

Dipeptidyl-Aminopeptidases 8 and 9 Regulate Autophagy and Tamoxifen Response in Breast Cancer Cells


Abstract
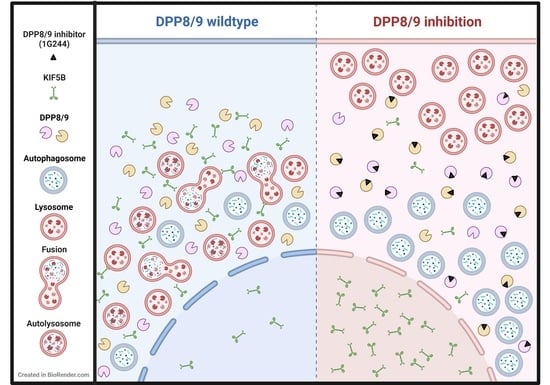
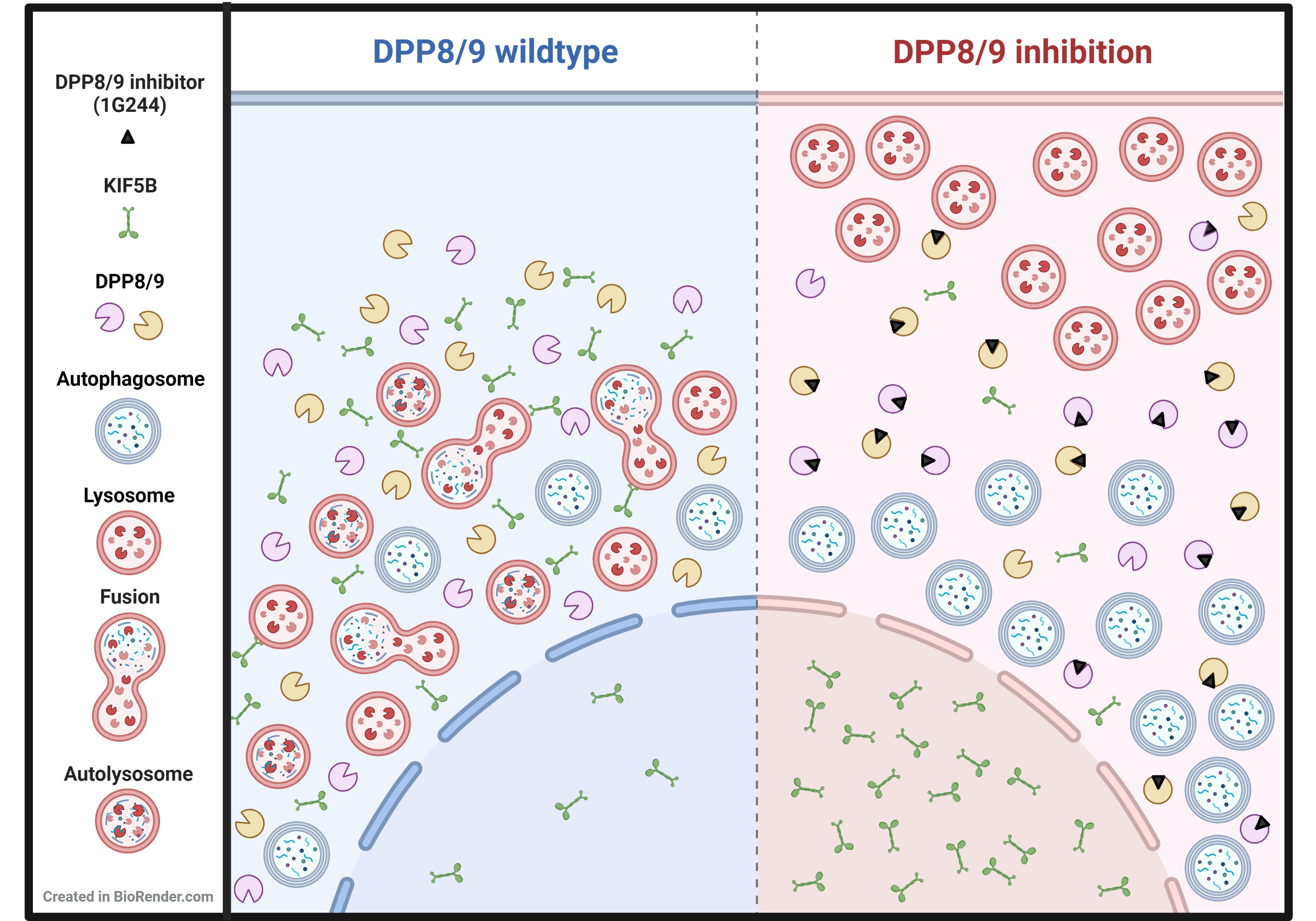
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Isolation and qRT-PCR
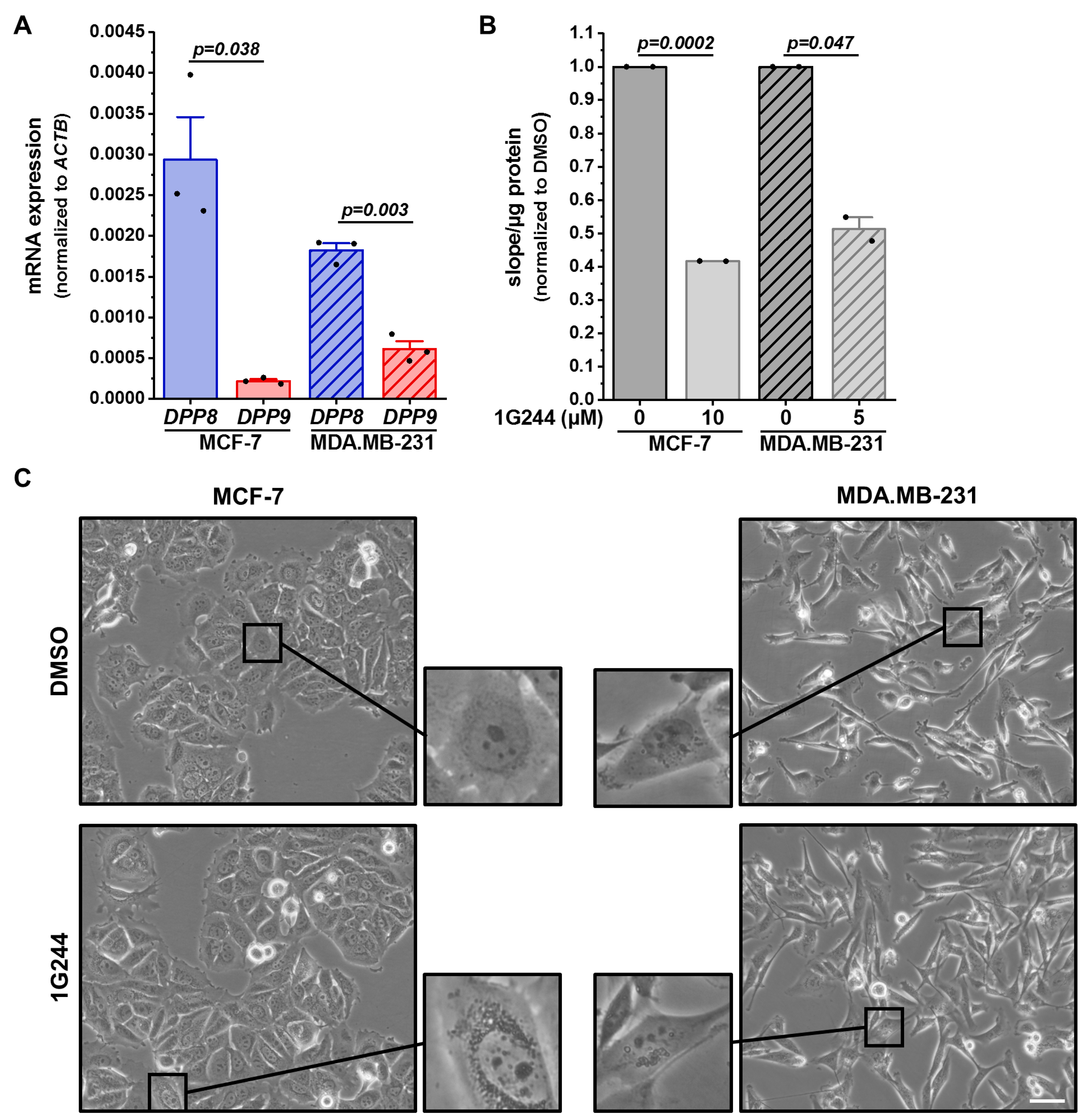
2.3. DPP8/9 Activity Assay
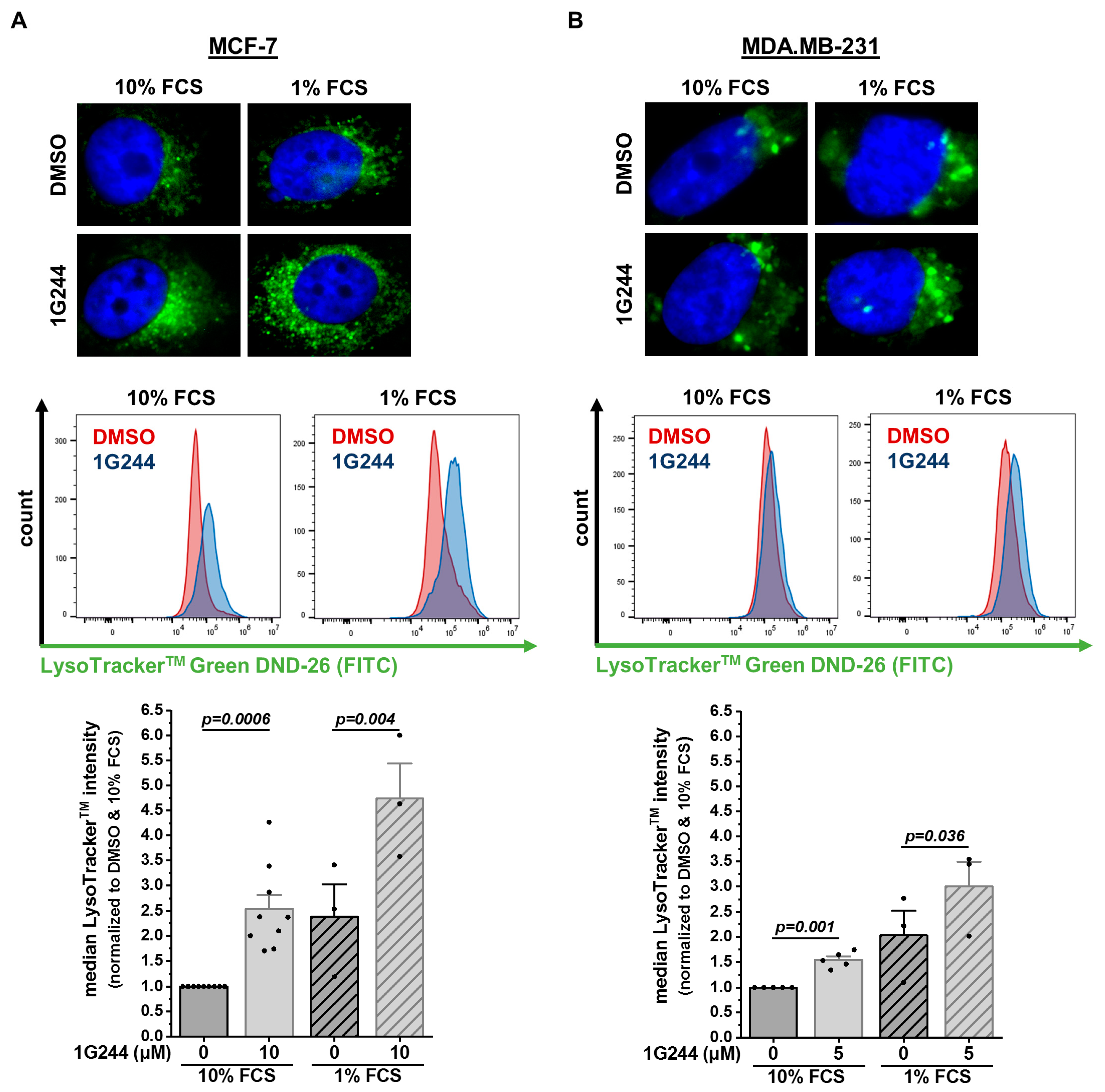
2.4. LysoTrackerTM Staining
2.5. Protein Isolation and Western Blot
2.6. Immunofluorescence
2.7. β-Galactosidase Staining
2.8. Plate Colony-Formation Assay
2.9. Trypan Blue Staining
2.10. FACS
2.11. Data Presentation and Statistics
3. Results
3.1. Inhibition of DPP8/9 Increases Acidic Endolysosomal Compartment in Different Breast Cancer Cell Lines
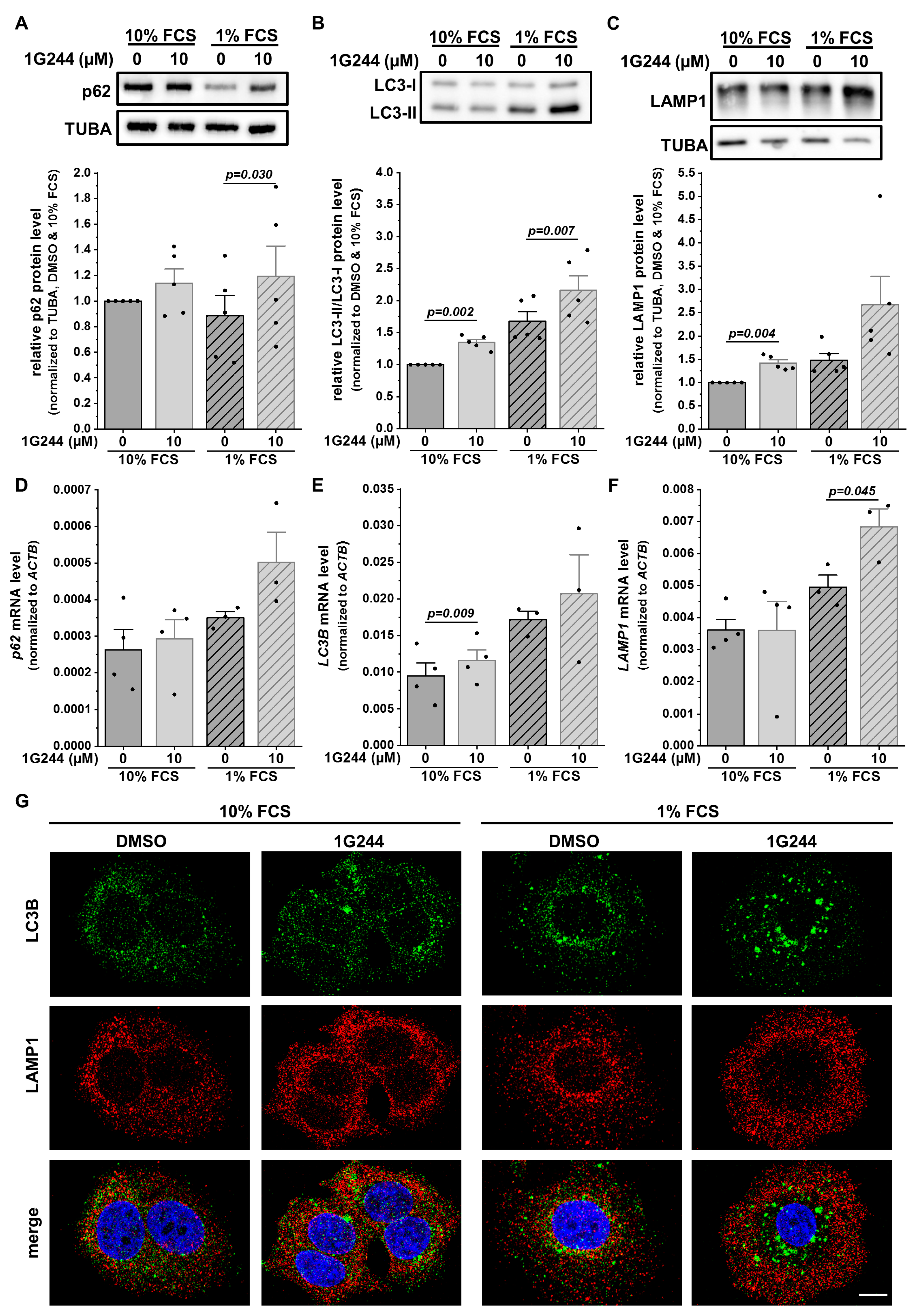
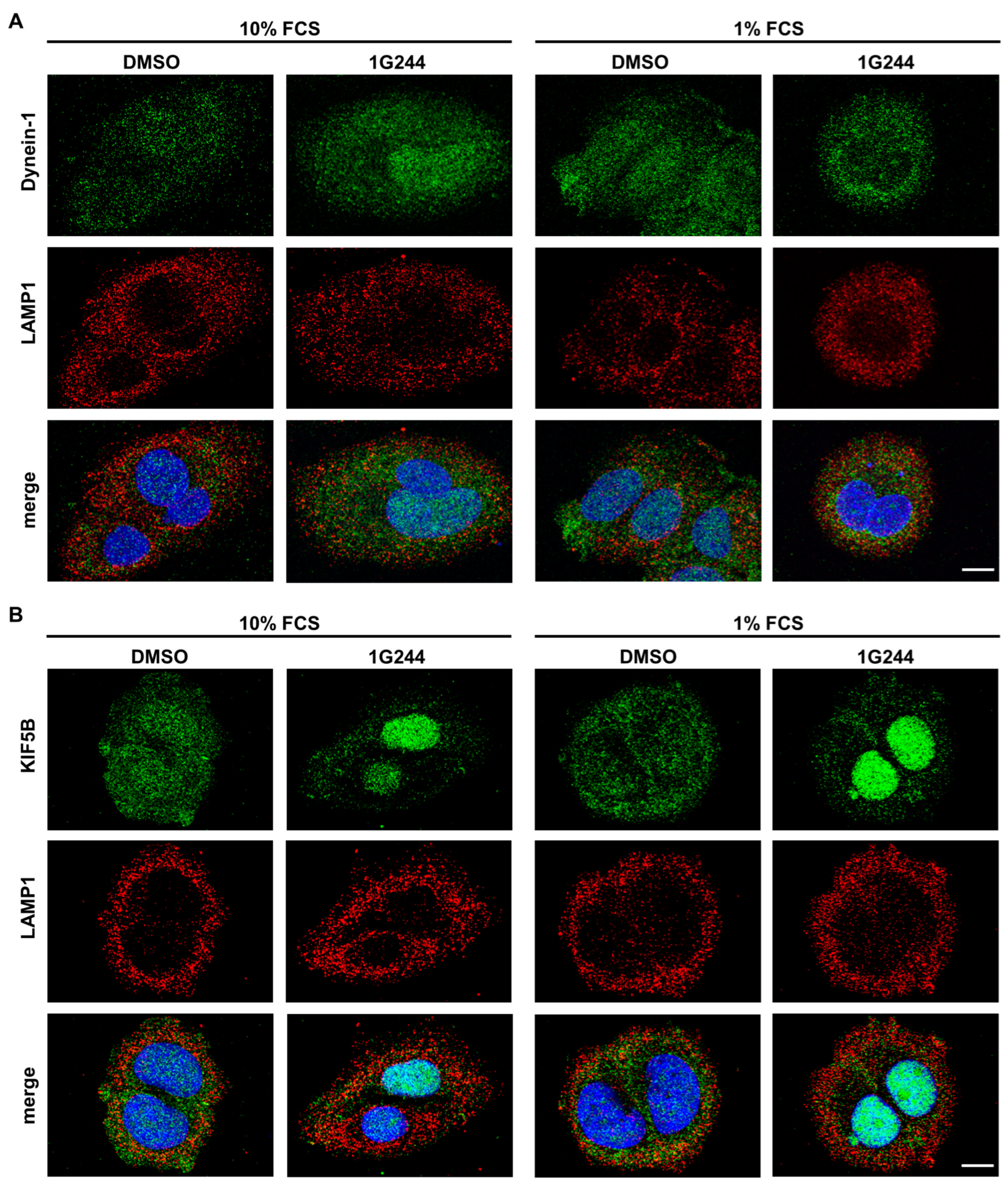
3.2. DPP8/9 Inhibition Dysregulates Lysosomal Positioning and Thereby Autophagy in Luminal Breast Cancer Cells
3.3. Inhibition of DPP8/9 Results in Nucelar Localization of Vesicle Transport Protein KIF5B
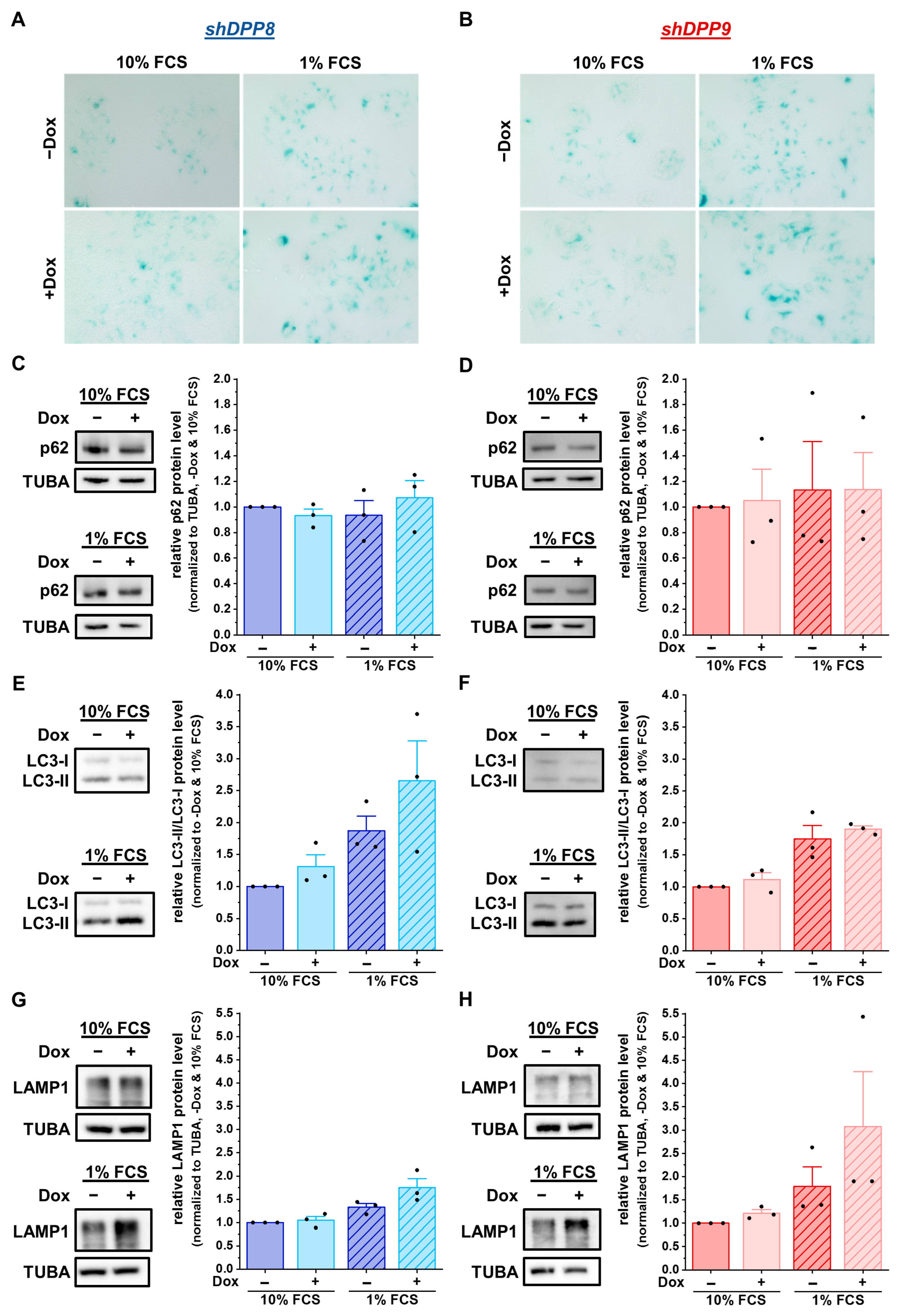
3.4. Knockdown of Either DPP8 or DPP9 Enhances Autophagy in Luminal Breast Cancer Cells
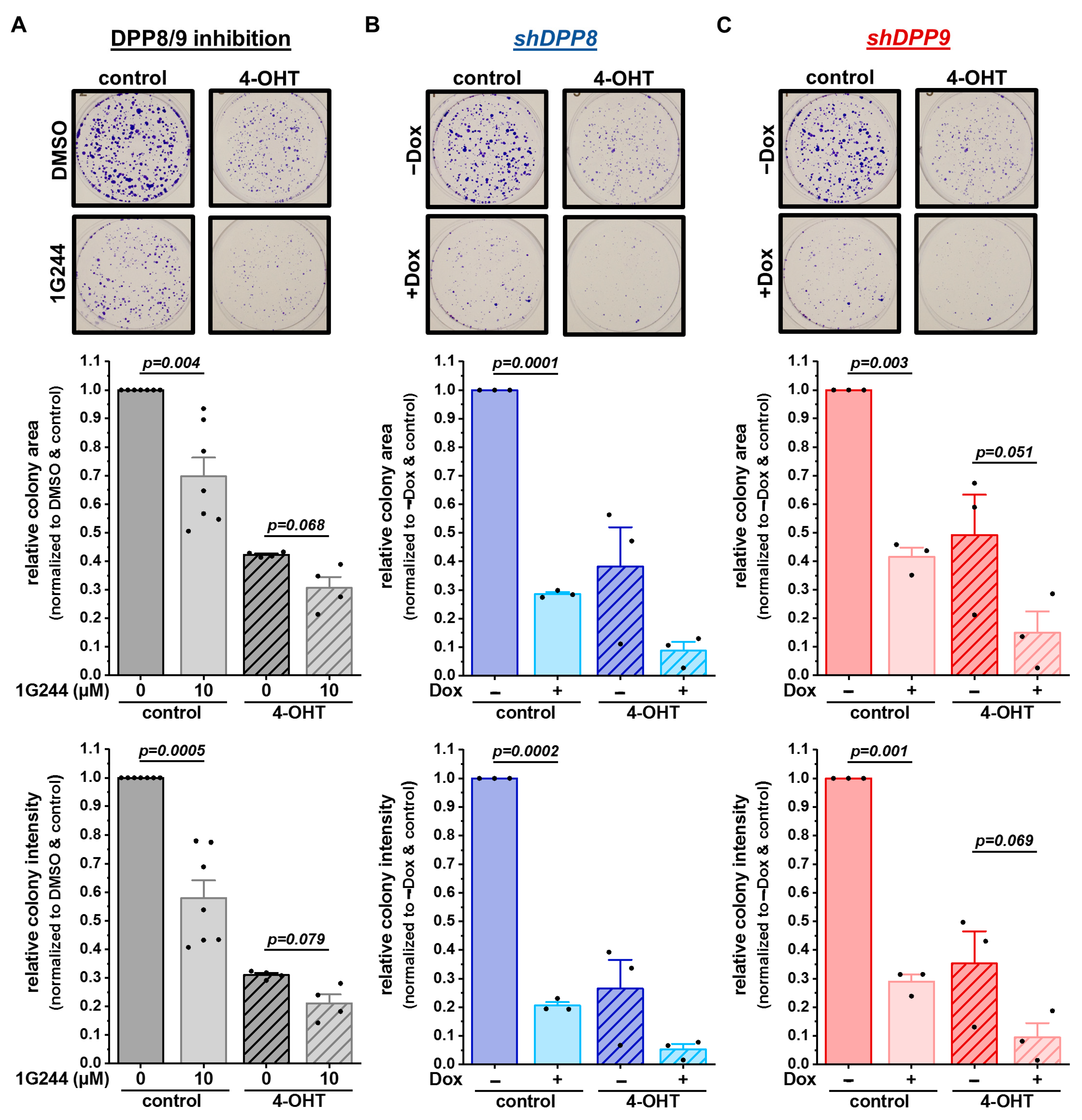
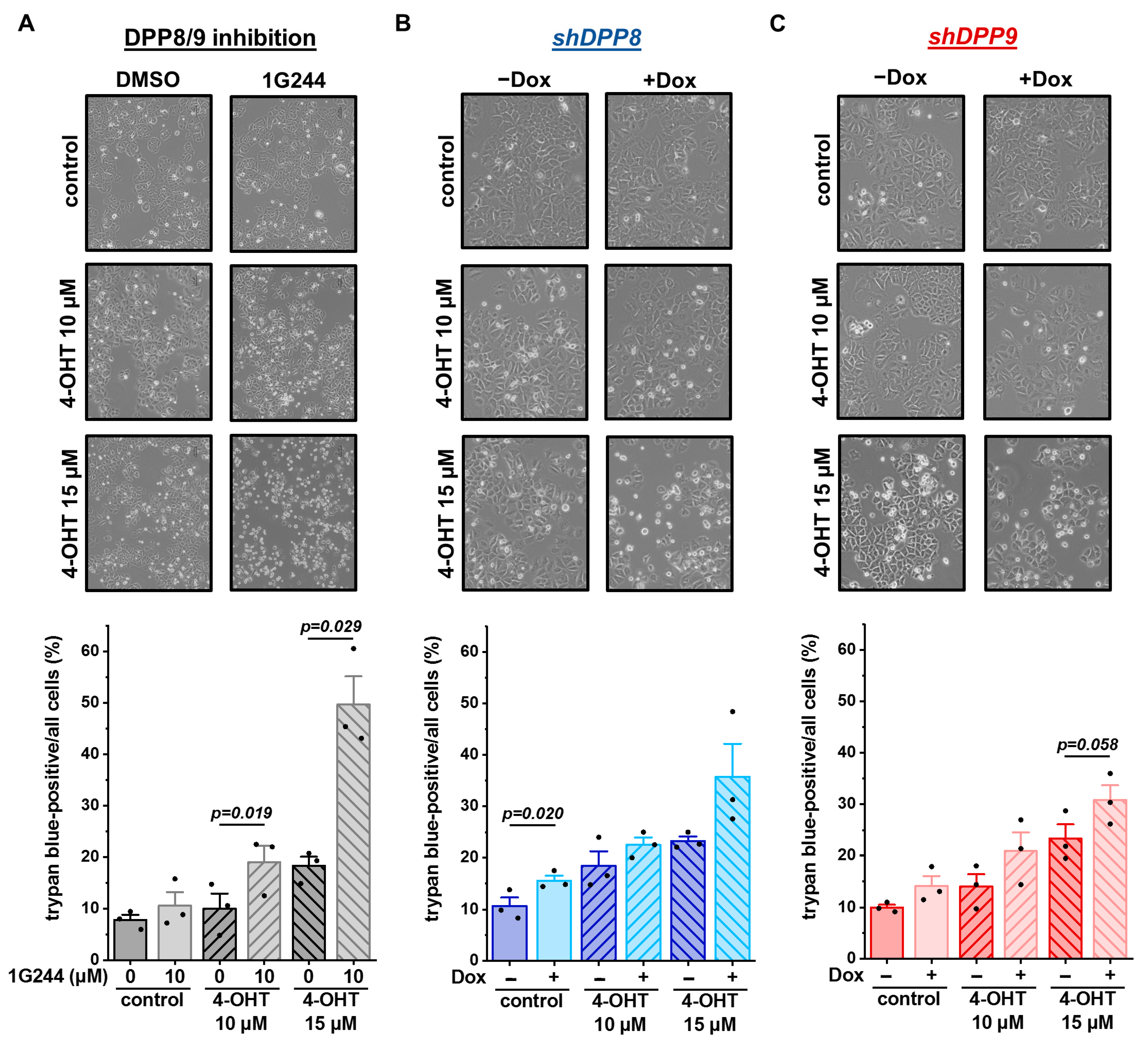
3.5. Inhibition and Single Knockdown of DPP8/9 Increases Responsiveness of Luminal Breast Cancer Cells to Tamoxifen
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bond, J.S. Proteases: History, Discovery, and Roles in Health and Disease. J. Biol. Chem. 2019, 294, 1643–1651. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Y.; Keane, F.M.; Gorrell, M.D. Advances in Understanding the Expression and Function of Dipeptidyl Peptidase 8 and 9. Mol. Cancer Res. 2013, 11, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Waumans, Y.; Baerts, L.; Kehoe, K.; Lambeir, A.