

Could a Non-Cellular Molecular Interactome in the Blood Circulation Influence Pathogens’ Infectivity?


Abstract
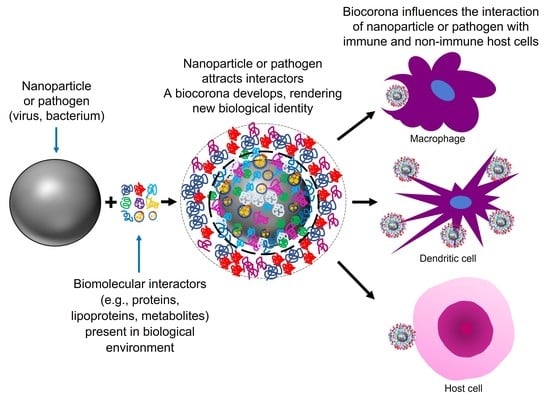
1. Introduction
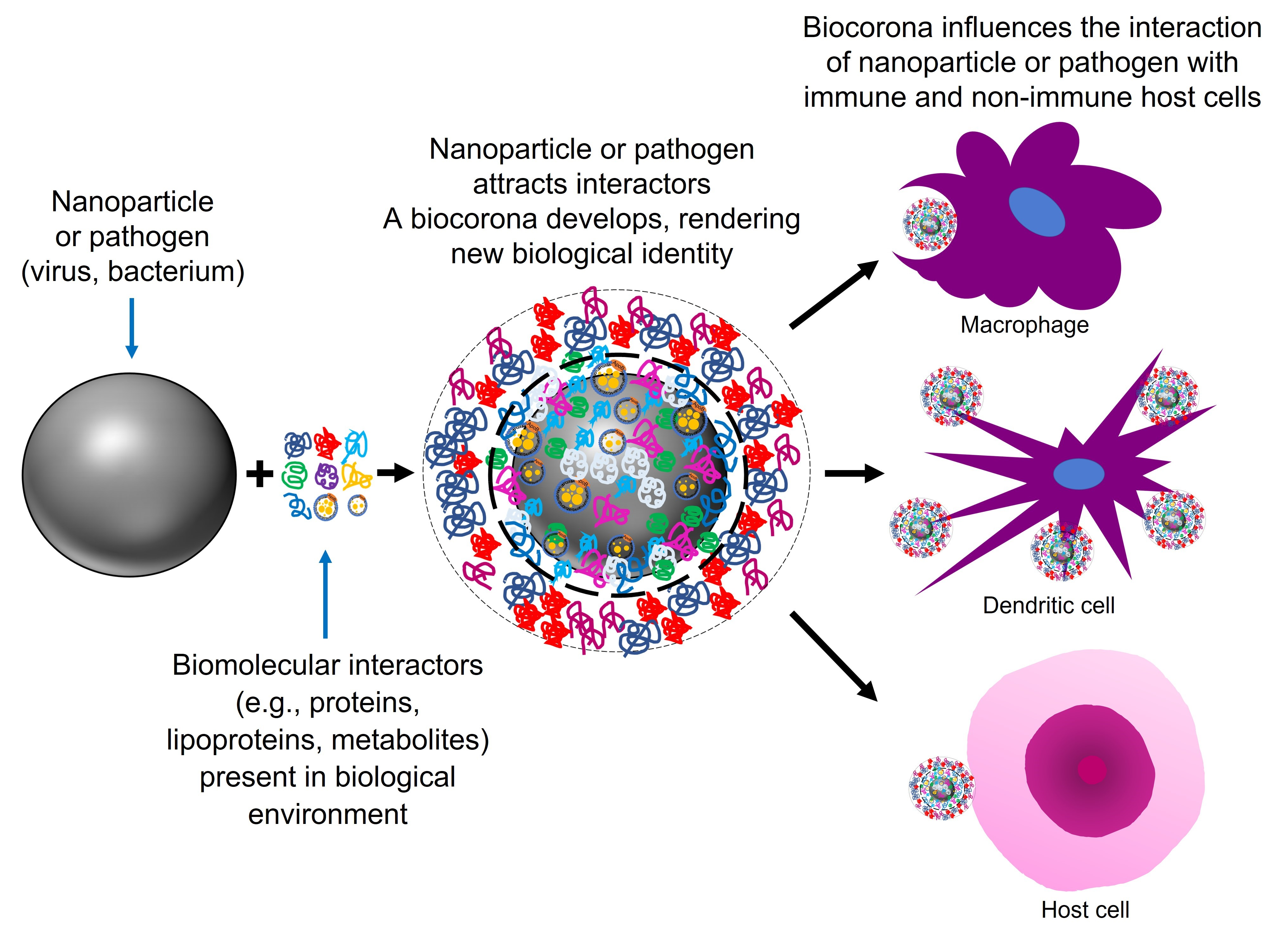
1.1. Artificial Nanoparticles and Their ‘Acquired Protein Coronas’
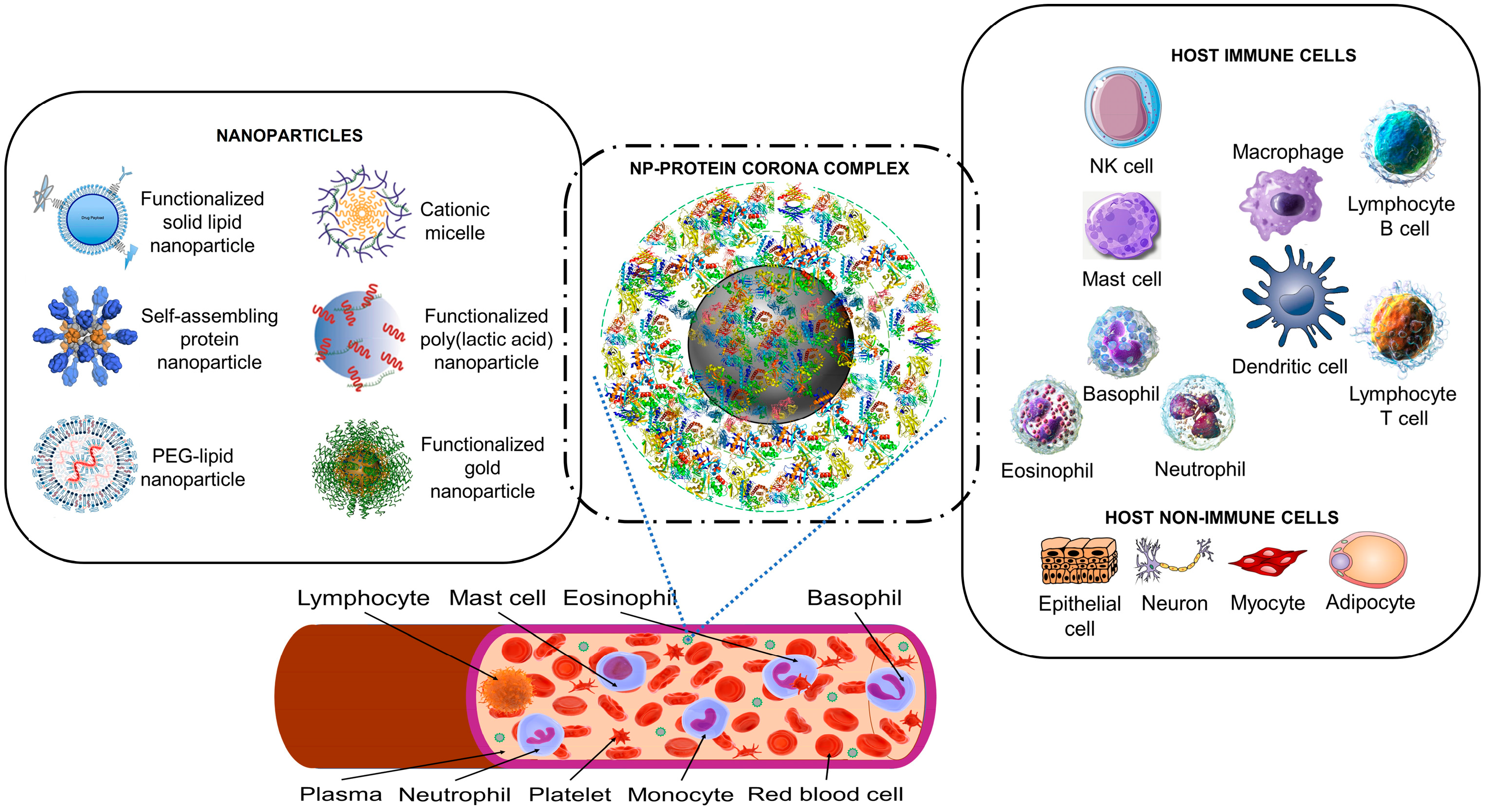
