
Ferroptosis in Cancer Progression

 and
and 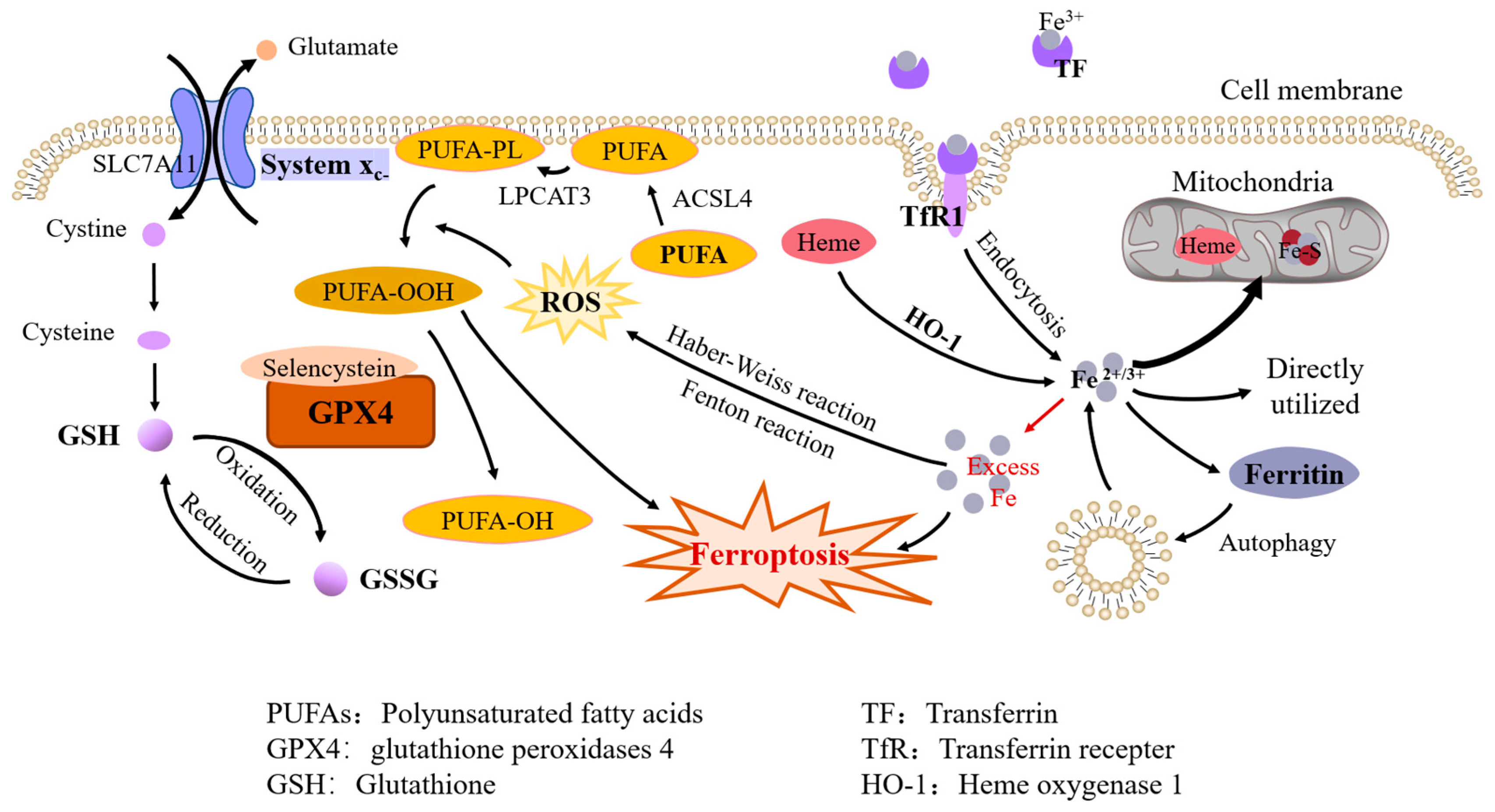{kind=link}
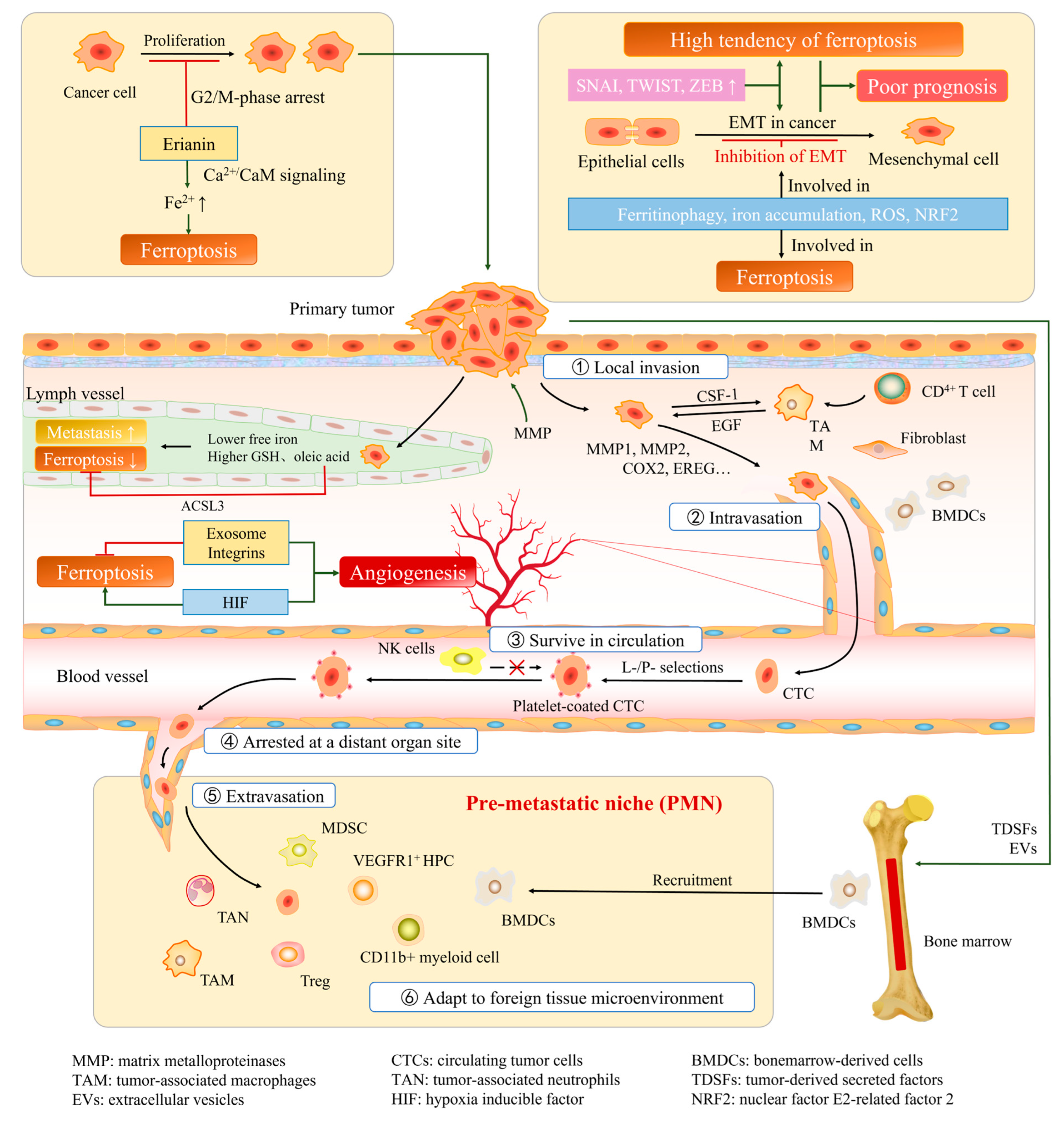{kind=link}
Abstract
1. Ferroptosis
1.1. Brief Description of Iron Metabolism
1.2. Mechanisms of Ferroptosis
1.2.1. Iron Overload
1.2.2. Lipid Peroxidation
1.2.3. Antioxidant Systems
2. Ferroptosis and EMT in Cancer
2.1. Characteristics and Mechanisms of EMT
2.2. Ferroptosis and EMT in Cancer
3. Ferroptosis and Cancer Invasion and Metastasis
3.1. Cancer Angiogenesis, Invasion, and Metastasis
3.1.1. Cancer Angiogenesis
3.1.2. Process of Cancer Invasion and Metastasis
3.2. Relationship between Ferroptosis and Cancer Angiogenesis, Invasion, and Metastasis
3.2.1. Factors That Promote Tumor Angiogenesis May Inhibit Ferroptosis
3.2.2. Ferroptosis Affects Different Steps of Cancer Invasion and Metastasis
3.2.3. Pathways Associated with Both Iron Death and Tumor Progression
4. Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal iron absorption. J. Trace Elem. Med. Biol. 2012, 26, 115–119. [Google Scholar] [CrossRef] [PubMed]
- West, A.R.; Oates, P.S. Mechanisms of heme iron absorption: Current questions and controversies. World J. Gastroenterol. 2008, 14, 4101–4110. [Google Scholar] [CrossRef]
- Dutt, S.; Hamza, I.; Bartnikas, T.B. Molecular Mechanisms of Iron and Heme Metabolism. Annu. Rev. Nutr. 2022, 42, 311–335. [Google Scholar] [CrossRef]
- Mleczko-Sanecka, K.; Silvestri, L. Cell-type-specific insights into iron regulatory processes. Am. J. Hematol. 2021, 96, 110–127. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Shapiro, J.S.; Geier, J.; Ardehali, H. Ironing out mechanisms of iron homeostasis and disorders of iron deficiency. J. Clin. Investig. 2021, 131, e148671. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J., 3rd; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef]
- Qi, X.; Zhang, Y.; Guo, H.; Hai, Y.; Luo, Y.; Yue, T. Mechanism and intervention measures of iron side effects on the intestine. Crit. Rev. Food Sci. Nutr. 2020, 60, 2113–2125. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, B.; Vandenabeele, P.; Vanden Berghe, T. Targeting Ferroptosis to Iron Out Cancer. Cancer Cell 2019, 35, 830–849. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Pratt, D.A. The chemical basis of ferroptosis. Nat. Chem. Biol. 2019, 15, 1137–1147. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 2018, 172, 409–422.e21. