

Anti-Cancer Prodrug Cyclophosphamide Exerts Thrombogenic Effects on Human Venous Endothelial Cells Independent of CYP450 Activation—Relevance to Thrombosis
 , , ,
, , ,  ,
, 
Abstract
1. Introduction
- ➢
- A reduced antithrombotic function of the endothelial cells in the blood vessel system;
- ➢
- Desquamation of endothelial cells by the dissolution of cell-substrate and/or cell-cell bonds;
- ➢
- By apoptosis/necrosis of endothelial cells (associated with the exposure of the thrombotic subendothelium);
- ➢
- Reduced regeneration capacity of endothelial cells, which finally also could lead to the exposure of the thrombotic subendothelium;
- ➢
- Changes in the glycocalix of the endothelial cells reduce the antithrombotic function.
2. Materials and Methods
2.1. Cell Culture
2.2. Determination of mRNA Expression Levels of Cytochrome P450 Enzymes (CYPs) and NADPH Cytochrome P450 Oxidoreductase (POR)
2.3. Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS)
2.4. Treatment of HUVEC with Cyclophosphamide
2.4.1. Detection of γH2AX Foci by Immunofluorescence
2.4.2. HUVEC Viability
2.4.3. Measurement of Metabolic Activity
2.4.4. Analysis of Eicosanoids
2.5. Statistics
3. Results
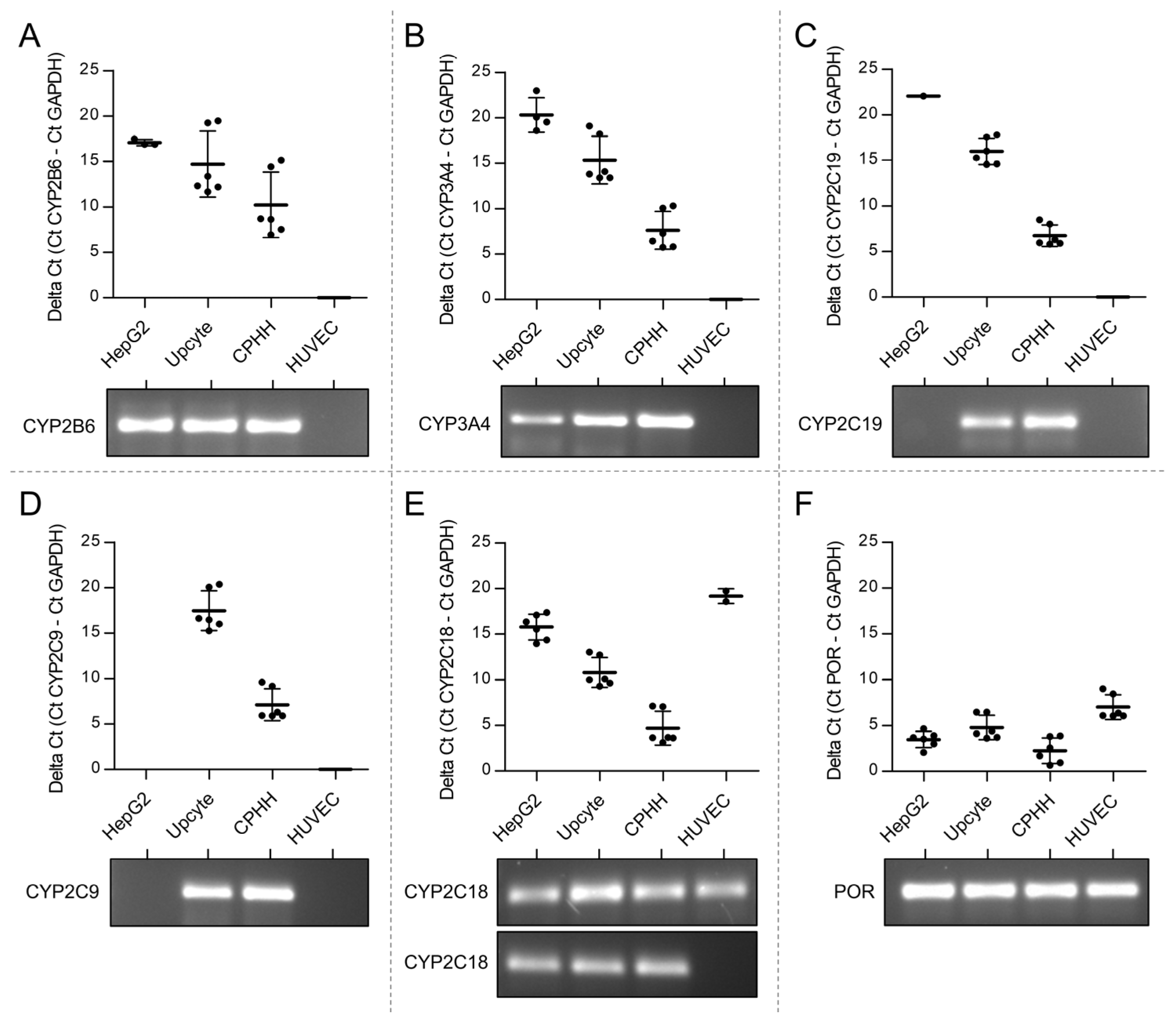
3.1. Lack of Expression of CPA Relevant CYPs in Venous Endothelial Cells
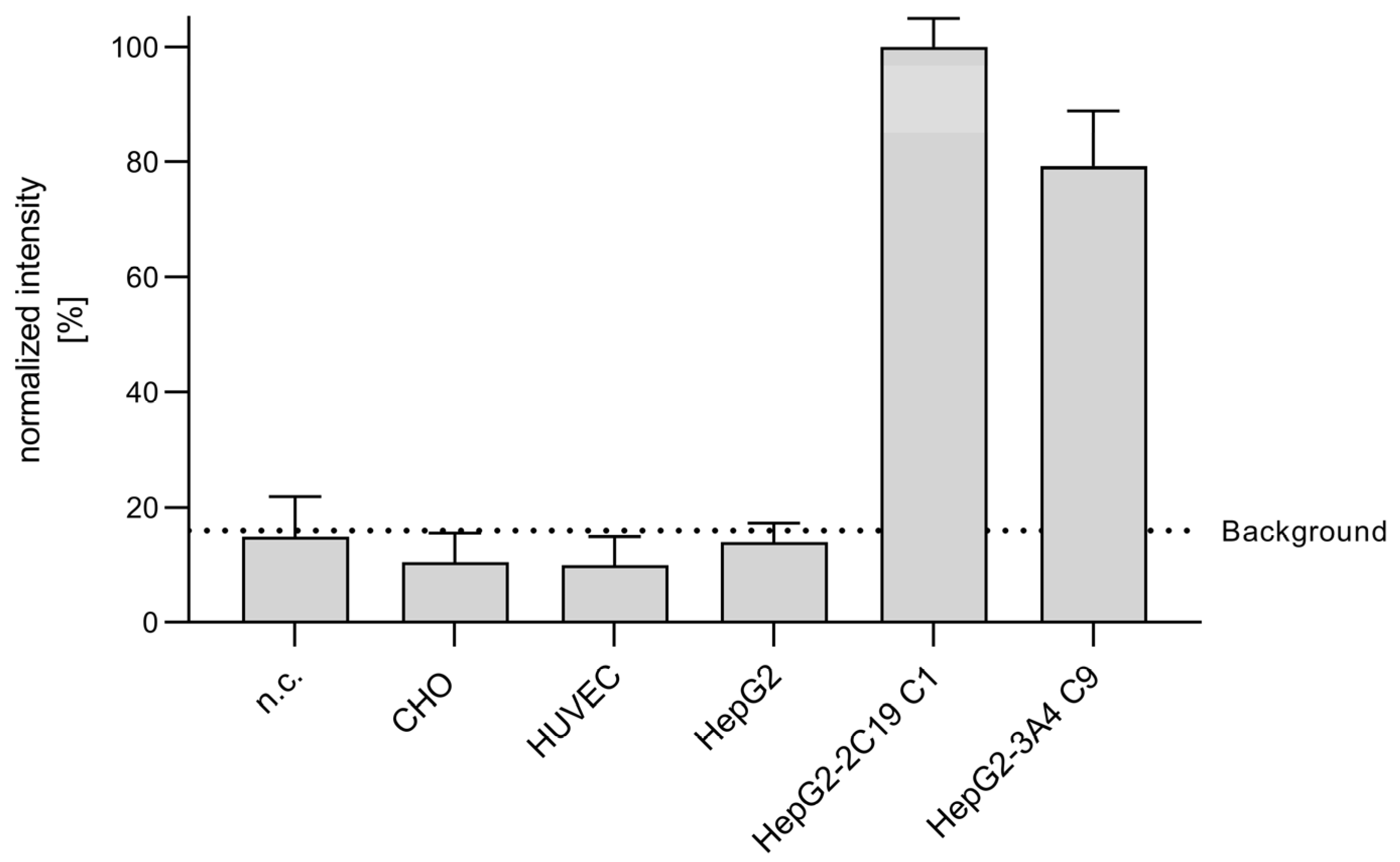
3.2. Metabolization of CPA in Endothelial Cells
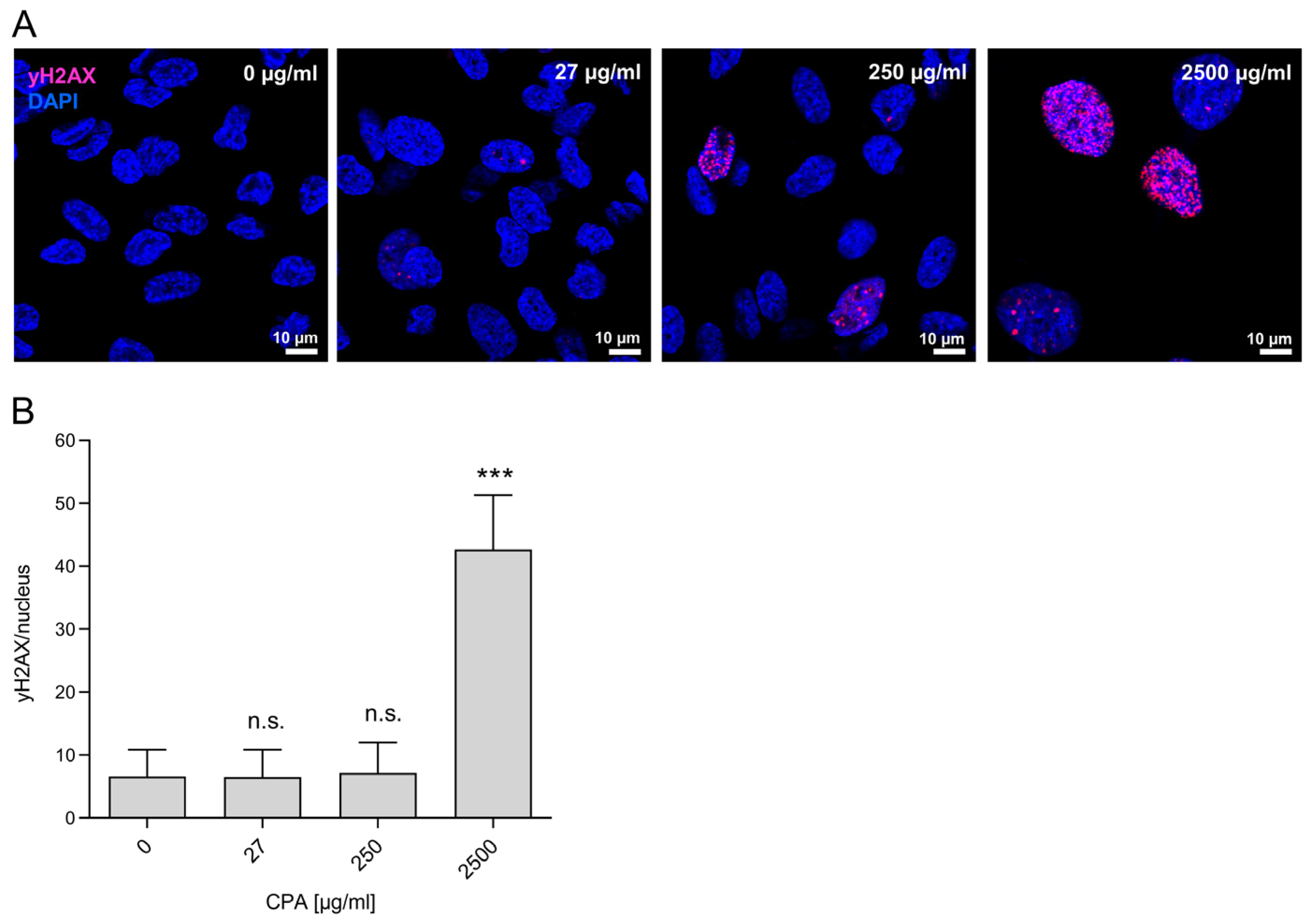
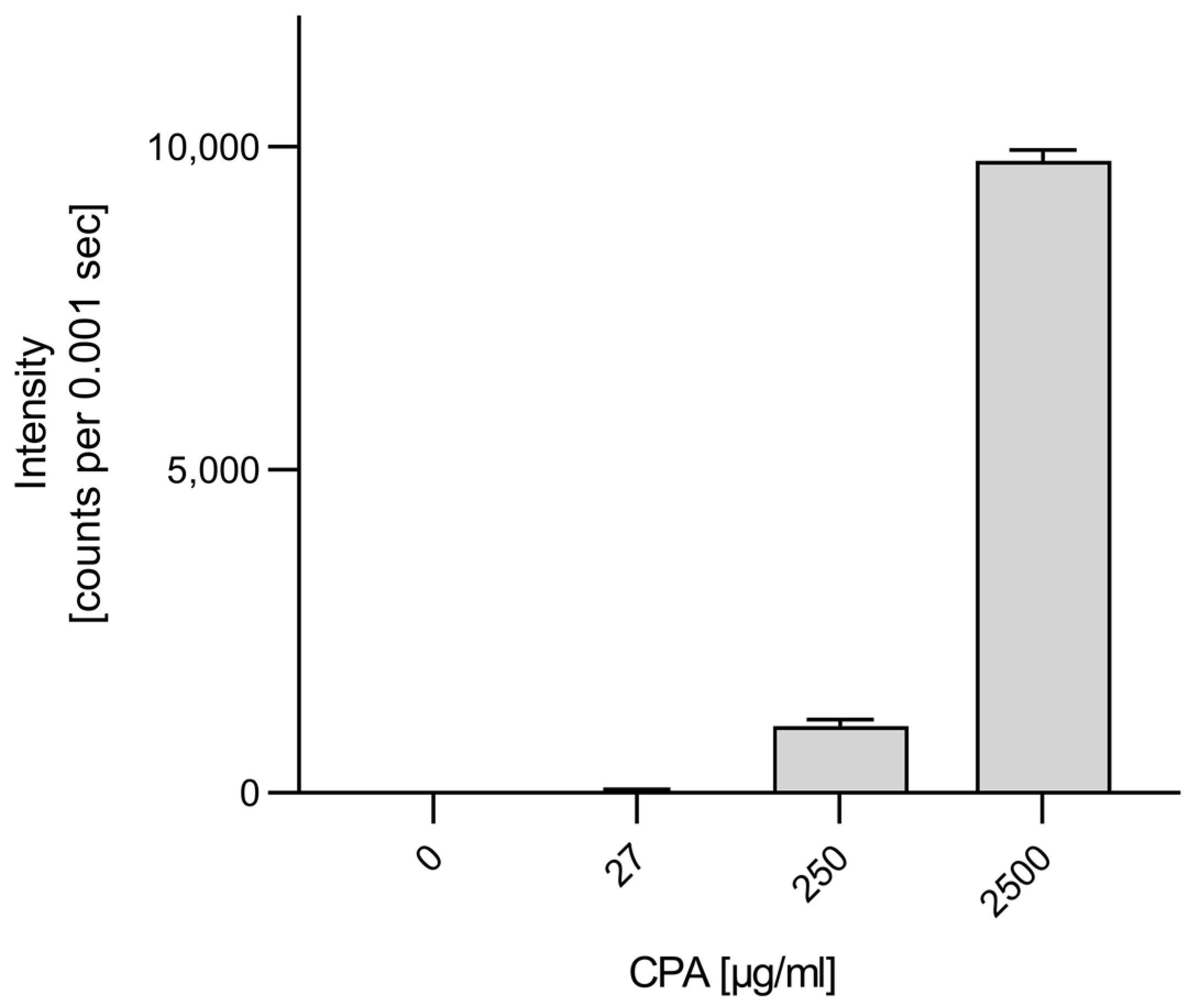
3.3. Genotoxicity of CPA in HUVEC
3.4. Influence of CPA on HUVEC Viability and Function
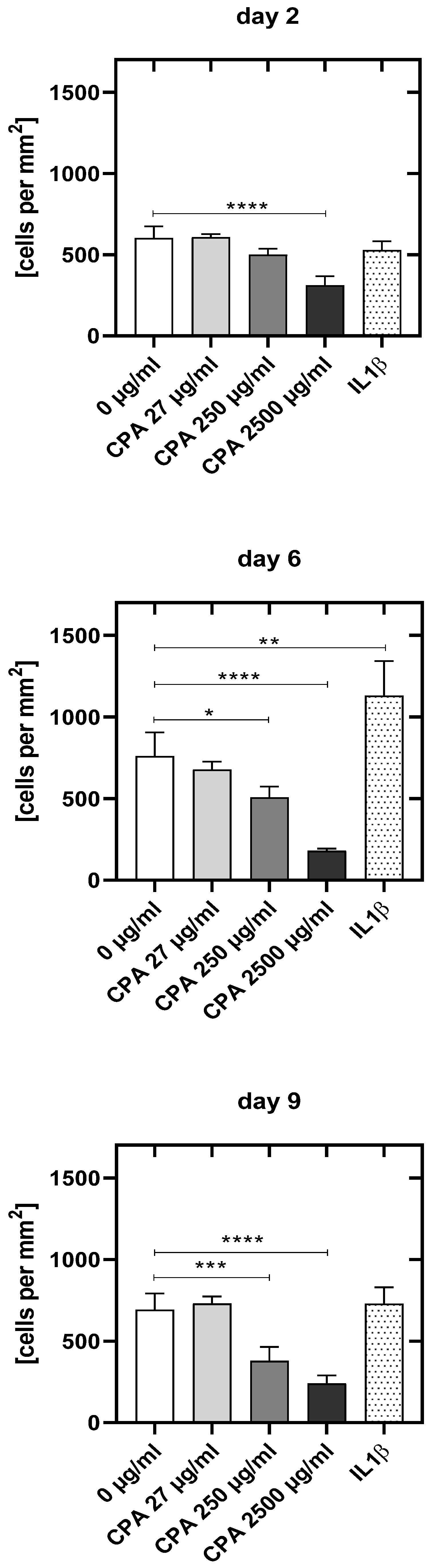
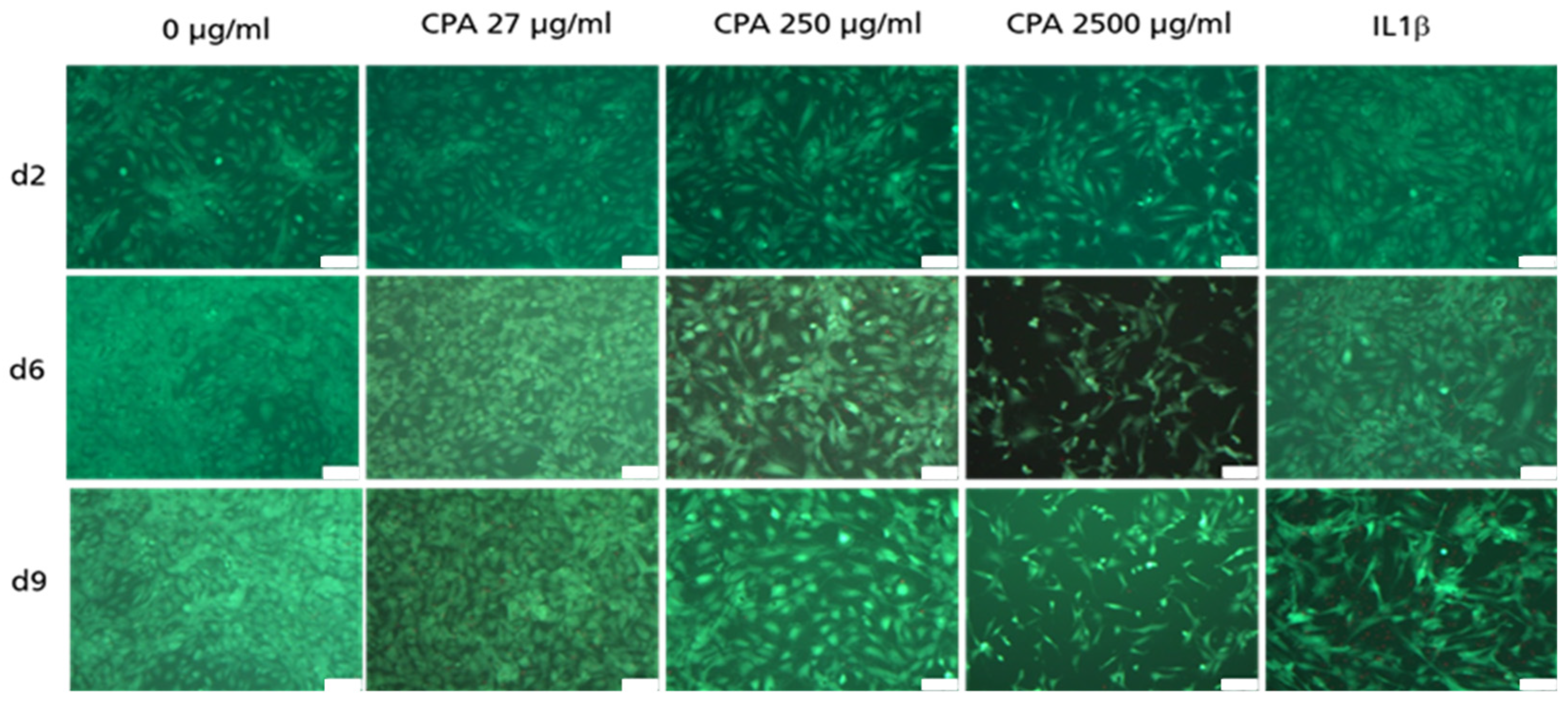
3.4.1. Decrease in HUVEC Density upon CPA Exposure
3.4.2. Adherent HUVEC with No Visible Decrease in Viability
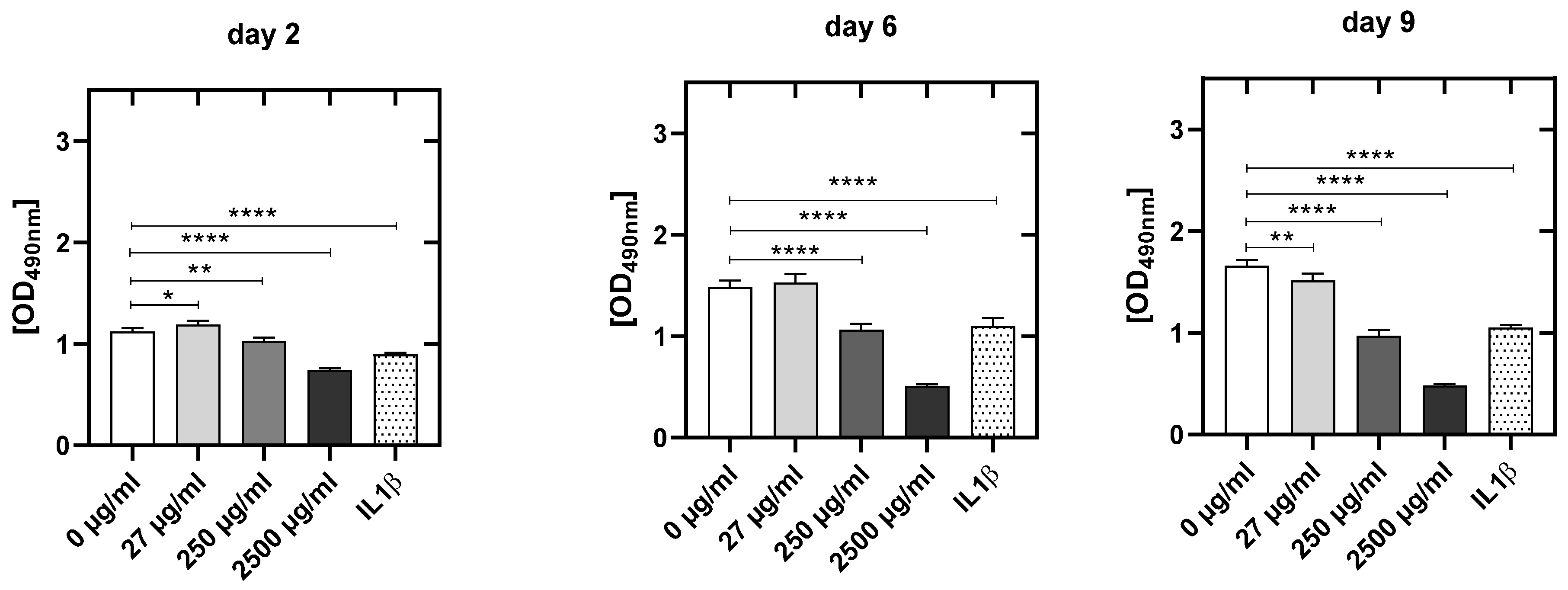
3.4.3. CPA Concentration-Dependent Decrease in Metabolic Activity in HUVEC
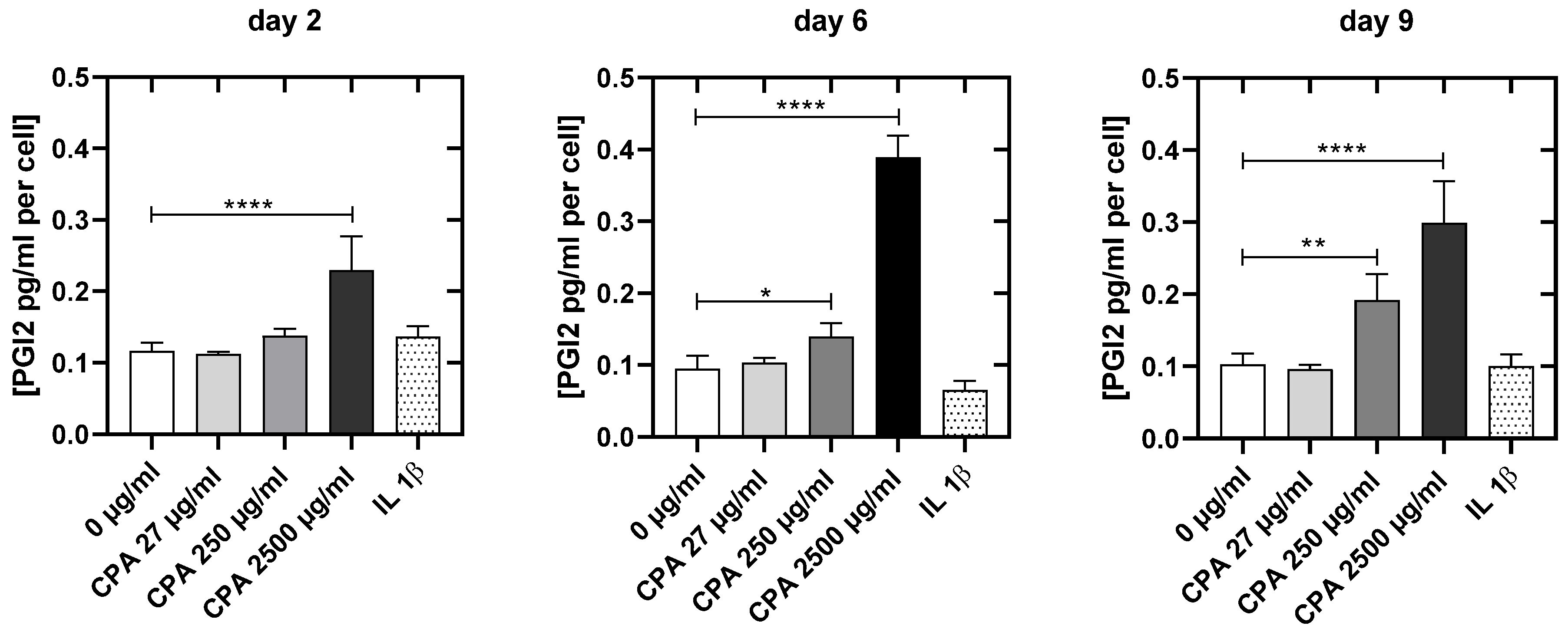
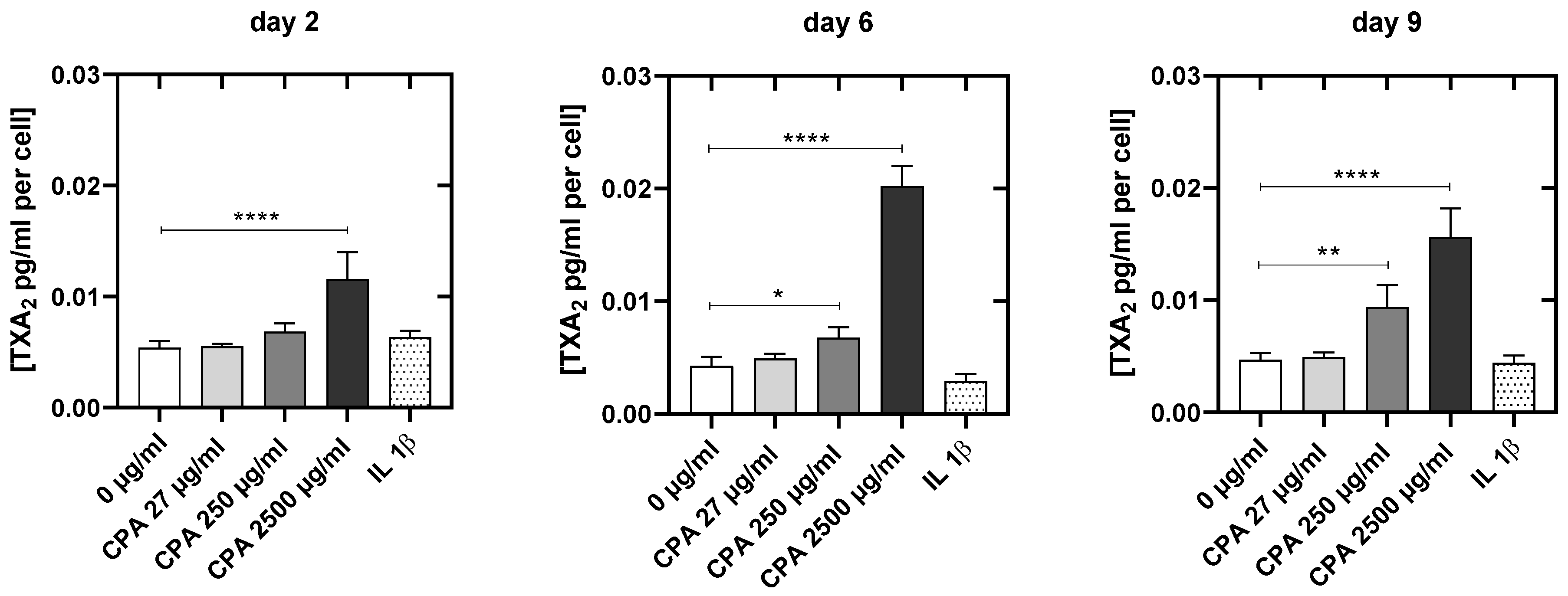
3.4.4. CPA Affects the Venous Endothelial Cell Function
4. Discussion
5. Conclusions
6. Limitations of the Study
- The CPA concentrations presented in the manuscript were selected in order to cover a large dose range. Here, the bolus administration after the injection should be simulated, in which even much higher intravascular CPA concentrations are present for a short time. However, since we are working in a static in vitro model, it is obvious that there is a significantly higher concentration of CPA in the cell culture than under dynamic conditions in vivo. Thus, the results obtained were also carefully and cautiously discussed and considered from these points of view. Naturally, continuous CPA metabolism cannot be simulated in vitro. This would require the time- and concentration-dependent addition of the active metabolites in vitro or investigations in animal models or in patients.
- The working group is aware that the selected time intervals are sometimes very long. The time points were selected to ensure that damage to the endothelium could also be recognized after multiple administrations. At the same time, the group wanted to enable the induction of CYPs in the cells through multiple administrations.