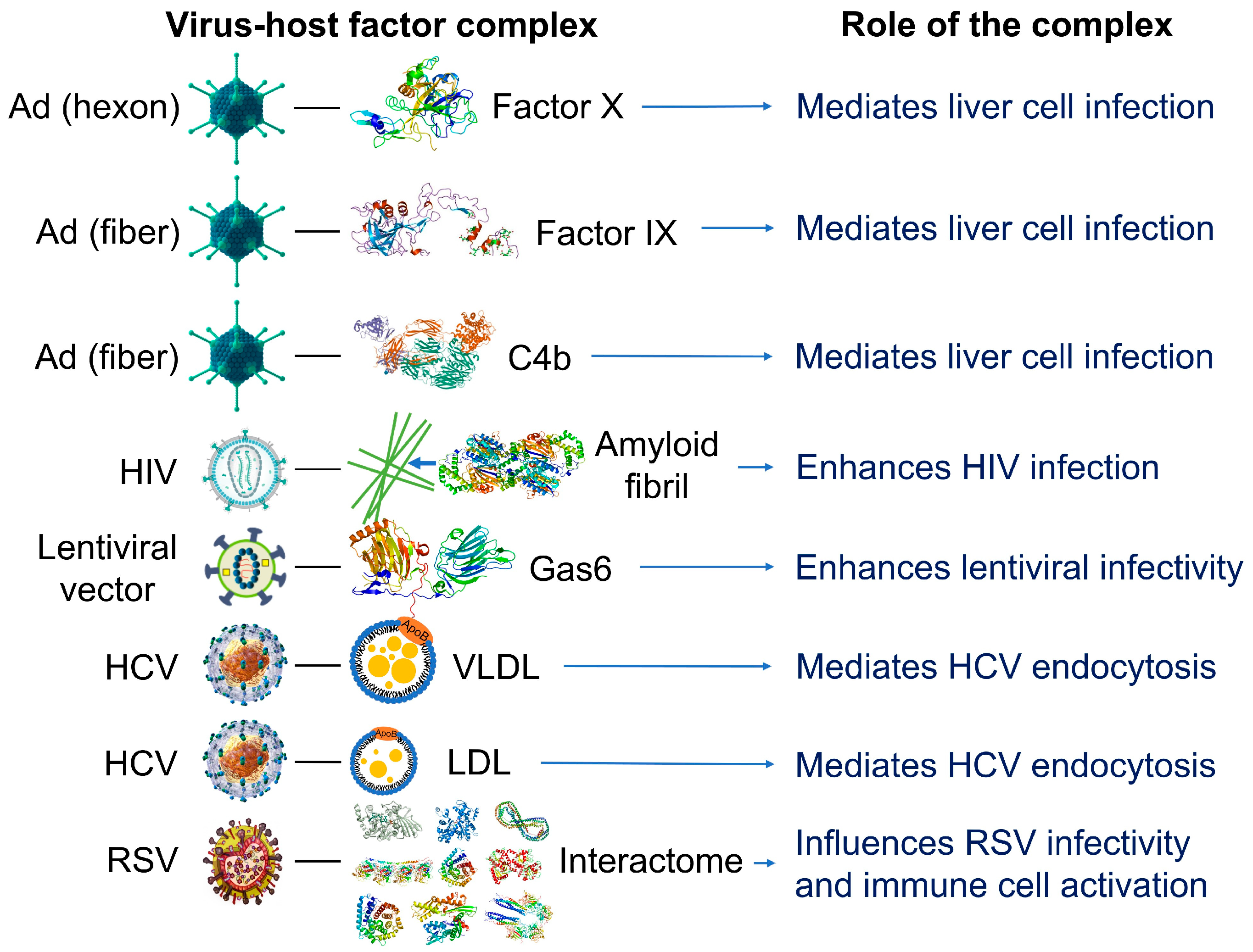
1.2. Nanoparticles Can Acquire Biocoronas: Do Viruses also Acquire a Biocorona?
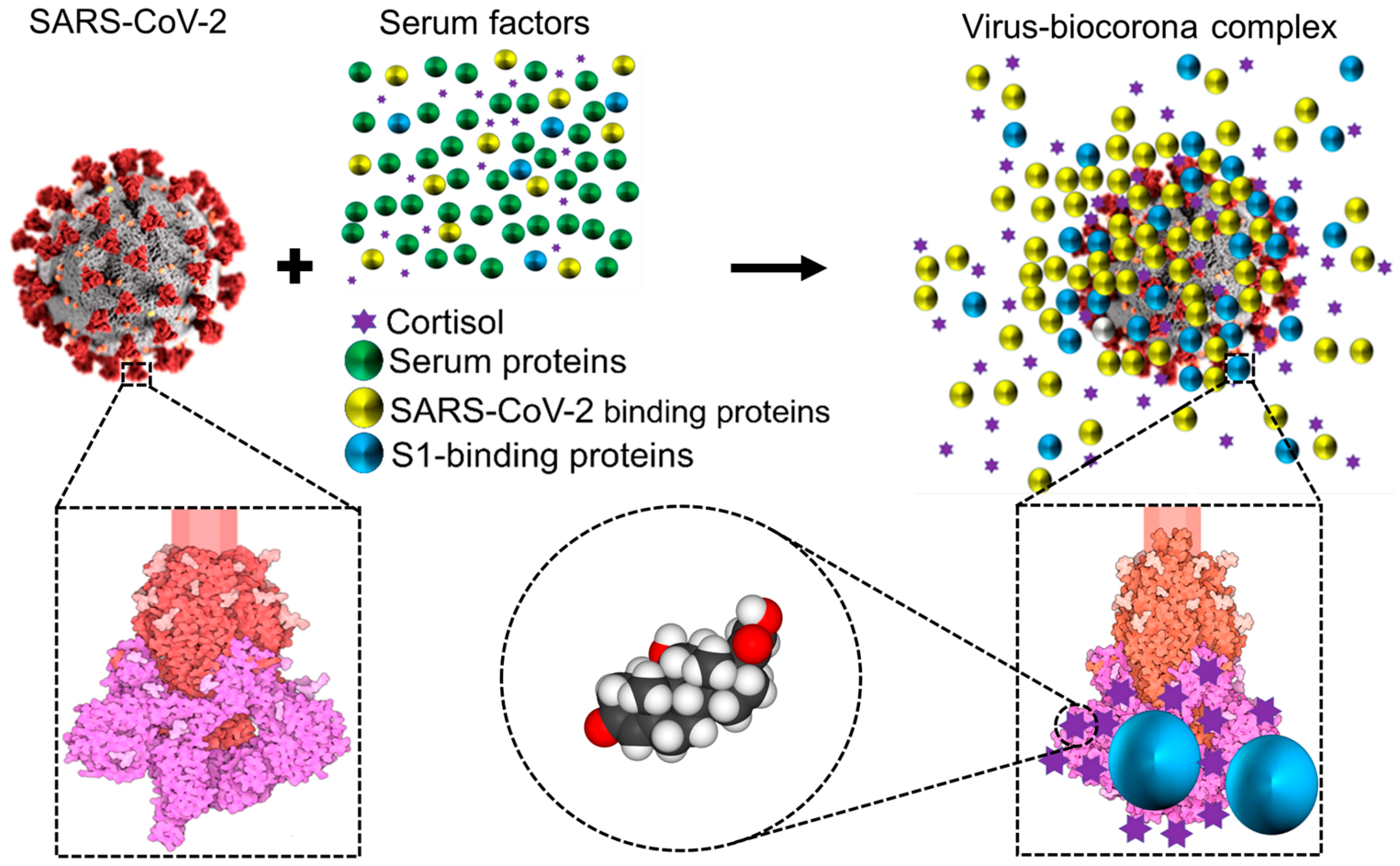
1.3. Cortisol and Dexamethasone Can Bind to Multiple Sites on SARS-CoV-2 S1: Could Glucocorticoids Be Components of Viral Biocoronas?
