

Ectopically Expressed Meiosis-Specific Cancer Testis Antigen HORMAD1 Promotes Genomic Instability in Squamous Cell Carcinomas

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Tissue Collection
2.2. Reagents
2.3. shRNA-Mediated Knockdown
2.4. LentiORF Overexpression
2.5. Immunofluorescence
2.6. Cytokinesis Block Micronucleus Assay (CBMN)
2.7. Chromatin Bridge Analysis
2.8. Western Blot Analysis
2.9. Cell Proliferation Assay
2.10. Immunohistochemistry
2.11. Colony Formation Assay
2.12. TCGA Data
2.13. RNA-Seq Data Analysis
2.14. Statistical Analysis
3. Results
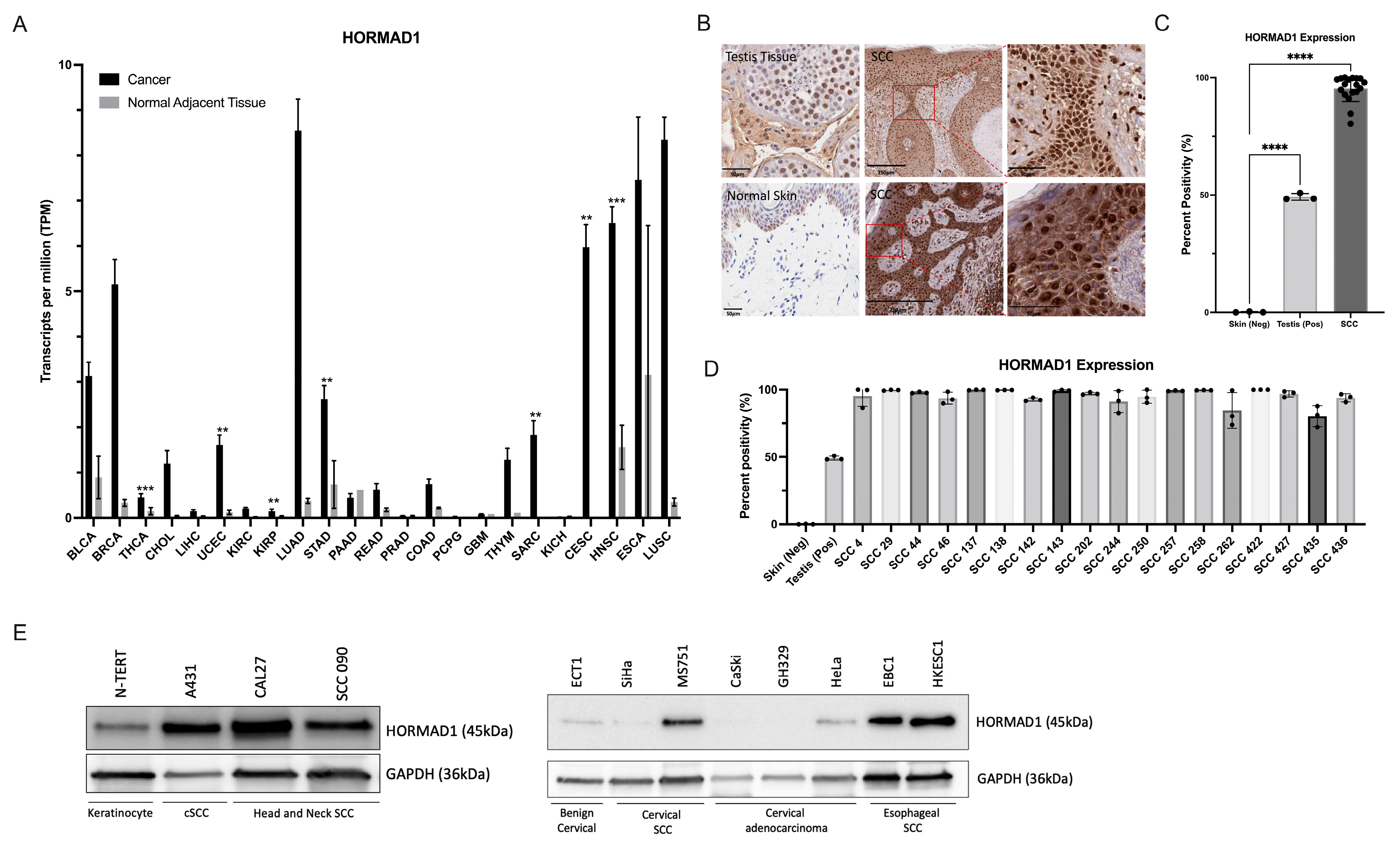
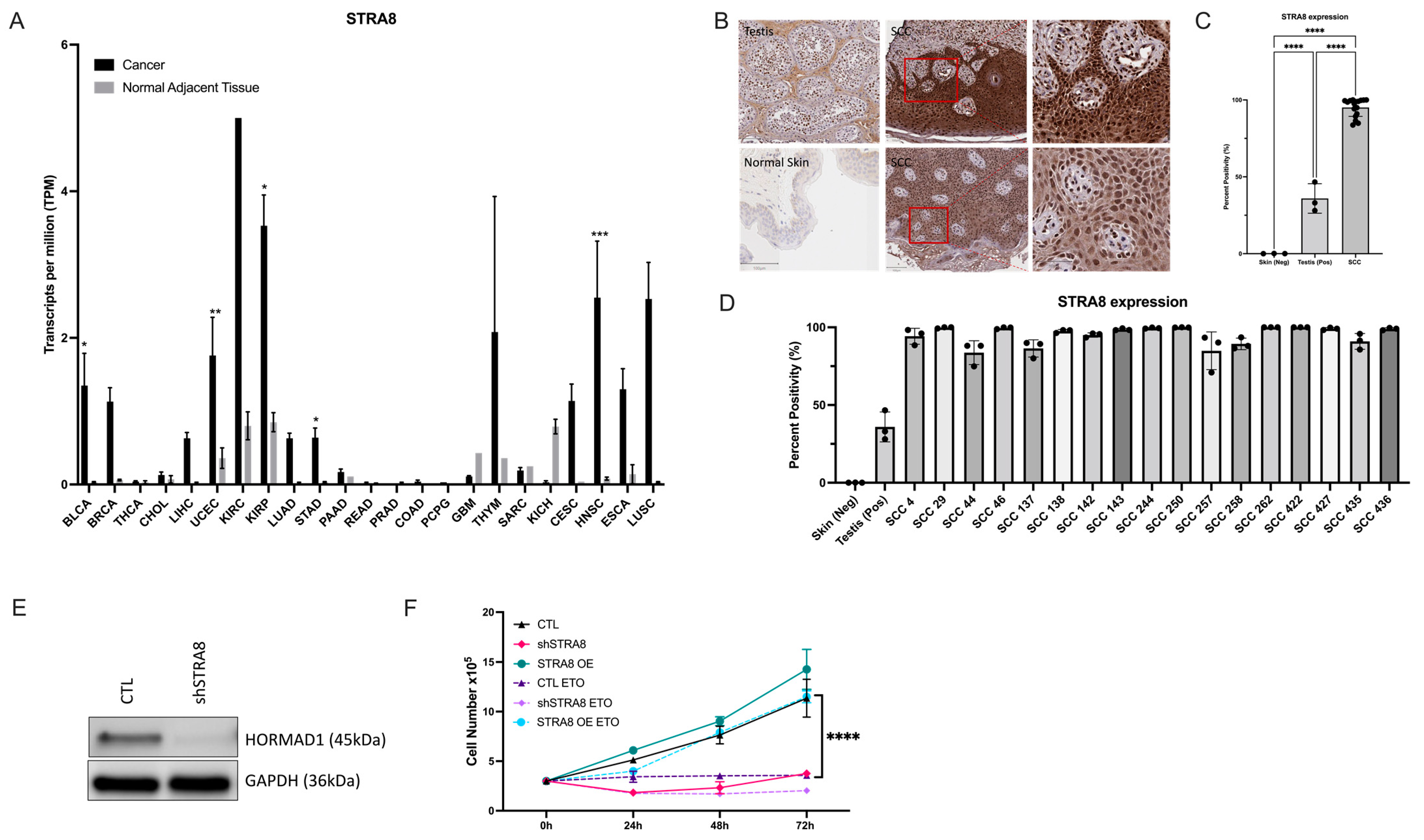
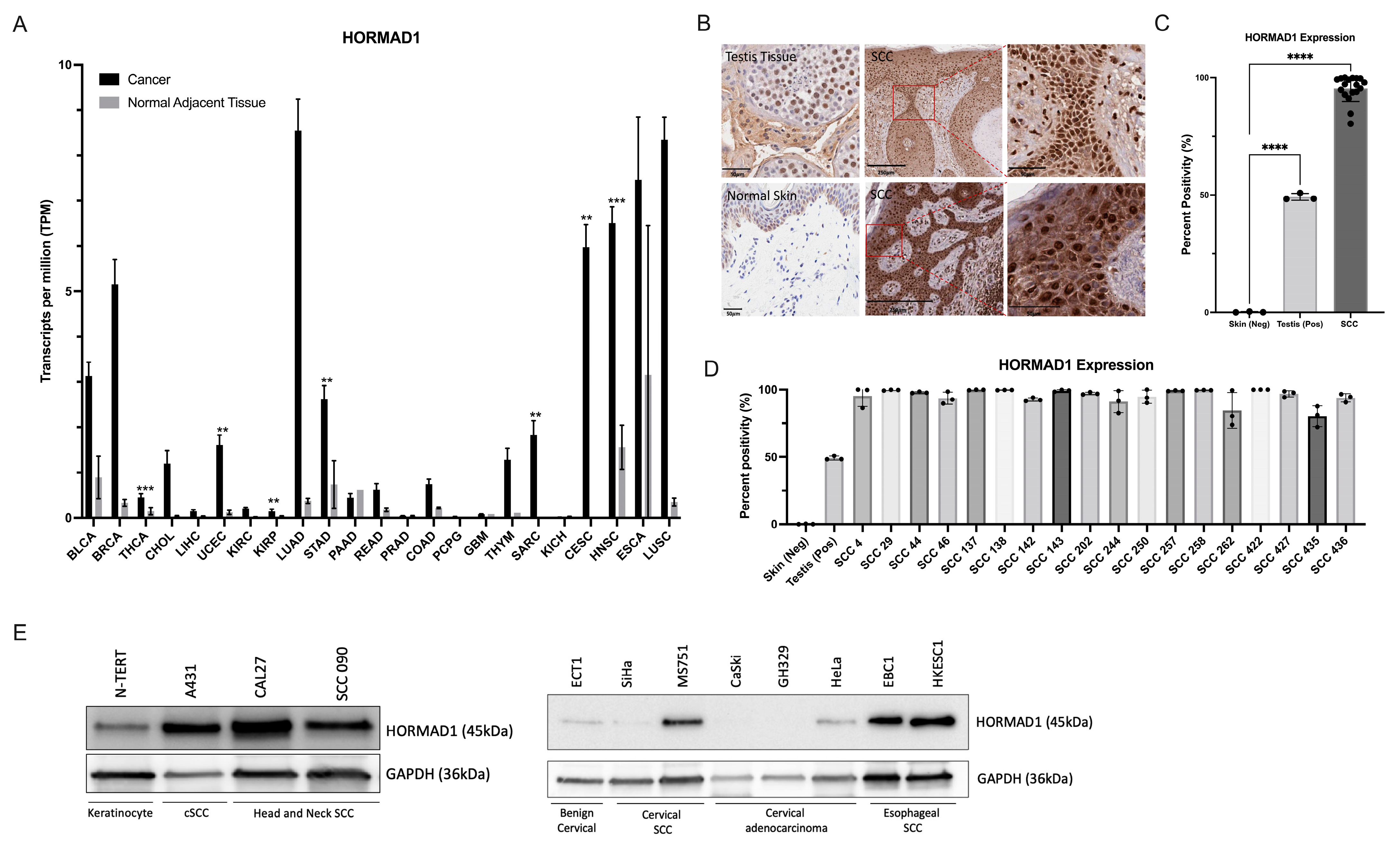
3.1. HORMAD1 Expression Is Significantly Increased in SCCs
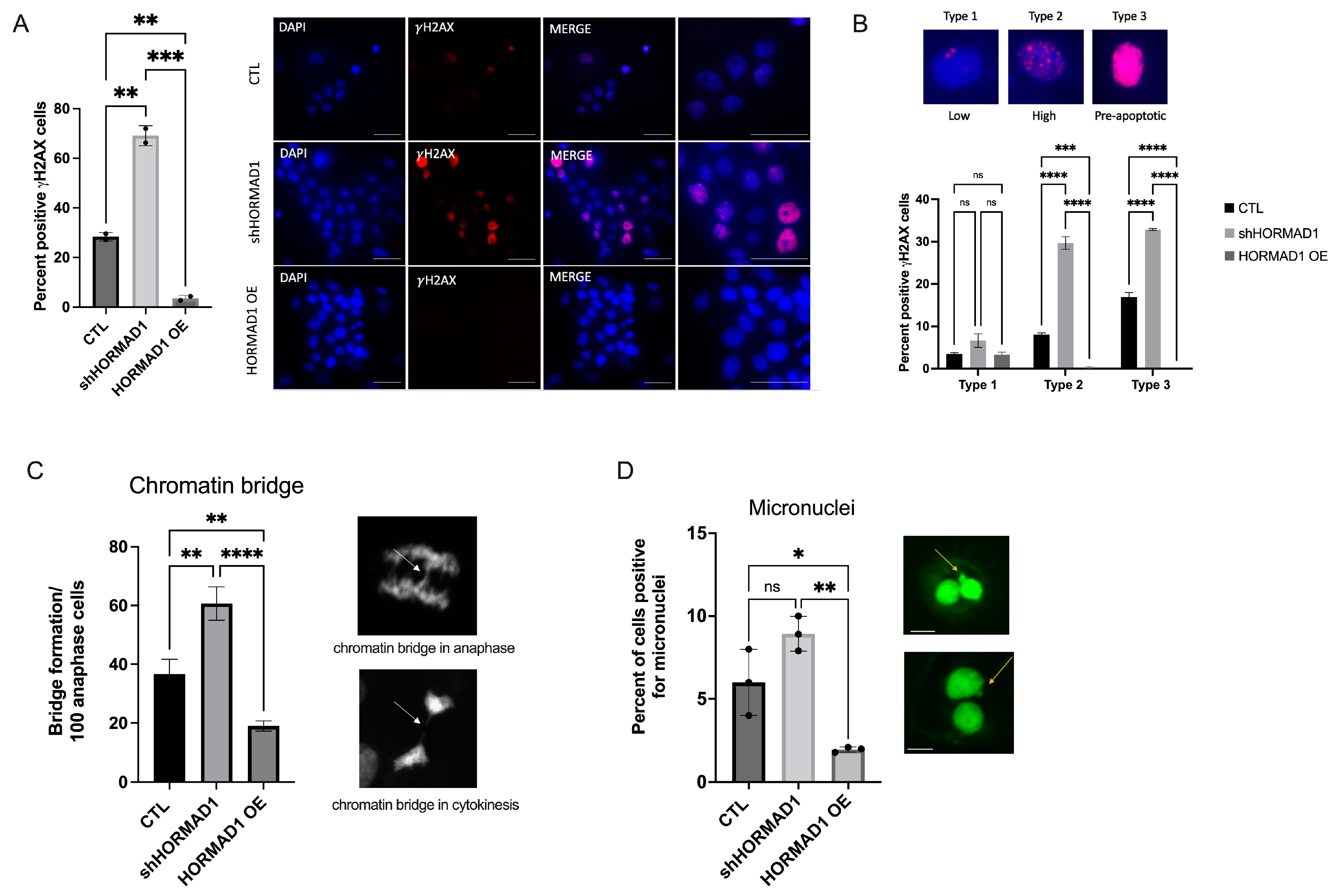
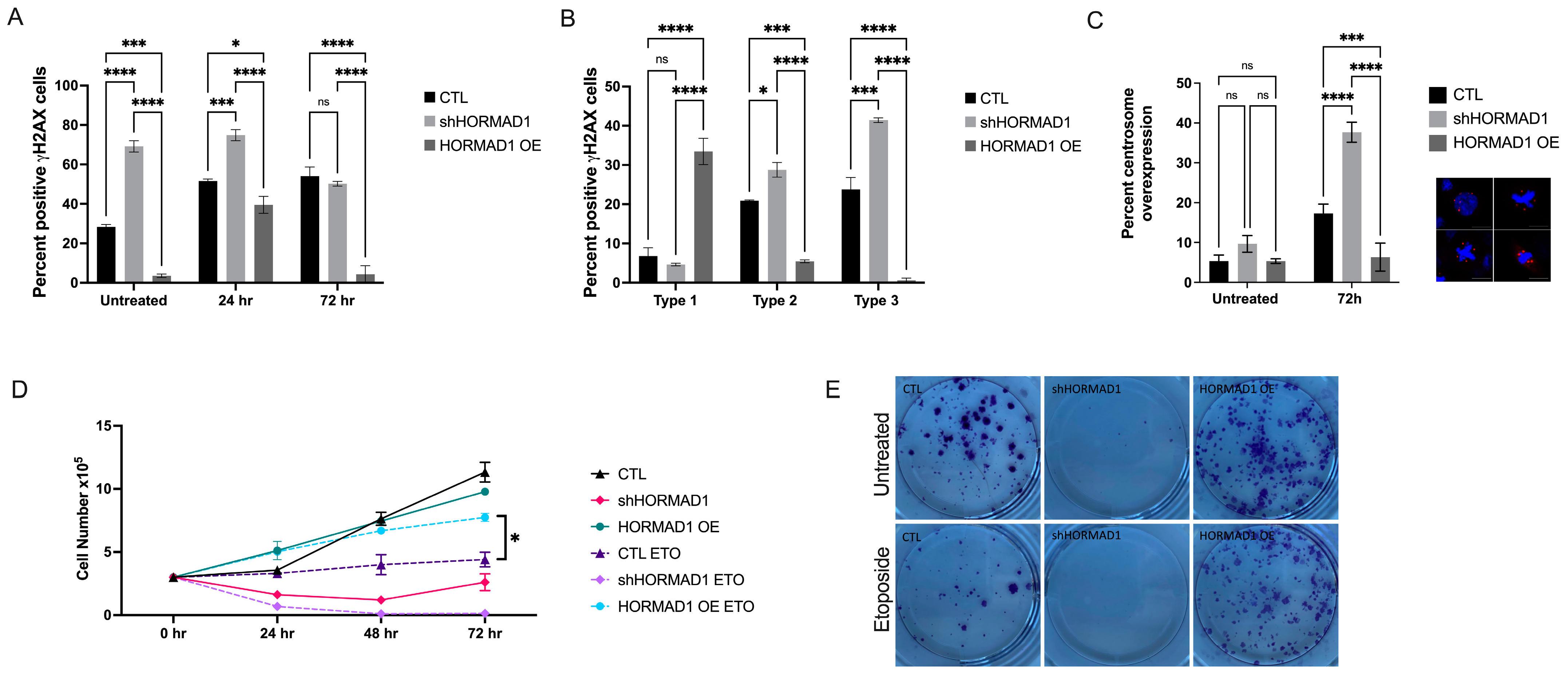
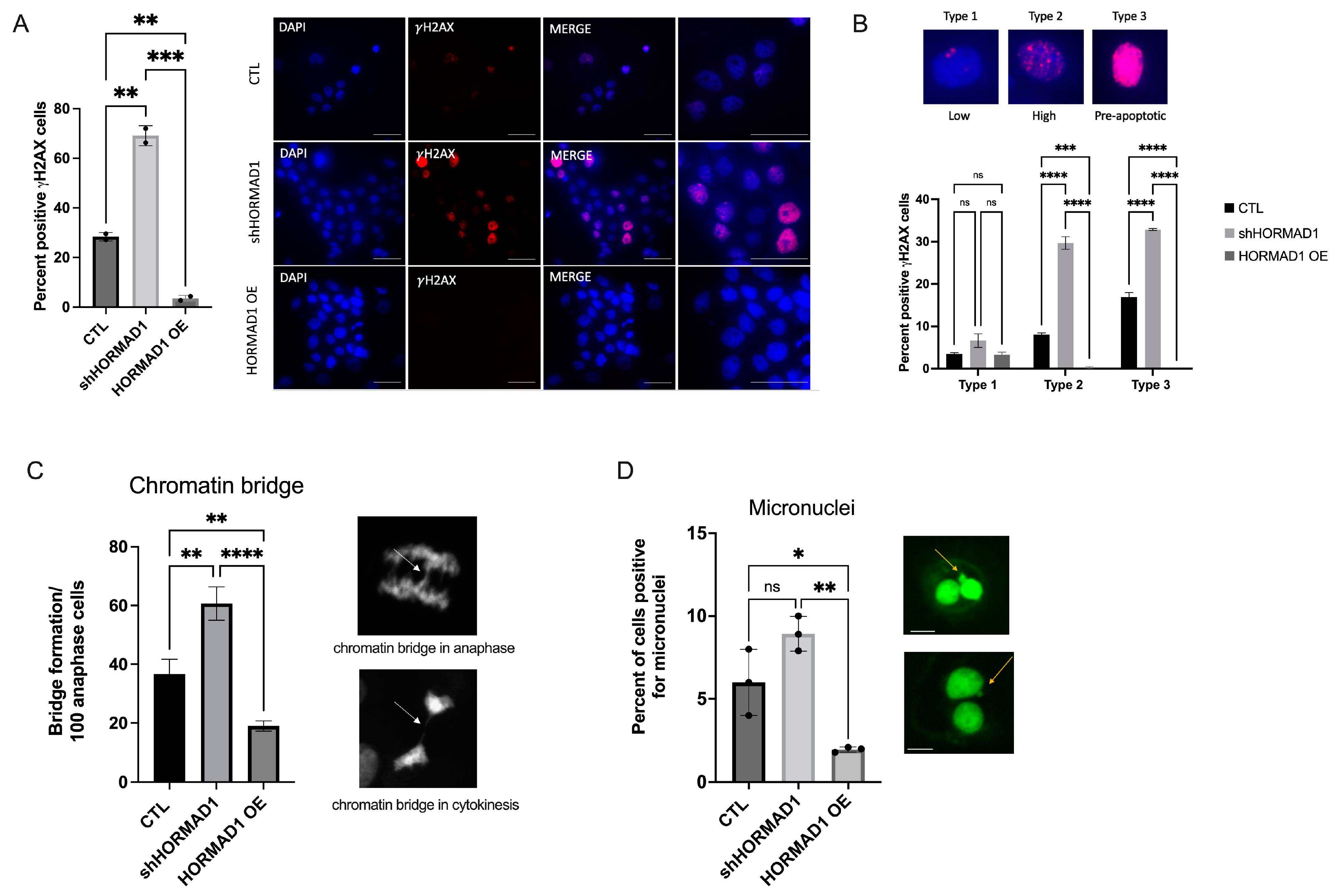
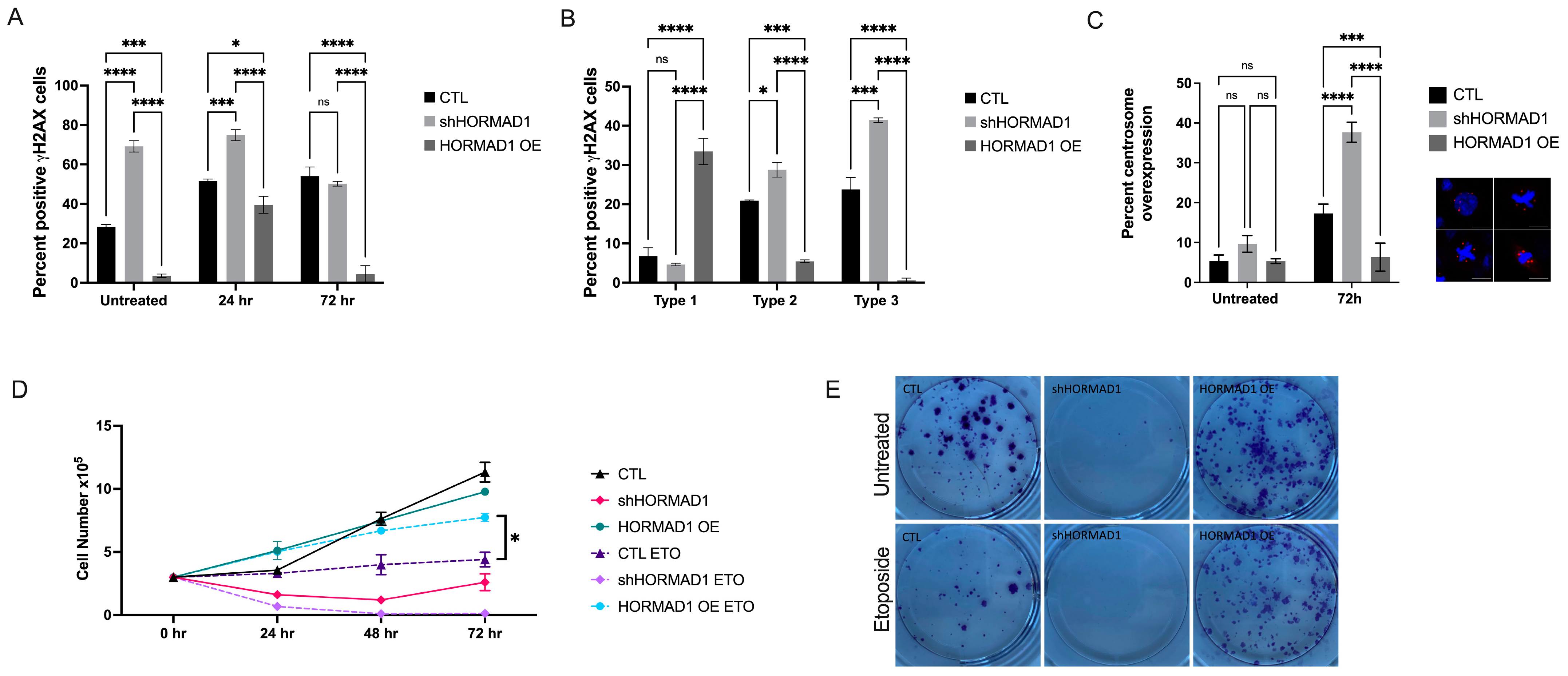
3.2. HORMAD1 Influences DNA Damage and Genomic Instability in SCC Cells
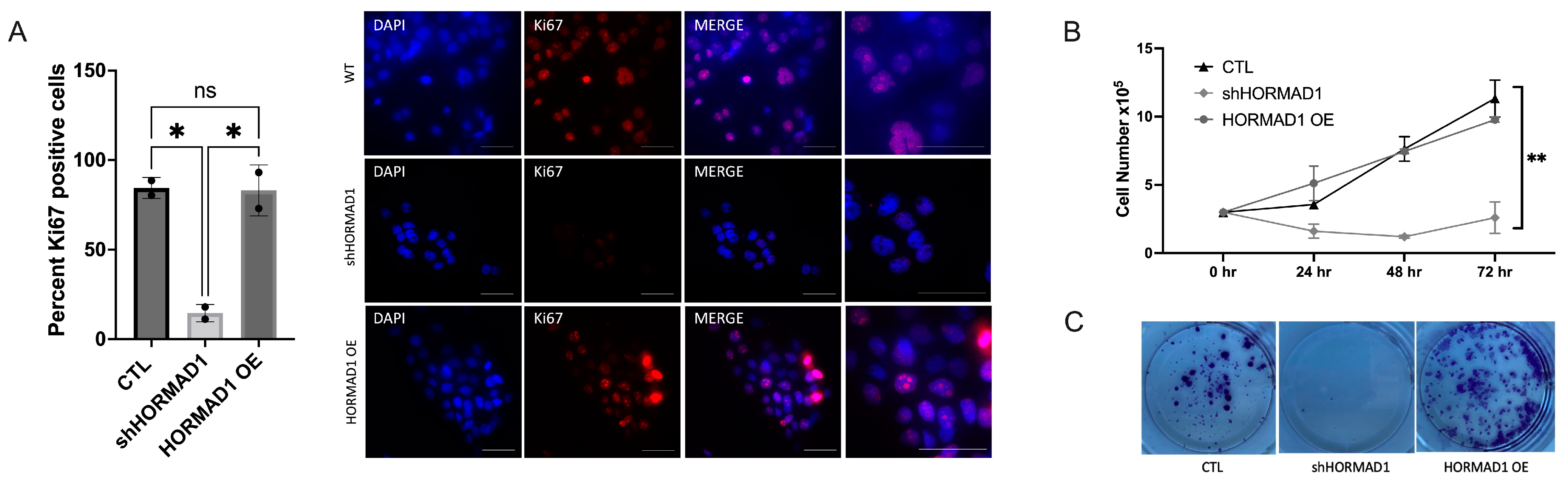
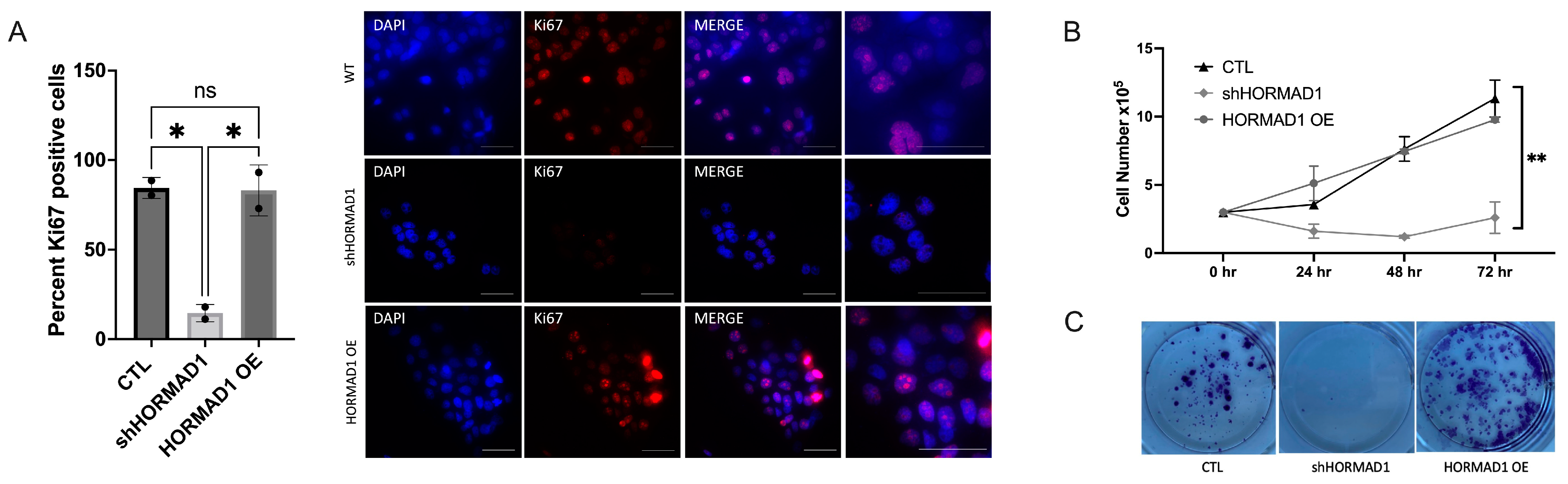
3.3. HORMAD1 Knockdown Leads to Reduced Proliferation and Survival in SCC Cells
3.4. HORMAD1 Expression Is Regulated by the Meiosis-Specific Transcription Factor STRA8 in SCC Cells
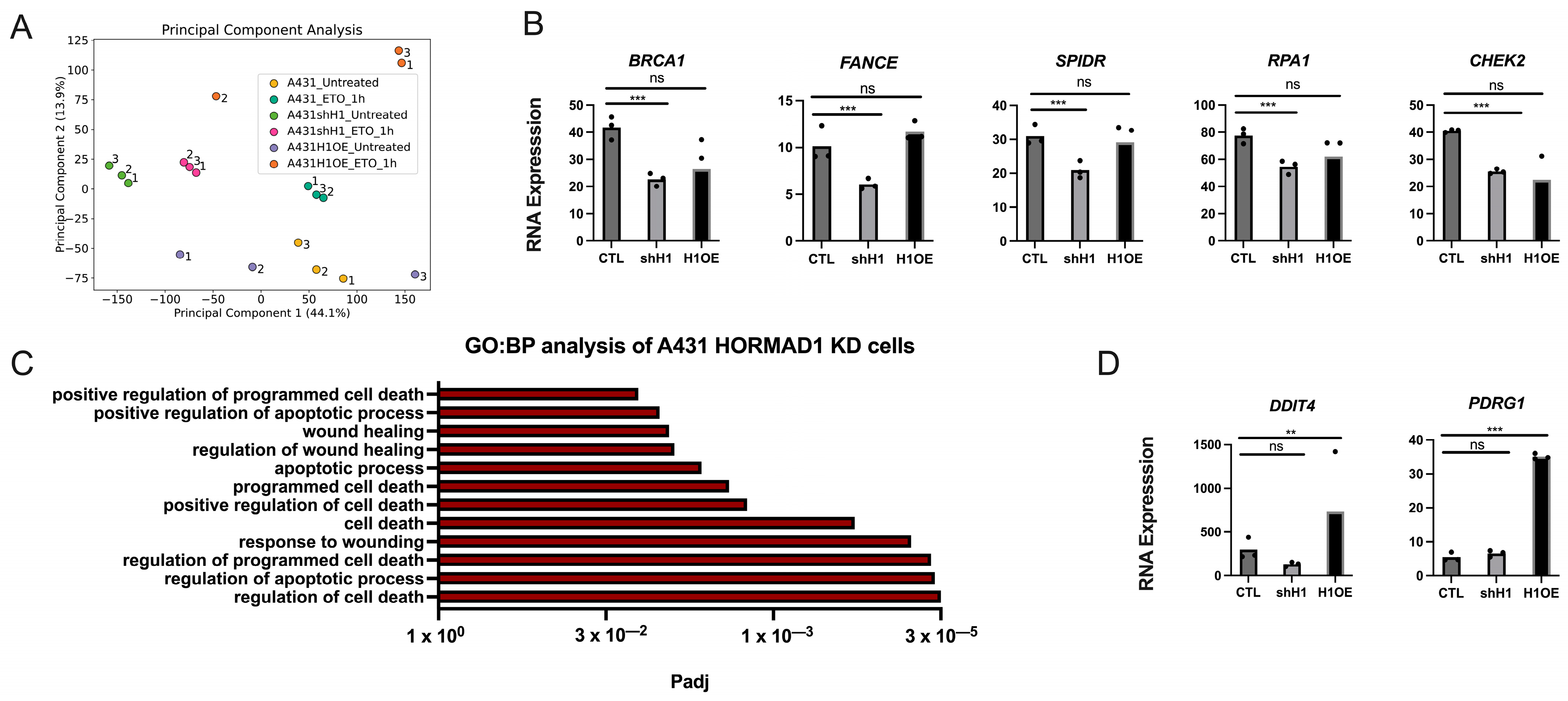
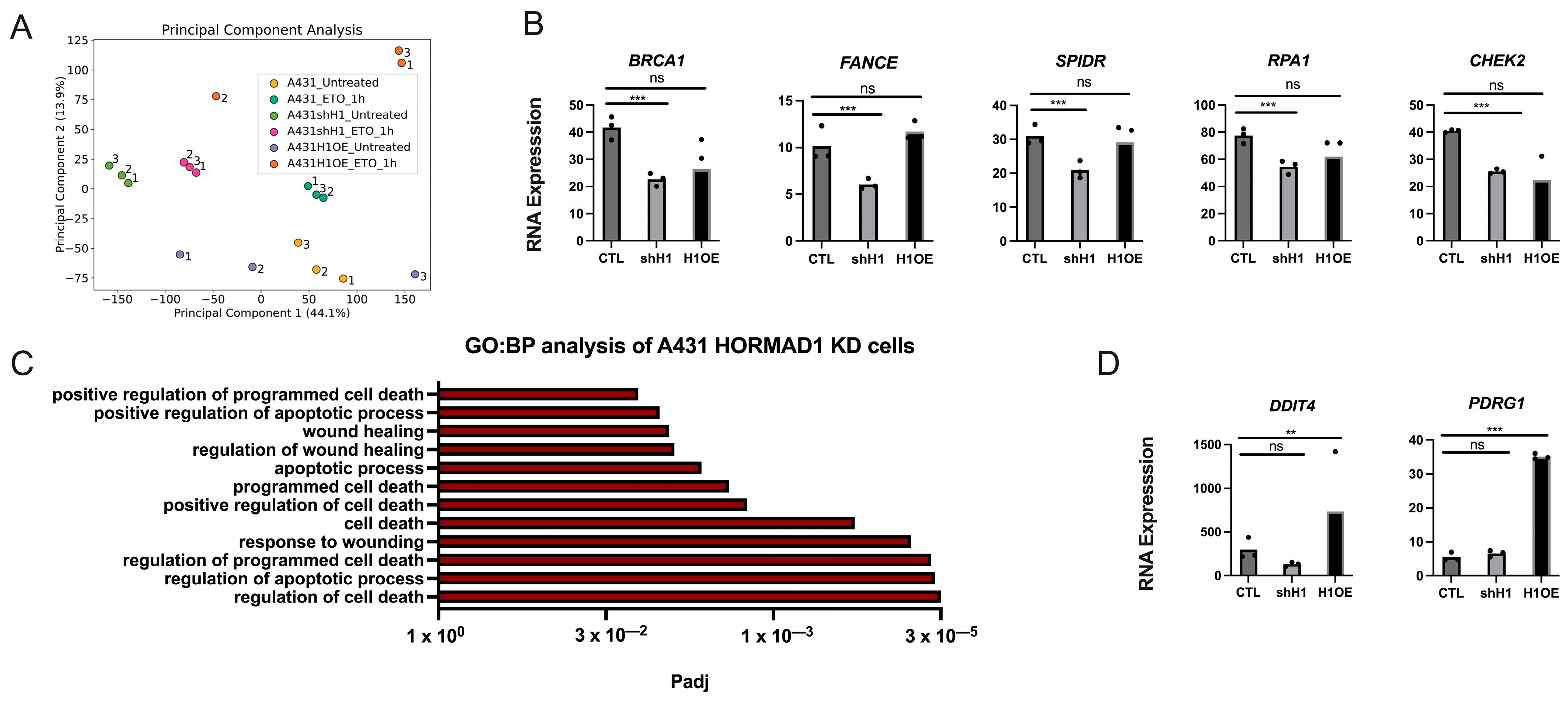
3.5. HORMAD1 Expression Leads to Changes in DNA Repair Gene Expression
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Nehal, K.S.; Bichakjian, C.K. Update on Keratinocyte Carcinomas. N. Engl. J. Med. 2018, 379, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P.; Rustgi, A.K. Squamous Cell Cancers: A Unified Perspective on Biology and Genetics. Cancer Cell 2016, 29, 622–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greve, K.B.; Lindgreen, J.N.; Terp, M.G.; Pedersen, C.B.; Schmidt, S.; Mollenhauer, J.; Kristensen, S.B.; Andersen, R.S.; Relster, M.M.; Ditzel, H.J.; et al. Ectopic expression of cancer/testis antigen SSX2 induces DNA damage and promotes genomic instability. Mol. Oncol. 2015, 9, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Tsang, M.; Gantchev, J.; Netchiporouk, E.; Moreau, L.; Ghazawi, F.M.; Glassman, S.; Sasseville, D.; Litvinov, I.V. A study of meiomitosis and novel pathways of genomic instability in cutaneous T-cell lymphomas (CTCL). Oncotarget 2018, 9, 37647–37661. [Google Scholar] [CrossRef] [Green Version]