- The working group is aware that in vitro conditions are not comparable with in vivo conditions. Thus, the results obtained here can only allow a first assessment. However, there are clear and significant changes in endothelial cell function toward a prothrombotic state even at the mean CPA concentration.
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Abdol Razak, N.B.; Jones, G.; Bhandari, M.; Berndt, M.C.; Metharom, P. Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers 2018, 10, 380. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Ren, M.; Li, R.; Deng, X.; Li, Y.; Yan, K.; Xiao, L.; Yang, Y.; Wang, L.; Luo, M.; et al. Bevacizumab promotes venous thromboembolism through the induction of PAI-1 in a mouse xenograft model of human lung carcinoma. Mol. Cancer 2015, 14, 140. [Google Scholar] [CrossRef] [PubMed]
- Heit, J.A.; Silverstein, M.D.; Mohr, D.N.; Petterson, T.M.; O’Fallon, W.M.; Melton, L.J., 3rd. Risk factors for deep vein thrombosis and pulmonary embolism: A population-based case-control study. Arch. Intern. Med. 2000, 160, 809–815. [Google Scholar] [CrossRef]
- Prandoni, P.; Falanga, A.; Piccioli, A. Cancer and venous thromboembolism. Lancet Oncol. 2005, 6, 401–410. [Google Scholar] [CrossRef]
- Noble, S.; Pasi, J. Epidemiology and pathophysiology of cancer-associated thrombosis. Br. J. Cancer 2010, 102, S2–S9. [Google Scholar] [CrossRef]
- Kyriazi, V. Breast cancer as an acquired thrombophilic state. J. Breast Cancer 2012, 15, 148–156. [Google Scholar] [CrossRef]
- Schuetze, S.M.; Zhao, L.; Chugh, R.; Thomas, D.G.; Lucas, D.R.; Metko, G.; Zalupski, M.M.; Baker, L.H. Results of a phase II study of sirolimus and cyclophosphamide in patients with advanced sarcoma. Eur. J. Cancer 2012, 48, 1347–1353. [Google Scholar] [CrossRef]
- Rickles, F.R.; Edwards, R.L. Activation of blood coagulation in cancer: Trousseau’s syndrome revisited. Blood 1983, 62, 14–31. [Google Scholar] [CrossRef]
- Warry, E.; Hansen, R.; Gustafson, D.; Lana, S. Pharmacokinetics of cyclophosphamide after oral and intravenous administration to dogs with lymphoma. J. Vet. Intern. Med. 2011, 25, 903–908. [Google Scholar] [CrossRef]
- Zwicker, J.I.; Liebman, H.A.; Neuberg, D.; Lacroix, R.; Bauer, K.A.; Furie, B.C.; Furie, B. Tumor-derived tissue factor–bearing microparticles are associated with venous thromboembolic events in malignancy. Clin. Cancer Res. 2009, 15, 6830–6840. [Google Scholar] [CrossRef]
- Falanga, A.; Marchetti, M.; Russo, L. The mechanisms of cancer-associated thrombosis. Thromb. Res. 2015, 135, S8–S11. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Jung, F.; Pietzsch, J. Human endothelial cell models in biomaterial research. Trends Biotechnol. 2017, 35, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Nasibullin, I.; Smirnov, I.; Ahmadi, P.; Vong, K.; Kurbangalieva, A.; Tanaka, K. Synthetic prodrug design enables biocatalytic activation in mice to elicit tumor growth suppression. Nat. Commun. 2022, 13, 39. [Google Scholar] [CrossRef]