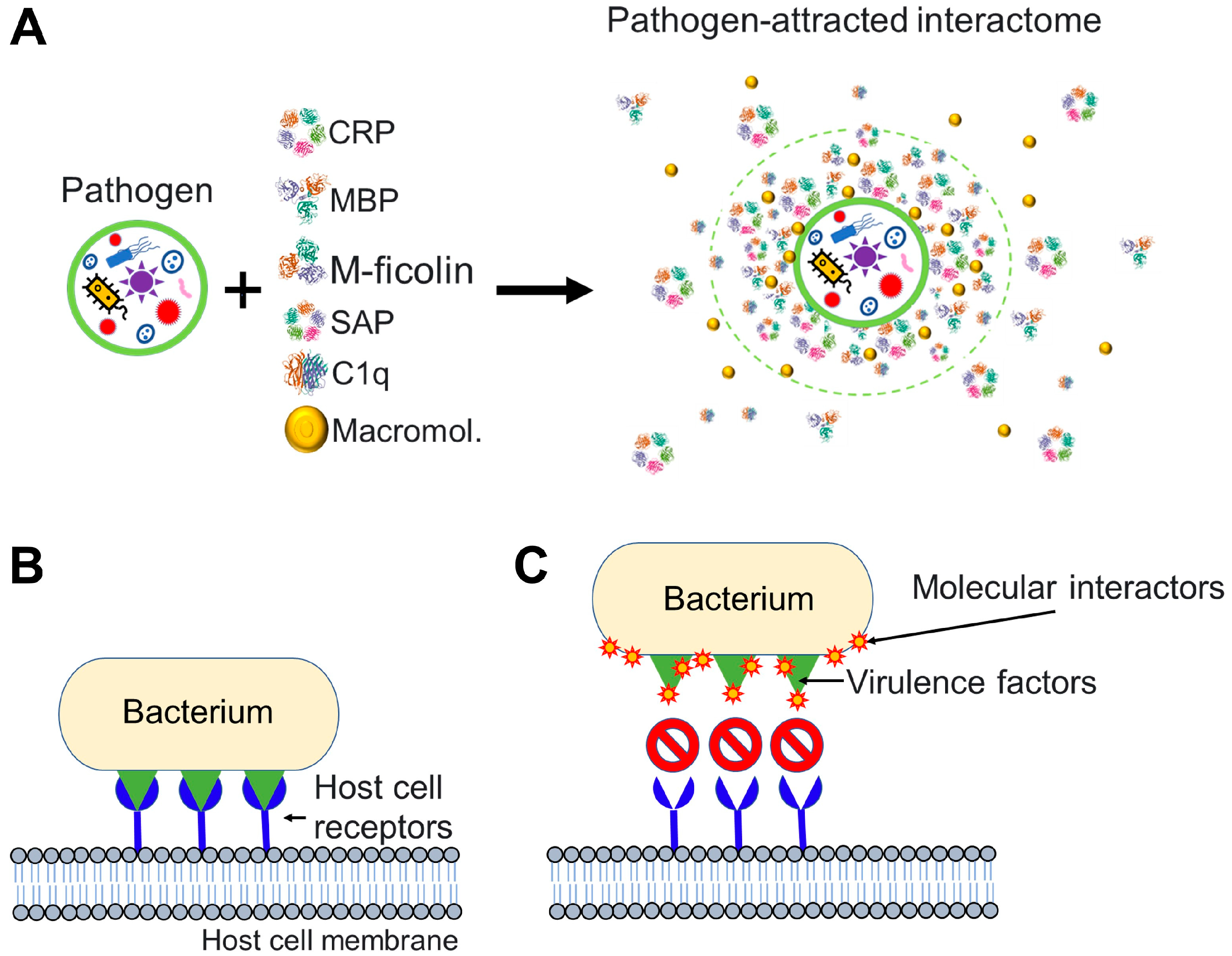
1.4. Nanoparticles and Many Different Viruses Can Acquire Biocoronas: Do Pathogenic Bacteria and Other Microorganisms Acquire a Biocorona?
2. Conclusions
3. Outlook
Several Questions Merit Investigation in Future Studies
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A.; Medzhitov, R. Innate Immune Recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C. Innate Immune Recognition: Mechanisms and Pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C. Innate Immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef]
- Paludan, S.R.; Pradeu, T.; Masters, S.L.; Mogensen, T.H. Constitutive Immune Mechanisms: Mediators of Host Defence and Immune Regulation. Nat. Rev. Immunol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Hardy, E.; Fernandez-Patron, C. Could Endogenous Glucocorticoids Influence SARS-CoV-2 Infectivity? Cells 2022, 11, 2955. [Google Scholar] [CrossRef] [PubMed]
- APTI (Air Pollution Training Institute). Module 3: Characteristics of Particles—Particle Size Categories. Basic Concepts in Environmental Sciences. USEPA (United States Environmental Protection Agency). “Less than 0.1 Microns”. Available online: https://web.archive.org/web/20101203205130/http://www.epa.gov/apti/bces/module3/category/category.htm#less0.1 (accessed on 10 April 2023).
- Vert, M.; Doi, Y.; Hellwich, K.H.; Hess, M.; Hodge, P.; Kubisa, P.; Rinaudo, M.; Schué, F. Terminology for Biorelated Polymers and Applications (IUPAC Recommendations 2012). Pure Appl. Chem. 2012, 84, 377–410. [Google Scholar] [CrossRef]
- ISO/TS 80004-2: 2015—Nanotechnologies—Vocabulary—Part 2: Nano-Objects. Available online: https://www.iso.org/standard/54440.html (accessed on 10 April 2023).
