
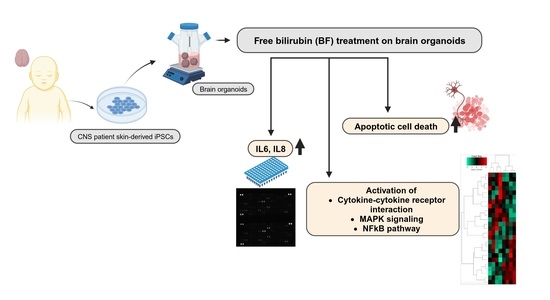
Free Bilirubin Induces Neuro-Inflammation in an Induced Pluripotent Stem Cell-Derived Cortical Organoid Model of Crigler-Najjar Syndrome



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
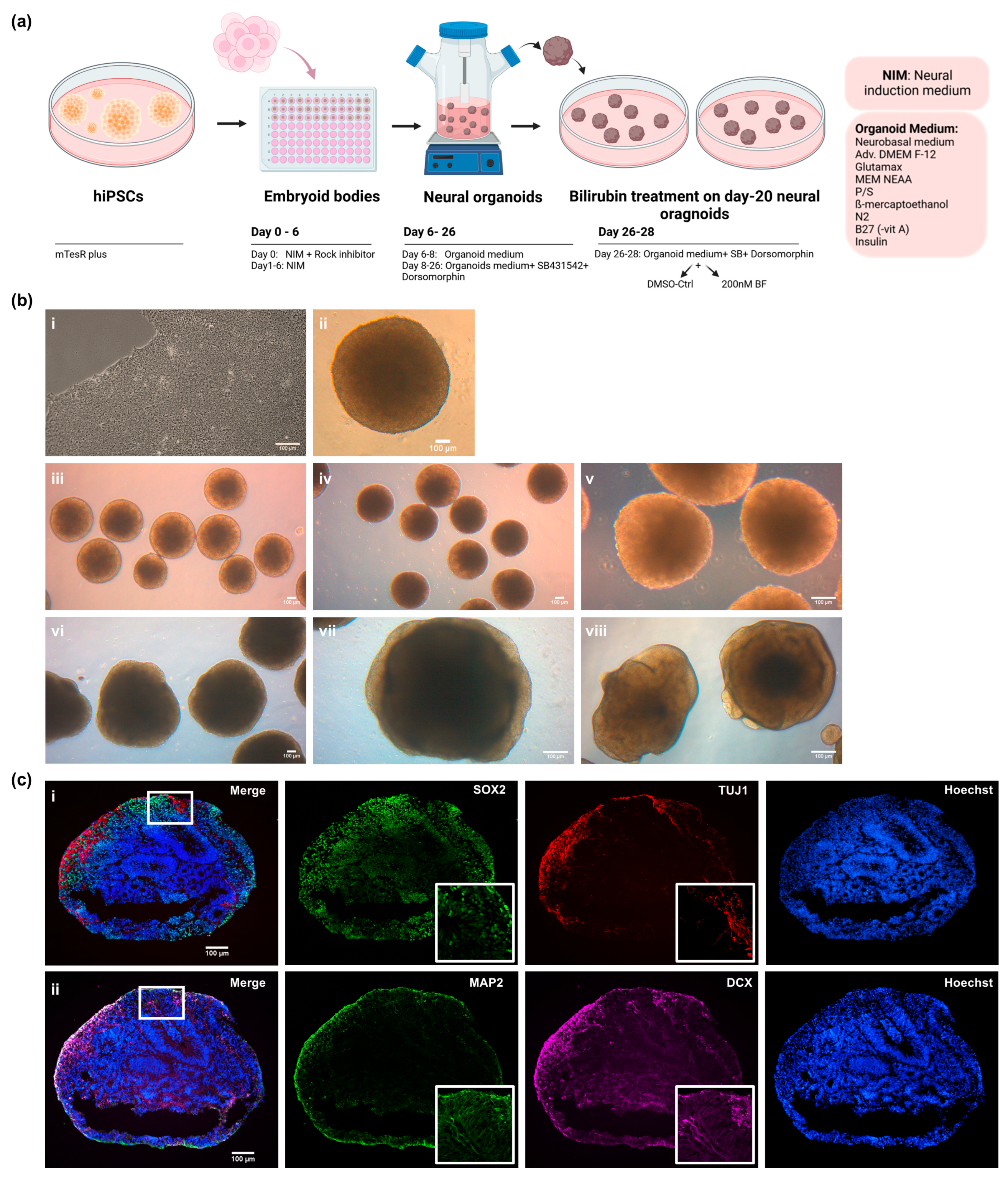
2.1. Cell Cultivation, Formation of Neural Cortical Organoids, and Bilirubin Treatment on Cortical Organoids
2.2. Cryosectioning
2.3. Immunocytochemistry
2.4. TUNEL Assay
2.5. Reverse Transcriptase PCR (RT-PCR)
2.6. Human XL Cytokine Assay
2.7. Image and Data Analysis of the Human XL Cytokine Array
2.8. Analysis of Gene Expression Data
2.9. Analysis of Pathways and Gene Ontologies (GOs)
2.10. Western Blotting
2.11. Statistical Analysis
2.12. Measurement of Cytochrome P450 Activity
3. Results
3.1. iPSC-Derived Cortical Organoids Show Typical Cortical Neuronal Features
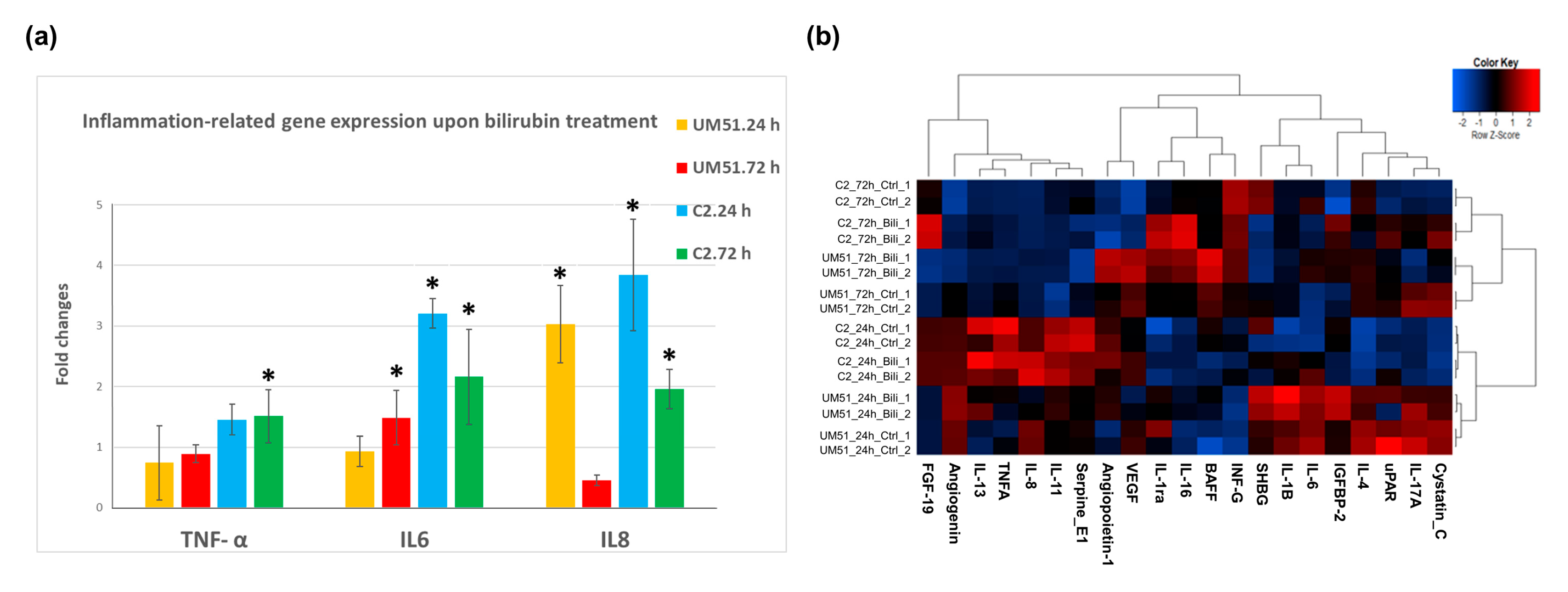
3.2. Bilirubin (BF)-Induced Neuro-Inflammation with Elevated Expression of Pro-Inflammatory Cytokines
3.3. Distinct Inflammation-Associated Pathways Are Activated by BF in Cortical Organoids
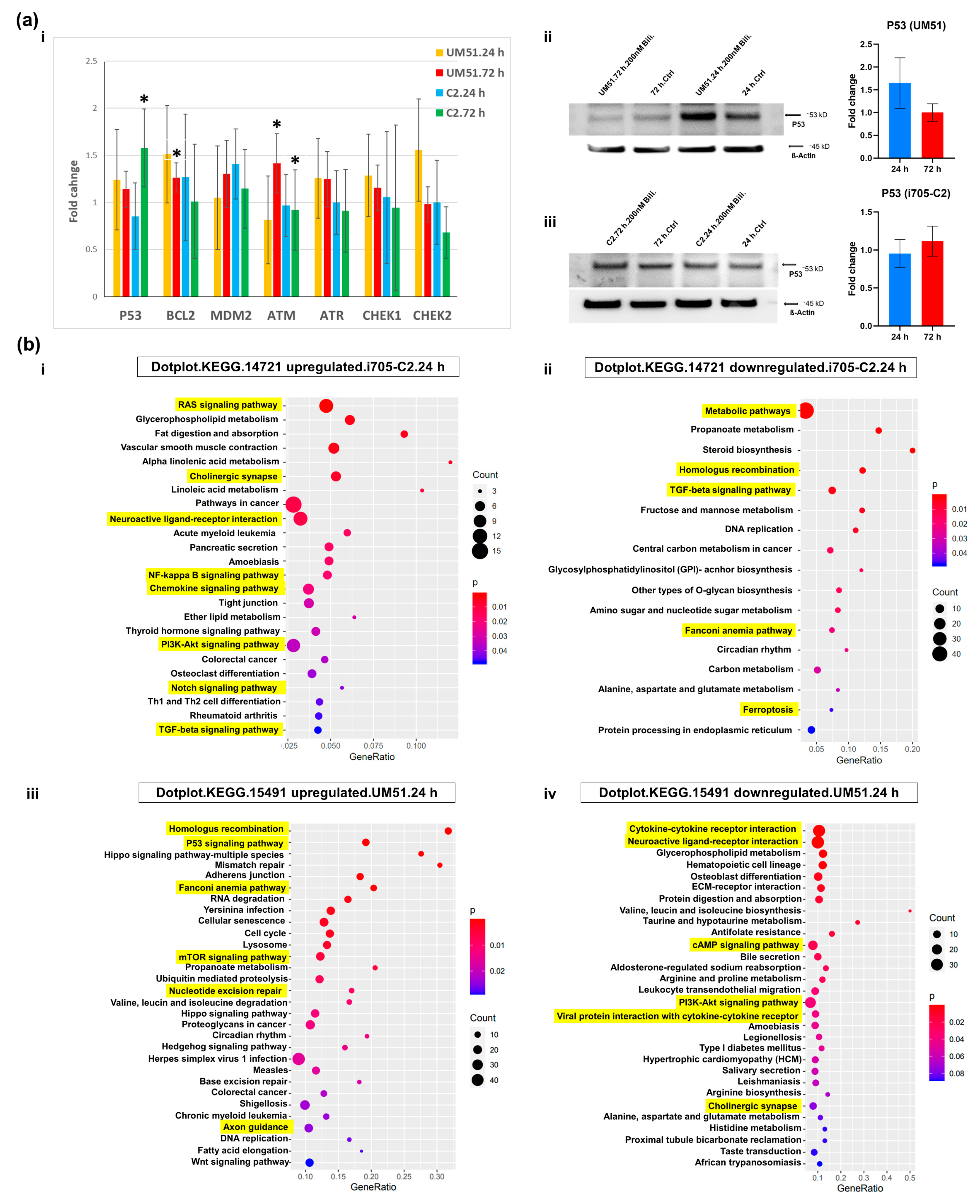
3.4. BF Treatment Differentially Regulates DNA Damage and Repairs-Related Pathways
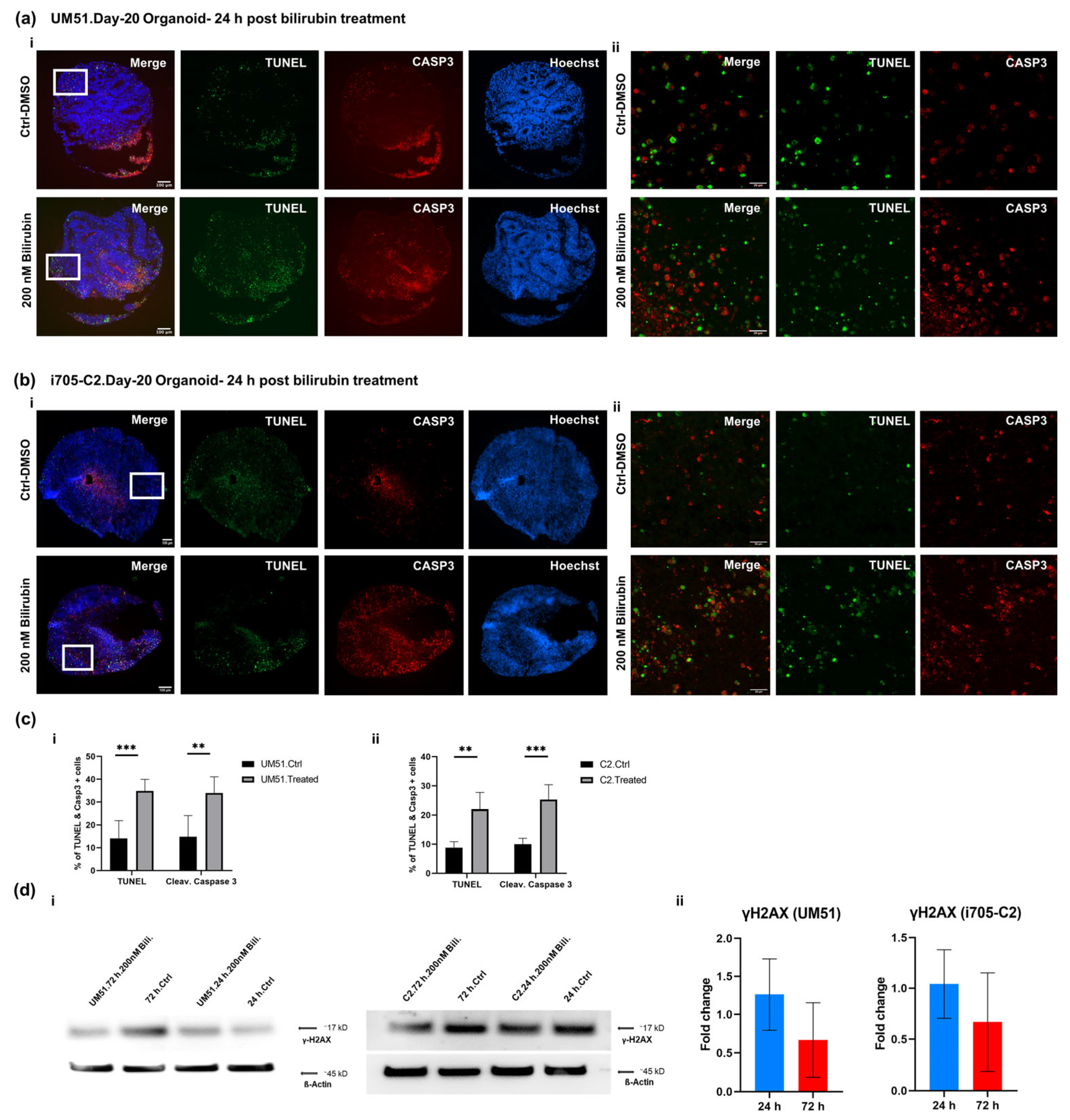
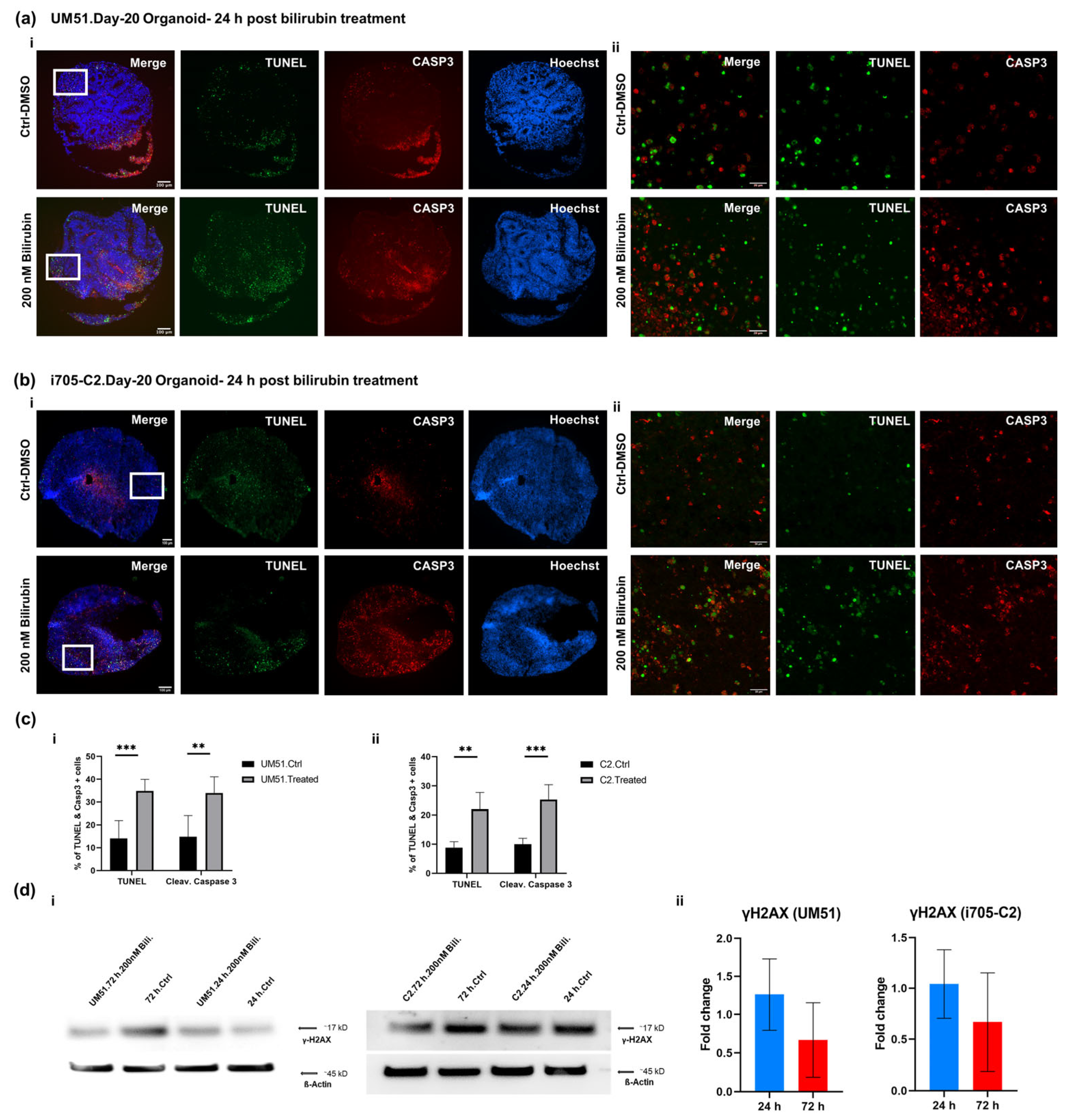
3.5. BF Induces Apoptotic Cell Death in Cortical Organoid
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Bortolussi, G.; Muro, A.F. Advances in understanding disease mechanisms and potential treatments for Crigler–Najjar syndrome. Expert Opin. Orphan Drugs 2018, 6, 425–439. [Google Scholar] [CrossRef]
- Watchko, J.F.; Tiribelli, C. Bilirubin-Induced Neurologic Damage-Mechanisms and Management Approaches. N. Engl. J. Med. 2013, 369, 2021–2030. [Google Scholar] [CrossRef] [PubMed]