- Scanlan, M.J.; Gure, A.O.; Jungbluth, A.A.; Old, L.J.; Chen, Y.T. Cancer/testis antigens: An expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002, 188, 22–32. [Google Scholar] [CrossRef]
- Simpson, A.J.; Caballero, O.L.; Jungbluth, A.; Chen, Y.T.; Old, L.J. Cancer/testis antigens, gametogenesis and cancer. Nat. Rev. Cancer 2005, 5, 615–625. [Google Scholar] [CrossRef]
- Feichtinger, J.; Aldeailej, I.; Anderson, R.; Almutairi, M.; Almatrafi, A.; Alsiwiehri, N.; Griffiths, K.; Stuart, N.; Wakeman, J.A.; Larcombe, L.; et al. Meta-analysis of clinical data using human meiotic genes identifies a novel cohort of highly restricted cancer-specific marker genes. Oncotarget 2012, 3, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Gantchev, J.; Martinez Villarreal, A.; Xie, P.; Lefrancois, P.; Gunn, S.; Netchiporouk, E.; Sasseville, D.; Litvinov, I.V. The Ectopic Expression of Meiosis Regulatory Genes in Cutaneous T-Cell Lymphomas (CTCL). Front. Oncol. 2019, 9, 429. [Google Scholar] [CrossRef]
- Gantchev, J.; Martinez Villarreal, A.; Gunn, S.; Zetka, M.; Odum, N.; Litvinov, I.V. The ectopic expression of meiCT genes promotes meiomitosis and may facilitate carcinogenesis. Cell Cycle 2020, 19, 837–854. [Google Scholar] [CrossRef]
- Grichnik, J.M. Melanoma, nevogenesis, and stem cell biology. J. Investig. Dermatol. 2008, 128, 2365–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey, S.F.; Byrnes, D.M.; Eller, M.S.; Rosa, A.M.; Dabas, N.; Escandon, J.; Grichnik, J.M. Potential role of meiosis proteins in melanoma chromosomal instability. J. Skin Cancer 2013, 2013, 190109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitehurst, A.W. Cause and consequence of cancer/testis antigen activation in cancer. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 251–272. [Google Scholar] [CrossRef] [PubMed]
