Wu, S.-H.; Miao, Y.; Zhu, X.-Z.; Li, G.-Y. [Assessment of the local blood supply when femoral neck fracture occurs:advances in the anatomy research and its clinical application]. Zhongguo Gu Shang 2023, 36, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Kinose, S.; Kato, K.; Sakai, T.; Ichimura, K. Anatomic Characterization of the Femoral Nutrient Artery: Application to Fracture and Surgery of the Femur. Clin. Anat. 2020, 33, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Rajani, S.J.; Ravat, M.K.; Rajani, J.K.; Bhedi, A.N. Cadaveric Study of Profunda Femoris Artery with Some Unique Variations. J. Clin. Diagn. Res. 2015, 9, AC01–AC03. [Google Scholar] [CrossRef]
- Zhao, D.; Qiu, X.; Wang, B.; Wang, Z.; Wang, W.; Ouyang, J.; Silva, R.M.; Shi, X.; Kang, K.; Xu, D.; et al. Epiphyseal Arterial Network and Inferior Retinacular Artery Seem Critical to Femoral Head Perfusion in Adults With Femoral Neck Fractures. Clin. Orthop. Relat. Res. 2017, 475, 2011–2023. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-J.; Jain, M.; Alimperti, S. Bone Microvasculature: Stimulus for Tissue Function and Regeneration. Tissue Eng. Part B Rev. 2021, 27, 313–329. [Google Scholar] [CrossRef]
- Grüneboom, A.; Kling, L.; Christiansen, S.; Mill, L.; Maier, A.; Engelke, K.; Quick, H.H.; Schett, G.; Gunzer, M. Next-Generation Imaging of the Skeletal System and Its Blood Supply. Nat. Rev. Rheumatol. 2019, 15, 533–549. [Google Scholar] [CrossRef] [PubMed]
- Grüneboom, A.; Hawwari, I.; Weidner, D.; Culemann, S.; Müller, S.; Henneberg, S.; Brenzel, A.; Merz, S.; Bornemann, L.; Zec, K.; et al. A Network of Trans-Cortical Capillaries as Mainstay for Blood Circulation in Long Bones. Nat. Metab. 2019, 1, 236–250. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of Angiogenesis and Osteogenesis by a Specific Vessel Subtype in Bone. Nature 2014, 507, 323–328. [Google Scholar] [CrossRef]
- Xu, Z.; Kusumbe, A.P.; Cai, H.; Wan, Q.; Chen, J. Type H Blood Vessels in Coupling Angiogenesis-Osteogenesis and Its Application in Bone Tissue Engineering. J. Biomed. Mater. Res. B Appl. Biomater. 2023, 111, 1434–1446. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, H.-Q.; Shen, B. Type H Vessels-a Bridge Connecting Subchondral Bone Remodelling and Articular Cartilage Degeneration in Osteoarthritis Development. Rheumatology 2023, 62, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.-S.; Qiu, X.; Wang, B.-J.; Zhao, D.-W. Relationship Between Blood Flow and Collapse of Nontraumatic Osteonecrosis of the Femoral Head. J. Bone Jt. Surg. Am. 2022, 104, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Chápuli, R.; Quesada, A.R.; Angel Medina, M. Angiogenesis and Signal Transduction in Endothelial Cells. Cell. Mol. Life Sci. 2004, 61, 2224–2243. [Google Scholar] [CrossRef] [PubMed]
- Kazerounian, S.; Lawler, J. Integration of Pro- and Anti-Angiogenic Signals by Endothelial Cells. J. Cell Commun. Signal. 2018, 12, 171–179. [Google Scholar] [CrossRef]
- Ding, H.; Gao, Y.-S.; Hu, C.; Wang, Y.; Wang, C.-G.; Yin, J.-M.; Sun, Y.; Zhang, C.-Q. HIF-1α Transgenic Bone Marrow Cells Can Promote Tissue Repair in Cases of Corticosteroid-Induced Osteonecrosis of the Femoral Head in Rabbits. PLoS ONE 2013, 8, e63628. [Google Scholar] [CrossRef]
- Felmeden, D.C.; Blann, A.D.; Lip, G.Y.H. Angiogenesis: Basic Pathophysiology and Implications for Disease. Eur. Heart J. 2003, 24, 586–603. [Google Scholar] [CrossRef]
- Han, Y.; You, X.; Xing, W.; Zhang, Z.; Zou, W. Paracrine and Endocrine Actions of Bone-the Functions of Secretory Proteins from Osteoblasts, Osteocytes, and Osteoclasts. Bone Res. 2018, 6, 16. [Google Scholar] [CrossRef]
- Dor, Y.; Keshet, E. Ischemia-Driven Angiogenesis. Trends Cardiovasc. Med. 1997, 7, 289–294. [Google Scholar] [CrossRef]
- Corrado, C.; Fontana, S. Hypoxia and HIF Signaling: One Axis with Divergent Effects. Int. J. Mol. Sci. 2020, 21, 5611. [Google Scholar] [CrossRef]
- Hu, K.; Olsen, B.R. The Roles of Vascular Endothelial Growth Factor in Bone Repair and Regeneration. Bone 2016, 91, 30–38. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Asahara, T.; Takahashi, T.; Masuda, H.; Kalka, C.; Chen, D.; Iwaguro, H.; Inai, Y.; Silver, M.; Isner, J.M. VEGF Contributes to Postnatal Neovascularization by Mobilizing Bone Marrow-Derived Endothelial Progenitor Cells. EMBO J. 1999, 18, 3964–3972. [Google Scholar] [CrossRef]
- Yang, Y.-Q.; Tan, Y.-Y.; Wong, R.; Wenden, A.; Zhang, L.-K.; Rabie, A.B.M. The Role of Vascular Endothelial Growth Factor in Ossification. Int. J. Oral Sci. 2012, 4, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Zelzer, E.; Mamluk, R.; Ferrara, N.; Johnson, R.S.; Schipani, E.; Olsen, B.R. VEGFA Is Necessary for Chondrocyte Survival during Bone Development. Development 2004, 131, 2161–2171. [Google Scholar] [CrossRef] [PubMed]
- Radke, S.; Battmann, A.; Jatzke, S.; Eulert, J.; Jakob, F.; Schütze, N. Expression of the Angiomatrix and Angiogenic Proteins CYR61, CTGF, and VEGF in Osteonecrosis of the Femoral Head. J. Orthop. Res. 2006, 24, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, Y.; Cornelia, R.; Swisher, S.; Kim, H. Regulation of VEGF Expression by HIF-1α in the Femoral Head Cartilage Following Ischemia Osteonecrosis. Sci. Rep. 2012, 2, 650. [Google Scholar] [CrossRef] [PubMed]
