

Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation

 , ,
, ,  ,
, 
Abstract

1. Introduction
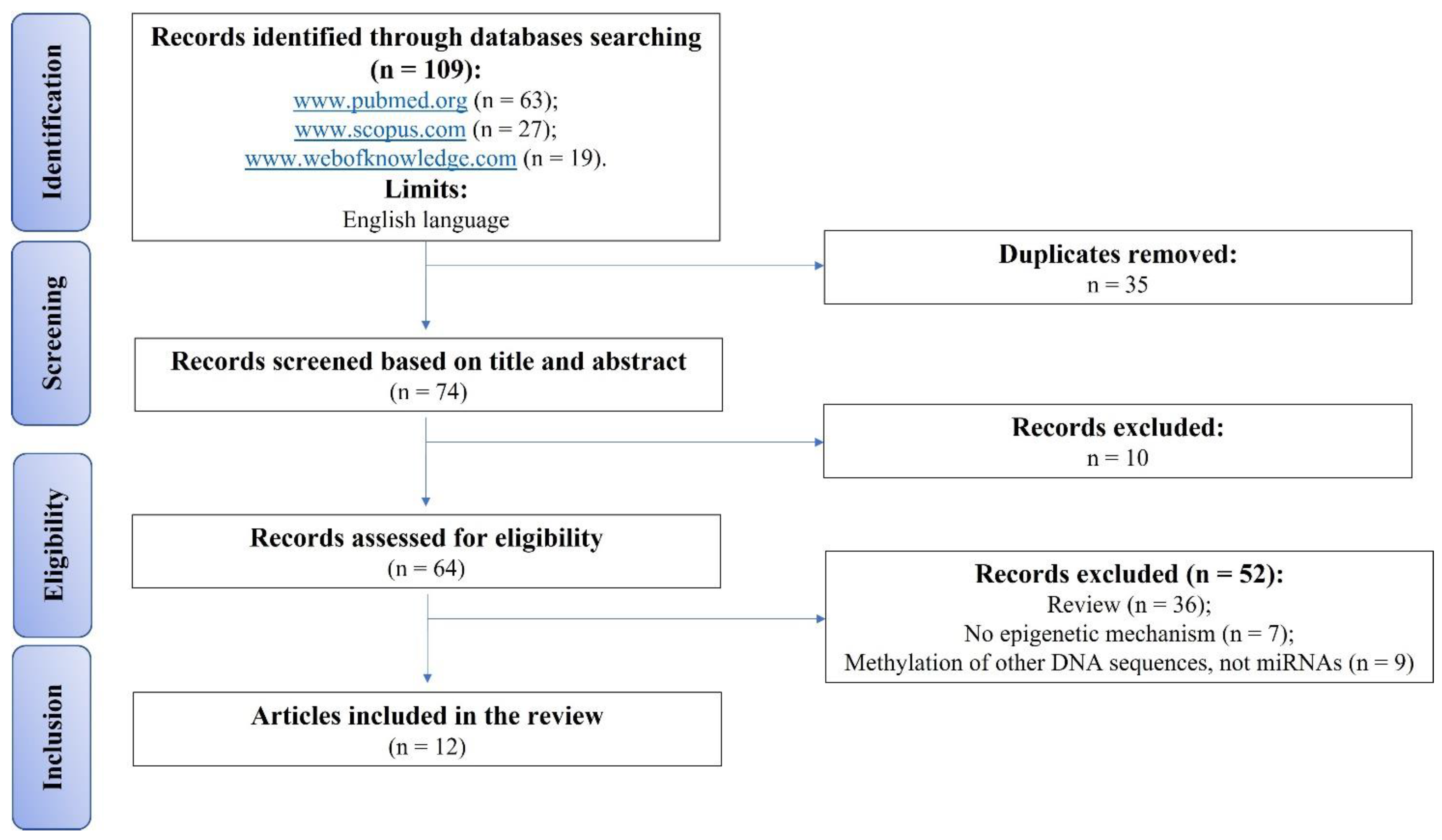
2. Materials and Methods
2.1. Eligibility Criteria
2.2. Search Strategy
2.3. Risk of Bias Assessments within Individual Studies
3. Results
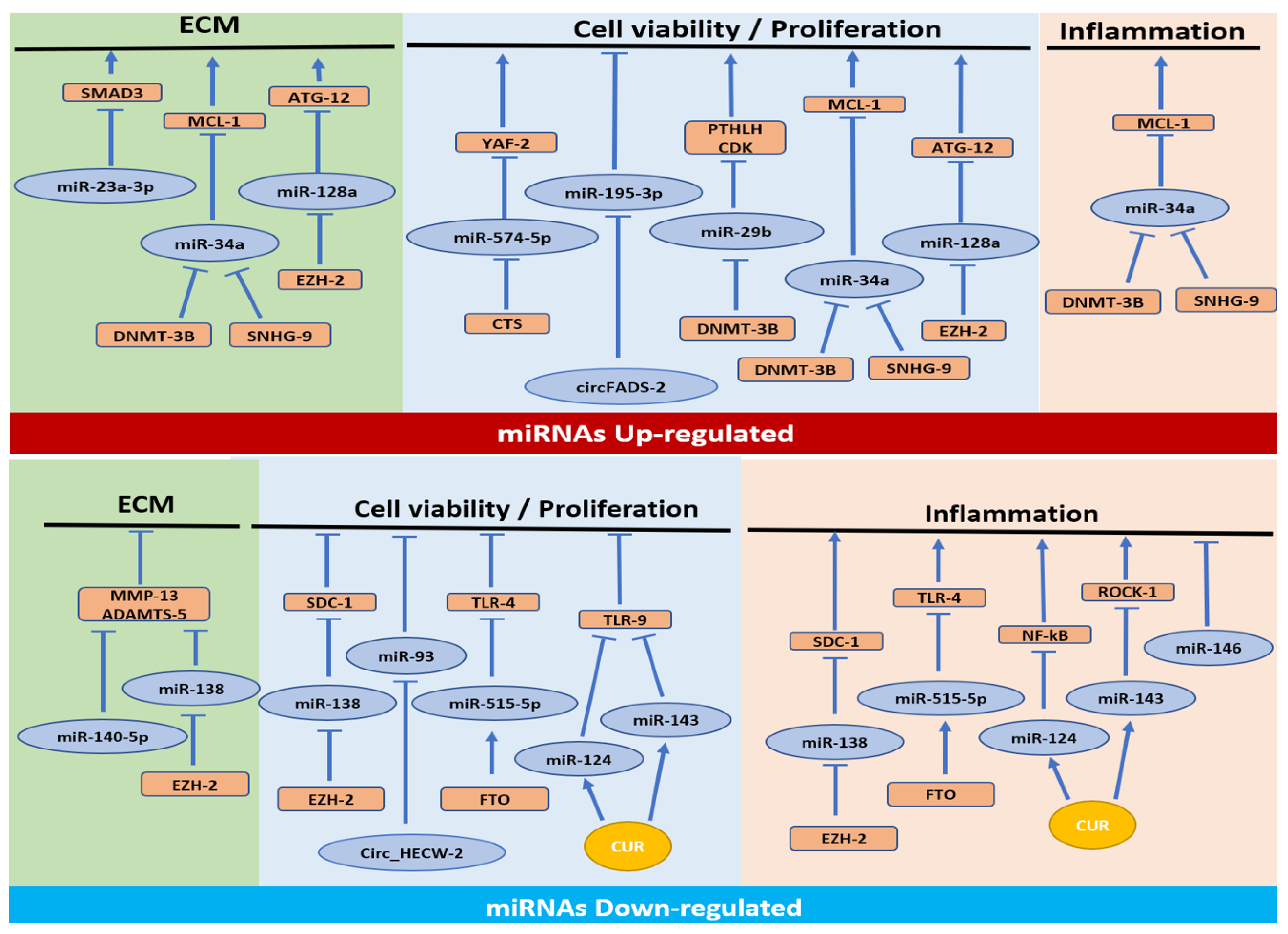
3.1. MiRNAs Upregulated in OA
3.1.1. Cell Proliferation/Apoptosis Pathway
3.1.2. Cell Proliferation/Apoptosis, ECM Synthesis and Inflammatory Pathways
3.1.3. Cell Proliferation/Apoptosis and ECM Synthesis Pathways
3.1.4. ECM Synthesis Pathway
3.2. MiRNAs Downregulated in OA
3.2.1. Cell Proliferation/Apoptosis and Inflammation Pathways
3.2.2. Inflammation Pathway
3.2.3. Cell Proliferation/Apoptosis Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, M.; Shen, J.; Jin, H.; Im, H.J.; Sandy, J.; Chen, D. Recent progress in understanding molecular mechanisms of cartilage degeneration during osteoarthritis. Ann. N. Y. Acad. Sci. 2011, 1240, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Yang, C.; Song, Y.; Liu, W.; Wang, K.; Li, S.; Zhang, Y. MicroRNA-23a-3p promotes the development of osteoarthritis by directly targeting SMAD3 in chondrocytes. Biochem. Biophys. Res. Commun. 2016, 478, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Safiri, S.; Kolahi, A.-A.; Smith, E.; Hill, C.; Bettampadi, D.; Mansournia, M.A.; Hoy, D.; Ashrafi-Asgarabad, A.; Sepidarkish, M.; Almasi-Hashiani, A.; et al. Global, regional and national burden of osteoarthritis 1990–2017: A systematic analysis of the Global Burden of Disease Study 2017. Ann. Rheum. Dis. 2020, 79, 819–828. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Grassel, S.; Zaucke, F.; Madry, H. Osteoarthritis: Novel molecular mechanisms increase our understanding of the disease pathology. J. Clin. Med. 2021, 10, 1938. [Google Scholar] [CrossRef]
- Maiese, K. Picking a bone with WISP1 (CCN4): New strategies against degenerative joint disease. J. Transl. Sci. 2016, 1, 83–85. [Google Scholar] [CrossRef]
- Wallis, J.A.; Barton, C.J.; Brusco, N.K.; Kemp, J.L.; Sherwood, J.; Young, K.; Jennings, S.; Trivett, A.; Ackerman, I.N. Exploring views of orthopaedic surgeons, rheumatologists and general practitioners about osteoarthritis management. Musculoskelet. Care 2021, 19, 524–532. [Google Scholar] [CrossRef]
- Carballo, C.B.; Nakagawa, Y.; Sekiya, I.; Rodeo, S.A. Basic Science of Articular Cartilage. Clin. Sports Med. 2017, 36, 413–425. [Google Scholar] [CrossRef]
- Sudirman, S.; Chang, H.W.; Chen, C.K.; Kong, Z.L. A dietary polysaccharide from Eucheuma cottonii downregulates proinflammatory cytokines and ameliorates osteoarthritis-associated cartilage degradation in obese rats. Food Funct. 2019, 10, 5697–5706. [Google Scholar] [CrossRef]
- Conaghan, P.G.; Cook, A.D.; Hamilton, J.A.; Tak, P.P. Therapeutic options for targeting inflammatory osteoarthritis pain. Nat. Rev. Rheumatol. 2019, 15, 355–363. [Google Scholar] [CrossRef]
- Watkins, L.R.; Chavez, R.A.; Landry, R.; Fry, M.; Green-Fulgham, S.M.; Coulson, J.D.; Collins, S.D.; Glover, D.K.; Rieger, J.; Forsayeth, J.R. Targeted interleukin-10 plasmid DNA therapy in the treatment of osteoarthritis: Toxicology and pain efficacy assessments. Brain Behav. Immun. 2020, 90, 155–166. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, X.; Xing, L.; Tian, F. Wnt signaling: A promising target for osteoarthritis therapy. Cell Commun. Signal. 2019, 17, 97. [Google Scholar] [CrossRef]
- Oliviero, A.; Della Porta, G.; Peretti, G.; Maffulli, N. MicroRNA in osteoarthritis: Physiopathology, diagnosis and therapeutic challenge. Br. Med. Bull. 2019, 130, 137–147. [Google Scholar] [CrossRef]
