

TGFβ Inhibitor A83-01 Enhances Murine HSPC Expansion for Gene Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. LSK SLAM Sorting and Expansion
2.2. MTT Assay
2.3. Flow Cytometry
2.4. Cytospin Analysis
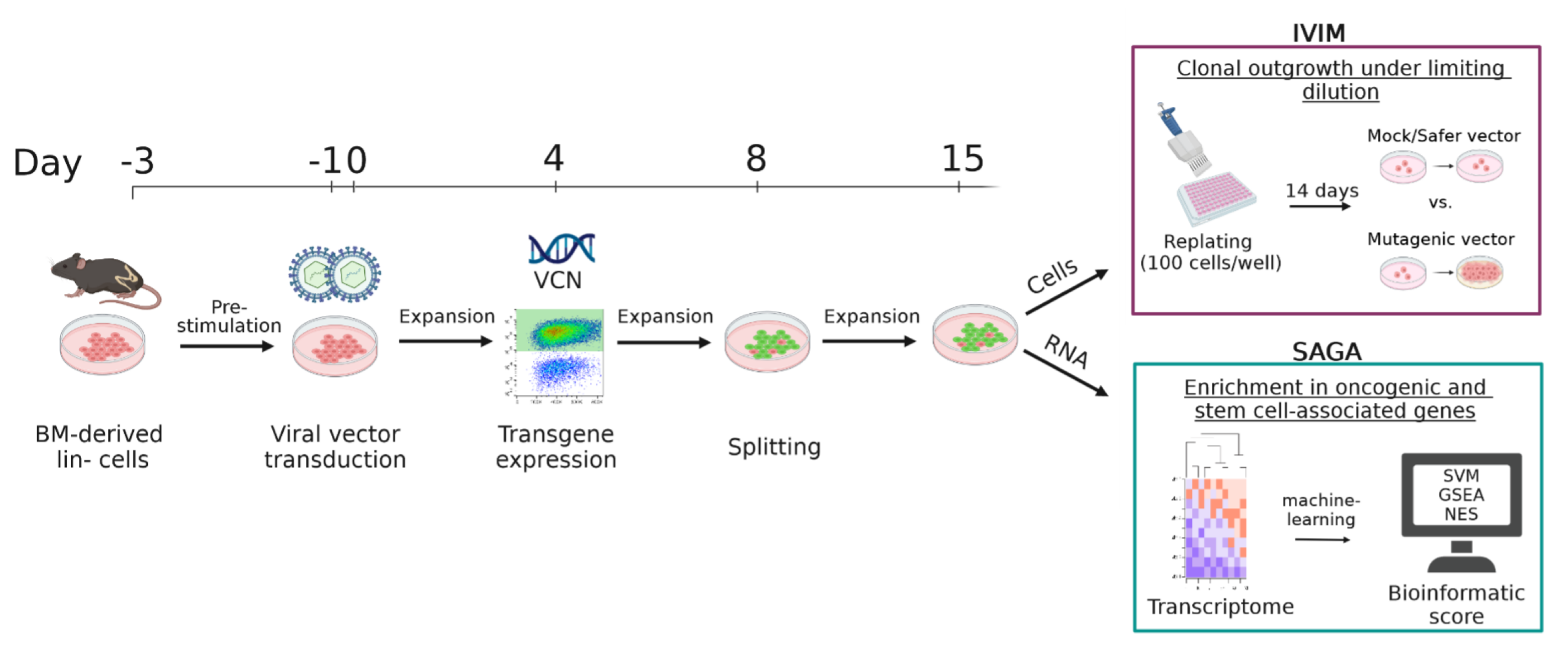
2.5. In Vitro Immortalization Assay
2.6. VCN Determination by qPCR
2.7. Surrogate Assay for Genotoxicity Assessment
2.8. Insertion Site Analysis with INSPIIRED
2.9. Transcriptome Analysis
2.10. Protein Extraction and Western Blot Analysis
3. Results
3.1. Massive Ex Vivo Expansion in S3F11+APU Medium after LSK SLAM Purification
3.2. A83-01 Is the Key Compound to Attenuate Mast Cell Differentiation
3.3. Transcriptomic Analysis Confirms the Inhibition of Multiple Differentiation Pathways by A83-01
3.4. Efficient Transduction of S3F11-APU-Exp Cells for IVIM Assay
3.5. Mutagenic Vector-Induced Differentiation Arrest of Lin- and Myeloid Progenitors in Fresh and S3F11+APU-Exp Cells
3.6. Mecom Integrations in Expanded Material Reproduce Clinical Observations and Previous IVIM Results
3.7. SAGA Detects an Oncogenic Gene Signature in Expanded Cells after Gammaretroviral Transduction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HSPCs | hematopoietic stem and progenitor cells |
| BM | bone marrow |
| APU | A83-01, pomalidomide, UM171 |
| lin- | ineage-negative |
| LSK SLAM | lin- Sca1+ cKit+ CD48- CD150+ |
| SCF | stem cell factor |
| IL | interleukin |
| FLT3-L | FMS-like tyrosine kinase 3 ligand |
| HSCs | hematopoietic stem cells |
| FcR1 | Fc epsilon receptor 1 alpha |
| S3F11 | SCF, IL-3, FLT3-L, IL-11 |
| TPO | thrombopoietin |
| ALK | activin receptor-like kinase |
| CRL4CRBN | CUL4-RBX1-DDB1-CRBN |
| IVIM assay | in vitro immortalization assay |
| SAGA | surrogate assay for genotoxicity |
| Exp | expanded |
| Mecom | MDS1 and EVI1 complex locus |
| EDTA | ethylenediaminetetraacetic acid |
| FACS | Ffuorescent-activated cell sorting |
| FCS | fetal calf serum |
| MTT | thiazolyl blue tetrazolium bromide |
| SDS | sodium dodecyl sulfate |
| RT | room temperature |
| VCN | vector copy number |
| WPRE | woodchuck hepatitis virus post-transcriptional regulatory element |
| PTBP2 | polypyrimidine tract binding protein 2 |
| NES | normalized enrichment score |
| INSPIIRED | integration site pipeline for paired-end reads |
| STIF | SCF, TPO, IGF2, FGF1 |
| IGF2 | insulin-like growth factor 2 |
| FGF1 | fibroblast growth factor 1 |
| DEG | differentially expressed genes |
| GSEA | gene set enrichment analysis |
| MSigDB | Mouse Molecular Signatures Database |
| RF | replating frequency |
| UIS | unique integration sites |
| SVM | support vector machine |
| FA | Fanconi Anemia |
References
- Meyts, I.; Bousfiha, A.; Duff, C.; Singh, S.; Lau, Y.L.; Condino-Neto, A.; Bezrodnik, L.; Ali, A.; Adeli, M.; Drabwell, J. Primary Immunodeficiencies: A Decade of Progress and a Promising Future. Front. Immunol. 2021, 11, 625753. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, L.D. Primary immunodeficiencies. J. Allergy Clin. Immunol. 2010, 125, S182–S194. [Google Scholar] [CrossRef]