- Caparica, R.; Lambertini, M.; de Azambuja, E. How I treat metastatic triple-negative breast cancer. ESMO Open 2019, 4, e000504. [Google Scholar] [CrossRef] [PubMed]
- Doloff, J.C.; Chen, C.-S.; Waxman, D.J. Anti-tumor innate immunity activated by intermittent metronomic cyclophosphamide treatment of 9L brain tumor xenografts is preserved by anti-angiogenic drugs that spare VEGF receptor 2. Mol. Cancer 2014, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Chang, T.; Yu, L.; Goldstein, J.A.; Waxman, D.J. Identification of the polymorphically expressed CYP2C19 and the wild-type CYP2C9-ILE359 allele as low-Km catalysts of cyclophosphamide and ifosfamide activation. Pharmacogenetics 1997, 7, 211–221. [Google Scholar] [CrossRef]
- Chang, T.K.; Yu, L.; Maurel, P.; Waxman, D.J. Enhanced cyclophosphamide and ifosfamide activation in primary human hepatocyte cultures: Response to cytochrome P-450 inducers and autoinduction by oxazaphosphorines. Cancer Res. 1997, 57, 1946–1954. [Google Scholar]
- Chang, T.K.; Weber, G.F.; Crespi, C.L.; Waxman, D.J. Differential activation of cyclophosphamide and ifosphamide by cytochromes P-450 2B and 3A in human liver microsomes. Cancer Res. 1993, 53, 5629–5637. [Google Scholar]
- Griskevicius, L.; Yasar, Ü.; Sandberg, M.; Hidestrand, M.; Eliasson, E.; Tybring, G.; Hassan, M.; Dahl, M.-L. Bioactivation of cyclophosphamide: The role of polymorphic CYP2C enzymes. Eur. J. Clin. Pharmacol. 2003, 59, 103–109. [Google Scholar] [CrossRef]
- Rodriguez-Antona, C.; Ingelman-Sundberg, M. Cytochrome P450 pharmacogenetics and cancer. Oncogene 2006, 25, 1679–1691. [Google Scholar] [CrossRef]
- Ren, S.; Yang, J.-S.; Kalhorn, T.F.; Slattery, J.T. Oxidation of cyclophosphamide to 4-hydroxycyclophosphamide and deschloroethylcyclophosphamide in human liver microsomes. Cancer Res. 1997, 57, 4229–4235. [Google Scholar]
- Schwab, M.; Marx, C.; Zanger, U.M.; Eichelbaum, M.; Fischer-Bosch, M. Pharmakogenetik der Zytochrom-P-450-Enzyme Bedeutung fur Wirkungen und Nebenwirkungen von Medikamenten. Dtsch. Ärzteblatt-Ärztliche Mitteilungen-Ausg. A 2002, 99, 497–503. [Google Scholar]
- Boddy, A.V.; Furtun, Y.; Sardas, S.; Sardas, O.; Idle, J.R. Individual variation in the activation and inactivation of metabolic pathways of cyclophosphamide. J. Natl. Cancer Inst. 1992, 84, 1744–1748. [Google Scholar] [CrossRef] [PubMed]
- Boddy, A.V.; Yule, S.M. Metabolism and pharmacokinetics of oxazaphosphorines. Clin. Pharmacokinet. 2000, 38, 291–304. [Google Scholar] [CrossRef]
- Pratt, W.B.; Pratt, W.B. The Anticancer Drugs, 2nd ed.; Oxford University Press: New York, NY, USA, 1994; 352p. [Google Scholar]
- Wagner, T.; Heydrich, D.; Voelcker, G.; Hohorst, H. Über Blutspiegel und urin-ausscheidung von aktiviertem cyclophosphamid und seinen deaktivierungsprodukten beim menschen. J. Cancer Res. Clin. Oncol. 1980, 96, 79–92. [Google Scholar] [CrossRef]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef]
- Yule, S.M.; Boddy, A.V.; Cole, M.; Price, L.; Wyllie, R.; Tasso, M.J.; Pearson, A.D.; Idle, J.R. Cyclophosphamide metabolism in children. Cancer Res. 1995, 55, 803–809. [Google Scholar] [PubMed]