- Adelaide, J.; Finetti, P.; Bekhouche, I.; Repellini, L.; Geneix, J.; Sircoulomb, F.; Charafe-Jauffret, E.; Cervera, N.; Desplans, J.; Parzy, D.; et al. Integrated profiling of basal and luminal breast cancers. Cancer Res. 2007, 67, 11565–11575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watkins, J.; Weekes, D.; Shah, V.; Gazinska, P.; Joshi, S.; Sidhu, B.; Gillett, C.; Pinder, S.; Vanoli, F.; Jasin, M.; et al. Genomic Complexity Profiling Reveals That HORMAD1 Overexpression Contributes to Homologous Recombination Deficiency in Triple-Negative Breast Cancers. Cancer Discov. 2015, 5, 488–505. [Google Scholar] [CrossRef] [Green Version]
- Daniel, K.; Lange, J.; Hached, K.; Fu, J.; Anastassiadis, K.; Roig, I.; Cooke, H.J.; Stewart, A.F.; Wassmann, K.; Jasin, M.; et al. Meiotic homologue alignment and its quality surveillance are controlled by mouse HORMAD1. Nat. Cell Biol. 2011, 13, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Wojtasz, L.; Daniel, K.; Roig, I.; Bolcun-Filas, E.; Xu, H.; Boonsanay, V.; Eckmann, C.R.; Cooke, H.J.; Jasin, M.; Keeney, S.; et al. Mouse HORMAD1 and HORMAD2, two conserved meiotic chromosomal proteins, are depleted from synapsed chromosome axes with the help of TRIP13 AAA-ATPase. PLoS Genet. 2009, 5, e1000702. [Google Scholar] [CrossRef] [Green Version]
- Boekhout, M.; Karasu, M.E.; Wang, J.; Acquaviva, L.; Pratto, F.; Brick, K.; Eng, D.Y.; Xu, J.; Camerini-Otero, R.D.; Patel, D.J.; et al. REC114 Partner ANKRD31 Controls Number, Timing, and Location of Meiotic DNA Breaks. Mol. Cell 2019, 74, 1053–1068.e8. [Google Scholar] [CrossRef]
- Stanzione, M.; Baumann, M.; Papanikos, F.; Dereli, I.; Lange, J.; Ramlal, A.; Trankner, D.; Shibuya, H.; de Massy, B.; Watanabe, Y.; et al. Meiotic DNA break formation requires the unsynapsed chromosome axis-binding protein IHO1 (CCDC36) in mice. Nat. Cell Biol. 2016, 18, 1208–1220. [Google Scholar] [CrossRef]
- Kogo, H.; Tsutsumi, M.; Ohye, T.; Inagaki, H.; Abe, T.; Kurahashi, H. HORMAD1-dependent checkpoint/surveillance mechanism eliminates asynaptic oocytes. Genes. Cells 2012, 17, 439–454. [Google Scholar] [CrossRef]
- Qiao, H.; Rao, H.; Yun, Y.; Sandhu, S.; Fong, J.H.; Sapre, M.; Nguyen, M.; Tham, A.; Van, B.W.; Chng, T.Y.H.; et al. Impeding DNA Break Repair Enables Oocyte Quality Control. Mol. Cell 2018, 72, 211–221.e3. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L.; Koonin, E.V. The HORMA domain: A common structural denominator in mitotic checkpoints, chromosome synapsis and DNA repair. Trends Biochem. Sci. 1998, 23, 284–286. [Google Scholar] [CrossRef]