- Taher, A.T.; Weatherall, D.J.; Cappellini, M.D. Thalassaemia. Lancet 2018, 391, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Scalone, L.; Mantovani, L.G.; Krol, M.; Rofail, D.; Ravera, S.; Grazia Bisconte, M.; Borgna-Pignatti, C.; Borsellino, Z.; Cianciulli, P.; Gallisai, D.; et al. Costs, quality of life, treatment satisfaction and compliance in patients with β-thalassemia major undergoing iron chelation therapy: The ITHACA study. Curr. Med. Res. Opin. 2008, 24, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Kohne, E. Hämoglobinopathien: Klinische erscheinungsbilder, diagnostische und therapeutische hinweise. Dtsch. Ärztebl. Int. 2011, 108, 532. [Google Scholar] [CrossRef]
- Dokal, I.; Vulliamy, T. Inherited aplastic anaemias/bone marrow failure syndromes. Blood Rev. 2008, 22, 141–153. [Google Scholar] [CrossRef]
- Dokal, I.; Vulliamy, T. Inherited bone marrow failure syndromes. Haematologica 2010, 95, 1236. [Google Scholar] [CrossRef]
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta-Mol. Cell Res. 2009, 1793, 684–696. [Google Scholar] [CrossRef]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef]
- Lui, W.C.; Chan, Y.F.; Chan, L.C.; Ng, R.K. Cytokine combinations on the potential for ex vivo expansion of murine hematopoietic stem cells. Cytokine 2014, 68, 127–132. [Google Scholar] [CrossRef][Green Version]
- Nitsche, A.; Junghahn, I.; Thulke, S.; Aumann, J.; Radoni, A.; Fichtner, I.; Siegert, W.; Radonic, A. Interleukin-3 Promotes Proliferation and Differentiation of Human Hematopoietic Stem Cells but Reduces Their Repopulation Potential in NOD/SCID Mice. Stem Cells 2003, 21, 236–244. [Google Scholar] [CrossRef]
- Ebina, W. Combinatorial Pathway Modulation toward Ex Vivo Maintenance and Propagation of Hematopoietic Stem Cells. Ph.D. Thesis, Harvard University, Cambridge, MA, USA, 2016. [Google Scholar]
- Li, W.; Wei, W.; Zhu, S.; Zhu, J.; Shi, Y.; Lin, T.; Hao, E.; Hayek, A.; Deng, H.; Ding, S. Generation of Rat and Human Induced Pluripotent Stem Cells by Combining Genetic Reprogramming and Chemical Inhibitors. Cell Stem Cell 2009, 4, 16–19. [Google Scholar] [CrossRef]
- Tojo, M.; Hamashima, Y.; Hanyu, A.; Kajimoto, T.; Saitoh, M.; Miyazono, K.; Node, M.; Imamura, T. The ALK-5 inhibitor A-83-01 inhibits Smad signaling and epithelial-to-mesenchymal transition by transforming growth factor-β. Cancer Sci. 2005, 96, 791–800. [Google Scholar] [CrossRef]
- Batard, P.; Monier, M.N.; Fortunel, N.; Ducos, K.; Sansilvestri-Morel, P.; Phan, T.H.; Hatzfeld, A.; Hatzfeld, J.A. TGF-β1 maintains hematopoietic immaturity by a reversible negative control of cell cycle and induces CD34 antigen up-modulation. J. Cell Sci. 2000, 113, 383–390. [Google Scholar] [CrossRef]
- Sitnicka, E.; Ruscetti, F.W.; Priestley, G.V.; Wolf, N.S.; Bartelmez, S.H. Transforming growth factor β1 directly and reversibly inhibits the initial cell divisions of long-term repopulating hematopoietic stem cells. Blood 1996, 88, 82–88. [Google Scholar] [CrossRef]