- Fadeel, B. Understanding the Immunological Interactions of Engineered Nanomaterials: Role of the Bio-Corona. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2022, 14, e1798. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, X.; Fan, L.; Xie, X.; Miao, Z.; Ma, Y.; He, T.; Zha, Z. Facile Synthesis of Monodisperse Chromogenic Amylose-Iodine Nanoparticles as an Efficient Broad-Spectrum Antibacterial Agent. J. Mater. Chem. B 2020, 8, 3010–3015. [Google Scholar] [CrossRef]
- Wang, X.; Shi, Q.; Zha, Z.; Zhu, D.; Zheng, L.; Shi, L.; Wei, X.; Lian, L.; Wu, K.; Cheng, L. Copper Single-Atom Catalysts with Photothermal Performance and Enhanced Nanozyme Activity for Bacteria-infected Wound Therapy. Bioact. Mater. 2021, 6, 4389. [Google Scholar] [CrossRef]
- Wu, K.; Zhu, D.; Dai, X.; Wang, W.; Zhong, X.; Fang, Z.; Peng, C.; Wei, X.; Qian, H.; Chen, X.; et al. Bimetallic Oxide Cu1.5Mn1.5O4 Cage-like Frame Nanospheres with Triple Enzyme-like Activities for Bacterial-Infected Wound Therapy. Nanotoday 2022, 43, 101380. [Google Scholar] [CrossRef]
- Amici, A.; Caracciolo, G.; Digiacomo, L.; Gambini, V.; Marchini, C.; Tilio, M.; Capriotti, A.L.; Colapicchioni, V.; Matassa, R.; Familiari, G.; et al. In Vivo Protein Corona Patterns of Lipid Nanoparticles. RSC Adv. 2017, 7, 1137–1145. [Google Scholar] [CrossRef]
- Al-Ahmady, Z.S.; Hadjidemetriou, M.; Gubbins, J.; Kostarelos, K. Formation of Protein Corona in Vivo Affects Drug Release from Temperature-Sensitive Liposomes. J. Control Release 2018, 276, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, D.; Caracciolo, G.; Capriotti, A.L.; Cavaliere, C.; Piovesana, S.; Colapicchioni, V.; Palchetti, S.; Riccioli, A.; Laganà, A. A Proteomics-Based Methodology to Investigate the Protein Corona Effect for Targeted Drug Delivery. Mol. Biosyst. 2014, 10, 2815–2819. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.H.; Lee, B.J. Protein Corona: A New Approach for Nanomedicine Design. Int. J. Nanomed. 2017, 12, 3137–3151. [Google Scholar] [CrossRef] [PubMed]
- Mohammad-Beigi, H.; Hayashi, Y.; Zeuthen, C.M.; Eskandari, H.; Scavenius, C.; Juul-Madsen, K.; Vorup-Jensen, T.; Enghild, J.J.; Sutherland, D.S. Mapping and Identification of Soft Corona Proteins at Nanoparticles and Their Impact on Cellular Association. Nat. Commun. 2020, 11, 4535. [Google Scholar] [CrossRef]
- Solorio-Rodríguez, A.; Escamilla-Rivera, V.; Uribe-Ramírez, M.; Chagolla, A.; Winkler, R.; García-Cuellar, C.M.; De Vizcaya-Ruiz, A. A Comparison of the Human and Mouse Protein Corona Profiles of Functionalized SiO2 Nanocarriers. Nanoscale 2017, 9, 13651–13660. [Google Scholar] [CrossRef]
- Pinals, R.L.; Yang, D.; Rosenberg, D.J.; Chaudhary, T.; Crothers, A.R.; Iavarone, A.T.; Hammel, M.; Landry, M.P. Quantitative Protein Corona Composition and Dynamics on Carbon Nanotubes in Biological Environments. Angew. Chem. Int. Ed. Engl. 2020, 59, 23668–23677. [Google Scholar] [CrossRef]
- Mekseriwattana, W.; Thiangtrongjit, T.; Reamtong, O.; Wongtrakoongate, P.; Katewongsa, K.P. Proteomic Analysis Reveals Distinct Protein Corona Compositions of Citrate- and Riboflavin-Coated SPIONs. ACS Omega 2022, 7, 37589–37599. [Google Scholar] [CrossRef]
- Faserl, K.; Chetwynd, A.J.; Lynch, I.; Thorn, J.A.; Lindner, H.H. Corona Isolation Method Matters: Capillary Electrophoresis Mass Spectrometry Based Comparison of Protein Corona Compositions Following On-Particle versus In-Solution or In-Gel Digestion. Nanomaterials 2019, 9, 898. [Google Scholar] [CrossRef]
- Whitwell, H.; Mackay, R.M.; Elgy, C.; Morgan, C.; Griffiths, M.; Clark, H.; Skipp, P.; Madsen, J. Nanoparticles in the Lung and Their Protein Corona: The Few Proteins That Count. Nanotoxicology 2016, 10, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Manzari, M.T.; Shamay, Y.; Kiguchi, H.; Rosen, N.; Scaltriti, M.; Heller, D.A. Targeted Drug Delivery Strategies for Precision Medicines. Nat. Rev. Mater. 2021, 6, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2020, 20, 101–124. [Google Scholar] [CrossRef] [PubMed]
- Monopoli, M.P.; Åberg, C.; Salvati, A.; Dawson, K.A. Biomolecular Coronas Provide the Biological Identity of Nanosized Materials. Nat. Nanotechnol. 2012, 7, 779–786. [Google Scholar] [CrossRef]
- Tenzer, S.; Docter, D.; Kuharev, J.; Musyanovych, A.; Fetz, V.; Hecht, R.; Schlenk, F.; Fischer, D.; Kiouptsi, K.; Reinhardt, C.; et al. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [Google Scholar] [CrossRef]
- Nel, A.E.; Mädler, L.; Velegol, D.; Xia, T.; Hoek, E.M.V.; Somasundaran, P.; Klaessig, F.; Castranova, V.; Thompson, M. Understanding Biophysicochemical Interactions at the Nano-Bio Interface. Nat. Mater. 2009, 8, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Champion, J.A.; Pustulka, S.M.; Ling, K.; Pish, S.L. Protein Nanoparticle Charge and Hydrophobicity Govern Protein Corona and Macrophage Uptake. ACS Appl. Mater. Interfaces 2020, 12, 48284–48295. [Google Scholar] [CrossRef]
- Corbo, C.; Molinaro, R.; Parodi, A.; Toledano Furman, N.E.; Salvatore, F.; Tasciotti, E. The Impact of Nanoparticle Protein Corona on Cytotoxicity, Immunotoxicity and Target Drug Delivery. Nanomedicine 2016, 11, 81–100. [Google Scholar] [CrossRef]
- Docter, D.; Strieth, S.; Westmeier, D.; Hayden, O.; Gao, M.; Knauer, S.K.; Stauber, R.H. No King without a Crown—Impact of the Nanomaterial-Protein Corona on Nanobiomedicine. Nanomedicine 2015, 10, 503–519. [Google Scholar] [CrossRef]
- Ke, P.C.; Lin, S.; Parak, W.J.; Davis, T.P.; Caruso, F. A Decade of the Protein Corona. ACS Nano 2017, 11, 11773–11776. [Google Scholar] [CrossRef]
- Docter, D.; Westmeier, D.; Markiewicz, M.; Stolte, S.; Knauer, S.K.; Stauber, R.H. The Nanoparticle Biomolecule Corona: Lessons Learned—Challenge Accepted? Chem. Soc. Rev. 2015, 44, 6094–6121. [Google Scholar] [CrossRef]
- Farrera, C.; Fadeel, B. It Takes Two to Tango: Understanding the Interactions between Engineered Nanomaterials and the Immune System. Eur. J. Pharm. Biopharm. 2015, 95, 3–12. [Google Scholar] [CrossRef]