- Qatrun Nada, D.; Masniza, M.L.; Abdullah, N.; Marlini, M.; Elias, M.H.; Pathmanathan, S.G.; Hayati, A.R.; Fadlul Azim, F.; Hamid, A.A.; Nur Fariha, M.M. Distinct microRNA expression pattern in breast cancer cells following anti-neoplastic treatment: A systematic review and functional analysis of microRNA target genes. Malays. J. Pathol. 2022, 44, 367–385. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Sayed, D.; Abdellatif, M.; Oikawa, S.; Lee, M.; Motohashi, N.; Maeda, S.; Akimoto, T.; Syed, M.; Ball, J.P.; Mathis, K.W.; et al. MicroRNAs in Development and Disease. Physiol. Rev. 2011, 91, 827–887. [Google Scholar] [CrossRef] [PubMed]
- Fathollahi, A.; Aslani, S.; Jamshidi, A.; Mahmoudi, M. Epigenetics in osteoarthritis: Novel spotlight. J. Cell. Physiol. 2019, 234, 12309–12324. [Google Scholar] [CrossRef] [PubMed]
- Swingler, T.E.; Niu, L.; Smith, P.; Paddy, P.; Le, L.; Barter, M.J.; Young, D.; Clark, I.M. The function of microRNAs in cartilage and osteoarthritis. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 120), 40–47. [Google Scholar] [PubMed]
- Díaz-Prado, S.; Cicione, C.; Muiños-López, E.; Hermida-Gómez, T.; Oreiro, N.; Fernández-López, C.; Blanco, F.J. Characterization of microRNA expression profiles in normal and osteoarthritic human chondrocytes. BMC Musculoskelet. Disord. 2012, 13, 144. [Google Scholar] [CrossRef]
- Trachana, V.; Ntoumou, E.; Anastasopoulou, L.; Tsezou, A. Studying microRNAs in osteoarthritis: Critical overview of different analytical approaches. Mech. Ageing Dev. 2018, 171, 15–23. [Google Scholar] [CrossRef]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Króliczewski, J.; Sobolewska, A.; Lejnowski, D.; Collawn, J.F.; Bartoszewski, R. microRNA single polynucleotide polymorphism influences on microRNA biogenesis and mRNA target specificity. Gene 2018, 640, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Simon, T.C.; Jeffries, M.A. The epigenomic landscape in osteoarthritis. Curr. Rheumatol. Rep. 2017, 19, 30. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; de Andrés, M.C.; Hashimoto, K.; Itoi, E.; Otero, M.; Goldring, M.B.; Oreffo, R.O.C. DNA methylation of the RUNX2 P1 promoter mediates MMP13 transcription in chondrocytes. Sci. Rep. 2017, 7, 7771. [Google Scholar] [CrossRef] [PubMed]
- van Wijnen, A.J.; Westendorf, J.J. Epigenetics as a New Frontier in Orthopedic Regenerative Medicine and Oncology. J. Orthop. Res. 2019, 37, 1465–1474. [Google Scholar] [CrossRef]
- Zhou, Y.; Chen, M.; O’Keefe, R.J.; Shen, J.; Li, Z.; Zhou, J.; Zhou, X.; Mao, J.J. Epigenetic and therapeutic implications of dnmt3b in temporomandibular joint osteoarthritis. Am. J. Transl. Res. 2019, 11, 1736–1747. [Google Scholar]
- Yue, S.; Su, X.; Teng, J.; Wang, J.; Guo, M. Cryptotanshinone interferes with chondrocyte apoptosis in osteoarthritis by inhibiting the expression of miR-574-5p. Mol. Med. Rep. 2021, 23, 424. [Google Scholar] [CrossRef]
- Zhang, H.; Li, J.; Shao, W.; Shen, N. LncRNA SNHG9 is downregulated in osteoarthritis and inhibits chondrocyte apoptosis by downregulating miR-34a through methylation. BMC Musculoskelet. Disord. 2020, 21, 511. [Google Scholar] [CrossRef]
- Zhang, H.; Ge, J.; Lu, X. CircFADS2 is downregulated in osteoarthritis and suppresses LPS-induced apoptosis of chondrocytes by regulating miR-195-5p methylation. Arch. Gerontol. Geriatr. 2021, 96, 104477. [Google Scholar] [CrossRef]