- Pajcini, K.V.; Speck, N.A.; Pear, W.S. Notch signaling in mammalian hematopoietic stem cells. Leukemia 2011, 25, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Lampreia, F.P.; Carmelo, J.G.; Anjos-Afonso, F. Notch Signaling in the Regulation of Hematopoietic Stem Cell. Curr. Stem Cell Rep. 2017, 3, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Kleinmann, E.; Geimer Le Lay, A.S.; Sellars, M.; Kastner, P.; Chan, S. Ikaros Represses the Transcriptional Response to Notch Signaling in T-Cell Development. Mol. Cell. Biol. 2008, 28, 7465–7475. [Google Scholar] [CrossRef] [PubMed]
- Kathrein, K.L.; Chari, S.; Winandy, S. Ikaros directly represses the notch target gene Hes1 in a leukemia T cell line: Implications for CD4 regulation. J. Biol. Chem. 2008, 283, 10476–10484. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.T.; Cimmino, L.; Hu, Y.; Trimarchi, T.; Tagoh, H.; McKenzie, M.D.; Best, S.A.; Tuohey, L.; Willson, T.A.; Nutt, S.L.; et al. Activated Notch counteracts Ikaros tumor suppression in mouse and human T-cell acute lymphoblastic leukemia. Leukemia 2015, 29, 1301–1311. [Google Scholar] [CrossRef]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef]
- Petzold, G.; Fischer, E.S.; Thomä, N.H. Structural basis of lenalidomide-induced CK1α degradation by the CRL4 CRBN ubiquitin ligase. Nature 2016, 532, 127–130. [Google Scholar] [CrossRef]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl Hydrocarbon Receptor Antagonists Promote the Expansion of Human Hematopoietic Stem Cells. Science 2010, 329, 1345–1348. [Google Scholar] [CrossRef]
- Chagraoui, J.; Girard, S.; Spinella, J.F.; Simon, L.; Bonneil, E.; Mayotte, N.; MacRae, T.; Coulombe-Huntington, J.; Bertomeu, T.; Moison, C.; et al. UM171 Preserves Epigenetic Marks that Are Reduced in Ex Vivo Culture of Human HSCs via Potentiation of the CLR3-KBTBD4 Complex. Cell Stem Cell 2021, 28, 48–62. [Google Scholar] [CrossRef]
- Cohen, S.; Roy, J.; Lachance, S.; Delisle, J.S.; Marinier, A.; Busque, L.; Roy, D.C.; Barabé, F.; Ahmad, I.; Bambace, N.; et al. Hematopoietic stem cell transplantation using single UM171-expanded cord blood: A single-arm, phase 1-2 safety and feasibility study. Lancet Haematol. 2019, 7, e134–e145. [Google Scholar] [CrossRef] [PubMed]
- Blank, U.; Karlsson, S. TGF-β signaling in the control of hematopoietic stem cells. Blood J. Am. Soc. Hematol. 2015, 125, 3542–3550. [Google Scholar] [CrossRef]
- Modlich, U.; Bohne, J.; Schmidt, M.; Von Kalle, C.; Knöss, S.; Schambach, A.; Baum, C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood 2006, 108, 2545–2553. [Google Scholar] [CrossRef]
- Schwarzer, A.; Talbot, S.R.; Selich, A.; Morgan, M.; Schott, J.W.; Dittrich-Breiholz, O.; Bastone, A.L.; Weigel, B.; Ha, T.C.; Dziadek, V.; et al. Predicting genotoxicity of viral vectors for stem cell gene therapy using gene expression-based machine learning. Mol. Ther. 2021, 29, 3383–3397. [Google Scholar] [CrossRef] [PubMed]