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Chen, D.; Tavana, O.; Chu, B.; Erber, L.; Chen, Y.; Baer, R.; Gu, W. NRF2 Is a Major Target of ARF in p53-Independent Tumor Suppression. Mol. Cell 2017, 68, 224–232.e4. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in cancer therapy: A novel approach to reversing drug resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef] [PubMed]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Stoker, M.; Perryman, M. An epithelial scatter factor released by embryo fibroblasts. J. Cell Sci. 1985, 77, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.-J.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Serrano-Gomez, S.J.; Maziveyi, M.; Alahari, S.K. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol. Cancer 2016, 15, 18. [Google Scholar] [CrossRef]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-Mesenchymal Transition (EMT): The Type-2 EMT in Wound Healing, Tissue Regeneration and Organ Fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.-N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2–YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; You, J.-H.; Kim, M.S.; Roh, J.-L. Epigenetic reprogramming of epithelial-mesenchymal transition promotes ferroptosis of head and neck cancer. Redox Biol. 2020, 37, 101697. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.; Hermann, E.; Lin, D.; Chowanadisai, W.; Hull, E.; Montgomery, M. HDAC inhibition induces EMT and alterations in cellular iron homeostasis to augment ferroptosis sensitivity in SW13 cells. Redox Biol. 2021, 47, 102149. [Google Scholar] [CrossRef]
- Yao, J.; Zhang, Y.; Li, M.; Sun, Z.; Liu, T.; Zhao, M.; Li, Z. Single-Cell RNA-Seq Reveals the Promoting Role of Ferroptosis Tendency During Lung Adenocarcinoma EMT Progression. Front. Cell Dev. Biol. 2021, 9, 822315. [Google Scholar] [CrossRef]
- Du, Y.; Miao, W.; Jiang, X.; Cao, J.; Wang, B.; Wang, Y.; Yu, J.; Wang, X.; Liu, H. The Epithelial to Mesenchymal Transition Related Gene Calumenin Is an Adverse Prognostic Factor of Bladder Cancer Correlated With Tumor Microenvironment Remodeling, Gene Mutation, and Ferroptosis. Front. Oncol. 2021, 11, 683951. [Google Scholar] [CrossRef]
- Shi, C.; Xie, Y.; Li, X.; Li, G.; Liu, W.; Pei, W.; Liu, J.; Yu, X.; Liu, T. Identification of Ferroptosis-Related Genes Signature Predicting the Efficiency of Invasion and Metastasis Ability in Colon Adenocarcinoma. Front. Cell Dev. Biol. 2021, 9, 815104. [Google Scholar] [CrossRef]