-M.; De Meester, I. The Dipeptidyl Peptidase Family, Prolyl Oligopeptidase, and Prolyl Carboxypeptidase in the Immune System and Inflammatory Disease, Including Atherosclerosis. Front. Immunol. 2015, 6, 387. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.M.T.; Yao, T.-W.; Chowdhury, S.; Nadvi, N.A.; Osborne, B.; Church, W.B.; McCaughan, G.W.; Gorrell, M.D. The Dipeptidyl Peptidase IV Family in Cancer and Cell Biology. FEBS J. 2010, 277, 1126–1144. [Google Scholar] [CrossRef] [PubMed]
- Justa-Schuch, D.; Silva-Garcia, M.; Pilla, E.; Engelke, M.; Kilisch, M.; Lenz, C.; Möller, U.; Nakamura, F.; Urlaub, H.; Geiss-Friedlander, R. DPP9 Is a Novel Component of the N-End Rule Pathway Targeting the Tyrosine Kinase Syk. elife 2016, 5, e16370. [Google Scholar] [CrossRef]
- Justa-Schuch, D.; Möller, U.; Geiss-Friedlander, R. The Amino Terminus Extension in the Long Dipeptidyl Peptidase 9 Isoform Contains a Nuclear Localization Signal Targeting the Active Peptidase to the Nucleus. Cell Mol. Life Sci. 2014, 71, 3611–3626. [Google Scholar] [CrossRef]
- Van Goethem, S.; Matheeussen, V.; Joossens, J.; Lambeir, A.-M.; Chen, X.; De Meester, I.; Haemers, A.; Augustyns, K.; Van der Veken, P. Structure–Activity Relationship Studies on Isoindoline Inhibitors of Dipeptidyl Peptidases 8 and 9 (DPP8, DPP9): Is DPP8-Selectivity an Attainable Goal? J. Med. Chem. 2011, 54, 5737–5746. [Google Scholar] [CrossRef]
- Finger, Y.; Habich, M.; Gerlich, S.; Urbanczyk, S.; van de Logt, E.; Koch, J.; Schu, L.; Lapacz, K.J.; Ali, M.; Petrungaro, C.; et al. Proteasomal Degradation Induced by DPP9-Mediated Processing Competes with Mitochondrial Import. EMBO J. 2020, 39, e103889. [Google Scholar] [CrossRef]