- Schambach, A.; Wodrich, H.; Hildinger, M.; Bohne, J.; Kräusslich, H.G.; Baum, C. Context dependence of different modules for posttranscriptional enhancement of gene expression from retroviral vectors. Mol. Ther. 2000, 2, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Schambach, A.; Mueller, D.; Galla, M.; Verstegen, M.M.; Wagemaker, G.; Loew, R.; Baum, C.; Bohne, J. Overcoming promoter competition in packaging cells improves production of self-inactivating retroviral vectors. Gene Ther. 2006, 13, 1524–1533. [Google Scholar] [CrossRef] [PubMed]
- Hildinger, M.; Abel, K.L.; Ostertag, W.; Baum, C. Design of 5′ Untranslated Sequences in Retroviral Vectors Developed for Medical Use. J. Virol. 1999, 73, 4083–4089. [Google Scholar] [CrossRef]
- Schambach, A.; Bohne, J.; Chandra, S.; Will, E.; Margison, G.P.; Williams, D.A.; Baum, C. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol. Ther. 2006, 13, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Dull, T.; Mandel, R.J.; Bukovsky, A.; Quiroz, D.; Naldini, L.; Trono, D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol. 1998, 72, 9873–9880. [Google Scholar] [CrossRef]
- Schwarzer, A.; Talbot, S.R.; Dittrich-Breiholz, O.; Thrasher, A.J.; Gaspar, B.; Modlich, U.; Baum, C.; Schambach, A.; Rothe, M. New Molecular Surrogate Assay for Genotoxicity Assessment of Gene Therapy Vectors (SAGA). Blood 2016, 128, 4710. [Google Scholar] [CrossRef]
- Berry, C.C.; Nobles, C.; Six, E.; Wu, Y.; Malani, N.; Sherman, E.; Dryga, A.; Everett, J.K.; Male, F.; Bailey, A.; et al. INSPIIRED: Quantification and Visualization Tools for Analyzing Integration Site Distributions. Mol. Ther.-Methods Clin. Dev. 2017, 4, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, A.C.; Ishida, R.; Kikuchi, M.; Sudo, K.; Morita, M.; Crisostomo, R.V.; Yamamoto, R.; Loh, K.M.; Nakamura, Y.; Watanabe, M.; et al. Long-term ex vivo haematopoietic-stem-cell expansion allows nonconditioned transplantation. Nature 2019, 571, 117–121. [Google Scholar] [CrossRef]
- Pejler, G.; Rönnberg, E.; Waern, I.; Wernersson, S. Mast cell proteases: Multifaceted regulators of inflammatory disease. Blood J. Am. Soc. Hematol. 2010, 115, 4981–4990. [Google Scholar] [CrossRef]
- Dai, H.; Korthuis, R.J. Mast cell proteases and inflammation. Drug Discov. Today Dis. Model. 2011, 8, 47–55. [Google Scholar] [CrossRef]
- Benditt, E.P.; Arase, M. An enzyme in mast cells with properties like chymotrypsin. J. Exp. Med. 1959, 110, 451–460. [Google Scholar] [CrossRef]
- Pejler, G. Novel Insight into the in vivo Function of Mast Cell Chymase: Lessons from Knockouts and Inhibitors. J. Innate Immun. 2020, 12, 357–372. [Google Scholar] [CrossRef]
- Glenner, G.G.; Cohen, L.A. Histochemical demonstration of a species-specific trypsin-like enzyme in mast cells. Nature 1960, 185, 846–847. [Google Scholar] [CrossRef]