- Brites, D. The Evolving Landscape of Neurotoxicity by Unconjugated Bilirubin: Role of Glial Cells and Inflammation. Front. Pharmacol. 2012, 3, 88. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Falcão, A.S.; Silva, R.F.M.; Gordo, A.C.; Gama, M.J.; Brito, M.A.; Brites, D. Inflammatory signalling pathways involved in astroglial activation by unconjugated bilirubin. J. Neurochem. 2006, 96, 1667–1679. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.M. Definition of the Clinical Spectrum of Kernicterus and Bilirubin-Induced Neurologic Dysfunction (BIND). J. Perinatol. 2004, 25, 54–59. [Google Scholar] [CrossRef]
- Canu, G.; Minucci, A.; Zuppi, C.; Capoluongo, E. Gilbert and Crigler Najjar syndromes: An update of the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene mutation database. Blood Cells Mol. Dis. 2013, 50, 273–280. [Google Scholar] [CrossRef]
- Memon, N.; I Weinberger, B.; Hegyi, T.; Aleksunes, L.M. Inherited disorders of bilirubin clearance. Pediatr. Res. 2015, 79, 378–386. [Google Scholar] [CrossRef]
- Pranty, A.I.; Shumka, S.; Adjaye, J. Bilirubin-Induced Neurological Damage: Current and Emerging iPSC-Derived Brain Organoid Models. Cells 2022, 11, 2647. [Google Scholar] [CrossRef]
- Singh, A.; Jialal, I. Unconjugated Hyperbilirubinemia. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Yueh, M.F.; Chen, S.; Nguyen, N.; Tukey, R.H. Developmental Onset of Bilirubin-induced Neurotoxicity Involves Toll-like Receptor 2-dependent Signaling in Humanized UDP-glucuronosyltransferase1 Mice. J. Biol. Chem. 2014, 289, 4699–4709. [Google Scholar] [CrossRef]
- Gidáli, J.; Poros, A.; Harsányi, V.; Fehér, I.; Mód, A.; Hollán, Z. Granulocyte-macrophage progenitor cells in allogenic bone marrow transplantation: Correlation of progenitor cell content and regeneration in the graft. Orvosi Hetil. 1989, 130, 2251–2254. [Google Scholar]
- Vodret, S.; Bortolussi, G.; Jašprová, J.; Vitek, L.; Muro, A.F. Inflammatory signature of cerebellar neurodegeneration during neonatal hyperbilirubinemia in Ugt1 -/- mouse model. J. Neuroinflamm. 2017, 14, 1–16. [Google Scholar] [CrossRef]