- Colvin, M.; Brundrett, R.B.; Kan, M.-N.N.; Jardine, I.; Fenselau, C. Alkylating properties of phosphoramide mustard. Cancer Res. 1976, 36, 1121–1126. [Google Scholar]
- Struck, R.F.; Kirk, M.C.; Witt, M.H.; Russell, W.L., Jr. Isolation and mass spectral identification of blood metabolities of cyclophosphamide: Evidence for phosphoramide mustard as the biologically active metabolite. Biomed. Mass Spectrom. 1975, 2, 46–52. [Google Scholar] [CrossRef]
- Chabner, B.A. Cancer Chemotherapy, 9th ed.; J. B. Lippincort: Philadelphia, PA, USA, 1990; pp. 276–283. [Google Scholar]
- Goodman, L.S.; Gilman, A. The Pharmacological Basis of Therapeutics, 9th ed.; Mc Graw-Hill: New York, NY, USA, 1995. [Google Scholar]
- Steinbrecht, S.; König, R.; Schmidtke, K.-U.; Herzog, N.; Scheibner, K.; Krüger-Genge, A.; Jung, F.; Kammerer, S.; Küpper, J.-H. Metabolic activity testing can underestimate acute drug cytotoxicity as revealed by HepG2 cell clones overexpressing cytochrome P450 2C19 and 3A4. Toxicology 2019, 412, 37–47. [Google Scholar] [CrossRef]
- Tsaryk, R.; Fabian, K.; Thacker, J.; Kaina, B. Xrcc2 deficiency sensitizes cells to apoptosis by MNNG and the alkylating anticancer drugs temozolomide, fotemustine and mafosfamide. Cancer Lett. 2006, 239, 305–313. [Google Scholar] [CrossRef]
- Farin, F.M.; Pohlman, T.H.; Omiecinski, C.J. Expression of cytochrome P450s and microsomal epoxide hydrolase in primary cultures of human umbilical vein endothelial cells. Toxicol. Appl. Pharmacol. 1994, 124, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Bayraktutan, U.; Draper, N.; Lang, D.; Shah, A.M. Expression of functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovasc. Res. 1998, 38, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Fisslthaler, B.; Popp, R.; Kiss, L.; Potente, M.; Harder, D.R.; Fleming, I.; Busse, R. Cytochrome P450 2C is an EDHF synthase in coronary arteries. Nature 1999, 401, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Fisslthaler, B.; Hinsch, N.; Chataigneau, T.; Popp, R.; Kiss, L.; Busse, R.; Fleming, I. Nifedipine increases cytochrome P4502C expression and endothelium-derived hyperpolarizing factor–mediated responses in coronary arteries. Hypertension 2000, 36, 270–275. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zeldin, D.C.; Foley, J.; Goldsworthy, S.M.; Cook, M.E.; Boyle, J.E.; Ma, J.; Moomaw, C.R.; Tomer, K.B.; Steenbergen, C.; Wu, S. CYP2J subfamily cytochrome P450s in the gastrointestinal tract: Expression, localization, and potential functional significance. Mol. Pharmacol. 1997, 51, 931–943. [Google Scholar] [CrossRef]
- Fleming, I. Cytochrome p450 and vascular homeostasis. Circ. Res. 2001, 89, 753–762. [Google Scholar] [CrossRef]
- Hoebel, B.G.; Steyrer, E.; Graier, W.F. Origin and function of epoxyeicosatrienoic acids in vascular endothelial cells: More than just endothelium-derived hyperpolarizing factor? Clin. Exp. Pharmacol. Physiol. 1998, 25, 826–830. [Google Scholar] [CrossRef]
- Elbekai, R.H.; El-Kadi, A.O. Cytochrome P450 enzymes: Central players in cardiovascular health and disease. Pharmacol. Ther. 2006, 112, 564–587. [Google Scholar] [CrossRef]