- Schwartz, L.B.; Metcalfe, D.D.; Miller, J.S.; Earl, H.; Sullivan, T. Tryptase Levels as an Indicator of Mast-Cell Activation in Systemic Anaphylaxis and Mastocytosis. N. Engl. J. Med. 1987, 316, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Atiakshin, D.; Kostin, A.; Trotsenko, I.; Samoilova, V.; Buchwalow, I.; Tiemann, M. Carboxypeptidase A3-A Key Component of the Protease Phenotype of Mast Cells. Cells 2022, 11, 570. [Google Scholar] [CrossRef]
- Ahmada, S.; Wrighta, K.N.; Sunb, X.; Grobanb, L.; Ferrarioa, C.M. Mast cell peptidases (carboxypeptidase A and chymase)- mediated hydrolysis of human angiotensin-(1–12) substrate Sarfaraz. Biochem. Biophys. Res. Commun. 2019, 176, 139–148. [Google Scholar] [CrossRef]
- Wernersson, S.; Pejler, G. Mast cell secretory granules: Armed for battle. Nat. Publ. Group 2014, 14, 478–494. [Google Scholar] [CrossRef]
- He, W.; Dorn, D.C.; Erdjument-Bromage, H.; Tempst, P.; Moore, M.A.; Massagué, J. Hematopoiesis Controlled by Distinct TIF1γ and Smad4 Branches of the TGFβ Pathway. Cell 2006, 125, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar] [CrossRef]
- Miller, D.G.; Adam, M.A.; Miller, A.D. Gene Transfer by Retrovirus Vectors Occurs Only in Cells That Are Actively Replicating at the Time of Infection. Mol. Cell. Biol. 1990, 10, 4239–4242. [Google Scholar]
- Ruck, P.; Horny, H.P.; Kaiserling, E. Immunoreactivity of human tissue mast cells: Nonspecific binding of primary antibodies against regulatory peptides by ionic linkage. J. Histochem. Cytochem. 1990, 38, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Schiltz, P.M.; Lieber, J.; Giorno, R.C.; Claman, H.N. Mast cell immunohistochemistry: Non-immunological immunostaining mediated by non-specific F(ab’)z-mast cell secretory granule interaction. Histochem. J. 1993, 25, 642–647. [Google Scholar] [CrossRef]
- Sherman, E.; Nobles, C.; Berry, C.C.; Six, E.; Wu, Y.; Dryga, A.; Malani, N.; Male, F.; Reddy, S.; Bailey, A.; et al. INSPIIRED: A Pipeline for Quantitative Analysis of Sites of New DNA Integration in Cellular Genomes. Mol. Ther.-Methods Clin. Dev. 2017, 4, 39–49. [Google Scholar] [CrossRef]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Krämer, A.; Schwäble, J.; Glimm, H.; et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef]
- Métais, J.Y.; Dunbar, C.E. The MDS1-EVI1 gene complex as a retrovirus integration site: Impact on behavior of hematopoietic cells and implications for gene therapy. Mol. Ther. 2008, 16, 439–449. [Google Scholar] [CrossRef]
- Braun, C.J.; Boztug, K.; Paruzynski, A.; Witzel, M.; Schwarzer, A.; Rothe, M.; Modlich, U.; Beier, R.; Göhring, G.; Steinemann, D.; et al. Gene therapy for Wiskott-Aldrich syndrome-long-term efficacy and genotoxicity. Sci. Transl. Med. 2014, 6, 227ra33. [Google Scholar] [CrossRef]
- Bluebird Bio, Inc. Bluebird bio Reports Second Quarter Financial Results and Provides Operational Update. Press Release. Available online: https://investor.bluebirdbio.com/news-releases/news-release-details/bluebird-bio-reports-second-quarter-financial-results-and (accessed on 25 April 2023).