- Geiss-Friedlander, R.; Parmentier, N.; Möller, U.; Urlaub, H.; Van den Eynde, B.J.; Melchior, F. The Cytoplasmic Peptidase DPP9 Is Rate-Limiting for Degradation of Proline-Containing Peptides. J. Biol. Chem. 2009, 284, 27211–27219. [Google Scholar] [CrossRef]
- Bolgi, O.; Silva-Garcia, M.; Ross, B.; Pilla, E.; Kari, V.; Killisch, M.; Spitzner, M.; Stark, N.; Lenz, C.; Weiss, K.; et al. Dipeptidyl Peptidase 9 Triggers BRCA2 Degradation and Promotes DNA Damage Repair. EMBO Rep. 2022, 23, e54136. [Google Scholar] [CrossRef]
- Ajami, K.; Pitman, M.R.; Wilson, C.H.; Park, J.; Menz, R.I.; Starr, A.E.; Cox, J.H.; Abbott, C.A.; Overall, C.M.; Gorrell, M.D. Stromal Cell-Derived Factors 1a and 1b, Inflammatory Protein-10 and Interferon-Inducible T Cell Chemo-Attractant Are Novel Substrates of Dipeptidyl Peptidase 8. FEBS Lett. 2008, 582, 819–825. [Google Scholar] [CrossRef]
- Matheeussen, V.; Waumans, Y.; Martinet, W.; Van Goethem, S.; Van Der Veken, P.; Scharpé, S.; Augustyns, K.; De Meyer, G.R.Y.; De Meester, I. Dipeptidyl Peptidases in Atherosclerosis: Expression and Role in Macrophage Differentiation, Activation and Apoptosis. Basic Res. Cardiol. 2013, 108, 350. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Chen, Y.; Yao, T.W.; Ajami, K.; Wang, X.M.; Popov, Y.; Schuppan, D.; Bertolino, P.; McCaughan, G.W.; Yu, D.M.T.; et al. Regulation of Dipeptidyl Peptidase 8 and 9 Expression in Activated Lymphocytes and Injured Liver. World J. Gastroenterol. 2013, 19, 2883–2893. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gall, M.G.; Zhang, H.; Keane, F.M.; McCaughan, G.W.; Yu, D.M.T.; Gorrell, M.D. Dipeptidyl Peptidase 9 Enzymatic Activity Influences the Expression of Neonatal Metabolic Genes. Exp. Cell Res. 2016, 342, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, M.; Holth, A.; Panagopoulos, I.; Staff, A.C.; Micci, F.; Davidson, B. Expression and Clinical Role of the Dipeptidyl Peptidases DPP8 and DPP9 in Ovarian Carcinoma. Virchows Arch. 2019, 474, 177–185. [Google Scholar] [CrossRef]