- Kliewer, S.A. The nuclear pregnane X receptor regulates xenobiotic detoxification. J. Nutr. 2003, 133, 2444S–2447S. [Google Scholar] [CrossRef]
- Bauer, B.; Hartz, A.M.; Fricker, G.; Miller, D.S. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol. Pharmacol. 2004, 66, 413–419. [Google Scholar] [PubMed]
- Chan, G.N.; Hoque, M.T.; Cummins, C.L.; Bendayan, R. Regulation of P-glycoprotein by orphan nuclear receptors in human brain microvessel endothelial cells. J. Neurochem. 2011, 118, 163–175. [Google Scholar] [CrossRef]
- Ghosh, C.; Hossain, M.; Solanki, J.; Najm, I.M.; Marchi, N.; Janigro, D. Overexpression of pregnane X and glucocorticoid receptors and the regulation of cytochrome P450 in human epileptic brain endothelial cells. Epilepsia 2017, 58, 576–585. [Google Scholar] [CrossRef]
- Ott, M.; Fricker, G.; Bauer, B. Pregnane X receptor (PXR) regulates P-glycoprotein at the blood-brain barrier: Functional similarities between pig and human PXR. J. Pharmacol. Exp. Ther. 2009, 329, 141–149. [Google Scholar] [CrossRef]
- Narang, V.S.; Fraga, C.; Kumar, N.; Shen, J.; Throm, S.; Stewart, C.F.; Waters, C.M. Dexamethasone increases expression and activity of multidrug resistance transporters at the rat blood-brain barrier. Am. J. Physiol.-Cell Physiol. 2008, 295, C440–C450. [Google Scholar] [CrossRef]
- Zastre, J.A.; Chan, G.N.; Ronaldson, P.T.; Ramaswamy, M.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Bendayan, M.; Bendayan, R. Up-regulation of P-glycoprotein by HIV protease inhibitors in a human brain microvessel endothelial cell line. J. Neurosci. Res. 2009, 87, 1023–1036. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Goodwin, B.; Willson, T.M. The nuclear pregnane X receptor: A key regulator of xenobiotic metabolism. Endocr. Rev. 2002, 23, 687–702. [Google Scholar] [CrossRef]
- Bertrand-Thiebault, C.; Masson, C.; Siest, G.; Batt, A.-M.; Visvikis-Siest, S. Effect of HMGCoA reductase inhibitors on cytochrome P450 expression in endothelial cell line. J. Cardiovasc. Pharmacol. 2007, 49, 306–315. [Google Scholar] [CrossRef]
- Zhou, T.; You, W.-T.; Ma, Z.-C.; Liang, Q.-D.; Tan, H.-l.; Xiao, C.-R.; Tang, X.-L.; Zhang, B.-L.; Wang, Y.-G.; Gao, Y. Ginkgolide B protects human umbilical vein endothelial cells against xenobiotic injuries via PXR activation. Acta Pharmacol. Sin. 2016, 37, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Miyahara, E.; Kurauchi, K.; Watanabe, E.; Ikawa, K.; Asaba, K.; Tanabe, T.; Okamoto, Y.; Kawano, Y. Mechanisms of fatal cardiotoxicity following high-dose cyclophosphamide therapy and a method for its prevention. PLoS ONE 2015, 10, e0131394. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Tondera, C.; Hauser, S.; Braune, S.; Görs, J.; Roch, T.; Klopfleisch, R.; Neffe, A.; Lendlein, A.; Pietzsch, J. Immunocompatibility and non-thrombogenicity of gelatin-based hydrogels. Clin. Hemorheol. Microcirc. 2021, 77, 335–350. [Google Scholar] [CrossRef]
- Krüger, A.; Fuhrmann, R.; Jung, F.; Franke, R. Influence of the coating with extracellular matrix and the number of cell passages on the endothelialization of a polystyrene surface. Clin. Hemorheol. Microcirc. 2015, 60, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, P.J.; Smith, J.R. Aging of endothelium in culture: Decrease in angiotensin-converting enzyme activity. Cell Biol. Int. Rep. 1982, 6, 379–384. [Google Scholar] [CrossRef]