- Qi, H.; Liu, S.; Guo, C.; Wang, J.; Greenaway, F.T.; Sun, M.Z. Role of annexin A6 in cancer (Review). Oncol. Lett. 2015, 10, 1947–1952. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Jones, T.; Abbasi, F.; Lee, H.; Stultz, B.; Hursh, D.A.; Mortin, M.A. Drosophila caliban, a nuclear export mediator, can function as a tumor suppressor in human lung cancer cells. Oncogene 2005, 24, 8229–8239. [Google Scholar] [CrossRef] [PubMed]
- Scanlan, M.J.; Chen, Y.T.; Williamson, B.; Gure, A.O.; Stockert, E.; Gordan, J.D.; Türeci, O.; Sahin, U.; Pfrendschuh, M.; Old, L.J. Characterization of human colon cancer antigens recognized by autologous antibodies. Int. J. Cancer 1998, 76, 652–658. [Google Scholar] [CrossRef]
- Chen, L.; Wu, Q.; Xu, X.; Yang, C.; You, J.; Chen, F.; Zeng, Y. Cancer/testis antigen LDHC promotes proliferation and metastasis by activating the PI3K/Akt/GSK-3β-signaling pathway and the in lung adenocarcinoma. Exp. Cell Res. 2021, 398, 112414. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Du, W.; Cui, Z.; Wang, L.; Yang, Z.; Zhang, H.; Lin, D. Expression of lactate dehydrogenase C in MDA-MB-231 cells and its role in tumor invasion and migration. Mol. Med. Rep. 2016, 13, 3533–3538. [Google Scholar] [CrossRef]
- O’Neal, J.; Gao, F.; Hassan, A.; Monahan, R.; Barrios, S.; Lee, I.; Chng, W.J.; Vij, R.; Tomasson, M.H. Neurobeachin (NBEA) is a target of recurrent interstitial deletions at 13q13 in patients with MGUS and multiple myeloma. Exp. Hematol. 2009, 37, 234–244. [Google Scholar] [CrossRef]
- Hattori, M.; Tsukamoto, N.; Nur-e Kamal, M.S.; Rubinfeld, B.; Iwai, K.; Kubota, H.; Maruta, H.; Minato, N. Molecular cloning of a novel mitogen-inducible nuclear protein with a Ran GTPase-activating domain that affects cell cycle progression. Mol. Cell. Biol. 1995, 15, 552–560. [Google Scholar] [CrossRef]
- Banerjee, R.; Mani, R.S.; Russo, N.; Scanlon, C.S.; Tsodikov, A.; Jing, X.; Cao, Q.; Palanisamy, N.; Metwally, T.; Inglehart, R.C.; et al. The tumor suppressor gene rap1GAP is silenced by miR-101-mediated EZH2 overexpression in invasive squamous cell carcinoma. Oncogene 2011, 30, 4339–4349. [Google Scholar] [CrossRef]
- Cui, X.; Song, L.; Bai, Y.; Wang, Y.; Wang, B.; Wang, W. Elevated IQGAP1 and CDC42 levels correlate with tumor malignancy of human glioma. Oncol. Rep. 2017, 37, 768–776. [Google Scholar] [CrossRef]
- White, C.D.; Brown, M.D.; Sacks, D.B. IQGAPs in cancer: A family of scaffold proteins underlying tumorigenesis. FEBS Lett. 2009, 583, 1817–1824. [Google Scholar] [CrossRef]
- Ma, B.B.; Zhang, T.J.; Wang, C.Z.; Xu, Z.J.; Zhou, J.D.; Gu, Y.; Ma, J.C.; Deng, Z.Q.; Lin, J.; Qian, J. Methylation-independent CRIP1 expression is a potential biomarker affecting prognosis in cytogenetically normal acute myeloid leukemia. Am. J. Transl. Res. 2020, 12, 4840–4852. [Google Scholar]
- Li, M.; Ma, Y.; Zhong, Y.; Liu, Q.; Chen, C.; Qiang, L.; Wang, X. KALRN mutations promote antitumor immunity and immunotherapy response in cancer. J. Immunother. Cancer 2020, 8, e000293. [Google Scholar] [CrossRef] [PubMed]
- Heckl, D.; Schwarzer, A.; Haemmerle, R.; Steinemann, D.; Rudolph, C.; Skawran, B.; Knoess, S.; Krause, J.; Li, Z.; Schlegelberger, B.; et al. Lentiviral vector induced insertional haploinsufficiency of EBF1 causes murine leukemia. Mol. Ther. 2012, 20, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Hirouchi, T.; Takabatake, T.; Yoshida, K.; Nitta, Y.; Nakamura, M.; Tanaka, S.; Ichinohe, K.; Oghiso, Y.; Tanaka, K. Upregulation of c-myc gene accompanied by PU.1 deficiency in radiation-induced acute myeloid leukemia in mice. Exp. Hematol. 2008, 36, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Paul, R.; Meynet, O.; Volturo, S.; Pinna, G.; Ricci, J.E. The E3 ligase UBR2 regulates cell death under caspase deficiency via Erk/MAPK pathway. Cell Death Dis. 2020, 11, 1041. [Google Scholar] [CrossRef]