- Zhou, Y.; Qian, W.; Li, X.; Wei, W. NF-κB Inhibitor Myrislignan Induces Ferroptosis of Glioblastoma Cells via Regulating Epithelial-Mesenchymal Transformation in a Slug-Dependent Manner. Oxidative Med. Cell. Longev. 2023, 2023, 7098313. [Google Scholar] [CrossRef]
- Zhang, H.; Shan, G.; Jin, X.; Yu, X.; Bi, G.; Feng, M.; Wang, H.; Lin, M.; Zhan, C.; Wang, Q.; et al. ARNTL2 is an indicator of poor prognosis, promotes epithelial-to-mesenchymal transition and inhibits ferroptosis in lung adenocarcinoma. Transl. Oncol. 2022, 26, 101562. [Google Scholar] [CrossRef]
- Li, H.; Zhou, W.; Wei, H.; Li, L.; Wang, X.; Li, Y.; Li, S.; Li, C. Ferritinophagic Flux Was a Driving Force in Determination of Status of EMT, Ferroptosis, and NDRG1 Activation in Action of Mechanism of 2-Pyridylhydrazone Dithiocarbamate S-Acetic Acid. J. Oncol. 2021, 2021, 3015710. [Google Scholar] [CrossRef]
- Guan, D.; Zhou, W.; Wei, H.; Wang, T.; Zheng, K.; Yang, C.; Feng, R.; Xu, R.; Fu, Y.; Li, C.; et al. Ferritinophagy-Mediated Ferroptosis and Activation of Keap1/Nrf2/HO-1 Pathway Were Conducive to EMT Inhibition of Gastric Cancer Cells in Action of 2,2′-Di-pyridineketone Hydrazone Dithiocarbamate Butyric Acid Ester. Oxidative Med. Cell. Longev. 2022, 2022, 3920664. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Y.; Dong, C.; Chen, T.; Dong, A.; Ren, J.; Li, W.; Shu, G.; Yang, J.; Shen, W.; et al. CST1 inhibits ferroptosis and promotes gastric cancer metastasis by regulating GPX4 protein stability via OTUB1. Oncogene 2023, 42, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314. [Google Scholar] [CrossRef]
- Zhang, Y.; Tan, Y.; Liu, S.; Yin, H.; Duan, J.; Fan, L.; Zhao, X.; Jiang, B. Implications of Withaferin A for the metastatic potential and drug resistance in hepatocellular carcinoma cells via Nrf2-mediated EMT and ferroptosis. Toxicol. Mech. Methods 2023, 33, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jiang, J.L.; Chen, Y.M.; Lu, W.Q. KLF2 inhibits colorectal cancer progression and metastasis by inducing ferroptosis via the PI3K/AKT signaling pathway. J. Pathol. Clin. Res. 2023. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Dhar, D.K.; Kubota, H.; Kotoh, T.; Tabara, H.; Watanabe, R.; Tachibana, M.; Kohno, H.; Nagasue, N. Tumor vascularity predicts recurrence in differentiated thyroid carcinoma. Am. J. Surg. 1998, 176, 442–447. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Arif, S.H.; Pity, I.S. P53 expression and micro-vessel density in relation with 5-year survival in patients with colorectal cancer. Ann. Med. Surg. 2020, 57, 311–314. [Google Scholar] [CrossRef]