- Francia, V.; Yang, K.; Deville, S.; Reker-Smit, C.; Nelissen, I.; Salvati, A. Corona Composition Can Affect the Mechanisms Cells Use to Internalize Nanoparticles. ACS Nano 2019, 13, 11107–11121. [Google Scholar] [CrossRef]
- Ren, J.; Andrikopoulos, N.; Velonia, K.; Tang, H.; Cai, R.; Ding, F.; Ke, P.C.; Chen, C. Chemical and Biophysical Signatures of the Protein Corona in Nanomedicine. J. Am. Chem. Soc. 2022, 144, 9184–9205. [Google Scholar] [CrossRef] [PubMed]
- Casals, E.; Pfaller, T.; Duschl, A.; Oostingh, G.J.; Puntes, V. Time Evolution of the Nanoparticle Protein Corona. ACS Nano 2010, 4, 3623–3632. [Google Scholar] [CrossRef] [PubMed]
- Dilliard, S.A.; Siegwart, D.J. Passive, Active and Endogenous Organ-Targeted Lipid and Polymer Nanoparticles for Delivery of Genetic Drugs. Nat. Rev. Mater. 2023, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Berardi, A.; Baldelli Bombelli, F. Oral Delivery of Nanoparticles—Let’s Not Forget about the Protein Corona. Expert Opin. Drug Deliv. 2019, 16, 563–566. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Zhou, Y.; Sun, J.; Wang, X.; Jia, Y.; Ge, K.; Yan, Y.; Dawson, K.A.; Guo, S.; Zhang, J.; et al. The Yin and Yang of the Protein Corona on the Delivery Journey of Nanoparticles. Nano Res. 2023, 16, 715–734. [Google Scholar] [CrossRef]
- Papini, E.; Tavano, R.; Mancin, F. Opsonins and Dysopsonins of Nanoparticles: Facts, Concepts, and Methodological Guidelines. Front. Immunol. 2020, 11, 567365. [Google Scholar] [CrossRef]
- Moein Moghimi, S.; Patel, H.M. Serum Opsonins and Phagocytosis of Saturated and Unsaturated Phospholipid Liposomes. BBA-Biomembr. 1989, 984, 384–387. [Google Scholar] [CrossRef]
- Tavano, R.; Gabrielli, L.; Lubian, E.; Fedeli, C.; Visentin, S.; Polverino De Laureto, P.; Arrigoni, G.; Geffner-Smith, A.; Chen, F.; Simberg, D.; et al. C1q-Mediated Complement Activation and C3 Opsonization Trigger Recognition of Stealth Poly(2-Methyl-2-Oxazoline)-Coated Silica Nanoparticles by Human Phagocytes. ACS Nano 2018, 12, 5834–5847. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, M.; Bianchi, M.; Sherry, B.; Sama, A.; Tracey, K.J. Fetuin (Alpha2-HS-Glycoprotein) Opsonizes Cationic Macrophagedeactivating Molecules. Proc. Natl. Acad. Sci. USA 1998, 95, 14429–14434. [Google Scholar] [CrossRef] [PubMed]
- Abarca-Cabrera, L.; Fraga-García, P.; Berensmeier, S. Bio-Nano Interactions: Binding Proteins, Polysaccharides, Lipids and Nucleic Acids onto Magnetic Nanoparticles. Biomater. Res. 2021, 25, 12. [Google Scholar] [CrossRef] [PubMed]
- Abarca-Cabrera, L.; Xu, L.; Berensmeier, S.; Fraga-García, P. Competition at the Bio-Nano Interface: A Protein, a Polysaccharide, and a Fatty Acid Adsorb onto Magnetic Nanoparticles. ACS Appl. Bio Mater. 2023, 6, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Lynch, I.; Cedervall, T.; Lundqvist, M.; Cabaleiro-Lago, C.; Linse, S.; Dawson, K.A. The Nanoparticle-Protein Complex as a Biological Entity; a Complex Fluids and Surface Science Challenge for the 21st Century. Adv. Colloid Interface Sci. 2007, 134–135, 167–174. [Google Scholar] [CrossRef]
- Fasoli, E. Protein Corona: Dr. Jekyll and Mr. Hyde of Nanomedicine. Biotechnol. Appl. Biochem. 2021, 68, 1139–1152. [Google Scholar] [CrossRef]
- Pellett, P.E.; Mitra, S.; Holland, T.C. Basics of Virology. Handb. Clin. Neurol. 2014, 123, 45. [Google Scholar] [CrossRef]
- Michen, B.; Graule, T. Isoelectric Points of Viruses. J. Appl. Microbiol. 2010, 109, 388–397. [Google Scholar] [CrossRef]
- Heffron, J.; Mayer, B.K. Virus Isoelectric Point Estimation: Theories and Methods. Appl. Environ. Microbiol. 2021, 87, e02319-20. [Google Scholar] [CrossRef]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral Mechanisms of Human Defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef]
- Hazrati, E.; Galen, B.; Lu, W.; Wang, W.; Ouyang, Y.; Keller, M.J.; Lehrer, R.I.; Herold, B.C. Human Alpha- and Beta-Defensins Block Multiple Steps in Herpes Simplex Virus Infection. J. Immunol. 2006, 177, 8658–8666. [Google Scholar] [CrossRef]
- Rapista, A.; Ding, J.; Benito, B.; Lo, Y.T.; Neiditch, M.B.; Lu, W.; Chang, T.L. Human Defensins 5 and 6 Enhance HIV-1 Infectivity through Promoting HIV Attachment. Retrovirology 2011, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zeng, L.; Yao, L.; Wang, Z.; Yang, X.; Shi, J.; Hu, L.; Liu, Q.; Chen, C.; Xia, T.; et al. Inherited and Acquired Corona of Coronavirus in the Host: Inspiration from the Biomolecular Corona of Nanoparticles. Nano. Today 2021, 39, 101161. [Google Scholar] [CrossRef] [PubMed]
- Chéneau, C.; Kremer, E.J. Adenovirus-Extracellular Protein Interactions and Their Impact on Innate Immune Responses by Human Mononuclear Phagocytes. Viruses 2020, 12, 1351. [Google Scholar] [CrossRef] [PubMed]
- Bottermann, M.; Foss, S.; Caddy, S.L.; Clift, D.; van Tienen, L.M.; Vaysburd, M.; Cruickshank, J.; O’Connell, K.; Clark, J.; Mayes, K.; et al. Complement C4 Prevents Viral Infection through Capsid Inactivation. Cell Host Microbe 2019, 25, 617–629.e7. [Google Scholar] [CrossRef]
- Doronin, K.; Flatt, J.W.; Di Paolo, N.C.; Khare, R.; Kalyuzhniy, O.; Acchione, M.; Sumida, J.P.; Ohto, U.; Shimizu, T.; Akashi-Takamura, S.; et al. Coagulation Factor X Activates Innate Immunity to Human Species C Adenovirus. Science 2012, 338, 795–798. [Google Scholar] [CrossRef]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.K.; Greig, J.A.; Denby, L.; et al. Adenovirus Serotype 5 Hexon Mediates Liver Gene Transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef]
- Castellano, L.M.; Shorter, J. The Surprising Role of Amyloid Fibrils in HIV Infection. Biology 2012, 1, 58–80. [Google Scholar] [CrossRef]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S.Y. The Soluble Serum Protein Gas6 Bridges Virion Envelope Phosphatidylserine to the TAM Receptor Tyrosine Kinase Axl to Mediate Viral Entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar] [CrossRef]