- Dou, P.; He, Y.; Yu, B.; Duan, J. Downregulation of microRNA-29b by DNMT3B decelerates chondrocyte apoptosis and the progression of osteoarthritis via PTHLH/CDK4/RUNX2 axis. Aging 2020, 13, 7676–7690. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Zhao, Y.; Xu, T. DNA methyltransferase 3 beta mediates the methylation of the microRNA-34a promoter and enhances chondrocyte viability in osteoarthritis. Bioengineered 2021, 12, 11138–11155. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.S.; Ko, J.Y.; Wu, R.W.; Sun, Y.C.; Chen, Y.S.; Wu, S.L.; Weng, L.H.; Jahr, H.; Wang, F.S. MicroRNA-128a represses chondrocyte autophagy and exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018, 9, 919. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, X.; Ding, X.; Huang, T.; Song, D.; Tao, H. EZH2 is associated with cartilage degeneration in osteoarthritis by promoting SDC1 expression via histone methylation of the microRNA-138 promoter. Lab. Investig. 2021, 101, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Zhang, J.; Yang, J.; Lv, Q.; Zhong, C. Overexpression of FTO alleviates osteoarthritis by regulating the processing of miR-515-5p and the TLR4/MyD88/NF-κB axis. Int. Immunopharmacol. 2023, 114, 109524. [Google Scholar] [CrossRef]
- Qiu, B.; Xu, X.; Yi, P.; Hao, Y. Curcumin reinforces MSC-derived exosomes in attenuating osteoarthritis via modulating the miR-124/NF-kB and miR-143/ROCK1/TLR9 signalling pathways. J. Cell Mol. Med. 2020, 24, 10855–10865. [Google Scholar] [CrossRef]
- Papathanasiou, I.; Trachana, V.; Mourmoura, E.; Tsezou, A. DNA methylation regulates miR-140-5p and miR-146a expression in osteoarthritis. Life Sci. 2019, 228, 274–284. [Google Scholar] [CrossRef]
- Zuo, J.; Chen, C.; Zhang, X.; Wu, J.; Li, C.; Huang, S.; He, P.; Wa, Q.; Zhang, W. Circ_HECW2 regulates LPS-induced apoptosis of chondrocytes via miR-93 methylation. Immunity Inflamm. Dis. 2021, 9, 943–949. [Google Scholar] [CrossRef]
- Nugent, M. MicroRNAs: Exploring new horizons in osteoarthritis. Osteoarthr. Cartil. 2016, 24, 573–580. [Google Scholar] [CrossRef]
- Jones, T.L.; Esa, M.S.; Li, K.H.C.; Krishnan, S.R.G.; Elgallab, G.M.; Pearce, M.S.; Young, D.A.; Birrell, F.N. Osteoporosis, fracture, osteoarthritis & sarcopenia: A systematic review of circulating microRNA association. Bone 2021, 152, 116068. [Google Scholar]
- Yan, Z.; Xiong, J.; Zhao, C.; Qin, C.; He, C. Decreasing cartilage damage in a rat model of osteoarthritis by intra-articular injection of deoxycholic acid. Int. J. Clin. Exp. Med. 2015, 8, 9038–9045. [Google Scholar] [PubMed]
- Wu, C.-J.; Liu, R.-X.; Huan, S.-W.; Tang, W.; Zeng, Y.-K.; Zhang, J.-C.; Yang, J.; Li, Z.-Y.; Zhou, Y.; Zha, Z.-G.; et al. Senescent skeletal cells cross-talk with synovial cells plays a key role in the pathogenesis of osteoarthritis. Arthritis Res. Ther. 2022, 24, 59. [Google Scholar] [CrossRef] [PubMed]
- Di Nicola, V. Degenerative osteoarthritis a reversible chronic disease. Regen. Ther. 2020, 15, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef]
- Thielen, N.; Neefjes, M.; Wiegertjes, R.; van den Akker, G.; Vitters, E.; van Beuningen, H.; Davidson, E.B.; Koenders, M.; van Lent, P.; van de Loo, F.; et al. Osteoarthritis-Related Inflammation Blocks TGF-β’s Protective Effect on Chondrocyte Hypertrophy via (de)Phosphorylation of the SMAD2/3 Linker Region. Int. J. Mol. Sci. 2021, 22, 8124. [Google Scholar] [CrossRef]
- Valdes, A.M.; Spector, T.D.; Tamm, A.; Kisand, K.; Doherty, S.A.; Dennison, E.M.; Mangino, M.; Tamm, A.; Kerna, I.; Hart, D.J.; et al. Genetic variation in the SMAD3 gene is associated with hip and knee osteoarthritis. Arthritis Rheum. 2010, 62, 2347–2352. [Google Scholar] [CrossRef]
- Sun, K.; Luo, J.; Guo, J.; Yao, X.; Jing, X.; Guo, F. The PI3K/AKT/mTOR signaling pathway in osteoarthritis: A narrative review. Osteoarthr. Cartil. 2020, 28, 400–409. [Google Scholar] [CrossRef]
- Li, Z.; Dai, A.; Yang, M.; Chen, S.; Deng, Z.; Li, L. p38MAPK Signaling Pathway in Osteoarthritis: Pathological and Therapeutic Aspects. J. Inflamm. Res. 2022, 15, 723–734. [Google Scholar] [CrossRef]