- Al-Ostoot, F.H.; Salah, S.; Khamees, H.A.; Khanum, S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021, 28, 100422. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, R.; Mutahir, Z. Cancer metastasis—Tricks of the trade. Bosn. J. Basic Med. Sci. 2017, 17, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Dirat, B.; Bochet, L.; Dabek, M.; Daviaud, D.; Dauvillier, S.; Majed, B.; Wang, Y.Y.; Meulle, A.; Salles, B.; Le Gonidec, S.; et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011, 71, 2455–2465. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Barreto, J.B.; Andreu, P.; Vasquez, L.; Tawfik, D.; Kolhatkar, N.; Coussens, L.M. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 2009, 16, 91–102. [Google Scholar] [CrossRef]
- Valastyan, S.; Weinberg, R.A. Tumor metastasis: Molecular insights and evolving paradigms. Cell 2011, 147, 275–292. [Google Scholar] [CrossRef]
- Carmeliet, P.; Jain, R.K. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat. Rev. Drug Discov. 2011, 10, 417–427. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell–mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.M.; Ruoslahti, E. Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 2004, 5, 365–374. [Google Scholar] [CrossRef]
- Al-Mehdi, A.B.; Tozawa, K.; Fisher, A.B.; Shientag, L.; Lee, A.; Muschel, R.J. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: A new model for metastasis. Nat. Med. 2000, 6, 100–102. [Google Scholar] [CrossRef]
- Gupta, G.P.; Nguyen, D.X.; Chiang, A.C.; Bos, P.D.; Kim, J.Y.; Nadal, C.; Gomis, R.R.; Manova-Todorova, K.; Massagué, J. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 2007, 446, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Padua, D.; Zhang, X.H.-F.; Wang, Q.; Nadal, C.; Gerald, W.L.; Gomis, R.R.; Massagué, J. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 2008, 133, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Psaila, B.; Lyden, D. The metastatic niche: Adapting the foreign soil. Nat. Rev. Cancer 2009, 9, 285–293. [Google Scholar] [CrossRef]