- Goldsmith, J.; McCormick, J.; Yen, A. Endothelial cell cycle kinetics. Changes in culture and correlation with endothelial properties. Lab Invest 1984, 51, 643–647. [Google Scholar] [PubMed]
- Herzog, N.; Katzenberger, N.; Martin, F.; Schmidtke, K.-U. Generation of cytochrome P450 3A4-overexpressing HepG2 cell clones for standardization of hepatocellular testosterone 6β-hydroxylation activity. J. Cell. Biotechnol. 2015, 1, 15–26. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Hauser, S.; Neffe, A.T.; Liu, Y.; Lendlein, A.; Pietzsch, J.; Jung, F. Response of endothelial cells to gelatin-based hydrogels. ACS Biomater. Sci. Eng. 2021, 7, 527–540. [Google Scholar] [CrossRef]
- Schulz, C.; Jung, F.; Küpper, J.-H. Inhibition of phase-1 biotransformation and cytostatic effects of diphenyleneiodonium on hepatoblastoma cell line HepG2 and a CYP3A4-overexpressing HepG2 cell clone. Clin. Hemorheol. Microcirc. 2021, 79, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecht, S.; Kammerer, S.; Küpper, J.-H. HepG2 cells with recombinant cytochrome P450 enzyme overexpression: Their use and limitation as in vitro liver model. J. Cell. Biotechnol. 2019, 5, 55–64. [Google Scholar] [CrossRef]
- Kuo, L.J.; Yang, L.-X. γ-H2AX-a novel biomarker for DNA double-strand breaks. In Vivo 2008, 22, 305–309. [Google Scholar]
- Zhou, C.; Li, Z.; Diao, H.; Yu, Y.; Zhu, W.; Dai, Y.; Chen, F.F.; Yang, J. DNA damage evaluated by γH2AX foci formation by a selective group of chemical/physical stressors. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2006, 604, 8–18. [Google Scholar] [CrossRef]
- Crook, T.R.; Souhami, R.L.; Mclean, A.E. Cytotoxicity, DNA cross-linking, and single strand breaks induced by activated cyclophosphamide and acrolein in human leukemia cells. Cancer Res. 1986, 46, 5029–5034. [Google Scholar] [PubMed]
- Jeggo, P.; Lobrich, M. DNA double-strand breaks: Their cellular and clinical impact? Oncogene 2007, 26, 7717–7720. [Google Scholar] [CrossRef] [PubMed]
- Krüger-Genge, A.; Steinbrecht, S.; Küpper, J.-H.; Lendlein, A.; Jung, F. Evidence for cytostatic effect of cyclophosphamide on human vein endothelial cells in cancer therapy: Preliminary in vitro results. Clin. Hemorheol. Microcirc. 2018, 69, 267–276. [Google Scholar] [CrossRef]
- Schulz, C.; Kammerer, S.; Küpper, J.-H. NADPH-cytochrome P450 reductase expression and enzymatic activity in primary-like human hepatocytes and HepG2 cells for in vitro biotransformation studies. Clin. Hemorheol. Microcirc. 2019, 73, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Juma, F.; Rogers, H.; Trounce, J. The pharmacokinetics of cyclophosphamide, phosphoramide mustard and nor-nitrogen mustard studied by gas chromatography in patients receiving cyclophosphamide therapy. Br. J. Clin. Pharmacol. 1980, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Shustova, O.N.; Antonova, O.A.; Golubeva, N.V.; Khaspekova, S.G.; Yakushkin, V.V.; Aksuk, S.A.; Alchinova, I.B.; Karganov, M.Y.; Mazurov, A.V. Differential procoagulant activity of microparticles derived from monocytes, granulocytes, platelets and endothelial cells: Impact of active tissue factor. Blood Coagul. Fibrinolysis 2017, 28, 373–382. [Google Scholar] [CrossRef]