- Rosenberg, S.C.; Corbett, K.D. The multifaceted roles of the HORMA domain in cellular signaling. J. Cell Biol. 2015, 211, 745–755. [Google Scholar] [CrossRef] [Green Version]
- West, A.M.V.; Komives, E.A.; Corbett, K.D. Conformational dynamics of the Hop1 HORMA domain reveal a common mechanism with the spindle checkpoint protein Mad2. Nucleic Acids Res. 2018, 46, 279–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.M.; Rosenberg, S.C.; Ur, S.N.; Lehmer, M.K.; Ye, Q.; Hagemann, G.; Caballero, I.; Uson, I.; MacQueen, A.J.; Herzog, F.; et al. A conserved filamentous assembly underlies the structure of the meiotic chromosome axis. eLife 2019, 8, e40372. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Tang, H.; Chen, X.; Zhang, G.; Wang, Y.; Xie, X.; Liao, N. Transcriptomic analyses identify key differentially expressed genes and clinical outcomes between triple-negative and non-triple-negative breast cancer. Cancer Manag. Res. 2019, 11, 179–190. [Google Scholar] [CrossRef] [Green Version]
- Nichols, B.A.; Oswald, N.W.; McMillan, E.A.; McGlynn, K.; Yan, J.; Kim, M.S.; Saha, J.; Mallipeddi, P.L.; LaDuke, S.A.; Villalobos, P.A.; et al. HORMAD1 Is a Negative Prognostic Indicator in Lung Adenocarcinoma and Specifies Resistance to Oxidative and Genotoxic Stress. Cancer Res. 2018, 78, 6196–6208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Kardos, J.; Yang, Y.; Tamir, T.Y.; Mutter-Rottmayer, E.; Weissman, B.; Major, M.B.; Kim, W.Y.; Vaziri, C. The Cancer/Testes (CT) Antigen HORMAD1 promotes Homologous Recombinational DNA Repair and Radioresistance in Lung adenocarcinoma cells. Sci. Rep. 2018, 8, 15304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Wang, Y.; Zhu, Q.; Li, P.; Chen, J.; Tang, Z.; Shen, Y.; Cheng, X.; Lu, L.Y.; Liu, Y. Aberrantly expressed HORMAD1 disrupts nuclear localization of MCM8-MCM9 complex and compromises DNA mismatch repair in cancer cells. Cell Death Dis. 2020, 11, 519. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Kupper, T.S.; Sasseville, D. The role of AHI1 and CDKN1C in cutaneous T-cell lymphoma progression. Exp. Dermatol. 2012, 21, 964–966. [Google Scholar] [CrossRef] [Green Version]