- Street, J.; Bao, M.; deGuzman, L.; Bunting, S.; Peale, F.V.; Ferrara, N.; Steinmetz, H.; Hoeffel, J.; Cleland, J.L.; Daugherty, A.; et al. Vascular Endothelial Growth Factor Stimulates Bone Repair by Promoting Angiogenesis and Bone Turnover. Proc. Natl. Acad. Sci. USA 2002, 99, 9656–9661. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Olsen, B.R. Osteoblast-Derived VEGF Regulates Osteoblast Differentiation and Bone Formation during Bone Repair. J. Clin. Investig. 2016, 126, 509–526. [Google Scholar] [CrossRef]
- Kapitsinou, P.P.; Rajendran, G.; Astleford, L.; Michael, M.; Schonfeld, M.P.; Fields, T.; Shay, S.; French, J.L.; West, J.; Haase, V.H. The Endothelial Prolyl-4-Hydroxylase Domain 2/Hypoxia-Inducible Factor 2 Axis Regulates Pulmonary Artery Pressure in Mice. Mol. Cell. Biol. 2016, 36, 1584–1594. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. VE-Cadherin and Claudin-5: It Takes Two to Tango. Nat. Cell Biol. 2008, 10, 883–885. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular Mechanisms and Clinical Applications of Angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Hutchings, H.; Ortega, N.; Plouët, J. Extracellular Matrix-Bound Vascular Endothelial Growth Factor Promotes Endothelial Cell Adhesion, Migration, and Survival through Integrin Ligation. FASEB J. 2003, 17, 1520–1522. [Google Scholar] [CrossRef] [PubMed]
- Phng, L.-K.; Potente, M.; Leslie, J.D.; Babbage, J.; Nyqvist, D.; Lobov, I.; Ondr, J.K.; Rao, S.; Lang, R.A.; Thurston, G.; et al. Nrarp Coordinates Endothelial Notch and Wnt Signaling to Control Vessel Density in Angiogenesis. Dev. Cell 2009, 16, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Song, S. The Role of Pericytes in Blood-Vessel Formation and Maintenance. Neuro-Oncology 2005, 7, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Noel, A.; Maillard, C.; Rocks, N.; Jost, M.; Chabottaux, V.; Sounni, N.E.; Maquoi, E.; Cataldo, D.; Foidart, J.M. Membrane Associated Proteases and Their Inhibitors in Tumour Angiogenesis. J. Clin. Pathol. 2004, 57, 577–584. [Google Scholar] [CrossRef]
- Cantatore, F.P.; Maruotti, N.; Corrado, A.; Ribatti, D. Angiogenesis Dysregulation in the Pathogenesis of Systemic Sclerosis. BioMed Res. Int. 2017, 2017, 5345673. [Google Scholar] [CrossRef]
- Almeida, I.; Oliveira Gomes, A.; Lima, M.; Silva, I.; Vasconcelos, C. Different Contributions of Angiostatin and Endostatin in Angiogenesis Impairment in Systemic Sclerosis: A Cohort Study. Clin. Exp. Rheumatol. 2016, 34 (Suppl. 100), 37–42. [Google Scholar]
- Manetti, M.; Guiducci, S.; Romano, E.; Ceccarelli, C.; Bellando-Randone, S.; Conforti, M.L.; Ibba-Manneschi, L.; Matucci-Cerinic, M. Overexpression of VEGF165b, an Inhibitory Splice Variant of Vascular Endothelial Growth Factor, Leads to Insufficient Angiogenesis in Patients with Systemic Sclerosis. Circ. Res. 2011, 109, e14–e26. [Google Scholar] [CrossRef]
- Bielecki, M.; Kowal, K.; Lapinska, A.; Chwiesko-Minarowska, S.; Chyczewski, L.; Kowal-Bielecka, O. Peripheral Blood Mononuclear Cells from Patients with Systemic Sclerosis Spontaneously Secrete Increased Amounts of Vascular Endothelial Growth Factor (VEGF) Already in the Early Stage of the Disease. Adv. Med. Sci. 2011, 56, 255–263. [Google Scholar] [CrossRef]
- Hirigoyen, D.; Burgos, P.I.; Mezzano, V.; Duran, J.; Barrientos, M.; Saez, C.G.; Panes, O.; Mezzano, D.; Iruretagoyena, M. Inhibition of Angiogenesis by Platelets in Systemic Sclerosis Patients. Arthritis Res. Ther. 2015, 17, 332. [Google Scholar] [CrossRef]
- Vosmaer, A.; Pereira, R.R.; Koenderman, J.S.; Rosendaal, F.R.; Cannegieter, S.C. Coagulation Abnormalities in Legg-Calvé-Perthes Disease. J. Bone Jt. Surg. Am. 2010, 92, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Dahlbäck, B.; Carlsson, M.; Svensson, P.J. Familial Thrombophilia Due to a Previously Unrecognized Mechanism Characterized by Poor Anticoagulant Response to Activated Protein C: Prediction of a Cofactor to Activated Protein C. Proc. Natl. Acad. Sci. USA 1993, 90, 1004–1008. [Google Scholar] [CrossRef] [PubMed]
- Svensson, P.J.; Dahlbäck, B. Resistance to Activated Protein C as a Basis for Venous Thrombosis. N. Engl. J. Med. 1994, 330, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Molino, D.; De Santo, N.G.; Marotta, R.; Anastasio, P.; Mosavat, M.; De Lucia, D. Plasma Levels of Plasminogen Activator Inhibitor Type 1, Factor VIII, Prothrombin Activation Fragment 1+2, Anticardiolipin, and Antiprothrombin Antibodies Are Risk Factors for Thrombosis in Hemodialysis Patients. Semin. Nephrol. 2004, 24, 495–501. [Google Scholar] [CrossRef]
- Zwaginga, J.J.; Ijsseldijk, M.J.; Beeser-Visser, N.; de Groot, P.G.; Vos, J.; Sixma, J.J. High von Willebrand Factor Concentration Compensates a Relative Adhesion Defect in Uremic Blood. Blood 1990, 75, 1498–1508. [Google Scholar] [CrossRef]
- Fortmann, S.P.; Marcovina, S.M. Lipoprotein(a), a Clinically Elusive Lipoprotein Particle. Circulation 1997, 95, 295–296. [Google Scholar] [CrossRef]
- Hughes, G.R. The Antiphospholipid Syndrome: Ten Years On. Lancet 1993, 342, 341–344. [Google Scholar] [CrossRef]
- Mont, M.A.; Jones, L.C.; Hungerford, D.S. Nontraumatic Osteonecrosis of the Femoral Head: Ten Years Later. J. Bone Jt. Surg. Am. 2006, 88, 1117–1132. [Google Scholar] [CrossRef]
- Dubois, E.L.; Cozen, L. Avascular (Aseptic) Bone Necrosis Associated with Systemic Lupus Erythematosus. JAMA 1960, 174, 966–971. [Google Scholar] [CrossRef]
- Ware, H.E.; Brooks, A.P.; Toye, R.; Berney, S.I. Sickle Cell Disease and Silent Avascular Necrosis of the Hip. J. Bone Jt. Surg. Br. 1991, 73, 947–949. [Google Scholar] [CrossRef]
- Sondag, D.; Verhoeven, S.; Löwik, D.W.P.M.; van Geffen, M.; Veer, C.V.; van Heerde, W.L.; Boltje, T.J.; Rutjes, F.P.J.T. Activity Sensing of Coagulation and Fibrinolytic Proteases. Chemistry 2023, 29, e202203473. [Google Scholar] [CrossRef] [PubMed]