- Vaz, A.R.; Silva, S.L.; Barateiro, A.; Falcão, A.S.; Fernandes, A.; Brito, M.A.; Brites, D. Selective vulnerability of rat brain regions to unconjugated bilirubin. Mol. Cell. Neurosci. 2011, 48, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Falcão, A.S.; Silva, R.F.M.; Vaz, A.R.; Gomes, C.; Fernandes, A.; Barateiro, A.; Tiribelli, C.; Brites, D. Cross-Talk Between Neurons and Astrocytes in Response to Bilirubin: Adverse Secondary Impacts. Neurotox. Res. 2013, 26, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Watchko, J.F. Kernicterus and the Molecular Mechanisms of Bilirubin-Induced CNS Injury in Newborns. NeuroMol. Med. 2006, 8, 513–530. [Google Scholar] [CrossRef] [PubMed]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res. 2003, 283, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Nirmala, J.G.; Lopus, M. Cell death mechanisms in eukaryotes. Cell Biol. Toxicol. 2019, 36, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Doré, S.; Takahashi, M.; Ferris, C.D.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef] [PubMed]
- Graffmann, N.; Martins, S.; Ljubikj, T.; Matte, J.C.; Bohndorf, M.; Wruck, W.; Adjaye, J. Generation of a Crigler-Najjar Syndrome Type I patient-derived induced pluripotent stem cell line CNS705 (HHUUKDi005-A). Stem Cell Res. 2021, 51, 102167. [Google Scholar] [CrossRef]
- Bohndorf, M.; Ncube, A.; Spitzhorn, L.-S.; Enczmann, J.; Wruck, W.; Adjaye, J. Derivation and characterization of integration-free iPSC line ISRM-UM51 derived from SIX2-positive renal cells isolated from urine of an African male expressing the CYP2D6 *4/*17 variant which confers intermediate drug metabolizing activity. Stem Cell Res. 2017, 25, 18–21. [Google Scholar] [CrossRef]
- Gabriel, E.; Gopalakrishnan, J. Generation of iPSC-derived Human Brain Organoids to Model Early Neurodevelopmental Disorders. J. Vis. Exp. 2017, e55372. [Google Scholar] [CrossRef]