- Wu, Q.Q.; Zhao, M.; Huang, G.Z.; Zheng, Z.N.; Chen, Y.; Zeng, W.S.; Lv, X.Z. Fibroblast Activation Protein (FAP) Overexpression Induces Epithelial–Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma by down-Regulating Dipeptidyl Peptidase 9 (DPP9). Onco Targets. Ther. 2020, 13, 2599–2611. [Google Scholar] [CrossRef]
- Tang, Z.; Li, J.; Shen, Q.; Feng, J.; Liu, H.; Wang, W.; Xu, L.; Shi, G.; Ye, X.; Ge, M.; et al. Contribution of Upregulated Dipeptidyl Peptidase 9 (DPP9) in Promoting Tumoregenicity, Metastasis and the Prediction of Poor Prognosis in Non-Small Cell Lung Cancer (NSCLC). Int. J. Cancer 2017, 140, 1620–1632. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast Cancer. Nat. Rev. Dis. Prim. 2019, 23, 66. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 533, 747–752. [Google Scholar] [CrossRef]
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast Cancer Intrinsic Subtype Classification, Clinical Use and Future Trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar] [PubMed]
- Bradley, R.; Burrett, J.; Clarke, M.; Davies, C.; Duane, F.; Evans, V.; Gettins, L.; Godwin, J.; Gray, R.; Liu, H.; et al. Aromatase Inhibitors versus Tamoxifen in Early Breast Cancer: Patient-Level Meta-Analysis of the Randomised Trials. Lancet 2015, 386, 1341–1352. [Google Scholar] [CrossRef]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; André, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. 5th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 5). Ann. Oncol. 2020, 31, 1623–1649. [Google Scholar] [CrossRef]