- Noubouossie, D.F.; Reeves, B.N.; Strahl, B.D.; Key, N.S. Neutrophils: Back in the Thrombosis Spotlight. Blood 2019, 133, 2186–2197. [Google Scholar] [CrossRef] [PubMed]
- Butenas, S.; Orfeo, T.; Mann, K.G. Tissue Factor in Coagulation: Which? Where? When? Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, T.; Bäuml, C.A.; Imhof, D. Inhibitors of Blood Coagulation Factor XIII. Anal. Biochem. 2020, 605, 113708. [Google Scholar] [CrossRef] [PubMed]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int. J. Hematol. Oncol. Stem Cell Res. 2017, 11, 319–327. [Google Scholar] [PubMed]
- Sachs, U.J.; Kirsch-Altena, A.; Müller, J. Markers of Hereditary Thrombophilia with Unclear Significance. Hamostaseologie 2022, 42, 370–380. [Google Scholar] [CrossRef]
- Meager, A. Cytokine Regulation of Cellular Adhesion Molecule Expression in Inflammation. Cytokine Growth Factor Rev. 1999, 10, 27–39. [Google Scholar] [CrossRef]
- Ito, T. PAMPs and DAMPs as Triggers for DIC. J. Intensive Care 2014, 2, 67. [Google Scholar] [CrossRef]
- Glueck, C.J.; Freiberg, R.A.; Wang, P. Role of Thrombosis in Osteonecrosis. Curr. Hematol. Rep. 2003, 2, 417–422. [Google Scholar]
- van Giezen, J.J.; Jansen, J.W. Correlation of in Vitro and in Vivo Decreased Fibrinolytic Activity Caused by Dexamethasone. Ann. N. Y. Acad. Sci. 1992, 667, 199–201. [Google Scholar] [CrossRef]
- Kerachian, M.A.; Séguin, C.; Harvey, E.J. Glucocorticoids in Osteonecrosis of the Femoral Head: A New Understanding of the Mechanisms of Action. J. Steroid Biochem. Mol. Biol. 2009, 114, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Glueck, C.J.; Fontaine, R.N.; Gruppo, R.; Stroop, D.; Sieve-Smith, L.; Tracy, T.; Wang, P. The Plasminogen Activator Inhibitor-1 Gene, Hypofibrinolysis, and Osteonecrosis. Clin. Orthop. Relat. Res. 1999, 366, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Pósán, E.; Hársfalvi, J.; Szepesi, K.; Gáspár, L.; Batár, P.; Udvardy, M. Increased Platelet Activation and Decreased Fibrinolysis in the Pathogenesis of Aseptic Necrosis of the Femoral Head. Platelets 1998, 9, 233–235. [Google Scholar] [CrossRef]
- Zhang, Q.; Lv, J.; Jin, L. Role of Coagulopathy in Glucocorticoid-Induced Osteonecrosis of the Femoral Head. J. Int. Med. Res. 2018, 46, 2141–2148. [Google Scholar] [CrossRef]
- Pacinella, G.; Ciaccio, A.M.; Tuttolomondo, A. Endothelial Dysfunction and Chronic Inflammation: The Cornerstones of Vascular Alterations in Age-Related Diseases. Int. J. Mol. Sci. 2022, 23, 15722. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.J.; Nordaby, R.A.; Vilariño, J.O.; Paragano, A.; Cacharrón, J.L.; Machado, R.A. Endothelial Dysfunction: A Comprehensive Appraisal. Cardiovasc. Diabetol. 2006, 5, 4. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef]
- Fan, T.; Song, Y.-J.; Liu, X.-L. Adenocarcinoma of the Lung with Concomitant ALK Fusion Gene and EGFR Gene Mutation: A Case Report and Literature Review. Mol. Clin. Oncol. 2016, 4, 203–205. [Google Scholar] [CrossRef]
- Simionescu, M.; Simionescu, N. Proatherosclerotic Events: Pathobiochemical Changes Occurring in the Arterial Wall before Monocyte Migration. FASEB J. 1993, 7, 1359–1366. [Google Scholar] [CrossRef]
- Davies, M.J. Stability and Instability: Two Faces of Coronary Atherosclerosis. The Paul Dudley White Lecture 1995. Circulation 1996, 94, 2013–2020. [Google Scholar] [CrossRef]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and Neutrophils Potentiate Endothelial Stress, Apoptosis and Detachment: Implications for Superficial Erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Ogita, H.; Liao, J. Endothelial Function and Oxidative Stress. Endothelium 2004, 11, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Vascular Oxidative Stress, Nitric Oxide and Atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef]
- Förstermann, U.; Sessa, W.C. Nitric Oxide Synthases: Regulation and Function. Eur. Heart J. 2012, 33, 829–837; 837a–837d. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Ognibene, F.P.; Pannell, L.K.; Nichols, J.S.; Pease-Fye, M.E.; Shelhamer, J.H.; Schechter, A.N. Relative Role of Heme Nitrosylation and Beta-Cysteine 93 Nitrosation in the Transport and Metabolism of Nitric Oxide by Hemoglobin in the Human Circulation. Proc. Natl. Acad. Sci. USA 2000, 97, 9943–9948. [Google Scholar] [CrossRef]
- Rochette, L.; Lorin, J.; Zeller, M.; Guilland, J.-C.; Lorgis, L.; Cottin, Y.; Vergely, C. Nitric Oxide Synthase Inhibition and Oxidative Stress in Cardiovascular Diseases: Possible Therapeutic Targets? Pharmacol. Ther. 2013, 140, 239–257. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Sobenin, I.A.; Orekhov, A.N.; Bobryshev, Y.V. Human MiR-221/222 in Physiological and Atherosclerotic Vascular Remodeling. BioMed Res. Int. 2015, 2015, 354517. [Google Scholar] [CrossRef]
- Qin, J.-Z.; Wang, S.-J.; Xia, C. MicroRNAs Regulate Nitric Oxide Release from Endothelial Cells by Targeting NOS3. J. Thromb. Thrombolysis 2018, 46, 275–282. [Google Scholar] [CrossRef]
- Parikh, S.; Gomez, O.; Davis, T.; Lyon, Z.; Corces, A. Avascular Necrosis as a Sequela of COVID-19: A Case Series. Cureus 2023, 15, e35368. [Google Scholar] [CrossRef]
- Agarwala, S.R.; Vijayvargiya, M.; Sawant, T. Secondary Osteonecrosis of the Knee as a Part of Long COVID-19 Syndrome: A Case Series. BMJ Case Rep. 2022, 15, e248583. [Google Scholar] [CrossRef]