- Graffmann, N.; Ring, S.; Kawala, M.-A.; Wruck, W.; Ncube, A.; Trompeter, H.-I.; Adjaye, J. Modeling Nonalcoholic Fatty Liver Disease with Human Pluripotent Stem Cell-Derived Immature Hepatocyte-Like Cells Reveals Activation of PLIN2 and Confirms Regulatory Functions of Peroxisome Proliferator-Activated Receptor Alpha. Stem Cells Dev. 2016, 25, 1119–1133. [Google Scholar] [CrossRef] [PubMed]
- Wruck, W.; Boima, V.; Erichsen, L.; Thimm, C.; Koranteng, T.; Kwakyi, E.; Antwi, S.; Adu, D.; Adjaye, J. Urine-Based Detection of Biomarkers Indicative of Chronic Kidney Disease in a Patient Cohort from Ghana. J. Pers. Med. 2022, 13, 38. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [PubMed]
- Du, P.; Kibbe, W.A.; Lin, S.M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [PubMed]
- Abu-Dawud, R.; Graffmann, N.; Ferber, S.; Wruck, W.; Adjaye, J. Pluripotent stem cells: Induction and self-renewal. Philos. Trans. R. Soc. B Biol. Sci. 2018, 373, 20170213. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Carvalho, B.S.; Irizarry, R.A. A framework for oligonucleotide microarray preprocessing. Bioinformatics 2010, 26, 2363–2367. [Google Scholar] [CrossRef]
- Chen, H.; Boutros, P.C. VennDiagram: A package for the generation of highly-customizable Venn and Euler diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- Warnes, G.R. Gplots: Various R Programming Tools for Plotting Data; ScienceOpen, Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Falcon, S.; Gentleman, R. Using GOstats to test gene lists for GO term association. Bioinformatics 2006, 23, 257–258. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef]