- Wrensch, F.; Crouchet, E.; Ligat, G.; Zeisel, M.B.; Keck, Z.Y.; Foung, S.K.H.; Schuster, C.; Baumert, T.F. Hepatitis C Virus (HCV)-Apolipoprotein Interactions and Immune Evasion and Their Impact on HCV Vaccine Design. Front. Immunol. 2018, 9, 1436. [Google Scholar] [CrossRef]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; et al. The Viral Protein Corona Directs Viral Pathogenesis and Amyloid Aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef]
- Croll, T.I.; Andersen, G.R. Re-Evaluation of Low-Resolution Crystal Structures via Interactive Molecular-Dynamics Flexible Fitting (IMDFF): A Case Study in Complement C4. Acta. Cryst. D Struct. Biol. 2016, 72, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. SARS-CoV-2 (COVID-19) by the Numbers. eLife 2020, 9, e57309. [Google Scholar] [CrossRef]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; MacAry, P.A.; Ng, L.F.P. The Trinity of COVID-19: Immunity, Inflammation and Intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Ye, D.; Liu, Q. Review of the 2019 Novel Coronavirus (SARS-CoV-2) Based on Current Evidence. Int. J. Antimicrob. Agents 2020, 55, 105948. [Google Scholar] [CrossRef] [PubMed]
- Stahn, C.; Buttgereit, F. Genomic and Nongenomic Effects of Glucocorticoids. Nat. Clin. Pract. Rheumatol. 2008, 4, 525–533. [Google Scholar] [CrossRef]
- Chrousos, G.P.; Kino, T. Glucocorticoid Action Networks and Complex Psychiatric and/or Somatic Disorders. Stress 2007, 10, 213–219. [Google Scholar] [CrossRef]
- Taves, M.D.; Gomez-Sanchez, C.E.; Soma, K.K. Extra-Adrenal Glucocorticoids and Mineralocorticoids: Evidence for Local Synthesis, Regulation, and Function. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E11–E24. [Google Scholar] [CrossRef]
- Shimba, A.; Ikuta, K. Control of Immunity by Glucocorticoids in Health and Disease. Semin. Immunopathol. 2020, 42, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Shimba, A.; Ikuta, K. Immune-Enhancing Effects of Glucocorticoids in Response to Day-Night Cycles and Stress. Int. Immunol. 2020, 32, 703–708. [Google Scholar] [CrossRef]
- Sarker, H.; Panigrahi, R.; Hardy, E.; Glover, J.N.M.; Elahi, S.; Fernandez-Patron, C. Glucocorticoids Bind to SARS-CoV-2 S1 at Multiple Sites Causing Cooperative Inhibition of SARS-CoV-2 S1 Interaction with ACE2. Front. Immunol. 2022, 13, 2833. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Wang, N.; Pallesen, J.; Wrapp, D.; Turner, H.L.; Cottrell, C.A.; Corbett, K.S.; Graham, B.S.; McLellan, J.S.; Ward, A.B. Stabilized Coronavirus Spikes Are Resistant to Conformational Changes Induced by Receptor Recognition or Proteolysis. Sci. Rep. 2018, 8, 15701. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ziebuhr, J. Molecular Biology of Severe Acute Respiratory Syndrome Coronavirus. Curr. Opin. Microbiol. 2004, 7, 412–419. [Google Scholar] [CrossRef]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.M.; Rottier, P.J.M. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801. [Google Scholar] [CrossRef]
- Yin, Y.; Sheng, Y.; Wang, M.; Ni, S.; Ding, H.; Ma, Y. Protein Corona Critically Affects the Bio-Behaviors of SARS-CoV-2. arXiv 2021, arXiv:2102.05440. [Google Scholar] [CrossRef]
- Iles, J.; Zmuidinaite, R.; Sadee, C.; Gardiner, A.; Lacey, J.; Harding, S.; Ule, J.; Roblett, D.; Heeney, J.; Baxendale, H.; et al. SARS-CoV-2 Spike Protein Binding of Glycated Serum Albumin-Its Potential Role in the Pathogenesis of the COVID-19 Clinical Syndromes and Bias towards Individuals with Pre-Diabetes/Type 2 Diabetes and Metabolic Diseases. Int. J. Mol. Sci. 2022, 23, 4126. [Google Scholar] [CrossRef]
- Shrive, A.K.; Cheetham, G.M.T.; Holden, D.; Myles, D.A.A.; Turnell, W.G.; Volanakis, J.E.; Pepys, M.B.; Bloomer, A.C.; Greenhough, T.J. Three Dimensional Structure of Human C-Reactive Protein. Nat. Struct. Biol. 1996, 3, 346–354. [Google Scholar] [CrossRef]
- Weis, W.I.; Drickamer, K. Trimeric Structure of a C-Type Mannose-Binding Protein. Structure 1994, 2, 1227–1240. [Google Scholar] [CrossRef]
- Tanio, M.; Kondo, S.; Sugio, S.; Kohno, T. Trivalent Recognition Unit of Innate Immunity System: Crystal Structure of Trimeric Human M-Ficolin Fibrinogen-like Domain. J. Biol. Chem. 2007, 282, 3889–3895. [Google Scholar] [CrossRef]
- Emsley, J.; White, H.E.; O’Hara, B.P.; Oliva, G.; Srinivasan, N.; Tickle, I.J.; Blundell, T.L.; Pepys, M.B.; Wood, S.P. Structure of Pentameric Human Serum Amyloid P Component. Nature 1994, 367, 338–345. [Google Scholar] [CrossRef]
- Gaboriaud, C.; Juanhuix, J.; Gruez, A.; Lacroix, M.; Darnault, C.; Pignol, D.; Verger, D.; Fontecilla-Camps, J.C.; Arlaud, G.J. The Crystal Structure of the Globular Head of Complement Protein C1q Provides a Basis for Its Versatile Recognition Properties. J. Biol. Chem. 2003, 278, 46974–46982. [Google Scholar] [CrossRef]
- Weiser, J.N. The Battle with the Host over Microbial Size. Curr. Opin. Microbiol. 2013, 16, 59–62. [Google Scholar] [CrossRef]
- Sohlenkamp, C.; Geiger, O. Bacterial Membrane Lipids: Diversity in Structures and Pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. [Google Scholar] [CrossRef]
- Bastos, P.; Trindade, F.; da Costa, J.; Ferreira, R.; Vitorino, R. Human Antimicrobial Peptides in Bodily Fluids: Current Knowledge and Therapeutic Perspectives in the Postantibiotic Era. Med. Res. Rev. 2018, 38, 101–146. [Google Scholar] [CrossRef]
- Bechinger, B.; Gorr, S.U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2017, 96, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Sun, L.C.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q.Y. The Antimicrobial Peptides and Their Potential Clinical Applications. Am. J. Transl. Res. 2019, 11, 3919. [Google Scholar] [PubMed]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Giangaspero, A.; Sandri, L.; Tossi, A. Amphipathic Alpha Helical Antimicrobial Peptides. Eur. J. Biochem. 2001, 268, 5589–5600. [Google Scholar] [CrossRef]
- Li, D.; Wu, M. Pattern Recognition Receptors in Health and Diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Farrar, C.A.; Sacks, S.H. Structural and Functional Diversity of Collectins and Ficolins and Their Relationship to Disease. Semin. Immunopathol. 2018, 40, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Pepys, M.B.; Hirschfield, G.M. C-Reactive Protein: A Critical Update. J. Clin. Investig. 2003, 111, 1805–1812. [Google Scholar] [CrossRef]