- Litvinov, I.V.; Zhou, Y.; Kupper, T.S.; Sasseville, D. Loss of BCL7A expression correlates with poor disease prognosis in patients with early-stage cutaneous T-cell lymphoma. Leuk Lymphoma 2013, 54, 653–654. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Cordeiro, B.; Huang, Y.; Zargham, H.; Pehr, K.; Dore, M.A.; Gilbert, M.; Zhou, Y.; Kupper, T.S.; Sasseville, D. Ectopic expression of cancer-testis antigens in cutaneous T-cell lymphoma patients. Clin. Cancer Res. 2014, 20, 3799–3808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litvinov, I.V.; Netchiporouk, E.; Cordeiro, B.; Zargham, H.; Pehr, K.; Gilbert, M.; Zhou, Y.; Moreau, L.; Woetmann, A.; Odum, N.; et al. Ectopic expression of embryonic stem cell and other developmental genes in cutaneous T-cell lymphoma. Oncoimmunology 2014, 3, e970025. [Google Scholar] [CrossRef] [PubMed]
- Gantchev, J.; Ramchatesingh, B.; Berman-Rosa, M.; Sikorski, D.; Raveendra, K.; Amar, L.; Xu, H.H.; Villarreal, A.M.; Ordaz, D.J.G.; Litvinov, I.V. Tools used to assay genomic instability in cancers and cancer meiomitosis. J. Cell Commun. Signal. 2021, 16, 159–177. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Cordeiro, B.; Fredholm, S.; Odum, N.; Zargham, H.; Huang, Y.; Zhou, Y.; Pehr, K.; Kupper, T.S.; Woetmann, A.; et al. Analysis of STAT4 expression in cutaneous T-cell lymphoma (CTCL) patients and patient-derived cell lines. Cell Cycle 2014, 13, 2975–2982. [Google Scholar] [CrossRef] [Green Version]
- Netchiporouk, E.; Gantchev, J.; Tsang, M.; Thibault, P.; Watters, A.K.; Hughes, J.M.; Ghazawi, F.M.; Woetmann, A.; Odum, N.; Sasseville, D.; et al. Analysis of CTCL cell lines reveals important differences between mycosis fungoides/Sezary syndrome vs. HTLV-1(+) leukemic cell lines. Oncotarget 2017, 8, 95981–95998. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Bizet, A.A.; Binamer, Y.; Jones, D.A.; Sasseville, D.; Philip, A. CD109 release from the cell surface in human keratinocytes regulates TGF-beta receptor expression, TGF-beta signalling and STAT3 activation: Relevance to psoriasis. Exp. Dermatol. 2011, 20, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef] [PubMed]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrancois, P.; Xie, P.; Gunn, S.; Gantchev, J.; Villarreal, A.M.; Sasseville, D.; Litvinov, I.V. In silico analyses of the tumor microenvironment highlight tumoral inflammation, a Th2 cytokine shift and a mesenchymal stem cell-like phenotype in advanced in basal cell carcinomas. J. Cell Commun. Signal. 2020, 14, 245–254. [Google Scholar] [CrossRef]