- Hassan, A.A.A.; Khalifa, A.A. Femoral Head Avascular Necrosis in COVID-19 Survivors: A Systematic Review. Rheumatol. Int. 2023, 43, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.-C.; Zhong, H.-M.; Lin, T.; Shi, J.-B. Comparison of Core Decompression and Conservative Treatment for Avascular Necrosis of Femoral Head at Early Stage: A Meta-Analysis. Int. J. Clin. Exp. Med. 2015, 8, 5207–5216. [Google Scholar] [PubMed]
- Tan, Y.; He, H.; Wan, Z.; Qin, J.; Wen, Y.; Pan, Z.; Wang, H.; Chen, L. Study on the Outcome of Patients with Aseptic Femoral Head Necrosis Treated with Percutaneous Multiple Small-Diameter Drilling Core Decompression: A Retrospective Cohort Study Based on Magnetic Resonance Imaging and Equivalent Sphere Model Analysis. J. Orthop. Surg. Res. 2020, 15, 264. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.-C.; Luo, R.-B.; Lin, T.; Zhong, H.-M.; Shi, J.-B. Efficacy of Alendronate for Preventing Collapse of Femoral Head in Adult Patients with Nontraumatic Osteonecrosis. BioMed Res. Int. 2014, 2014, 716538. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, T.; Liao, J.; Gu, H.; Lin, X.; Jiang, Q.; Bulsara, M.K.; Zheng, M.; Zheng, Q. Efficacy of Autologous Bone Marrow Buffy Coat Grafting Combined with Core Decompression in Patients with Avascular Necrosis of Femoral Head: A Prospective, Double-Blinded, Randomized, Controlled Study. Stem Cell Res. Ther. 2014, 5, 115. [Google Scholar] [CrossRef]
- Tang, H.-Y.; Zhao, Y.; Li, Y.-Z.; Wang, T.-S. Effectiveness of Extracorporeal Shock Wave Monotherapy for Avascular Necrosis of Femoral Head: A Systematic Review Protocol of Randomized Controlled Trial. Medicine 2019, 98, e15119. [Google Scholar] [CrossRef]
- Tu, Y.; Chen, Z.; Lineaweaver, W.C.; Zhang, F. Different Recipient Vessels for Free Microsurgical Fibula Flaps in the Treatment of Avascular Necrosis of the Femoral Head: A Systematic Review and Meta-Analysis. Ann. Plast. Surg. 2017, 79, 583–589. [Google Scholar] [CrossRef]
- Li, M.; Wang, F.; Chen, X.; Cao, S.; Zhou, Y.; Ou, X.; He, M.; Cai, H.; Dai, W.; Yuan, D.; et al. Therapeutic Assessment of Crystalloid Fluid Resuscitation in Experimental Military Injury. Curr. Pharm. Biotechnol. 2023, 25, 93–101. [Google Scholar] [CrossRef]
- Chen, C.; Fu, L.; Luo, Y.; Zeng, W.; Qi, X.; Wei, Y.; Chen, L.; Zhao, X.; Li, D.; Tian, M.; et al. Engineered Exosome-Functionalized Extracellular Matrix-Mimicking Hydrogel for Promoting Bone Repair in Glucocorticoid-Induced Osteonecrosis of the Femoral Head. ACS Appl. Mater. Interfaces 2023, 15, 28891–28906. [Google Scholar] [CrossRef]
- Guo, M.; Zhang, J. Vitamin B2 Prevents Glucocorticoid-Caused Damage of Blood Vessels in Osteonecrosis of the Femoral Head. BioMed Res. Int. 2022, 2022, 4006184. [Google Scholar] [CrossRef]
- Ren, P.; Lu, L.; Cai, S.; Chen, J.; Lin, W.; Han, F. Alternative Splicing: A New Cause and Potential Therapeutic Target in Autoimmune Disease. Front. Immunol. 2021, 12, 713540. [Google Scholar] [CrossRef] [PubMed]
- Sarsenova, M.; Issabekova, A.; Abisheva, S.; Rutskaya-Moroshan, K.; Ogay, V.; Saparov, A. Mesenchymal Stem Cell-Based Therapy for Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 11592. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Hamlet, S.; Vaquette, C.; Petcu, E.B.; Ramamurthy, P.; Ivanovski, S. Local Delivery of Hydrogel Encapsulated Vascular Endothelial Growth Factor for the Prevention of Medication-Related Osteonecrosis of the Jaw. Sci. Rep. 2021, 11, 23371. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Li, L.; Peng, Y.; Zhuang, A.; Wei, W.; Zhang, D.; Pang, Y.; Bi, X. Biomimetic Nanofibrous Hybrid Hydrogel Membranes with Sustained Growth Factor Release for Guided Bone Regeneration. Biomater. Sci. 2021, 9, 1256–1271. [Google Scholar] [CrossRef] [PubMed]
- Martín-Hernández, P.; Gutiérrez-Leonard, H.; Quintana, A.R.; Ojeda-Delgado, J.L.; Montes-Bautista, C.; Valdéz-Becerril, G.; Aguirre-Alvarado, A.; Hernández-Jiménez, L. Hyperbaric Oxygen Therapy Following Percutaneous Coronary Intervention for ST-Segment Elevation Myocardial Infarction. Cardiovasc. Revasc. Med. 2021, 27, 14–19. [Google Scholar] [CrossRef]
- Lee, S.S.; Kim, J.H.; Jeong, J.; Kim, S.H.L.; Koh, R.H.; Kim, I.; Bae, S.; Lee, H.; Hwang, N.S. Sequential Growth Factor Releasing Double Cryogel System for Enhanced Bone Regeneration. Biomaterials 2020, 257, 120223. [Google Scholar] [CrossRef]
- Zuo, R.; Kong, L.; Wang, M.; Wang, W.; Xu, J.; Chai, Y.; Guan, J.; Kang, Q. Exosomes Derived from Human CD34+ Stem Cells Transfected with MiR-26a Prevent Glucocorticoid-Induced Osteonecrosis of the Femoral Head by Promoting Angiogenesis and Osteogenesis. Stem Cell Res. Ther. 2019, 10, 321. [Google Scholar] [CrossRef]
- Elbaz-Greener, G.; Sud, M.; Tzuman, O.; Leitman, M.; Vered, Z.; Ben-Dov, N.; Oron, U.; Blatt, A. Adjunctive Laser-Stimulated Stem-Cells Therapy to Primary Reperfusion in Acute Myocardial Infarction in Humans: Safety and Feasibility Study. J. Interv. Cardiol. 2018, 31, 711–716. [Google Scholar] [CrossRef]
- Kuttappan, S.; Mathew, D.; Jo, J.-I.; Tanaka, R.; Menon, D.; Ishimoto, T.; Nakano, T.; Nair, S.V.; Nair, M.B.; Tabata, Y. Dual Release of Growth Factor from Nanocomposite Fibrous Scaffold Promotes Vascularisation and Bone Regeneration in Rat Critical Sized Calvarial Defect. Acta Biomater. 2018, 78, 36–47. [Google Scholar] [CrossRef]
- Zhang, H.; Kot, A.; Lay, Y.-A.E.; Fierro, F.A.; Chen, H.; Lane, N.E.; Yao, W. Acceleration of Fracture Healing by Overexpression of Basic Fibroblast Growth Factor in the Mesenchymal Stromal Cells. Stem Cells Transl. Med. 2017, 6, 1880–1893. [Google Scholar] [CrossRef]