- Palmela, I.; Cardoso, F.L.; Bernas, M.; Correia, L.; Vaz, A.R.; Silva, R.F.M.; Fernandes, A.; Kim, K.S.; Brites, D.; Brito, M.A. Elevated levels of bilirubin and long-term exposure impair human brain microvascular endothelial cell integrity. Curr. Neurovascular Res. 2011, 8, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Allan, S.M.; Rothwell, N.J. Cytokines and acute neurodegeneration. Nat. Rev. Neurosci. 2001, 2, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Smith, C.J.; Van Eldik, L.J. Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia 2003, 45, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, N.J. Cytokines—Killers in the brain? J. Physiol. 1999, 514, 3–17. [Google Scholar] [CrossRef]
- Morandi, A.; Hughes, C.G.; Girard, T.D.; McAuley, D.F.; Ely, E.W.; Pandharipande, P.P. Statins and Brain Dysfunction: A Hypothesis to Reduce the Burden of Cognitive Impairment in Patients Who Are Critically Ill. Chest 2011, 140, 580–585. [Google Scholar] [CrossRef]
- Daulatzai, M.A. Dysfunctional Nucleus Tractus Solitarius: Its Crucial Role in Promoting Neuropathogentic Cascade of Alzheimer’s Dementia—A Novel Hypothesis. Neurochem. Res. 2012, 37, 846–868. [Google Scholar] [CrossRef]
- Gazzin, S.; Tiribelli, C. Bilirubin-induced neurological damage. J. Matern. Neonatal Med. 2011, 24, 154–155. [Google Scholar] [CrossRef] [PubMed]
- Jeanjean, A.; Moussaoui, S.; Maloteaux, J.-M.; Laduron, P. Interleukin-1β induces long-term increase of axonally transported opiate receptors and substance P. Neuroscience 1995, 68, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, A.; Feige, U.; Fontana, A.; Müller, K.; Dinarello, C.A. Interleukin-1 enhances pain reflexes. Mediation through increased prostaglandin E2 levels. Inflamm. Res. 1988, 25, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, S.; Zhao, Z.; Luo, Y.; Hou, Y.; Li, H.; He, L.; Zhou, L.; Wu, W. Effect of VEGF on Inflammatory Regulation, Neural Survival, and Functional Improvement in Rats following a Complete Spinal Cord Transection. Front. Cell. Neurosci. 2017, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, Y.; Li, D.; Liu, Z.; Zhao, Z.; Han, D.; Yuan, Y.; Bi, J.; Mei, X. VEGF inhibits the inflammation in spinal cord injury through activation of autophagy. Biochem. Biophys. Res. Commun. 