- Rugo, H.S.; Rumble, R.B.; Macrae, E.; Barton, D.L.; Connolly, H.K.; Dickler, M.N.; Fallowfield, L.; Fowble, B.; Ingle, J.N.; Jahanzeb, M.; et al. Endocrine Therapy for Hormone Receptor-Positive Metastatic Breast Cancer: American Society of Clinical Oncology Guideline. J. Clin. Oncol. 2016, 34, 3069–3103. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Tilan, J.U.; Everhart, L.; Czarnecka, M.; Soldin, S.J.; Mendu, D.R.; Jeha, D.; Hanafy, J.; Lee, C.K.; Sun, J.; et al. Dipeptidyl Peptidases as Survival Factors in Ewing Sarcoma Family of Tumors: Implications for Tumor Biology and Therapy. J. Biol. Chem. 2011, 286, 27494–27505. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, F.; Wu, K.; Wu, W.; Wu, H.; Zhang, W. Targeting Dipeptidyl Peptidase 8 Genes Inhibits Proliferation, Migration and Invasion by Inhibition of Cyclin D1 and MMP2MMP9 Signal Pathway in Cervical Cancer. J. Gene Med. 2018, 20, e3056. [Google Scholar] [CrossRef]
- Wilson, C.H.; Abbott, C.A. Expression Profiling of Dipeptidyl Peptidase 8 and 9 in Breast and Ovarian Carcinoma Cell Lines. Int. J. Oncol. 2012, 41, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Hölzen, L.; Mitschke, J.; Schönichen, C.; Hess, M.E.; Ehrenfeld, S.; Boerries, M.; Miething, C.; Brummer, T.; Reinheckel, T. RNA Interference Screens Discover Proteases as Synthetic Lethal Partners of PI3K Inhibition in Breast Cancer Cells. Theranostics 2022, 12, 4348–4373. [Google Scholar] [CrossRef]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ Plugin to Automatically Quantify Colony Formation in Clonogenic Assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the Lysosome: A Control Centre for Cellular Clearance and Energy Metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296. [Google Scholar] [CrossRef]