- Mackman, N. The many faces of tissue factor. J. Thromb. Haemost. 2009, 7, 136–139. [Google Scholar] [CrossRef]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Franke, R.; Fuhrmann, R.; Hiebl, B.; Jung, F. Influence of radiographic contrast media (iodixanol und iomeprol) on the morphology of human arterial and venous endothelial cells on extracellular matrix in vitro. Clin. Hemorheol. Microcirc. 2011, 48, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Holland, J.A.; Pritchard, K.; Rogers, N.; Stemerman, M. Perturbation of cultured human endothelial cells by atherogenic levels of low-density lipoprotein. Am. J. Pathol. 1988, 132, 474. [Google Scholar] [PubMed]
- Mitchell, J.A.; Ali, F.; Bailey, L.; Moreno, L.; Harrington, L.S. Role of nitric oxide and prostacyclin as vasoactive hormones released by the endothelium. Exp. Physiol. 2008, 93, 141–147. [Google Scholar] [CrossRef]
- Franke, R.; Fuhrmann, R.; Schnittler, H.; Petrow, W.; Simons, G. Humane Endothelzellen in vitro unter hydrodynamischer Scherbelastung: Pharmakologische Einflüsse auf Haftfähigkeit und Nonthrombogenität der Gefäßinnenwandzellen. VASA 1988, 24, 11–16. [Google Scholar] [PubMed]
- Nawroth, P.P.; Stern, D.M.; Kaplan, K.L.; Nossel, H.L. Prostacyclin production by perturbed bovine aortic endothelial cells in culture. Blood 1984, 64, 801–806. [Google Scholar] [CrossRef]
- Moncada, S. Generation of Prostacyclin and Endothelium-Derived Relaxing Factor from Endothelial Cells. Rev. Clin. Espanola 1986, 179, 323–328. [Google Scholar]
- Schnittler, H.; Franke, R.; Fuhrmann, R.; Mittermayer, C.; Drenckhahn, D. Development of stressfibers (sf) and microfilaments in human umbilical venous endothelial-cells (huvec) in dependence of varying culture periods within one passage. Eur. J. Cell Biol. 1986, 44, 18. [Google Scholar]
- Paubert-Braquet, M.; Braquet, P.; Demling, B.; Fletcher, R.; Foegh, M. Lipid Mediators in the Immunology of Shock; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; Volume 139. [Google Scholar]
- Wang, Y.; Gallant, R.C.; Ni, H. Extracellular matrix proteins in the regulation of thrombus formation. Curr. Opin. Hematol. 2016, 23, 280–287. [Google Scholar] [CrossRef]
- Sellers, M.M.; Stallone, J.N. Sympathy for the devil: The role of thromboxane in the regulation of vascular tone and blood pressure. Am. J. Physiol.-Heart Circ. Physiol. 2008, 294, H1978–H1986. [Google Scholar] [CrossRef]
- Moncada, S.; Gryglewski, R.; Bunting, S.; Vane, J. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 1976, 263, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Ally, A.; Horrobin, D. Thromboxane A2 in blood vessel walls and its physiological significance: Relevance to thrombosis and hypertension. Prostaglandins Med. 1980, 4, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Braune, S.; Küpper, J.-H.; Jung, F. Effect of prostanoids on human platelet function: An overview. Int. J. Mol. Sci. 2020, 21, 9020. [Google Scholar] [CrossRef] [PubMed]