- Liu, X.; Li, Q.; Niu, X.; Hu, B.; Chen, S.; Song, W.; Ding, J.; Zhang, C.; Wang, Y. Exosomes Secreted from Human-Induced Pluripotent Stem Cell-Derived Mesenchymal Stem Cells Prevent Osteonecrosis of the Femoral Head by Promoting Angiogenesis. Int. J. Biol. Sci. 2017, 13, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Özdel, A.; Sarısözen, B.; Yalçınkaya, U.; Demirağ, B. The Effect of HIF Stabilizer on Distraction Osteogenesis. Acta Orthop. Traumatol. Turc. 2015, 49, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Gomes, R.S.M.; das Neves, R.P.; Cochlin, L.; Lima, A.; Carvalho, R.; Korpisalo, P.; Dragneva, G.; Turunen, M.; Liimatainen, T.; Clarke, K.; et al. Efficient Pro-Survival/Angiogenic MiRNA Delivery by an MRI-Detectable Nanomaterial. ACS Nano 2013, 7, 3362–3372. [Google Scholar] [CrossRef]
- Kumar, S.; Wan, C.; Ramaswamy, G.; Clemens, T.L.; Ponnazhagan, S. Mesenchymal Stem Cells Expressing Osteogenic and Angiogenic Factors Synergistically Enhance Bone Formation in a Mouse Model of Segmental Bone Defect. Mol. Ther. 2010, 18, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, J.O.; Onikepe, A.O.; MacKrell, J.; Einhorn, T.; Bradica, G.; Lynch, S.; Hart, C.E. Accelerated Fracture Healing in the Geriatric, Osteoporotic Rat with Recombinant Human Platelet-Derived Growth Factor-BB and an Injectable Beta-Tricalcium Phosphate/Collagen Matrix. J. Orthop. Res. 2008, 26, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Holstein, J.H.; Menger, M.D.; Scheuer, C.; Meier, C.; Culemann, U.; Wirbel, R.J.; Garcia, P.; Pohlemann, T. Erythropoietin (EPO): EPO-Receptor Signaling Improves Early Endochondral Ossification and Mechanical Strength in Fracture Healing. Life Sci. 2007, 80, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Usas, A.; Olshanski, A.; Ho, A.M.; Gearhart, B.; Cooper, G.M.; Huard, J. VEGF Improves, Whereas SFlt1 Inhibits, BMP2-Induced Bone Formation and Bone Healing through Modulation of Angiogenesis. J. Bone Miner. Res. 2005, 20, 2017–2027. [Google Scholar] [CrossRef]
- Ballara, S.C.; Miotla, J.M.; Paleolog, E.M. New Vessels, New Approaches: Angiogenesis as a Therapeutic Target in Musculoskeletal Disorders. Int. J. Exp. Pathol. 1999, 80, 235–250. [Google Scholar] [CrossRef]
- Liu, X.; Virk, S.; Fedorova, T.; Oo, W.M.; Hunter, D.J. The Effect of Pentosan Polysulfate Sodium for Improving Dyslipidaemia and Knee Pain in People with Knee Osteoarthritis: A Pilot Study. Osteoarthr. Cartil. Open. 2023, 5, 100343. [Google Scholar] [CrossRef]
- Song, H.-X.; Zhang, B.; Liu, S.; Shi, Z.-C.; Wang, Z.-Y.; Lu, H.-L.; Yao, J.; Chen, J. Efficacy and Safety of Low Dose Aspirin plus Clopidogrel in the Treatment of Elderly Patients with Symptomatic Intracranial Artery Stenosis. Front. Neurol. 2023, 14, 1060733. [Google Scholar] [CrossRef]
- Wang, Z.; Ji, K.; Fang, Q. Low-Dose vs. Standard-Dose Intravenous Alteplase for Acute Ischemic Stroke with Unknown Time of Onset. Front. Neurol. 2023, 14, 1165237. [Google Scholar] [CrossRef] [PubMed]
- Tashani, M.; Stevens, R.A.; de Souza Goncalves, B.; Lakhani, H.V.; Jones, S.E.; Given, L.; Sicking, R.; Dougherty, T.; Thompson, E.; Sodhi, K.; et al. Transitioning to Unfractionated Heparin in Treatment of Non-ST-Segment Elevation Myocardial Infarction Patients on Direct Oral Anti-Xa Inhibitors. Cell. Mol. Biol. 2023, 69, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhao, Q.; Qin, J.; Guo, Y.; Zhang, C.; Li, Y. Urokinase Loaded Black Phosphorus Nanosheets for Sequential Thrombolysis and Reactive Oxygen Species Scavenging in Ischemic Stroke Treatment. Biomater. Sci. 2022, 10, 4656–4666. [Google Scholar] [CrossRef] [PubMed]
- Tsivgoulis, G.; Katsanos, A.H.; Christogiannis, C.; Faouzi, B.; Mavridis, D.; Dixit, A.K.; Palaiodimou, L.; Khurana, D.; Petruzzellis, M.; Psychogios, K.; et al. Intravenous Thrombolysis with Tenecteplase for the Treatment of Acute Ischemic Stroke. Ann. Neurol. 2022, 92, 349–357. [Google Scholar] [CrossRef]
- Adik-Pathak, L.; Shirodkar, S.; Gupta, A. Rivaroxaban, a New Molecule with Potential to Balance Bleeding Risk and Ischemic Events in Patients with Chronic Coronary Syndrome. J. Assoc. Physicians India 2022, 70, 11–12. [Google Scholar] [CrossRef]
- Khan, M.Y.; Ponde, C.K.; Kumar, V.; Gaurav, K. Fondaparinux: A Cornerstone Drug in Acute Coronary Syndromes. World J. Cardiol. 2022, 14, 40–53. [Google Scholar] [CrossRef]
- Koh, H.P.; Md Redzuan, A.; Mohd Saffian, S.; Nagarajah, J.R.; Ross, N.T.; Hassan, H. The Outcomes of Reperfusion Therapy with Streptokinase versus Tenecteplase in ST-Elevation Myocardial Infarction (STEMI): A Propensity-Matched Retrospective Analysis in an Asian Population. Int. J. Clin. Pharm. 2022, 44, 641–650. [Google Scholar] [CrossRef]
- Tu, L.; Zhao, M.; Wang, X.; Kong, Q.; Chen, Z.; Wei, Q.; Li, Q.; Yu, Q.; Ye, Z.; Cao, S.; et al. Etanercept/Celecoxib on Improving MRI Inflammation of Active Ankylosing Spondylitis: A Multicenter, Open-Label, Randomized Clinical Trial. Front. Immunol. 2022, 13, 967658. [Google Scholar] [CrossRef]
- Okada, K.; Nishioka, M.; Kaji, H. Roles of Fibrinolytic Factors in the Alterations in Bone Marrow Hematopoietic Stem/Progenitor Cells during Bone Repair. Inflamm. Regen. 2020, 40, 22. [Google Scholar] [CrossRef]
- Haydock, M.M.; Elhamdani, S.; Alsharedi, M. Long-Term Direct Oral Anticoagulation in Primary Osteonecrosis with Elevated Plasminogen Activation Inhibitor. SAGE Open Med. Case Rep. 2019, 7, 2050313X19827747. [Google Scholar] [CrossRef]