- Seranova, E.; Connolly, K.J.; Zatyka, M.; Rosenstock, T.R.; Barrett, T.; Tuxworth, R.I.; Sarkar, S. Dysregulation of Autophagy as a Common Mechanism in Lysosomal Storage Diseases. Essays Biochem. 2017, 61, 733–749. [Google Scholar] [CrossRef] [PubMed]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between Autophagy Receptors and Ubiquitin-like Proteins Form the Molecular Basis for Selective Autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S. Regulation of Autophagy by MTOR-Dependent and MTOR-Independent Pathways: Autophagy Dysfunction in Neurodegenerative Diseases and Therapeutic Application of Autophagy Enhancers. Biochem. Soc. Trans. 2013, 41, 1103–1130. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin Superfamily Motor Proteins and Intracellular Transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Mulcahy Levy, J.M.; Thorburn, A. Autophagy in Cancer: Moving from Understanding Mechanism to Improving Therapy Responses in Patients. Cell Death Differ. 2020, 27, 843–857. [Google Scholar] [CrossRef]
- He, C.; Klionsky, D.J. Regulation Mechanisms and Signaling Pathways of Autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal Positioning Coordinates Cellular Nutrient Responses. Nat. Cell Biol. 2011, 13, 453–462. [Google Scholar] [CrossRef]
- Lőrincz, P.; Juhász, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef]
- Cardoso, C.M.P.; Groth-Pedersen, L.; Høyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jä, M.; Nylandsted, J. Depletion of Kinesin 5B Affects Lysosomal Distribution and Stability and Induces Peri-Nuclear Accumulation of Autophagosomes in Cancer Cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef]