- Middleton, J.D.; Sivakumar, S.; Hai, T. Chemotherapy-Induced Changes in the Lung Microenvironment: The Role of MMP-2 in Facilitating Intravascular Arrest of Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 10280. [Google Scholar] [CrossRef]
- Kobayashi, M.; Wada, H.; Fukui, S.; Mizutani, H.; Ichikawa, Y.; Shiraki, K.; Moritani, I.; Inoue, H.; Shimaoka, M.; Shimpo, H. A Clot Waveform Analysis Showing a Hypercoagulable State in Patients with Malignant Neoplasms. J. Clin. Med. 2021, 10, 5352. [Google Scholar] [CrossRef]
- Yu, L.; Guo, Y.; Chang, Z.; Zhang, D.; Zhang, S.; Pei, H.; Pang, J.; Zhao, Z.J.; Chen, Y. Bidirectional Interaction Between Cancer Cells and Platelets Provides Potential Strategies for Cancer Therapies. Front. Oncol. 2021, 11, 764119. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primer 5′-3′ | Sequence |
|---|---|---|
| CYP2B6 | Forward | CTCTCCATGACCCACACTAC |
| Reverse | TGTTGGGGGTATTTTGCCCA | |
| CYP3A4 | Forward | GTGGGGCTTTTATGATGGTCA |
| Reverse | GCCTCAGATTTCTCACCAACACA | |
| CYP2C19 | Forward | TTGACCCTCGTCACTTTCTG |
| Reverse | GTTGTGTCAAGGTCCTTTGG | |
| CYP2C9 | Forward | CCAACCCAGAGATGTTTGAC |
| Reverse | GAATGAAGCACAGCTGGTAG | |
| CYP2C18 | Forward | GAGACAACGAGCACCACTCTGA |
| Reverse | ACCACAGCATCTGTGTAGGGCA | |
| POR | Forward | AAGGCGGTGCCCACATCTAC |
| Reverse | TAGCGGCCCTTGGTCATCAG | |
| GAPDH | Forward | TGCACCACCAACTGCTTAGC |
| Reverse | GGCATGGACTGTGGTCATGAG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krüger-Genge, A.; Köhler, S.; Laube, M.; Haileka, V.; Lemm, S.; Majchrzak, K.; Kammerer, S.; Schulz, C.; Storsberg, J.; Pietzsch, J.; et al. Anti-Cancer Prodrug Cyclophosphamide Exerts Thrombogenic Effects on Human Venous Endothelial Cells Independent of CYP450 Activation—Relevance to Thrombosis. Cells 2023, 12, 1965. https://doi.org/10.3390/cells12151965
Krüger-Genge A, Köhler S, Laube M, Haileka V, Lemm S, Majchrzak K, Kammerer S, Schulz C, Storsberg J, Pietzsch J, et al. Anti-Cancer Prodrug Cyclophosphamide Exerts Thrombogenic Effects on Human Venous Endothelial Cells Independent of CYP450 Activation—Relevance to Thrombosis. Cells. 2023; 12(15):1965. https://doi.org/10.3390/cells12151965
Chicago/Turabian StyleKrüger-Genge, Anne, Susanne Köhler, Markus Laube, Vanessa Haileka, Sandy Lemm, Karolina Majchrzak, Sarah Kammerer, Christian Schulz, Joachim Storsberg, Jens Pietzsch, and et al. 2023. "Anti-Cancer Prodrug Cyclophosphamide Exerts Thrombogenic Effects on Human Venous Endothelial Cells Independent of CYP450 Activation—Relevance to Thrombosis" Cells 12, no. 15: 1965. https://doi.org/10.3390/cells12151965
APA StyleKrüger-Genge, A., Köhler, S., Laube, M., Haileka, V., Lemm, S., Majchrzak, K., Kammerer, S., Schulz, C., Storsberg, J., Pietzsch, J., Küpper, J.-H., & Jung, F. (2023). Anti-Cancer Prodrug Cyclophosphamide Exerts Thrombogenic Effects on Human Venous Endothelial Cells Independent of CYP450 Activation—Relevance to Thrombosis. Cells, 12(15), 1965. https://doi.org/10.3390/cells12151965