- Rajagopal, K.; Ramesh, S.; Madhuri, V. Early Addition of Parathyroid Hormone–Related Peptide Regulates the Hypertrophic Differentiation of Mesenchymal Stem Cells. Cartilage 2021, 13, 143S–152S. [Google Scholar] [CrossRef] [PubMed]
- Gambari, L.; Cellamare, A.; Grassi, F.; Grigolo, B.; Panciera, A.; Ruffilli, A.; Faldini, C.; Desando, G. Overview of Anti-Inflammatory and Anti-Nociceptive Effects of Polyphenols to Halt Osteoarthritis: From Preclinical Studies to New Clinical Insights. Int. J. Mol. Sci. 2022, 23, 15861. [Google Scholar] [CrossRef] [PubMed]
- Tew, S.R.; Hardingham, T.E. Regulation of SOX9 mRNA in human articular chondrocytes involving p38 MAPK activation and mRNA stabilization. J. Biol. Chem. 2006, 281, 39471–39479. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Jia, Y.; Liu, H.; He, M.; Yang, Y.; Xiao, W.; Li, Y. RhoA/ROCK pathway: Implication in osteoarthritis and therapeutic targets. Am. J. Transl. Res. 2019, 11, 5324–5331. [Google Scholar]
- Segarra-Queralt, M.; Piella, G.; Noailly, J. Network-based modelling of mechano-inflammatory, chondrocyte regulation in early osteoarthritis. Front. Bioeng. Biotechnol. 2023, 11, 1006066. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Aim | In Vitro Model | Analyses | Results | Conclusions in OA | Ref. |
|---|---|---|---|---|---|
| Evaluation of CTS on miR-574-5p methylation in chondrocyte apoptosis during OA | Chondrocytes from mice (4 days) treated with IL1β. (1) Cells+CTS; (2) Cells+CTS+5-aza-CdR; (3) Cells+CTS and transfected with: miR-574-5p mimic; oe-YAF2 | RT-qPCR (miR-574-5p and Yaf2); WB (Bax, Bcl-2); Flow cytometry (cell apoptosis); CCK-8 assay (cell proliferation); Dual-luciferase reporter assay (relationship between miR-574-5p and Yaf2); MSP (miR-574-5p methylation) | Cells: ↑ miR-574-5p, Bax, apoptosis; ↓ Yaf2, Bcl-2, cell proliferation, ↓miR-574-5p promoter methylation. Cells+CTS: ↓ miR-574-5p, Bax, apoptosis; ↑ Yaf2, Bcl-2, cell proliferation, miR-574-5p promoter methylation. Cells+CTS+5-aza-CdR: ↓ miR-574-5p promoter methylation. Cells+CTS+miR-574-5p mimic: ↑ miR-574-5p, Bax, apoptosis; ↓ Yaf2, Bcl2, cell proliferation. Cells+CTS+miR-574-5p mimic +oe-YAF2: ↑ Yaf2, cell proliferation; ↓ Bax, cell apoptosis | - miR-574-5p upregulated. - Bax is upregulated, while Yaf2 and Bcl-2 are downregulated. - Negative correlation between miR-574-5p and Yaf2 and Bcl-2. - Positive correlation between miR-574-5p and Bax. - CTS reduces miR-574-5p and increases miR-574-5p promoter methylation. - CTS could affect chondrocyte proliferation and apoptosis by regulating the expression of miR-574-5p and then interfering with Yaf2. | Yue 2021 [28] |
| Evaluation of the interaction between SNHG9 and miR-34a methylation in chondrocyte apoptosis during OA | (1)SF from OA pz (n = 60). (2) Chondrocytes from OA (n = 60) or healthy donors (n = 60) and transfected with: oe-SNHG9; miR-34a mimic | RT-qPCR (SNHG9, miR-34a); ELISA (CASPASE-3); Flow cytometry (cell apoptosis); MSP (miR-34a methylation) | OA SF: ↓ SNHG9; ↑ miR-34a, CASPASE-3, cell apoptosis. OA cells+oe-SNHG9: ↓ miR-34a, apoptosis, CASPASE-3; ↑ miR-34a methylation. OA cells+miR-34a mimic: ↑ apoptosis, CASPASE-3. Healthy cells+oe-SNHG9: no changes in miR-34a, miR-34a methylation, apoptosis, CASPASE-3. Healthy cells+miR-34a mimic: no changes in SNHG9, apoptosis, CASPASE-3. | - miR-34a upregulated. - Negative correlation between SNHG9 and miR-34a. - Overexpression of SNHG9 decreases miR-34a through methylation. - Overexpression of SNHG9 decreases chondrocyte apoptosis through miR-34a | Zhang 2020 [29] |