2015, 464, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Kushiyama, A.; Sakoda, H.; Fujishiro, M.; Yamamotoya, T.; Nakatsu, Y.; Kikuchi, T.; Kaneko, S.; Tanaka, H.; Asano, T. Protective Effect of Sex Hormone-Binding Globulin against Metabolic Syndrome: In Vitro Evidence Showing Anti-Inflammatory and Lipolytic Effects on Adipocytes and Macrophages. Mediat. Inflamm. 2018, 2018, 3062319. [Google Scholar] [CrossRef] [PubMed]
- Brites, D. Bilirubin Injury to Neurons and Glial Cells: New Players, Novel Targets, and Newer Insights. Semin. Perinatol. 2011, 35, 114–120. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kang, Y.-J.; Kim, W.-J.; Woo, D.-K.; Lee, Y.; Kim, D.-I.; Park, Y.B.; Kwon, B.S.; Park, J.-E.; Lee, W.-H. TWEAK Can Induce Pro-Inflammatory Cytokines and Matrix Metalloproteinase-9 in Macrophages. Circ. J. 2004, 68, 396–399. [Google Scholar] [CrossRef]
- Bayer, A.L.; Alcaide, P. MyD88: At the heart of inflammatory signaling and cardiovascular disease. J. Mol. Cell. Cardiol. 2021, 161, 75–85. [Google Scholar] [CrossRef]
- Vorkapic, E.; Lundberg, A.M.; Mäyränpää, M.I.; Eriksson, P.; Wågsäter, D. TRIF adaptor signaling is important in abdominal aortic aneurysm formation. Atherosclerosis 2015, 241, 561–568. [Google Scholar] [CrossRef]
- Fukasaka, M.; Ori, D.; Kawagoe, T.; Uematsu, S.; Maruyama, K.; Okazaki, T.; Kozaki, T.; Imamura, T.; Tartey, S.; Mino, T.; et al. Critical Role of AZI2 in GM-CSF–Induced Dendritic Cell Differentiation. J. Immunol. 2013, 190, 5702–5711. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.; Zheng, Y.; Xu, J.; Sun, Q. AZI2 positively regulates the induction of type I interferon in influenza-trigger pediatric pneumonia. Pathog. Dis. 2022, 80, ftac016. [Google Scholar] [CrossRef] [PubMed]