- Mao, J.; Liang, Z.; Zhang, B.; Yang, H.; Li, X.; Fu, H.; Zhang, X.; Yan, Y.; Xu, W.; Qian, H. UBR2 Enriched in p53 Deficient Mouse Bone Marrow Mesenchymal Stem Cell-Exosome Promoted Gastric Cancer Progression via Wnt/β-Catenin Pathway. Stem Cells 2017, 35, 2267–2279. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rivière, I. Genetic Engineering and Manufacturing of Hematopoietic Stem Cells. Mol. Ther.-Methods Clin. Dev. 2017, 5, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Tajer, P.; Canté-Barrett, K.; Naber, B.A.; Vloemans, S.A.; van Eggermond, M.C.; van der Hoorn, M.L.; Pike-Overzet, K.; Staal, F.J. IL3 Has a Detrimental Effect on Hematopoietic Stem Cell Self-Renewal in Transplantation Settings. Int. J. Mol. Sci. 2022, 23, 2736. [Google Scholar] [CrossRef] [PubMed]
- Ro<i>β</i>manith, T.; Schröder, B.; Bug, G.; Müller, P.; Klenner, T.; Knaus, R.; Hoelzer, D.; Ottmann, O.G.; Bug, G. Interleukin 3 Improves the Ex Vivo Expansion of Primitive Human Cord Blood Progenitor Cells and Maintains the Engraftment Potential of SCID Repopulating Cells. Stem Cells 2001, 19, 313–320. [Google Scholar] [CrossRef]
- Uchida, N.; Hsieh, M.M.; Hayakawa, J.; Madison, C.; Washington, K.N.; Tisdale, J.F. Optimal conditions for lentiviral transduction of engrafting human CD34 + cells. Gene Ther. 2011, 18, 1078–1086. [Google Scholar] [CrossRef]
- Hatzfeld, J.; Li, M.L.; Brown, E.L.; Sookdeo, H.; Levesque, J.P.; O’toole, T.; Gurney, C.; Clark, S.C.; Hatzfeld, A. Release of Early Human Hematopoietic Progenitors from Quiescence by Antisense Transforming Growth Factorβ1 or Rb Oligonucleotides. J. Exp. Med. 1991, 174, 925–929. [Google Scholar] [CrossRef]
- Thompson, N.L.; Flanders, K.C.; Smith, J.M.; Ellingsworth, L.R.; Roberts, A.B.; Sporn, M.B. Expression of transforming growth factor-β1 in specific cells and tissues of adult and neonatal mice. J. Cell Biol. 1989, 108, 661–669. [Google Scholar] [CrossRef]
- Yamazaki, S.; Iwama, A.; Takayanagi, S.I.; Eto, K.; Ema, H.; Nakauchi, H. TGF-b as a candidate bone marrow niche signal to induce hematopoietic stem cell hibernation. Blood 2009, 113, 1250–1256. [Google Scholar] [CrossRef]
- Challen, G.A.; Boles, N.C.; Chambers, S.M.; Goodell, M.A. Distinct Hematopoietic Stem Cell Subtypes Are Differentially Regulated by TGF-β1. Cell Stem Cell 2010, 6, 265–278. [Google Scholar] [CrossRef]
- Fortunel, N.; Batard, P.; Hatzfeld, A.; Monier, M.N.; Panterne, B.; Lebkowski, J.; Hatzfeld, J. High Proliferative Potential-Quiescent cells: A working model to study primitive quiescent hematopoietic cells. J. Cell Sci. 1998, 111, 1867–1875. [Google Scholar] [CrossRef]
- Soma, T.; Yu, J.M.; Dunbar, C.E. Maintenance of Murine Long-Term Repopulating Stem Cells in Ex Vivo Culture Is Affected by Modulation of Transforming Growth Factor-/3 But Not Macrophage Inflammatory Protein—1 a Activities. Blood 2015, 87, 4561–4567. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Parmar, K.; Mouly, E.; Delord, M.; Kim, J.M.; Regairaz, M.; Pla, M.; Vasquez, N.; Zhang, Q.S.; Pondarre, C.; et al. Bone marrow failure in fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell 2012, 11, 36–49. [Google Scholar] [CrossRef]
- Río, P.; Navarro, S.; Wang, W.; Sánchez-Domínguez, R.; Pujol, R.M.; Segovia, J.C.; Bogliolo, M.; Merino, E.; Wu, N.; Salgado, R.; et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019, 25, 1396–1401. [Google Scholar] [CrossRef]
- Zhang, H.; Kozono, D.E.; O’Connor, K.W.; Vidal-Cardenas, S.; Rousseau, A.; Hamilton, A.; Moreau, L.; Gaudiano, E.F.; Greenberger, J.; Bagby, G.; et al. TGF-β inhibition rescues hematopoietic stem cell defects and bone marrow failure in Fanconi anemia. Cell Stem Cell 2016, 18, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Yang, C.; Furutani, E.; García de Teresa, B.; Velázquez, M.; Filiatrault, J.; Sambel, L.A.; Phan, T.; Flores-Guzmán, P.; Sánchez, S.; et al. Inhibition of TGFβ1 and TGFβ3 promotes hematopoiesis in Fanconi anemia. Exp. Hematol. 2021, 93, 70–84. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Guidance for Industry Preclinical Assessment of Investigational Cellular and Gene Therapy Products Additional; Food and Drug Administration: Rockville, MD, USA, 2013. Available online: https://www.fda.gov/media/87564/download (accessed on 22 March 2023).