- Litvinov, I.V.; Xie, P.; Gunn, S.; Sasseville, D.; Lefrancois, P. The transcriptional landscape analysis of basal cell carcinomas reveals novel signalling pathways and actionable targets. Life Sci. Alliance 2021, 4, e202000651. [Google Scholar] [CrossRef]
- Carofiglio, F.; Sleddens-Linkels, E.; Wassenaar, E.; Inagaki, A.; van Cappellen, W.A.; Grootegoed, J.A.; Toth, A.; Baarends, W.M. Repair of exogenous DNA double-strand breaks promotes chromosome synapsis in SPO11-mutant mouse meiocytes, and is altered in the absence of HORMAD1. DNA Repair. 2018, 63, 25–38. [Google Scholar] [CrossRef]
- Rinaldi, V.D.; Bolcun-Filas, E.; Kogo, H.; Kurahashi, H.; Schimenti, J.C. The DNA Damage Checkpoint Eliminates Mouse Oocytes with Chromosome Synapsis Failure. Mol. Cell 2017, 67, 1026–1036.e2. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Zhang, Y.; Kidane, Y.; Feiveson, A.; Stodieck, L.; Karouia, F.; Ramesh, G.; Rohde, L.; Wu, H. Cellular responses and gene expression profile changes due to bleomycin-induced DNA damage in human fibroblasts in space. PLoS ONE 2017, 12, e0170358. [Google Scholar] [CrossRef] [Green Version]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar] [CrossRef]
- Finardi, A.; Massari, L.F.; Visintin, R. Anaphase Bridges: Not All Natural Fibers Are Healthy. Genes 2020, 11, 902. [Google Scholar] [CrossRef]
- Kwon, M.; Leibowitz, M.L.; Lee, J.H. Small but mighty: The causes and consequences of micronucleus rupture. Exp. Mol. Med. 2020, 52, 1777–1786. [Google Scholar] [CrossRef]
- Fenech, M. Cytokinesis-block micronucleus cytome assay. Nat. Protoc. 2007, 2, 1084–1104. [Google Scholar] [CrossRef] [Green Version]
- Negrini, S.; Gorgoulis, V.G.; Halazonetis, T.D. Genomic instability--an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.L.; de Rooij, D.G.; Page, D.C. Amplification of a broad transcriptional program by a common factor triggers the meiotic cell cycle in mice. eLife 2019, 8, e43738. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, F.; Saeednejad Zanjani, L.; Habibi Shams, Z.; Kiani, J.; Mehrazma, M.; Najafi, M.; Madjd, Z. High expression of DNA damage-inducible transcript 4 (DDIT4) is associated with advanced pathological features in the patients with colorectal cancer. Sci. Rep. 2021, 11, 13626. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Nielsen, A.Y.; Gjerstorff, M.F. Ectopic Expression of Testis Germ Cell Proteins in Cancer and Its Potential Role in Genomic Instability. Int. J. Mol. Sci. 2016, 17, 890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oulad-Abdelghani, M.; Bouillet, P.; Decimo, D.; Gansmuller, A.; Heyberger, S.; Dolle, P.; Bronner, S.; Lutz, Y.; Chambon, P. Characterization of a premeiotic germ cell-specific cytoplasmic protein encoded by Stra8, a novel retinoic acid-responsive gene. J. Cell Biol. 1996, 135, 469–477. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Li, Y.; Nie, R.; Friel, P.; Mitchell, D.; Evanoff, R.M.; Pouchnik, D.; Banasik, B.; McCarrey, J.R.; Small, C.; et al. Expression of stimulated by retinoic acid gene 8 (Stra8) and maturation of murine gonocytes and spermatogonia induced by retinoic acid in vitro. Biol. Reprod. 2008, 78, 537–545. [Google Scholar] [CrossRef]
- Zhou, Q.; Nie, R.; Li, Y.; Friel, P.; Mitchell, D.; Hess, R.A.; Small, C.; Griswold, M.D. Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoic acid: An in vivo study in vitamin A-sufficient postnatal murine testes. Biol. Reprod. 2008, 79, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Everts, H.B.; Silva, K.A.; Schmidt, A.N.; Opalenik, S.; Duncan, F.J.; King, L.E., Jr.; Sundberg, J.P.; Ong, D.E. Estrogen regulates the expression of retinoic acid synthesis enzymes and binding proteins in mouse skin. Nutr. Res. 2021, 94, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tan, Y.; Cao, X.; Kim, J.A.; Chen, T.; Hu, Y.; Wexler, M.; Wang, X. Epigenetic activation of HORMAD1 in basal-like breast cancer: Role in Rucaparib sensitivity. Oncotarget 2018, 9, 30115–30127. [Google Scholar] [CrossRef] [Green Version]
- Zong, B.; Sun, L.; Peng, Y.; Wang, Y.; Yu, Y.; Lei, J.; Zhang, Y.; Guo, S.; Li, K.; Liu, S. HORMAD1 promotes docetaxel resistance in triple negative breast cancer by enhancing DNA damage tolerance. Oncol. Rep. 2021, 46, 138. [Google Scholar] [CrossRef]