- Kaltschmidt, B.; Kaltschmidt, C. NF-B in the Nervous System. Cold Spring Harb. Perspect. Biol. 2009, 1, a001271. [Google Scholar] [CrossRef] [PubMed]
- Teng, F.Y.H.; Tang, B.L. NF-κΒ Signaling in Neurite Growth and Neuronal Survival. Rev. Neurosci. 2010, 21, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, H.; Davies, A.M. Regulation of neural process growth, elaboration and structural plasticity by NF-κB. Trends Neurosci. 2011, 34, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Yakovleva, T.; Bazov, I.; Watanabe, H.; Hauser, K.F.; Bakalkin, G. Transcriptional control of maladaptive and protective responses in alcoholics: A role of the NF-κB system. Brain Behav. Immun. 2011, 25, S29–S38. [Google Scholar] [CrossRef]
- Jiménez, A.; Lu, D.; Kalocsay, M.; Berberich, M.J.; Balbi, P.; Jambhekar, A.; Lahav, G. Time-series transcriptomics and proteomics reveal alternative modes to decode p53 oscillations. Mol. Syst. Biol. 2022, 18, e10588. [Google Scholar] [CrossRef] [PubMed]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Bassani, T.B.; Vital, M.A.; Rauh, L.K. Neuroinflammation in the pathophysiology of Parkinson’s disease and therapeutic evidence of anti-inflammatory drugs. Arq. Neuro-Psiquiatria 2015, 73, 616–623. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, X.; Gao, X. Taurine protects against bilirubin-induced neurotoxicity in vitro. Brain Res. 2010, 1320, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Nie, Q.; Gao, M.; Yang, L.; Xiang, J.-W.; Xiao, Y.; Liu, F.-Y.; Gong, X.-D.; Fu, J.-L.; Wang, Y.; et al. The transcription factor CREB acts as an important regulator mediating oxidative stress-induced apoptosis by suppressing αB-crystallin expression. Aging 2020, 12, 13594–13617. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Singh, K.; Almasan, A. Histone H2AX Phosphorylation: A Marker for DNA Damage. Methods Mol. Biol. 2012, 920, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Vucic, D. XIAP at the crossroads of cell death and inflammation. Oncotarget 2018, 9, 27319–27320. [Google Scholar] [CrossRef] [PubMed]