- Erler, J.T.; Bennewith, K.L.; Cox, T.R.; Lang, G.; Bird, D.; Koong, A.; Le, Q.-T.; Giaccia, A.J. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 2009, 15, 35–44. [Google Scholar] [CrossRef]
- Khan, I.; Steeg, P.S. Endocytosis: A pivotal pathway for regulating metastasis. Br. J. Cancer 2021, 124, 66–75. [Google Scholar] [CrossRef]
- Liu, S.J.; Dang, H.X.; Lim, D.A.; Feng, F.Y.; Maher, C.A. Long noncoding RNAs in cancer metastasis. Nat. Rev. Cancer 2021, 21, 446–460. [Google Scholar] [CrossRef]
- Wang, B.; Guo, H.; Yu, H.; Chen, Y.; Xu, H.; Zhao, G. The Role of the Transcription Factor EGR1 in Cancer. Front. Oncol. 2021, 11, 642547. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef] [PubMed]
- Lugano, R.; Ramachandran, M.; Dimberg, A. Tumor angiogenesis: Causes, consequences, challenges and opportunities. Cell. Mol. Life Sci. 2020, 77, 1745–1770. [Google Scholar] [CrossRef]
- Jiang, Y.; Mao, C.; Yang, R.; Yan, B.; Shi, Y.; Liu, X.; Lai, W.; Liu, Y.; Wang, X.; Xiao, D.; et al. EGLN1/c-Myc Induced Lymphoid-Specific Helicase Inhibits Ferroptosis through Lipid Metabolic Gene Expression Changes. Theranostics 2017, 7, 3293–3305. [Google Scholar] [CrossRef]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.-Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Strzyz, P. Iron expulsion by exosomes drives ferroptosis resistance. Nat. Rev. Mol. Cell Biol. 2020, 21, 4–5. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Ning, T.; Yang, H.; Liu, D.; Zhang, Q.; Lin, D.; Ge, S.; Bai, M.; et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol. Cancer 2020, 19, 43. [Google Scholar] [CrossRef]
- Brown, C.W.; Amante, J.J.; Goel, H.L.; Mercurio, A.M. The α6β4 integrin promotes resistance to ferroptosis. J. Cell Biol. 2017, 216, 4287–4297. [Google Scholar] [CrossRef]
- Chen, P.; Wu, Q.; Feng, J.; Yan, L.; Sun, Y.; Liu, S.; Xiang, Y.; Zhang, M.; Pan, T.; Chen, X.; et al. Erianin, a novel dibenzyl compound in Dendrobium extract, inhibits lung cancer cell growth and migration via calcium/calmodulin-dependent ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 51. [Google Scholar] [CrossRef]
- Nassar, Z.D.; Mah, C.Y.; Dehairs, J.; Burvenich, I.J.; Irani, S.; Centenera, M.M.; Helm, M.; Shrestha, R.K.; Moldovan, M.; Don, A.S.; et al. Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. eLife 2020, 9, e54166. [Google Scholar] [CrossRef]