- Pepys, M.B.; Booth, D.R.; Hutchinson, W.L.; Gallimore, J.R.; Collins, P.M.; Hohenester, E. Amyloid P Component. A Critical Review. J. Protein Fold. Disord. 1997, 4, 274–295. [Google Scholar] [CrossRef]
- Clos, T.W. Du Pentraxins: Structure, Function, and Role in Inflammation. ISRN Inflamm. 2013, 2013, 379040. [Google Scholar] [CrossRef]
- Schwalbe, R.A.; Nelsestuen, G.L.; Dahlbáck, B.; Coe, J.E. Pentraxin Family of Proteins Interact Specifically with Phosphorylcholine and/or Phosphorylethanolamine. Biochemistry 1992, 31, 4907–4915. [Google Scholar] [CrossRef]
- Dommett, R.M.; Klein, N.; Turner, M.W. Mannose-Binding Lectin in Innate Immunity: Past, Present and Future. Tissue Antigens 2006, 68, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Kingma, P.; Jobe, A.H. The Surfactant System. In Kendig’s Disorders of the Respiratory Tract in Children; Elsevier: Amsterdam, The Netherlands, 2019; pp. 57–62.e2. [Google Scholar] [CrossRef]
- Bidula, S.; Sexton, D.W.; Schelenz, S. Ficolins and the Recognition of Pathogenic Microorganisms: An Overview of the Innate Immune Response and Contribution of Single Nucleotide Polymorphisms. J. Immunol. Res. 2019, 2019, 3205072. [Google Scholar] [CrossRef] [PubMed]
- Reid, K.B.M. Complement Component C1q: Historical Perspective of a Functionally Versatile, and Structurally Unusual, Serum Protein. Front. Immunol. 2018, 9, 764. [Google Scholar] [CrossRef]
- Kishore, U.; Ghai, R.; Greenhough, T.J.; Shrive, A.K.; Bonifati, D.M.; Gadjeva, M.G.; Waters, P.; Kojouharova, M.S.; Chakraborty, T.; Agrawal, A. Structural and Functional Anatomy of the Globular Domain of Complement Protein C1q. Immunol. Lett. 2004, 95, 113–128. [Google Scholar] [CrossRef]
- Elsevier. Complement. In Immunology for Pharmacy; Elsevier: Amsterdam, The Netherlands, 2012; pp. 87–96. [Google Scholar] [CrossRef]
- Mantovani, A.; Garlanda, C. Humoral Innate Immunity and Acute-Phase Proteins. N. Engl. J. Med. 2023, 388, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Calvert, M.B.; Jumde, V.R.; Titz, A. Pathoblockers or Antivirulence Drugs as a New Option for the Treatment of Bacterial Infections. Beilstein J. Org. Chem. 2018, 14, 2607. [Google Scholar] [CrossRef] [PubMed]
- García-Fernández, E.; Koch, G.; Wagner, R.M.; Fekete, A.; Stengel, S.T.; Schneider, J.; Mielich-Süss, B.; Geibel, S.; Markert, S.M.; Stigloher, C.; et al. Membrane Microdomain Disassembly Inhibits MRSA Antibiotic Resistance. Cell 2017, 171, 1354–1367.e20. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Mattsson, K.; Rissler, J.; Karlsson, H.M.; Svensson, C.R.; Gudmundsson, A.; Lindh, C.H.; Jönsson, B.A.G.; Cedervall, T.; Kåredal, M. Analysis of Nanoparticle–Protein Coronas Formed in Vitro between Nanosized Welding Particles and Nasal Lavage Proteins. Nanotoxicology 2016, 10, 226. [Google Scholar] [CrossRef]
- Bai, X.; Wang, J.; Mu, Q.; Su, G. In Vivo Protein Corona Formation: Characterizations, Effects on Engineered Nanoparticles’ Biobehaviors, and Applications. Front. Bioeng. Biotechnol. 2021, 9, 646708. [Google Scholar]
- Zhang, T.; Zhu, G.; Lu, B.; Qian, Z.; Peng, Q. Protein Corona Formed in the Gastrointestinal Tract and Its Impacts on Oral Delivery of Nanoparticles. Med. Res. Rev. 2020, 41, 1835–1850. [Google Scholar] [CrossRef]
- Keselowsky, B.G.; Acharya, A.; Lewis, J.S. Innate and Adaptive Immunity: The Immune Response to Foreign Materials. In Biomaterials Science; Elsevier: Amsterdam, The Netherlands, 2020; pp. 747–775. [Google Scholar] [CrossRef]
- Corbo, C.; Molinaro, R.; Tabatabaei, M.; Farokhzad, O.C.; Mahmoudi, M. Personalized Protein Corona on Nanoparticles and Its Clinical Implications. Biomater. Sci. 2017, 5, 378–387. [Google Scholar] [CrossRef]
- Hajipour, M.J.; Laurent, S.; Aghaie, A.; Rezaee, F.; Mahmoudi, M. Personalized Protein Coronas: A “Key” Factor at the Nanobiointerface. Biomater. Sci. 2014, 2, 1210–1221. [Google Scholar] [CrossRef]
- Deng, Z.J.; Liang, M.; Monteiro, M.; Toth, I.; Minchin, R.F. Nanoparticle-Induced Unfolding of Fibrinogen Promotes Mac-1 Receptor Activation and Inflammation. Nat. Nanotechnol. 2011, 6, 39–44. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Zhou, P.; Huang, J.; Tian, F. Specific Noncovalent Interactions at Protein-Ligand Interface: Implications for Rational Drug Design. Curr. Med. Chem. 2012, 19, 226–238. [Google Scholar] [CrossRef]
- Hadjidemetriou, M.; Al-Ahmady, Z.; Mazza, M.; Collins, R.F.; Dawson, K.; Kostarelos, K. In Vivo Biomolecule Corona around Blood-Circulating, Clinically Used and Antibody-Targeted Lipid Bilayer Nanoscale Vesicles. ACS Nano 2015, 9, 8142–8156. [Google Scholar] [CrossRef]
- Sakulkhu, U.; Maurizi, L.; Mahmoudi, M.; Motazacker, M.; Vries, M.; Gramoun, A.; Ollivier Beuzelin, M.G.; Vallée, J.P.; Rezaee, F.; Hofmann, H. Ex Situ Evaluation of the Composition of Protein Corona of Intravenously Injected Superparamagnetic Nanoparticles in Rats. Nanoscale 2014, 6, 11439–11450. [Google Scholar] [CrossRef]
- Simon, J.; Kuhn, G.; Fichter, M.; Gehring, S.; Landfester, K.; Mailänder, V. Unraveling the In Vivo Protein Corona. Cells 2021, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Gudelj, I.; Lauc, G.; Pezer, M. Immunoglobulin G Glycosylation in Aging and Diseases. Cell Immunol. 2018, 333, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Kelly, P.M.; Mahon, E.; Stöckmann, H.; Rudd, P.M.; Caruso, F.; Dawson, K.A.; Yan, Y.; Monopoli, M.P. The “Sweet” Side of the Protein Corona: Effects of Glycosylation on Nanoparticle-Cell Interactions. ACS Nano 2015, 9, 2157–2166. [Google Scholar] [CrossRef] [PubMed]
- Tavakol, M.; Montazeri, A.; Naghdabadi, R.; Hajipour, M.J.; Zanganeh, S.; Caracciolo, G.; Mahmoudi, M. Disease-Related Metabolites Affect Protein-Nanoparticle Interactions. Nanoscale 2018, 10, 7108–7115. [Google Scholar] [CrossRef]