- Fukuta, T.; Ishii, T.; Asai, T.; Oku, N. Applications of Liposomal Drug Delivery Systems to Develop Neuroprotective Agents for the Treatment of Ischemic Stroke. Biol. Pharm. Bull. 2019, 42, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhu, Y.; Zheng, W.; Qian, T.; Wang, H.; Hou, X. Antagonism of NK-1R Using Aprepitant Suppresses Inflammatory Response in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-G.; Shen, L.; Yang, Y.-P.; Xu, X.-J.; Shuai, B.; Ma, C. Effects of Modified Qing’e Pill (加味青娥丸) on Expression of Adiponectin, Bone Morphogenetic Protein 2 and Coagulation-Related Factors in Patients with Nontraumatic Osteonecrosis of Femoral Head. Chin. J. Integr. Med. 2017, 23, 183–189. [Google Scholar] [CrossRef]
- Li, X.; Ling, L.; Li, C.; Ma, Q. Efficacy and Safety of Desmoteplase in Acute Ischemic Stroke Patients: A Systematic Review and Meta-Analysis. Medicine 2017, 96, e6667. [Google Scholar] [CrossRef] [PubMed]
- Özlüer, Y.E.; Avcil, M. Providing Full Recovery with Single-Dose Intravenous Reteplase in a Patient Presented to Emergency Department with Acute Ischemic Stroke. Clin. Case Rep. 2017, 5, 598–600. [Google Scholar] [CrossRef]
- Li, X.I.; Dong, Z.; Zhang, F.; Dong, J.; Zhang, Y. Vitamin E Slows down the Progression of Osteoarthritis. Exp. Ther. Med. 2016, 12, 18–22. [Google Scholar] [CrossRef]
- Dou, C.; Han, X.; Xie, H.; Liao, H.; Xiao, X.; Huang, Z.; Luo, G.; Zhang, X.; Yao, W. Protective Role of Nitric Oxide Donors on Endothelium in Ischemia-Reperfusion Injury: A Meta-Analysis of Randomized Controlled Trials. BMC Anesthesiol. 2023, 23, 189. [Google Scholar] [CrossRef]
- Shao, W.; Li, Z.; Wang, B.; Gong, S.; Wang, P.; Song, B.; Chen, Z.; Feng, Y. Dimethyloxalylglycine Attenuates Steroid-Associated Endothelial Progenitor Cell Impairment and Osteonecrosis of the Femoral Head by Regulating the HIF-1α Signaling Pathway. Biomedicines 2023, 11, 992. [Google Scholar] [CrossRef]
- Peng, P.; He, W.; Zhang, Y.-X.; Liu, X.-H.; Chen, Z.-Q.; Mao, J.-G. CircHIPK3 Promotes Bone Microvascular Endothelial Cell Proliferation, Migration and Angiogenesis by Targeting MiR-7 and KLF4/VEGF Signaling in Steroid-Induced Osteonecrosis of the Femoral Head. Adv. Clin. Exp. Med. 2023, 32, 43–55. [Google Scholar] [CrossRef]
- Tsofack Ngueguim, F.; Kamkumo Gounoue, R.; Hubert Donfack, J.; Manefen Simo, S.; Jouonzo, J.; Ngapout Fifen, R.; Djomeni Dzeufiet, P.D.; Dimo, T. Chromolaena odorata (L.) R. M. King and H. Robinson Leaves Aqueous Extract Improves the Femoral Head in Ethanol-Induced Osteonecrosis in Rats. Evid. Based Complement. Alternat. Med. 2023, 2023, 5436771. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, T.; Li, Z.; Lu, J.; Wu, X.; Gao, F.; Sun, W. Autocrine Activity of Extracellular Vesicles Induced by Icariin and Its Effectiveness in Glucocorticoid-Induced Injury of Bone Microvascular Endothelial Cells. Cells 2022, 11, 1921. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lin, L.; Liu, K.; Jiang, Y.; Zhou, Z. Effects of Simvastatin on Cartilage Homeostasis in Steroid-Induced Osteonecrosis of Femoral Head by Inhibiting Glucocorticoid Receptor. Cells 2022, 11, 3945. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.R.; Howard, M.T.; Wang, S.; Berger, A.G.; Hammond, P.T. Oxidation-Responsive, Tunable Growth Factor Delivery from Polyelectrolyte-Coated Implants. Adv. Healthc. Mater. 2021, 10, e2001941. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Yu, S.; Jing, X.; Guo, J.; Sun, K.; Guo, F.; Ye, Y. PTEN Inhibitor VO-OHpic Attenuates GC-Associated Endothelial Progenitor Cell Dysfunction and Osteonecrosis of the Femoral Head via Activating Nrf2 Signaling and Inhibiting Mitochondrial Apoptosis Pathway. Stem Cell Res. Ther. 2020, 11, 140. [Google Scholar] [CrossRef]
- Gambardella, J.; Khondkar, W.; Morelli, M.B.; Wang, X.; Santulli, G.; Trimarco, V. Arginine and Endothelial Function. Biomedicines 2020, 8, 277. [Google Scholar] [CrossRef]
- Agidigbi, T.S.; Kim, C. Reactive Oxygen Species in Osteoclast Differentiation and Possible Pharmaceutical Targets of ROS-Mediated Osteoclast Diseases. Int. J. Mol. Sci. 2019, 20, 3576. [Google Scholar] [CrossRef]
- Deng, G.; Dai, C.; Chen, J.; Ji, A.; Zhao, J.; Zhai, Y.; Kang, Y.; Liu, X.; Wang, Y.; Wang, Q. Porous Se@SiO2 Nanocomposites Protect the Femoral Head from Methylprednisolone-Induced Osteonecrosis. Int. J. Nanomed. 2018, 13, 1809–1818. [Google Scholar] [CrossRef]
- Sprague, S.; Slobogean, G.P.; Bogoch, E.; Petrisor, B.; Garibaldi, A.; O’Hara, N.; Bhandari, M.; FAITH Investigators. Vitamin D Use and Health Outcomes After Surgery for Hip Fracture. Orthopedics 2017, 40, e868–e875. [Google Scholar] [CrossRef]
- Radenkovic, M.; Stojanović, M.; Nešić, I.M.; Prostran, M. Angiotensin Receptor Blockers & Endothelial Dysfunction: Possible Correlation & Therapeutic Implications. Indian J. Med. Res. 2016, 144, 154–168. [Google Scholar] [CrossRef]
- Sun, J.-Y.; Zhai, L.; Li, Q.-L.; Ye, J.-X.; Kang, L.-N.; Xie, J.; Xu, B. Effects of ACE Inhibition on Endothelial Progenitor Cell Mobilization and Prognosis after Acute Myocardial Infarction in Type 2 Diabetic Patients. Clinics 2013, 68, 665–673. [Google Scholar] [CrossRef]
- Wang, D.S.; Miura, M.; Demura, H.; Sato, K. Anabolic Effects of 1,25-Dihydroxyvitamin D3 on Osteoblasts Are Enhanced by Vascular Endothelial Growth Factor Produced by Osteoblasts and by Growth Factors Produced by Endothelial Cells. Endocrinology 1997, 138, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
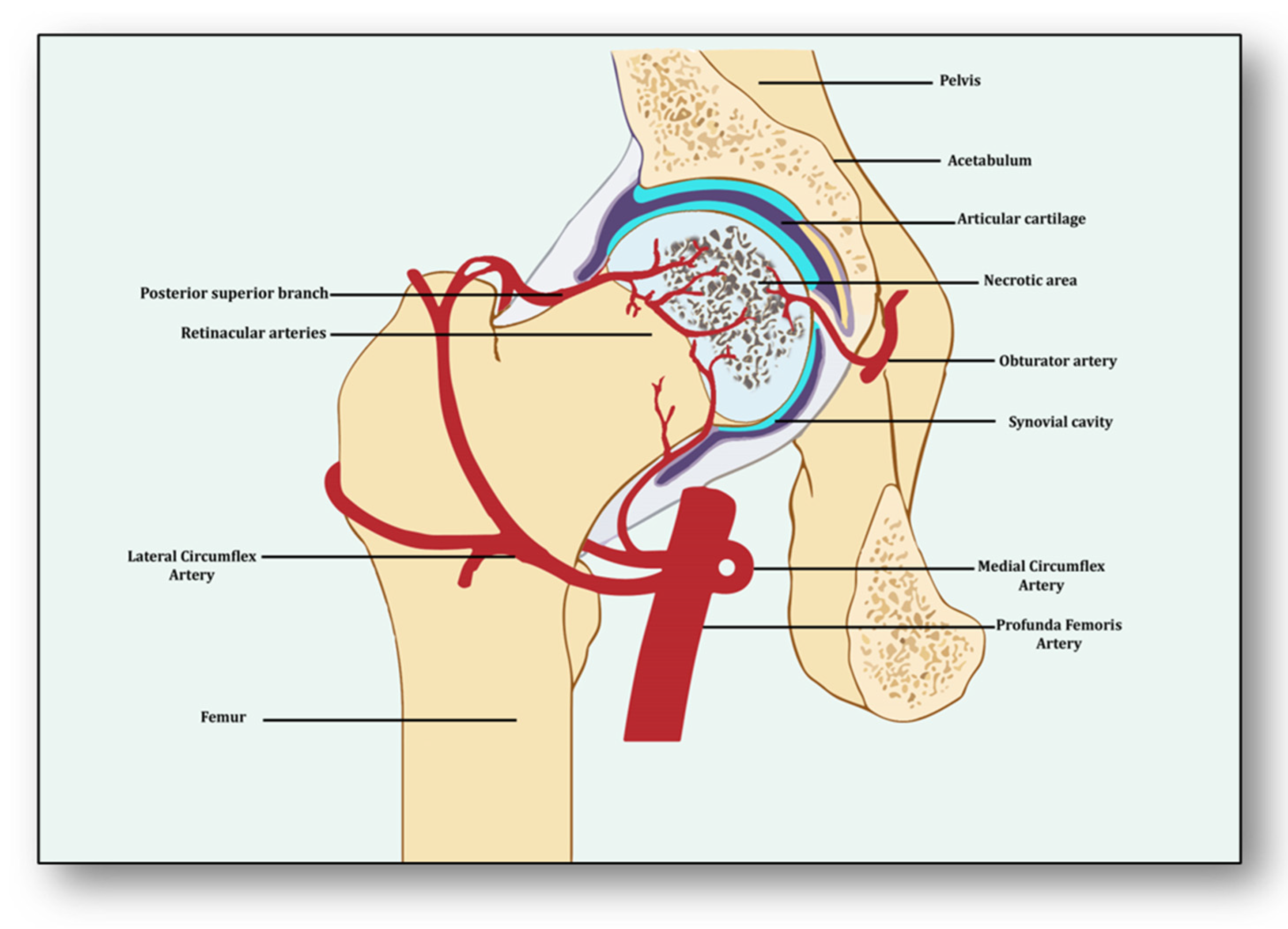
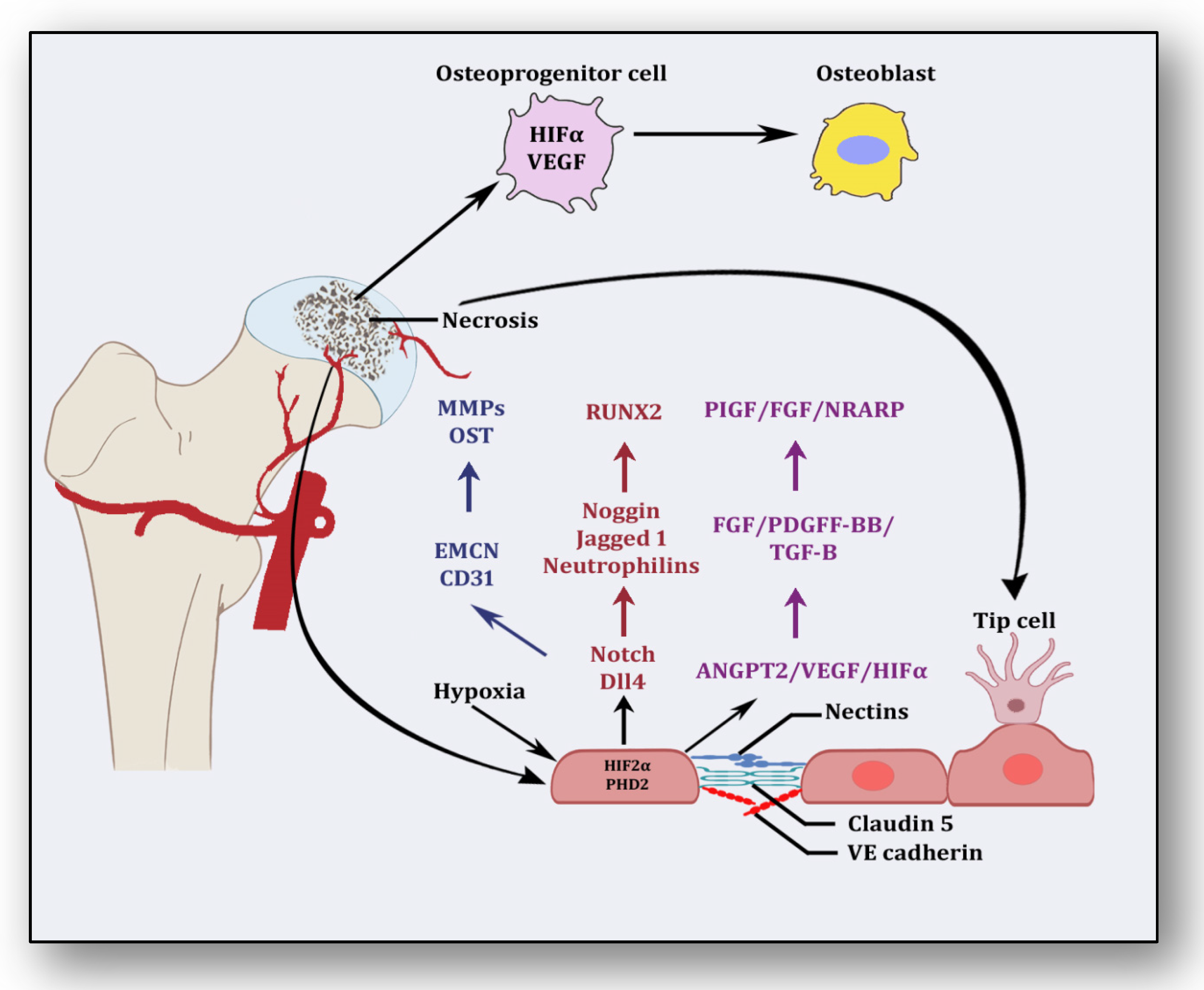

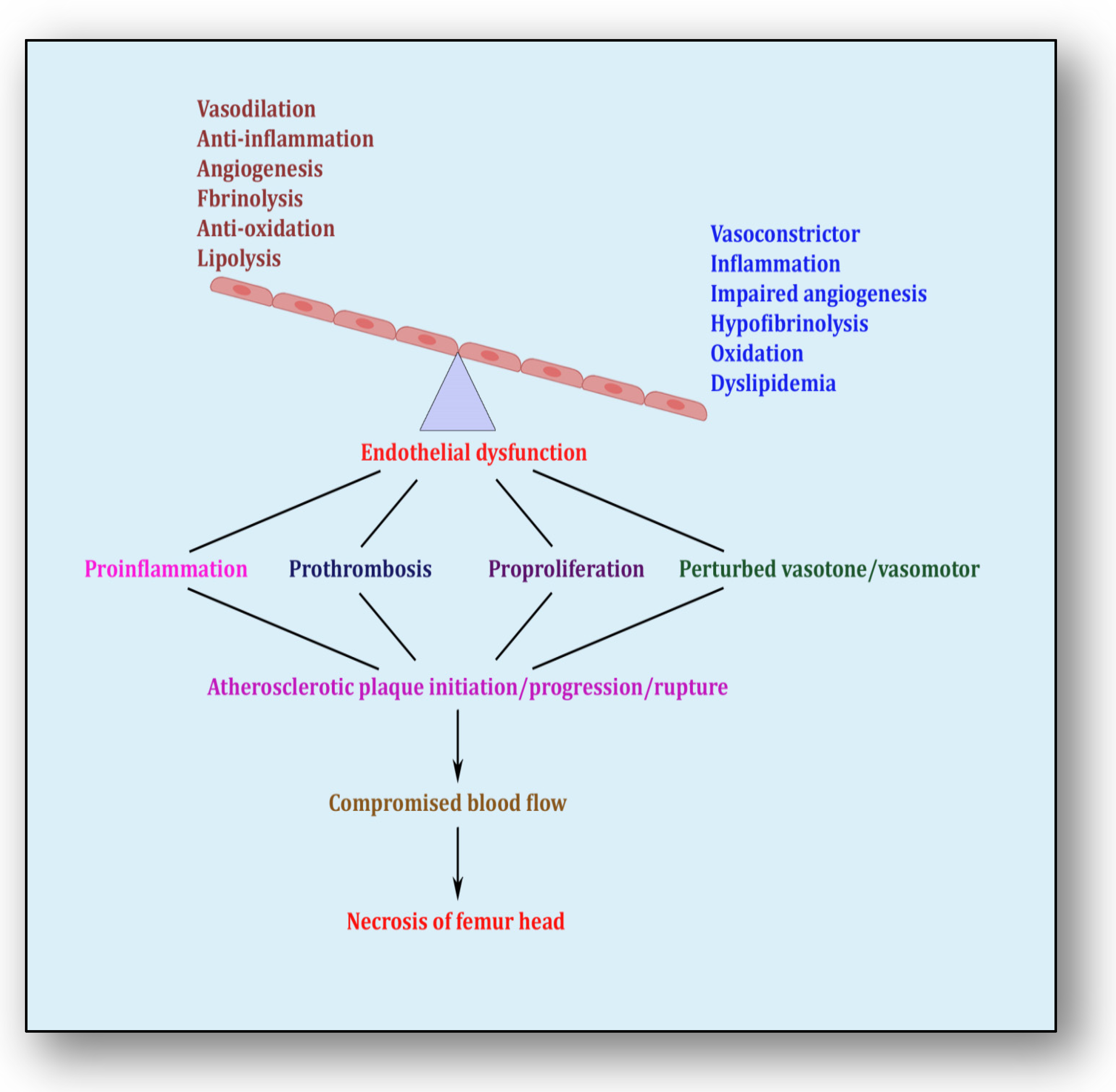