- Blomme, A.; Ford, C.A.; Mui, E.; Patel, R.; Ntala, C.; Jamieson, L.E.; Planque, M.; McGregor, G.H.; Peixoto, P.; Hervouet, E.; et al. 2,4-dienoyl-CoA reductase regulates lipid homeostasis in treatment-resistant prostate cancer. Nat. Commun. 2020, 11, 2508. [Google Scholar] [CrossRef]
- You, Y.; Fan, Q.; Huang, J.; Wu, Y.; Lin, H.; Zhang, Q. Ferroptosis-Related Gene Signature Promotes Ovarian Cancer by Influencing Immune Infiltration and Invasion. J. Oncol. 2021, 2021, 9915312. [Google Scholar] [CrossRef] [PubMed]
- Ubellacker, J.M.; Tasdogan, A.; Ramesh, V.; Shen, B.; Mitchell, E.C.; Martin-Sandoval, M.S.; Gu, Z.; McCormick, M.L.; Durham, A.B.; Spitz, D.R.; et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature 2020, 585, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Roh, W.; Sullivan, R.J.; Wong, K.H.K.; Wittner, B.S.; Guo, H.; Dubash, T.D.; Sade-Feldman, M.; Wesley, B.; Horwitz, E.; et al. The Lipogenic Regulator SREBP2 Induces Transferrin in Circulating Melanoma Cells and Suppresses Ferroptosis. Cancer Discov. 2021, 11, 678–695. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Yang, Z.; Xia, Q.; Gao, S.; Sun, S.; Luo, X.; Li, Z.; Zhang, X.; Li, X. ACADSB regulates ferroptosis and affects the migration, invasion, and proliferation of colorectal cancer cells. Cell Biol. Int. 2020, 44, 2334–2343. [Google Scholar] [CrossRef]
- Nwosu, Z.C.; Battello, N.; Rothley, M.; Piorońska, W.; Sitek, B.; Ebert, M.P.; Hofmann, U.; Sleeman, J.; Wölfl, S.; Meyer, C.; et al. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J. Exp. Clin. Cancer Res. 2018, 37, 211. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Wang, Z.; Xu, R.; Hu, X.; Sun, Y.; Wang, Q.; Ju, F.; Ren, S.; Zhang, C.; et al. The promising novel biomarkers and candidate small molecule drugs in kidney renal clear cell carcinoma: Evidence from bioinformatics analysis of high-throughput data. Mol. Genet. Genom. Med. 2019, 7, e607. [Google Scholar] [CrossRef]
- Arden, K.C.; Viars, C.S.; Fu, K.; Rozen, R. Localization of short/branched chain acyl-CoA dehydrogenase (ACADSB) to human chromosome 10. Genomics 1995, 25, 743–745. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ge, Z.; Wang, Z.; Gao, Y.; Wang, Y.; Qu, X. Circular RNA RHOT1 promotes progression and inhibits ferroptosis via mir-106a-5p/STAT3 axis in breast cancer. Aging 2021, 13, 8115–8126. [Google Scholar] [CrossRef]