- Tarantino, D.; Walker, C.; Weekes, D.; Pemberton, H.; Davidson, K.; Torga, G.; Frankum, J.; Mendes-Pereira, A.M.; Prince, C.; Ferro, R.; et al. Functional screening reveals HORMAD1-driven gene dependencies associated with translesion synthesis and replication stress tolerance. Oncogene 2022, 41, 3969–3977. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Lain, S.; Verma, C.S.; Fersht, A.R.; Lane, D.P. Awakening guardian angels: Drugging the p53 pathway. Nat. Rev. Cancer 2009, 9, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Li, J.Q.; Li, H.Z.; Song, H.; Wei, C.S.; Zhang, S.Q. PDRG1 gene silencing contributes to inhibit the growth and induce apoptosis of gastric cancer cells. Pathol. Res. Pract. 2019, 215, 152567. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gantchev, J.; Messina-Pacheco, J.; Martínez Villarreal, A.; Ramchatesingh, B.; Lefrançois, P.; Xie, P.; Amar, L.; Xu, H.H.; Raveendra, K.; Sikorski, D.; et al. Ectopically Expressed Meiosis-Specific Cancer Testis Antigen HORMAD1 Promotes Genomic Instability in Squamous Cell Carcinomas. Cells 2023, 12, 1627. https://doi.org/10.3390/cells12121627
Gantchev J, Messina-Pacheco J, Martínez Villarreal A, Ramchatesingh B, Lefrançois P, Xie P, Amar L, Xu HH, Raveendra K, Sikorski D, et al. Ectopically Expressed Meiosis-Specific Cancer Testis Antigen HORMAD1 Promotes Genomic Instability in Squamous Cell Carcinomas. Cells. 2023; 12(12):1627. https://doi.org/10.3390/cells12121627
Chicago/Turabian StyleGantchev, Jennifer, Julia Messina-Pacheco, Amelia Martínez Villarreal, Brandon Ramchatesingh, Philippe Lefrançois, Pingxing Xie, Laetitia Amar, Hong Hao Xu, Keerthenan Raveendra, Daniel Sikorski, and et al. 2023. "Ectopically Expressed Meiosis-Specific Cancer Testis Antigen HORMAD1 Promotes Genomic Instability in Squamous Cell Carcinomas" Cells 12, no. 12: 1627. https://doi.org/10.3390/cells12121627
APA StyleGantchev, J., Messina-Pacheco, J., Martínez Villarreal, A., Ramchatesingh, B., Lefrançois, P., Xie, P., Amar, L., Xu, H. H., Raveendra, K., Sikorski, D., Guerra Ordaz, D. J., Gill, R. P. K., Lambert, M., & Litvinov, I. V. (2023). Ectopically Expressed Meiosis-Specific Cancer Testis Antigen HORMAD1 Promotes Genomic Instability in Squamous Cell Carcinomas. Cells, 12(12), 1627. https://doi.org/10.3390/cells12121627