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Therapy | Methodology | Functional Output | Authors |
|---|---|---|---|---|
| 1. | Reperfusion therapy | Crystalloid fluid resuscitation. | Reperfusion therapy enhanced angiogenesis in a rat model of hemorrhagic shock. | Li et al. [108] |
| 2. | BMSC-derived Li-exosome therapy | Surgical implantation of extracellular matrix-mimicking hydrogels infused engineered exosome. | BMSC-derived Li exosomes increased osteogenesis and angiogenesis in rat models of GIONFH. | Chen et al. [109] |
| 3. | Vitamin B2 therapy | Intramuscular injection. | Vitamin B2 promoted angiogenesis in a rat model of GIONFH. | Guo et al. [110] |
| 4. | Gene therapy | Targeted delivery of pro-angiogenic factors via plasmids. | Gene therapy induced angiogenesis in patients of RA and SLE. | Ren et al. [111] |
| 5. | Stem-cell therapy | Targeted delivery of marrow-derived and genetically modified stem cells. | MSCs triggered angiogenesis in various pre-clinical and clinical phases of RA. | Sarsenova et al. [112] |
| 6. | Hydrogel-based VEGF therapy | Intraperitoneal injection. | VEGF initiated angiogenesis in a rat model of MRONJ. | Sharma et al. [113] |
| 7. | BD-2 therapy | Targeted implantation of BC-ALG-BD2 hydrogel membranes. | BD-2 prompted angiogenesis in a rat model of a calvarial defect. | Yuan et al. [114] |
| 8. | HBOT | Oxygen administration at a pressure greater than atmospheric pressure. | HBOT developed angiogenesis in a randomized clinical trial of patients with STEMI. | Martin-Hernandez et al. [115] |
| 9. | Growth factor therapy | Targeted implantation of cryogels infused with VEGF and BMP-4. | Coupled growth factor therapy initiated angiogenesis in a mouse model of cranial defect. | Lee et al. [116] |
| 10. | CD34+ stem-cell-derived exosome therapy | Intravenous injection. | CD34+ stem-cell-derived exosomes triggered angiogenesis in a rat model of ONFH. | Zuo et al. [117] |
| 11. | LLLT/PBM | Electromagnetic beam of a particular frequency and wavelength. | LLLT started angiogenesis in a randomized clinical trial of patients with STEMI. | Elbaz-Greener et al. [118] |
| 12. | Combined growth factor therapy | Subcutaneous implantation of VEGF-BMP-2- and FGF-2-BMP-2-loaded composite scaffolds. | Combined growth factor therapy prompted angiogenesis in a rat model of a calvarial defect. | Kuttapan et al. [119] |
| 13. | b-FGF therapy | Targeted intravenous infusion. | b-FGF initiated angiogenesis for fracture repair of the femur in a mouse model. | Zhang et al. [120] |
| 14. | iPS-MSC-Exo therapy | Intravenous infusion. | iPS-MSC-Exo stimulated angiogenesis in a rat model of ONFH. | Liu et al. [121] |
| 15. | Butyl 10-undecenoate therapy | Oral administration. | Butyl 10-undecenoate therapy triggered angiogenesis in a distraction osteogenesis rat model. | Ozdel et al. [122] |
| 16. | miRNA therapy | Targeted intravenous infusions. | miR-132 induced angiogenesis in a hind–limb ischemia mouse model. | Gomes et al. [123] |
| 17. | BMP-2 therapy | Targeted intravenous administration. | BMP-2 promoted angiogenesis in a rat model of a bone segmental defect. | Kumar et al. [124] |
| 18. | PDGF therapy | Targeted intravenous injection. | PDGF prompted angiogenesis for fracture repair of the tibia in a rat model. | Hollinger et al. [125] |
| 19. | Erythropoietin therapy | Targeted intravenous administration. | Erythropoietin enhanced angiogenesis for fracture repair of the right femur in a mouse model. | Holstein et al. [126] |
| 20. | Dual growth factor therapy | Targeted delivery of BMP-2 and VEGF via retroviral vectors. | Dual growth factor therapy promoted angiogenesis in a mouse model of calvarial defects. | Peng et al. [127] |
| 21. | VEGF therapy | Targeted intravenous infusion. | Targeted VEGF therapy induced angiogenesis in RA patients. | Ballara et al. [128] |
| 22. | PPS therapy | Oral administration. | PPS initiated fibrinolysis in a non-randomized trial of patients with knee osteoarthritis. | Liu et al. [129] |
| 23. | Dual-antiplatelet therapy | Oral administration of clopidogrel combined with aspirin. | Dual-antiplatelet therapy enhanced thrombolysis in a randomized trial of elderly patients with symptomatic ICAS. | Song et al. [130] |
| 24. | Alteplase | Intravenous infusion. | Standard-dose alteplase increased fibrinolysis in acute-ischemic stroke patients in a clinical trial. | Wang et al. [131] |
| 24. | Heparin therapy | Oral and intravenous administration. | Unfractioned heparin helped to induce thrombolysis in non-STEMI patients. | Tashani et al. [132] |
| 25. | Urokinase | Black phosphorous nanosheet-loaded intravenous infusion. | Urokinase helped to enhance fibrinolysis in a mouse model of middle-cerebral artery occlusion. | Wang et al. [133] |
| 26. | Tenecteplase therapy | Intravenous administration. | Tenecteplase treatment helped to promote thrombolysis in acute ischemic stroke patients. | Tsivgoulis et al., 2022, [134] |
| 27. | Rivaroxaban therapy | Oral administration. | Rivarobaxan promoted thrombolysis in a randomized trial of chronic coronary syndrome patients. | Adik-Pathak et al. [135] |
| 28. | Fondaparinux therapy | Subcutaneous injection. | Fondaparinux enhanced thrombolysis in acute coronary syndrome patients. | Khan et al. [136] |
| 29. | Streptokinase therapy | Intravenous administration. | Streptokinase promoted thrombolysis in STEMI patients. | Koh et al. [137] |