- Cao, F.; Luo, A.; Yang, C. G6PD inhibits ferroptosis in hepatocellular carcinoma by targeting cytochrome P450 oxidoreductase. Cell. Signal. 2021, 87, 110098. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, A.; Ouellette, R.J.; Surette, M.E. 17β-estradiol-induced ACSL4 protein expression promotes an invasive phenotype in estrogen receptor positive mammary carcinoma cells. Carcinogenesis 2017, 38, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. Biophys. Acta 2012, 1826, 370–384. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.; Wang, S.-J.; Li, D.; Chu, B.; Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 2016, 113, E6806–E6812. [Google Scholar] [CrossRef] [PubMed]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef]
- Xie, Y.; Zhu, S.; Song, X.; Sun, X.; Fan, Y.; Liu, J.; Zhong, M.; Yuan, H.; Zhang, L.; Billiar, T.R.; et al. The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep. 2017, 20, 1692–1704. [Google Scholar] [CrossRef]
- Xu, F.; Guan, Y.; Xue, L.; Zhang, P.; Li, M.; Gao, M.; Chong, T. The roles of ferroptosis regulatory gene SLC7A11 in renal cell carcinoma: A multi-omics study. Cancer Med. 2021, 10, 9078–9096. [Google Scholar] [CrossRef]
- Wang, M.-E.; Chen, J.; Lu, Y.; Bawcom, A.R.; Wu, J.; Ou, J.; Asara, J.M.; Armstrong, A.J.; Wang, Q.; Li, L.; et al. RB1-deficient prostate tumor growth and metastasis are vulnerable to ferroptosis induction via the E2F/ACSL4 axis. J. Clin. Investig. 2023, 133, e166647. [Google Scholar] [CrossRef]
- Xie, W.; Agarwal, S.; Yu, J. Ferroptosis: The vulnerability within a cancer monster. J. Clin. Investig. 2023, 133, e170027. [Google Scholar] [CrossRef]
- Lu, B.; Chen, X.B.; Ying, M.D.; He, Q.J.; Cao, J.; Yang, B. The Role of Ferroptosis in Cancer Development and Treatment Response. Front. Pharmacol. 2017, 8, 992. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, R.; Chen, J.; Wang, S.; Zhang, W.; Zheng, Q.; Cai, R. Ferroptosis in Cancer Progression. Cells 2023, 12, 1820. https://doi.org/10.3390/cells12141820
Zhang R, Chen J, Wang S, Zhang W, Zheng Q, Cai R. Ferroptosis in Cancer Progression. Cells. 2023; 12(14):1820. https://doi.org/10.3390/cells12141820
Chicago/Turabian StyleZhang, Rongyu, Jinghong Chen, Saiyang Wang, Wenlong Zhang, Quan Zheng, and Rong Cai. 2023. "Ferroptosis in Cancer Progression" Cells 12, no. 14: 1820. https://doi.org/10.3390/cells12141820
APA StyleZhang, R., Chen, J., Wang, S., Zhang, W., Zheng, Q., & Cai, R. (2023). Ferroptosis in Cancer Progression. Cells, 12(14), 1820. https://doi.org/10.3390/cells12141820