| Evaluation of the interaction between circFADS2 and miR-195-5p methylation in chondrocyte apoptosis during OA | (1) SF from OA pz (n = 63); (2) Purchased human OA chondrocytes transfected with: oe-circFADS2; miR-195-5p mimic | RT-qPCR (circFADS2, miR-195-5p); Flow cytometry (cell apoptosis); MSP (miR-195-5p methylation). | OA SF: ↓ circFADS2; ↑ miR-195-5p. Cells+oe-circFADS2: ↓ miR-195-5p, apoptosis; ↑ miR-195-5p methylation. Cells+miR-195-5p mimic: ↑ apoptosis; no changes in circFADS2 expression | - miR-195-5p upregulated. - Negative correlation between CircFADS2 and miR-195-5p. - Overexpression of circFADS2 decreases miR-195-5p expression through methylation. - Overexpression of circFADS2 decrases chondrocytes apoptosis through miR-195-5p | Zhang 2021 [30] |
| Evaluation of the relationship between DNMT3B and miR-29b methylation in cell apoptosis, ECM synthesis and inflammation in OA | (1) Cartilage or chondrocytes from OA pz (n = 46) treated with IL1β and transfected with: sh-DNMT3B; sh-CDK4; oe-DNMT3B; oe-PTHLH; miR-29b mimic; miR-29b inhibitor | RT-qPCR (DNMT3B, miR-29b, PTHLH, CDK4, RUNX2); WB (DNMT3B, PTHLH, CDK4, Ki67, Aggrecan, MMP3, MMP13); MTT (cell viability); Flow cytometry (cell apoptosis); IHF (Ki67, Aggrecan); MSP (miR-29b promoter methylation); Dual-luciferase reporter gene assay (relationship between miR-29b and PTHLH). | OA cartilage: ↓ DNMT3B, PTHLH, CDK4, miR-29b promoter methylation; ↑ miR-29b. Cells: ↓ miR-29b methylation; ↑ miR-29b expression. Cells+oe-DNMT3B: ↑ miR-29b methylation, PTHLH, CDK4, ubiquitinated RUNX2, cell viability, Ki67, Aggrecan; ↓ miR-29b expression, apoptosis, MMP3, MMP13. Cells+miR-29b mimic: ↓ luciferase activity of PTHLH. Cells+miR-29b inhibitor: ↑ PTHLH. Cells+sh-DNMT3B: ↓ PTHLH. Cells+oe-PTHLH: ↑ CDK4, cell viability, Ki67 and Aggrecan; ↓ apoptosis, MMP3 and MMP13. Cells+oe-PTHLH+sh-CDK4: ↓ cell viability, Ki67, Aggrecan; ↑ apoptosis, MMP3 and MMP13 | - miR-29b upregulated. - Negative correlation between DNMT3B and miR-29b. - Overexpression of DNMT3B decreases miR-29b through methylation of the miR-29b promoter. - DNMT3B increases PTHLH expression by inhibiting miR-29b. - PTHLH impedes the apoptosis of OA chondrocytes by elevating CDK4 | Dou 2021 [31] |
| Evaluation of the role of miR-34a epigenetic mechanism in the degradation of ECM, cell viability and inflammatory response in OA | (1) Cartilage or chondrocytes from OA pz (n = 55) transfected or not with: miR-34a inhibitor; sh-DNMT3B; sh-MCL1. | RT-qPCR (miR-34a, DNMT3B, MCL1, MMP3, MMP13, COL2A1, iNOS, COX2, TNFα, IL6, PG); In situ hybridization (miR-34a expression in tissues); CCK-8 (cell viability); Flow cytometry (Cell apoptosis); WB (PI3K/AKT); MSP (miR-34a methylation); Dual-luciferase reporter gene assay (relationship between MCL1 and miR-34a). | OA cartilage and cells: ↑ miR-34a, MMP3, MMP13, iNOS, COX2, TNFα, IL6; ↓ COL2A1, MCL1, DNMT3B, miR-34a methylation. Cells+sh-DNMT3B: ↑ miR-34a; ↓ MCL1, miR-34a methylation. Cells+miR-34a inhibitor: ↓ MMP3, MMP13, apoptosis, iNOS, COX2, TNFα, IL6; ↑ cell viability, COL2A1, PG, PI3K/AKT pathway activity, MCL1 luciferase activity. Cells+sh-DNMT3B+sh-MCL1: ↓ cell viability, PG, PI3K/AKT pathway activity; ↑ apoptosis, iNOS, COX2, TNFα, IL6. | - miR-34a upregulated. - Positive correlation between miR-34a and MMP3, MMP13, iNOS, COX2, TNFα and IL6. - Negative correlation between miR-34a and COL2A1, MCL1 and DNMT3B. - DNMT3B suppresses miR-34 through an epigenetic mechanism. - miR-34a targets and inhibits MCL1 expression. - DNMT3B/miR-34a/MCL1 axis regulates chondrocyte viability. - DNMT3B/miR-34a/MCL1 axis regulates ECM degradation. - DNMT3B/miR-34a/MCL1 axis regulates the inflammatory response. - DNMT3B/miR-34a/MCL1 axis affects chondrocyte viability and OA progression via the PI3K/AKT pathway. | Xiong 2021 [32] |