- Wautier, J.L.; Schmidt, A.M. Protein Glycation. Circ. Res. 2004, 95, 233–238. [Google Scholar] [CrossRef]
- Tan, C.S.H.; Go, K.D.; Bisteau, X.; Dai, L.; Yong, C.H.; Prabhu, N.; Ozturk, M.B.; Lim, Y.T.; Sreekumar, L.; Lengqvist, J.; et al. Nordlund, Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science 2018, 359, 1170–1177. [Google Scholar] [CrossRef]
- Dai, L.; Prabhu, N.; Yu, L.Y.; Bacanu, S.; Ramos, A.D.; Nordlund, P. Horizontal Cell Biology: Monitoring Global Changes of Protein Interaction States with the Proteome-Wide Cellular Thermal Shift Assay (CETSA). Annu. Rev. Biochem. 2019, 88, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Teitz, J.; Sander, J.; Sarker, H.; Fernandez-Patron, C. Potential of dissimilarity measure-based computation of protein thermal stability data for determining protein interactions. Brief. Bioinform. 2023, 24, bbad143. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPs c | Synthesis Material | Use | Top 10 Most-Abundant Constituents | Ref. |
|---|---|---|---|---|
| Liposomes | HSPC, DSPG, Chol | Drug delivery (e.g., liposomal amphotericin B) | Blood coagulation: Coagulation factor XIII B chain, coagulation factor XIII A chain, fibrinogen beta chain, fibrinogen gamma chain, alpha-1-antitrypsin 1-3 Adaptive immunity: Ig kappa chain C region, fibrinogen beta chain Complement pathway: C4b-binding protein Others: Serum albumin, actin (cytoplasmic 2), fibronectin | [14] |
| Lysolipid-containing TSL | DPPC, MSPC, HSPC, DSPE-PEG2000, Chol, Dox | Drug delivery (e.g., cancer therapy) | Acute phase: Alpha-2-macroglobulin Lipid transport: Apolipoprotein C-III, apolipoprotein E Others: Beta-globin (A8DUK0 [+2]), Beta-globin (A8DUK4), Beta-2-globin (fragment), Beta-globin OS, Alpha-globin 1 (Q91V88 [+2]), Alpha-globin A8DUV1, Ig mu chain region | [15] |
| PEGylated cationic liposomes | DOTAP, DC-Chol, DOPC, DOPE, and DOPE-PEG 2000 | Potential vehicle to target cancer cells | Adaptive immunity: Ig kappa chain C region, Ig mu chain C region, Ig lambda-2 chain C regions Complement pathway: Complement C3, complement C1q subcomponent subunit A, complement C1q subcomponent subunit B, complement C4-B Lipid transport: Apolipoprotein C-III, apolipoprotein E Others: Serum albumin | [16] |
| Silica NPs modified with surface NH2 | Silicon dioxide | Targeting drug delivery | Blood coagulation: Coagulation factor V Complement pathway: Complement C3 Complement alternate pathway: Complement factor H, complement C1r subcomponent Lipid transport: Apolipoprotein B100, apolipoprotein A Others: Fibronectin, gelsolin, thrombospondin, inter α trypsin inhibitor heavy chain H4 | [17] |
| Negatively charged hydrophilic silica NPs | Silicon dioxide | Studies on nano–bio interfaces | Blood coagulation: Plasminogen Lipid transport: Apolipoprotein A-I Others: Serum albumin, hemoglobin fetal subunit beta, hemoglubin subunit alpha, alpha-1 antiproteinase, tetranectin, alpha-2-HS-glycoprotein, beta-2-glycoprotein 1, serotransferrin | [18] |
| Silica NPs bioconjugated with PEG and transferrin | Silicon dioxide, PEG, and transferrin | Active targeting | Adaptive immunity: Immunoglobulin kappa constant, immunoglobulin heavy constant mu Complement pathway: Immunoglobulin lambda-like polypeptide 5, complement C3 Lectin complement pathway: Ficolin-3 Lipid transport: Apolipoprotein A-I Others: Albumin, actin cytoplasmic 1, hemoglobin subunit beta, serotransferrin | [19] |
| Carbon nanotubes (ssDNA-SWCNTs) | Bioimaging, molecular sensing, delivery | Blood coagulation: Histidine-rich glycoprotein, kininogen-1, prothrombin Adaptive immunity: Ig heavy constant gamma Immunity: Haptoglobin Complement pathway: Clusterin, complement C3 Complement alternate pathway: Complement factor H, complement C1r subcomponent Lipid transport: Aapolipoprotein A-I Cell adhesion: Vitronectin Others: A disintegrin and metalloproteinase with thrombospondin motifs 12 | [20] | |
| Riboflavin-coated SPIONs | Cores made of iron oxides (e.g., magnetite or maghemite) | Theranostic applications | Complement pathway: Complement C4 (fragments) Complement alternate pathway: Complement factor H Lipid transport: Apolipoprotein E, apolipoprotein A-I Others: Hemoglobin fetal subunit beta, hemoglubin subunit alpha, serum albumin, peptidyl-prolyl cis-trans isomerase A, tetranectin, α-2-HS-glycoprotein | [21] |
| Carboxylated polystyrene-NPs | Polystyrene, surface carboxyl groups | Drug delivery and diagnostic fields | Blood coagulation: Fibrinogen, histidine-rich glycoprotein, kininogen-1, plasma kallikrein Adaptive immunity: Immunoglobulin Complement pathway: Complement components, clusterin Lipid transport: Apolipoproteins Cell adhesion: Vitronectin Others: Serum albumin, trypsin inhibitor heavy chains, beta-2-glycoprotein 1 | [22] |
| TiO2 NPs | Titanium dioxide | Nanoparticle toxicity studies | Host–virus interaction: Moesin, annexin A2, keratin (type II cytoskeletal 8) Autophagy: Ras-related protein Rab-8A Others: Pulmonary surfactant-associated protein A1, actin (cytoplasmic 1), L-lactate dehydrogenase A-like 6A, alpha-actinin-4, POTE ankyrin domain family member E, serum albumin | [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hardy, E.; Sarker, H.; Fernandez-Patron, C. Could a Non-Cellular Molecular Interactome in the Blood Circulation Influence Pathogens’ Infectivity? Cells 2023, 12, 1699. https://doi.org/10.3390/cells12131699
Hardy E, Sarker H, Fernandez-Patron C. Could a Non-Cellular Molecular Interactome in the Blood Circulation Influence Pathogens’ Infectivity? Cells. 2023; 12(13):1699. https://doi.org/10.3390/cells12131699
Chicago/Turabian StyleHardy, Eugenio, Hassan Sarker, and Carlos Fernandez-Patron. 2023. "Could a Non-Cellular Molecular Interactome in the Blood Circulation Influence Pathogens’ Infectivity?" Cells 12, no. 13: 1699. https://doi.org/10.3390/cells12131699
APA StyleHardy, E., Sarker, H., & Fernandez-Patron, C. (2023). Could a Non-Cellular Molecular Interactome in the Blood Circulation Influence Pathogens’ Infectivity? Cells, 12(13), 1699. https://doi.org/10.3390/cells12131699