| 30. | Coupled anticoagulant therapy | Oral administration of etanercept combined with celecoxib. | Coupled anticoagulant therapy helps to induce thrombolysis in a randomized trial of patients with ankylosing spondylitis. | Tu et al. [138] |
| 31. | Fibrinolytic factor therapy | Targeted delivery of different fibrinolytic factors. | Fibrinolytic factor therapy promoted fibrinolysis in mouse models of various bone-diseases. | Okada et al. [139] |
| 32. | Enoxaparin therapy | Direct oral administration. | Enoxaparin reduced hypofibrinolysis in a case report of a patient with ONFH. | Haydock et al. [140] |
| 33. | tPA therapy | Intravenous infusions, hydrogels, liposome systems. | tPA administered via liposomal drug delivery systems induced thrombolysis in ischemic stroke patients. | Fukuta et al. [141] |
| 34. | NK1R antagonists | Oral administration. | Aprepitant stimulated fibrinolysis in patients with RA. | Liu et al. [142] |
| 35. | MQEP therapy | Oral administration. | MQEP helped to induce fibrinolysis in patients with non-traumatic ONFH. | Li et al. [143] |
| 36. | Desmoteplase therapy | Intravenous infusion. | Desmoteplase helped to promote thrombolysis in acute ischemic stroke patients. | Li et al. [144] |
| 37. | Reteplase therapy | Intravenous injection. | Reteplase increased thrombolysis in acute ischemic stroke patients. | Ozluer et al. [145] |
| 38. | Vitamin E therapy | Oral and intravenous delivery. | Vitamin E helped to start fibrinolysis in osteoarthritis patients. | Li et al. [146] |
| 39. | NO donors | Oral, sublingual and intravenous administration. | NO donors helped to increase endothelial function in ischemia-reperfusion injury in multiple randomized clinical trials. | Dou et al. [147] |
| 40. | Dimethyloxalylglycine | Intravenous infusion. | Dimethyloxalylglycine enhanced endothelial function in a rat model of ONFH. | Shao et al. [148] |
| 41. | CircHIPK3 therapy | Targeted intravenous injection. | CircHIPK3 improved endothelial function in patients with ONFH. | Peng et al. [149] |
| 42. | Chromolaena odarata therapy | Oral administration of aqueous extract. | Chromolaena odarata extract helped to induce endothelial function in a rat model of ONFH. | Nguenum et al. [150] |
| 43. | Icariin therapy | Oral administration. | Icariin helped to increase endothelial function in osteonecrosis and osteoporosis patients. | Zhang et al. [151] |
| 44. | Statins | Oral administration. | Statins improved endothelial function in ONFH patients and in vivo studies. | Yu et al. [152] |
| 45. | Tissue regeneration therapy | Targeted delivery of BMP-2 via PEM-coated scaffolds. | Tissue regeneration therapy enhanced endothelial function in a rat model of calvarial defects. | Martin et al. [153] |
| 46. | PTEN inhibitors | Intravenous infusion. | VO-OHpic reduced endothelial dysfunction in an in vivo study of an ONFH animal model. | Yao et al. [154] |
| 47. | L-Arg therapy | Oral and intravenous administration. | L-Arg promoted endothelial function in ischemic diseases in various clinical trials. | Gamberdella et al. [155] |
| 48. | ROS inhibitors | Oral and intravenous infusion. | ROS inhibitors attenuated endothelial dysfunction in multiple bone disorders. | Agidigbi et al. [156] |
| 49. | Se@SiO2 nanocomposites therapy | Intraperitoneal injection. | Se@SiO2 nanocomposites lessened endothelial dysfunction in rat models of ONFH. | Deng et al. [157] |
| 50. | Antioxidant therapy | Oral and intravenous administration. | Antioxidant therapy improved endothelial function in hip fracture patients. | Sprague et al. [158] |
| 51. | ARBs | Oral and intravenous infusion. | ARBs helped to reduce endothelial dysfunction in various clinical trials of different cardiovascular diseases. | Radenkovic et al. [159] |
| 52. | ACE inhibitors | Oral administration. | ACE inhibitors increased endothelial function in a randomized controlled trial of T2DM patients with myocardial infarction. | Sun et al. [160] |
| 53. | ET-1 therapy | Intravenous injection. | ET-1 improved endothelial function in in vivo studies using recombinant endothelial progenitor cells and osteoblasts. | Wang et al. [161] |
 Indicates pro-angiogenic therapies Indicates pro-angiogenic therapies  Indicates fibrinolytic therapies Indicates fibrinolytic therapies  Therapies for improving endothelial function. Therapies for improving endothelial function. | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, M.; Singh, B.; Sharma, K.; Kumar, N.; Mastana, S.; Singh, P. A Molecular Troika of Angiogenesis, Coagulopathy and Endothelial Dysfunction in the Pathology of Avascular Necrosis of Femoral Head: A Comprehensive Review. Cells 2023, 12, 2278. https://doi.org/10.3390/cells12182278
Singh M, Singh B, Sharma K, Kumar N, Mastana S, Singh P. A Molecular Troika of Angiogenesis, Coagulopathy and Endothelial Dysfunction in the Pathology of Avascular Necrosis of Femoral Head: A Comprehensive Review. Cells. 2023; 12(18):2278. https://doi.org/10.3390/cells12182278
Chicago/Turabian StyleSingh, Monica, Baani Singh, Kirti Sharma, Nitin Kumar, Sarabjit Mastana, and Puneetpal Singh. 2023. "A Molecular Troika of Angiogenesis, Coagulopathy and Endothelial Dysfunction in the Pathology of Avascular Necrosis of Femoral Head: A Comprehensive Review" Cells 12, no. 18: 2278. https://doi.org/10.3390/cells12182278
APA StyleSingh, M., Singh, B., Sharma, K., Kumar, N., Mastana, S., & Singh, P. (2023). A Molecular Troika of Angiogenesis, Coagulopathy and Endothelial Dysfunction in the Pathology of Avascular Necrosis of Femoral Head: A Comprehensive Review. Cells, 12(18), 2278. https://doi.org/10.3390/cells12182278