| Evaluation of the miR-128a methylation effects on chondrocyte survival and articular cartilage anabolism during OA | (1) Cartilage from OA pz (n = 28). 2) 293T cells treated with IL1β transfected with: oe-miR-128; Sh-miR-128. (3) Chondrocytes from 7 days old rats and transfected with: oe-EZH2; EZH2 Rnai; (4) Chondrocytes from 7 days old rats, cultured in micromasses and transfected with: oe-miR-128a; sh-miR-128a | TUNEL staining and flow cytometry (Cell apoptosis); qRT-PCR (ATG4, ATG12, p62, BECLIN, COL2A1, AGGRECAN, SOX9, IL1β, CXCL9); WB (LC3, BAX, BCL2,CASPASE-3, EZH2, H3K27me1, H3K27me2, H3K27me3); Dual-Luciferase reporter assay (relationship between ATG12 and miR-128a); ChIP (EZH2 and H3K27me3 binding on miR-138 promoter). | OA cartilage and cells: ↑ miR-128a expression; ↓ ATG12 expression, LC3, H3K27. Cells+oe-miR-128: ↓ 3′-UTR luciferase activity of ATG12, ATG12 expression, LC3 concentration. Cells+sh-miR-128: ↑ 3′-UTR luciferase activity of ATG12, ATG12 expression, LC3 concentration. Cells+oe-EZH2: ↑ methylated H3K27 and H3K27me2 enrichment in the proximal region of the miR-128a promoter, ATG12 and LC3; ↓ miR-128a expression. Cells+EZH2 Rnai: ↓ methylated H3K27, H3K27me2 enrichment to the miR-128a promoter region, ATG12, LC3; ↑ miR-128a expression. Cell micromasses+oe-miR-128a: ↑ BAX, BCL2, and cleaved caspase-3, annexin-V, apoptosis; ↓ Alcian blue staining, SOX9, COL2A1, and AGGRECAN. Cell micromasses+sh-miR-128a: ↓ Bax, Bcl-2, cleaved caspase-3, apoptosis; ↑SOX9, COL2A1, and AGGRECAN. | - miR-128a upregulated. - Negative correlation between miR-128a and ATG12, EZH2 and H3K27. - Overexpression of EZH2 decreases miR-128a through methylation of histone H3K27. - miR-128a binds directly to the 3′-UTR of ATG12. - miR-128a reduces survival and cartilage formation capacity of chondrocytes | Lian 2018 [33] |
| Evaluation of the biological effects of miR-23a-3p methylation in ECM synthesis in OA | (1) Cartilage from OA pz (n = 10). (2) Purchased SW1353 cells transfected with: miR-23a-3p mimic; miR-23a-3p inhibitor; sh-SMAD3 | qRT-PCR (SMAD3, COL2A1, AGGRECAN, miR-23a-3p); WB (SMAD3, COL2A1, AGGRECAN); MSP (miR-23a-3p promoter methylation); Dual-luciferase reporter assay (relationship between SMAD3 and miR-23a-3p) | OA cartilage: ↑ miR-23a-3p; ↓ SMAD3, miR-23a-3p promoter methylation. Cells+miR-23a-3p mimics: ↓ luciferase activity of SMAD3,, COL2A1, AGGRECAN. Cells+miR-23a-3p inhibitor: ↑ SMAD3, COL2A1, AGGRECAN. Cells+sh-SMAD3: ↓ SMAD3. Cells+ miR-23a-3p inhibitor+sh-SMAD3: ↓ SMAD3, COL2A1, AGGRECAN. | - miR-23a-3p upregulated. - Negative correlation between miR-23a-3p and SMAD3. - Hypomethylation of the miR-23a-3p promoter explains the increased expression of miR-23a-3p. - SMAD3 is a target of miR-23a-3p. - miR-23a-3p overexpression suppresses ECM synthesis. - SMAD3 is essential in the miR-23a-3p-induced downregulation of AGGRECAN and COL2A1. | Kang 2016 [2] |
| Evaluation of the molecular mechanism of EZH2/miR-138/SDC1 and epigenetic regulation in cell apoptosis and inflammation in OA | (1) Cartilage and chondrocytes from OA pz (n = 25). (2) Chondrocytes from healthy donors treated with IL1β and transfected or not with: sh-EZH2; miR-138 mimic; oe-EZH2; oe-EZH2 | RT-qPCR (EZH2, miR-138, SDC1, MMP13, ADAMTS4, ADAMTS5); WB (EZH2, SDC1); Flow Cytometry (cell apoptosis); Dual-luciferase reporter assay (relationship between SDC1 and miR-138); ChIP (EZH2 and H3K27me3 binding on miR-138 promoter). | OA cartilage and chondrocytes: ↑ EZH2, SDC1; ↓ miR-138. Cells from healthy donors+IL1β: ↑ EZH2, SDC1, apoptosis, MMP13, ADAMTS4, ADAMTS5, miR-138 promoter histone methylation; ↓ miR-138. Cells+sh-EZH2: ↓ EZH2, apoptosis, MMP13, ADAMTS-4, ADAMTS-5, miR-138 promoter histone methylation; ↑ miR-138. Cells+oe-EZH2: ↑ EZH2, SDC1, apoptosis, MMP13, ADAMTS4, ADAMTS5, miR-138 promoter histone methylation; ↓ miR-138. Cells+miR-138 mimic: ↓ SDC1, SDC1 luciferase activity. Cells+oe-EZH2+mir-138 mimic: ↑ miR-138; ↓ SDC1, apoptosis, MMP13, ADAMTS4, ADAMTS5. | - miR-138 deregulated. - EZH2 is upregulated and promotes OA progression. - Negative correlation between EZH2 and SDC1 with miR-138. - EZH2 inhibits miR-138 expression by increasing histone methylation at its promoter. - SDC1 is a target of miR-138. - EZH2 induces cartilage catabolism-related factors by regulating miR-138/SDC1 signalling | Wang 2021 [34] |
| Evaluation of the relationship between FTO and miR-515-5p in m6A-dependent manner on cell apoptosis and inflammation in OA | (1) Purchased human OA C28/I2 chondrocytes treated with LPS transfected or not with: oe-FTO; sh-FTO; miR-515-5p inhibitor; oe-TLR4 | CCK-8 (cell viability); Flow cytometry (cell apoptosis); WB (BCL-2, BAX, CLEAVED-CASPASE-3, FTO, TLR4, MYD88, P/T-P65, P/T-IΚBα); ELISA (IL-6, IL-1β, and TNF-α); m6A RNA methylation level; RT-qPCR (COX-2, iNOS, FTO, miR-515-5p and pri-miR-515-5p); MeRIP (m6A RNA immunoprecipitation); Dual-luciferase reporter assay (relationship between pri-miR-515-5p and DGCR8). | Cells: ↓ cell viability, BCL-2, miR-515-5p expression, FTO; ↑ apoptosis, BAX, CLEAVED-CASPASE-3, COX-2 and iNOS expression, IL-6, IL-1β, and TNF-α, COX-2, pri-miR-515-5p m6A methylation level, TLR4, MyD88, p/t-p65, and p/t-IκBα. Cells+oe-FTO: ↓ cell apotosis, BAX, CLEAVED-CASPASE-3, IL-6, IL-1β, and TNF-α, COX-2, iNOS, pri-miR-515-5p m6A methylation level, TLR4, MYD88, P/T-P65, AND P/T-IΚBα; ↑ cell viability, BCL-2, miR-515-5p expression, pri-miR-515-5p binding level to DGCR8. Cells+sh-FTO: ↓ FTO expression, miR-515-5p expression; ↑ pri-miR-515-5p m6A methylation level. Cells+oe-FTO+miR-515-5p inhibitor: ↓ miR-515-5p, cell viability, Bcl-2; ↑ apoptosis, Bax, cleaved-caspase-3, IL-6, IL-1β, TNF-α, COX-2, iNOS, TLR4. Cells+oe-FTO+oe-TLR4: ↑ MyD88, p/t-p65, and p/t-IκBα | - miR-515-5p deregulated. - FTO reduced. - FTO interacts with DGCR8 and modulates the pri-miR-515-5p processing in an m6A dependent manner. - FTO alleviates OA injury by regulating miR-515-5p. - miR-515-5p inhibits the MyD88/NF-κB pathway by targeting TLR4. - FTO might inhibit TLR4 levels by targeting miR-515-5p. | Cai 2023 [35] |
| Evaluation of the miR-143 methylation underlying the role of CUR in OA treatment | (1) Purchased primary chondrocytes treated with IL1β with or without: BMSC-Exos; BMSC-Exos+CUR. (2) Primary chondrocytes+CUR. (3)Primary chondrocytes transfected with: miR-143 mimics; miR-124 mimics | qRT-PCR (miR-124, miR-143, Rock1, NF-kB); CCK-8 (cell proliferation); Annexin V assay (Cell apoptosis); WB (Rock1, Tlr9, Nf-kB); Bisulphite sequencing; Dual-luciferase reporter assay (relationship between NF-kB and miR-124 or ROCK1 and miR-143) | Cells: ↓ cell viability, miR-124, miR-143; ↑ cell apoptosis, Rock1, Tlr9, Nf-kB. Cells+BMSC-Exos and cells+BMSC-Exos+CUR: ↑ cell viability, miR-124, miR-143; ↓ cell apoptosis, Rock1, Tlr9, Nf-kB. Cells+CUR: ↓ methylation of miR-143 and miR-124 promoters; ↑ miR-143 and miR-124. Cells+miR-143 mimics: ↓ 3′-UTR luciferase activity of Nf-kB. Cells+miR-124 mimics: ↓ 3′-UTR luciferase activity of Rock1. | - miR-124 and miR-143 deregulated. - Exosomes derived from CUR treated MSCs maintain the viability of chondrocytes and protects chondrocytes against IL-1β-induced apoptosis. - Exosomes derived from CUR treated MSCs restore the expression of miRNAs and genes related to OA. - CUR up-regulates miR-143 and miR-124 expression by reducing the DNA methylation of their promoters. - miR-143 and miR-124 act by inhibiting Rock1 and Nf-kB gene target. | Qiu 2020 [36] |