- Zhang, H.; Maqsudi, S.; Rainczuk, A.; Duffield, N.; Lawrence, J.; Keane, F.M.; Justa-Schuch, D.; Geiss-Friedlander, R.; Gorrell, M.D.; Stephens, A.N. Identification of Novel Dipeptidyl Peptidase 9 Substrates by Two-Dimensional Differential in-Gel Electrophoresis. FEBS J. 2015, 282, 3737–3757. [Google Scholar] [CrossRef]
- Noda, T.; Ohsumi, Y. Tor, a Phosphatidylinositol Kinase Homologue, Controls Autophagy in Yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Wang, M.; Yu, M.; Zhang, J.; Bristow, R.G.; Hill, R.P.; Tannock, I.F. Role of Autophagy as a Survival Mechanism for Hypoxic Cells in Tumors. Neoplasia 2016, 18, 347–355. [Google Scholar] [CrossRef]
- Sato, K.; Tsuchihara, K.; Fujii, S.; Sugiyama, M.; Goya, T.; Atomi, Y.; Ueno, T.; Ochiai, A.; Esumi, H. Autophagy Is Activated in Colorectal Cancer Cells and Contributes to the Tolerance to Nutrient Deprivation. Cancer Res. 2007, 67, 9677–9684. [Google Scholar] [CrossRef]
- Wu, Q.; Zhao, M.; Huang, G.; Zheng, Z.; Zeng, W.; Lv, X. FAP Overexpression Induce Epithelial-Mesenchymal Transition (EMT) in Oral Squamous Cell Carcinoma by down-Regulating DPP9 Gene. bioRxiv 2019. [Google Scholar] [CrossRef]
- Yao, T.-W.; Kim, W.-S.; Yu, D.M.; Sharbeen, G.; McCaughan, G.W.; Choi, K.-Y.; Xia, P.; Gorrell, M.D. A Novel Role of Dipeptidyl Peptidase 9 in Epidermal Growth Factor Signaling. Mol. Cancer Res. 2011, 9, 948–959. [Google Scholar] [CrossRef]
- Green, K.A.; Carroll, J.S. Oestrogen-Receptor-Mediated Transcription and the Influence of Co-Factors and Chromatin State. Nat. Rev. Cancer 2007, 7, 713–722. [Google Scholar] [CrossRef]
- Yao, J.; Deng, K.; Huang, J.; Zeng, R.; Zuo, J. Progress in the Understanding of the Mechanism of Tamoxifen Resistance in Breast Cancer. Front. Pharmacol. 2020, 11, 592912. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Q.; Wang, Q.; Cao, J.; Sun, J.; Zhu, Z. Mechanisms of Resistance to Estrogen Receptor Modulators in ER+/HER2− Advanced Breast Cancer. Cell Mol. Life Sci. 2020, 77, 559–572. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Actis, C.; Muzio, G.; Autelli, R. Autophagy Triggers Tamoxifen Resistance in Human Breast Cancer Cells by Preventing Drug-Induced Lysosomal Damage. Cancers 2021, 13, 1252. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yue, W.; Chen, H. The Correlation between Autophagy and Tamoxifen Resistance in Breast Cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 2066–2074. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Sequence (5′–3′) |
|---|---|
| DPP8 | TGCTGTTGACAGTGAGCGCACGGTTTGTGGTAGTAATCTATAGTGAAGCCACAGATGTATAGATTACTACCACAAACCGTATGCCTACTGCCTCGGA |
| DPP9 | TGCTGTTGACAGTGAGCGCCCACGGCTTCCTGGACGAAAATAGTGAAGCCACAGATGTATTTTCGTCCAGGAAGCCGTGGATGCCTACTGCCTCGGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bettecken, A.; Heß, L.; Hölzen, L.; Reinheckel, T. Dipeptidyl-Aminopeptidases 8 and 9 Regulate Autophagy and Tamoxifen Response in Breast Cancer Cells. Cells 2023, 12, 2031. https://doi.org/10.3390/cells12162031
Bettecken A, Heß L, Hölzen L, Reinheckel T. Dipeptidyl-Aminopeptidases 8 and 9 Regulate Autophagy and Tamoxifen Response in Breast Cancer Cells. Cells. 2023; 12(16):2031. https://doi.org/10.3390/cells12162031
Chicago/Turabian StyleBettecken, Aaron, Lisa Heß, Lena Hölzen, and Thomas Reinheckel. 2023. "Dipeptidyl-Aminopeptidases 8 and 9 Regulate Autophagy and Tamoxifen Response in Breast Cancer Cells" Cells 12, no. 16: 2031. https://doi.org/10.3390/cells12162031
APA StyleBettecken, A., Heß, L., Hölzen, L., & Reinheckel, T. (2023). Dipeptidyl-Aminopeptidases 8 and 9 Regulate Autophagy and Tamoxifen Response in Breast Cancer Cells. Cells, 12(16), 2031. https://doi.org/10.3390/cells12162031