- Cornetta, K.; Lin, T.Y.; Pellin, D.; Kohn, D.B. Meeting FDA Guidance recommendations for replication-competent virus and insertional oncogenesis testing. Mol. Ther.-Methods Clin. Dev. 2023, 28, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Yuasa, H.; Oike, Y.; Iwama, A.; Nishikata, I.; Sugiyama, D.; Perkins, A.; Mucenski, M.L.; Suda, T.; Morishita, K. Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J. 2005, 24, 1976–1987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Stehling-Sun, S.; Lezon-Geyda, K.; Juneja, S.C.; Coillard, L.; Chatterjee, G.; Wuertzer, C.A.; Camargo, F.; Perkins, A.S. PR-domain—Containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood 2011, 118, 3853–3861. [Google Scholar] [CrossRef]
- Kataoka, K.; Sato, T.; Yoshimi, A.; Goyama, S.; Tsuruta, T.; Kobayashi, H.; Shimabe, M.; Arai, S.; Nakagawa, M.; Imai, Y.; et al. Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J. Exp. Med. 2011, 208, 2403–2416. [Google Scholar] [CrossRef] [PubMed]
- Voit, R.A.; Tao, L.; Yu, F.; Cato, L.D.; Cohen, B.; Fleming, T.J.; Antoszewski, M.; Liao, X.; Fiorini, C.; Nandakumar, S.K.; et al. nature immunology A genetic disorder reveals a hematopoietic stem cell regulatory network co-opted in leukemia. Nat. Immunol. 2023, 24, 69–83. [Google Scholar] [CrossRef]
- Birdwell, C.; Fiskus, W.; Kadia, T.M.; DiNardo, C.D.; Mill, C.P.; Bhalla, K.N. EVI1 dysregulation: Impact on biology and therapy of myeloid malignancies. Blood Cancer J. 2021, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.C.; Caruso, M. A nuclear localization signal in the matrix of spleen necrosis virus (SNV) does not allow efficient gene transfer into quiescent cells with SNV-derived vectors. Virology 2005, 338, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Roe, T.; Reynolds, T.C.; Yu, G.; Brown, P.O. Integration of murine leukemia virus DNA depends on mitosis. EMBO J. 1993, 12, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Schwieger, M.; Lange, C.; Kraunus, J.; Sun, H.; van den Akker, E.; Modlich, U.; Serinsöz, E.; Will, E.; von Laer, D.; et al. Predictable and efficient retroviral gene transfer into murine bone marrow repopulating cells using a defined vector dose. Exp. Hematol. 2003, 31, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.C.; Lodish, H.F. Murine hematopoietic stem cells change their surface phenotype during ex vivo expansion. Blood 2005, 105, 4314–4320. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fleischauer, J.; Bastone, A.L.; Selich, A.; John-Neek, P.; Weisskoeppel, L.; Schaudien, D.; Schambach, A.; Rothe, M. TGFβ Inhibitor A83-01 Enhances Murine HSPC Expansion for Gene Therapy. Cells 2023, 12, 1978. https://doi.org/10.3390/cells12151978
Fleischauer J, Bastone AL, Selich A, John-Neek P, Weisskoeppel L, Schaudien D, Schambach A, Rothe M. TGFβ Inhibitor A83-01 Enhances Murine HSPC Expansion for Gene Therapy. Cells. 2023; 12(15):1978. https://doi.org/10.3390/cells12151978
Chicago/Turabian StyleFleischauer, Jenni, Antonella Lucia Bastone, Anton Selich, Philipp John-Neek, Luisa Weisskoeppel, Dirk Schaudien, Axel Schambach, and Michael Rothe. 2023. "TGFβ Inhibitor A83-01 Enhances Murine HSPC Expansion for Gene Therapy" Cells 12, no. 15: 1978. https://doi.org/10.3390/cells12151978
APA StyleFleischauer, J., Bastone, A. L., Selich, A., John-Neek, P., Weisskoeppel, L., Schaudien, D., Schambach, A., & Rothe, M. (2023). TGFβ Inhibitor A83-01 Enhances Murine HSPC Expansion for Gene Therapy. Cells, 12(15), 1978. https://doi.org/10.3390/cells12151978