| Evaluation of the role of miR-140-5p and miR-146a methylation in OA | (1)Chondrocytes and synoviocytes from OA pz (n = 20) treated or not with 5-AzadC and trasfected or not with: sh-SMAD3; miR-140-5p inhibitor; sh-NF-kB; miR-146a inhibitor. | RT-qPCR (miR-140, miR-146a, MMP13, ADAMTS5, TRAF-6, IRAK-1, IL6, IL1β, TNFA); MSP (specific regions of miR-140 and miR-146a promoter methylation); Bisulfite sequencing (miR-146a promoter methylation); ChIP (NF-kB or SMAD3 binding on miR-146a promoter). | Chondrocytes: ↓ miR-140-5p binding affinity of SMAD3; ↑ miR-140 regulatory region methylation. Synoviocytes: ↓ miR-146a, NF-kB binding on miR-146a promoter; ↑ miR-146a promoter methylation. Chondrocytes+5-AzadC: ↓ miR-140 regulatory region methylation, MMP13, ADAMTS5; ↑ miR-140-5p. Synoviocytes+5-AzadC: ↓ miR-146a promoter methylation, IRAK-1, IL1β, IL6; ↑ miR-146a. Chondrocytes+5-AzadC+sh-SMAD3: ↓ miR-140-5p. Chondrocytes+5-AzadC+miR-140-5p inhibitor: ↑ MMP13, ADAMTS5. Synoviocytes+5-AzadC+sh-NF-kB: ↓ miR-146a. Synoviocytes+5-AzadC+miR-146a inhibitor: ↑ IRAK-1, IL1β, IL6. Synoviocytes+miR-146a: ↓ IRAK-1, IL1β, IL6. | - miR-140-5p and miR-146a deregulated. - miR-140 regulatory region methylation in chondrocytes. - Negative correlation between miR-140-5p expression and miR-140 regulatory region methylation in chondrocytes. - miR-146a promoter methylation in synoviocytes. - Negative correlation between miR-146a expression and miR-146a promoter methylation in synoviocytes. - Methylation impairs SMAD3 binding affinity on miR-140 regulatory region. - MiR-146a promoter methylation impairs the binding affinity of NF-kB. - Methylation mediates downregulation of miR-140-5p on MMP-13 and ADAMTS5 expression levels. - Methylation mediates miR-146a on the expression of inflammatory factors involved in OA | Papathanasiou 2019 [37] |
| Evaluation of the role of Circ_HECW2 and miR-93 methylation in cell apoptosis in OA | (1) SF from OA pz (n = 64). (2) Purchased human OA chondrocytes transfected with: oe-Circ_HECW2; miR-93 mimic | RT-qPCR (Circ_HECW2, miR-93); Flow cytometry (cell apoptosis); MSP (miR-93 methylation) | OA SF and cells: ↑ Circ_HECW2; ↓ miR-93. Cells+oe-Circ_HECW2: ↓ miR-93; ↑ miR-93 methylation, apoptosis. Cells+miR-93 mimic: ↓ apoptosis; no changes in Circ_HECW2 | - miR-93 deregulated. - Circ_HECW2 upregulated. - Negative correlation between Circ_HECW2 and miR-93. - Circ_HECW2 overexpression decreases miR-93 expression through methylation. - Circ_HECW2 overexpression increases chondrocyte apoptosis via miR-93 | Zuo 2021 [38] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veronesi, F.; Costa, V.; Bellavia, D.; Basoli, V.; Giavaresi, G. Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation. Cells 2023, 12, 1821. https://doi.org/10.3390/cells12141821
Veronesi F, Costa V, Bellavia D, Basoli V, Giavaresi G. Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation. Cells. 2023; 12(14):1821. https://doi.org/10.3390/cells12141821
Chicago/Turabian StyleVeronesi, Francesca, Viviana Costa, Daniele Bellavia, Valentina Basoli, and Gianluca Giavaresi. 2023. "Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation" Cells 12, no. 14: 1821. https://doi.org/10.3390/cells12141821
APA StyleVeronesi, F., Costa, V., Bellavia, D., Basoli, V., & Giavaresi, G. (2023). Epigenetic Modifications of MiRNAs in Osteoarthritis: A Systematic Review on Their Methylation Levels and Effects on Chondrocytes, Extracellular Matrix and Joint Inflammation. Cells, 12(14), 1821. https://doi.org/10.3390/cells12141821