- Shiozaki, E.N.; Chai, J.; Rigotti, D.J.; Riedl, S.J.; Li, P.; Srinivasula, S.M.; Alnemri, E.S.; Fairman, R.; Shi, Y. Mechanism of XIAP-Mediated Inhibition of Caspase-9. Mol. Cell 2003, 11, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Lalani, A.I.; Zhu, S.; Gokhale, S.; Jin, J.; Xie, P. TRAF Molecules in Inflammation and Inflammatory Diseases. Curr. Pharmacol. Rep. 2017, 4, 64–90. [Google Scholar] [CrossRef] [PubMed]
- Perner, F.; Ernst, T.; Heidel, F.H. Roles of JAK2 in Aging, Inflammation, Hematopoiesis and Malignant Transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef]
- Duodu, P.; Sosa, G.; Canar, J.; Chhugani, O.; Gamero, A.M. Exposing the Two Contrasting Faces of STAT2 in Inflammation. J. Interf. Cytokine Res. 2022, 42, 467–481. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pranty, A.I.; Wruck, W.; Adjaye, J. Free Bilirubin Induces Neuro-Inflammation in an Induced Pluripotent Stem Cell-Derived Cortical Organoid Model of Crigler-Najjar Syndrome. Cells 2023, 12, 2277. https://doi.org/10.3390/cells12182277
Pranty AI, Wruck W, Adjaye J. Free Bilirubin Induces Neuro-Inflammation in an Induced Pluripotent Stem Cell-Derived Cortical Organoid Model of Crigler-Najjar Syndrome. Cells. 2023; 12(18):2277. https://doi.org/10.3390/cells12182277
Chicago/Turabian StylePranty, Abida Islam, Wasco Wruck, and James Adjaye. 2023. "Free Bilirubin Induces Neuro-Inflammation in an Induced Pluripotent Stem Cell-Derived Cortical Organoid Model of Crigler-Najjar Syndrome" Cells 12, no. 18: 2277. https://doi.org/10.3390/cells12182277
APA StylePranty, A. I., Wruck, W., & Adjaye, J. (2023). Free Bilirubin Induces Neuro-Inflammation in an Induced Pluripotent Stem Cell-Derived Cortical Organoid Model of Crigler-Najjar Syndrome. Cells, 12(18), 2277. https://doi.org/10.3390/cells12182277