
Ferroptosis-Mediated Cell Death Induced by NCX4040, The Non-Steroidal Nitric Oxide Donor, in Human Colorectal Cancer Cells: Implications in Therapy


 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods and Materials
2.1. Cell Culture
2.2. Cytotoxicity Studies
2.3. Flow Cytometric Analysis of Mitochondrial ROS
2.4. Flow Cytometric Analysis for Intracellular Glutathione
2.5. Lipid Peroxidation Assay
2.6. Real Time RT-PCR
2.7. Western Blot Assay
2.8. Metabolomics Studies
2.9. Statistical Analysis
3. Results
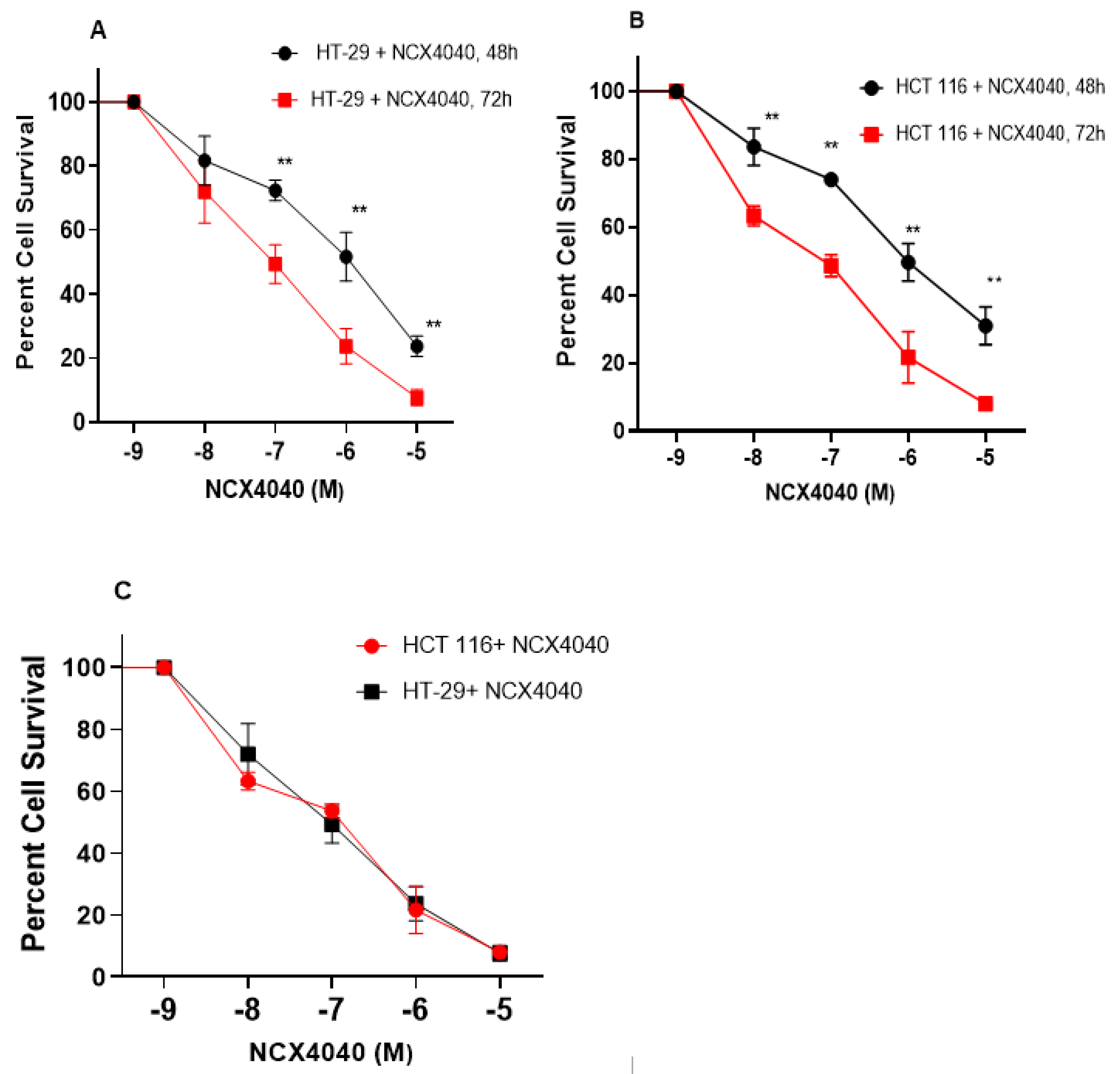
3.1. Cytotoxicity Studies with NCX4040
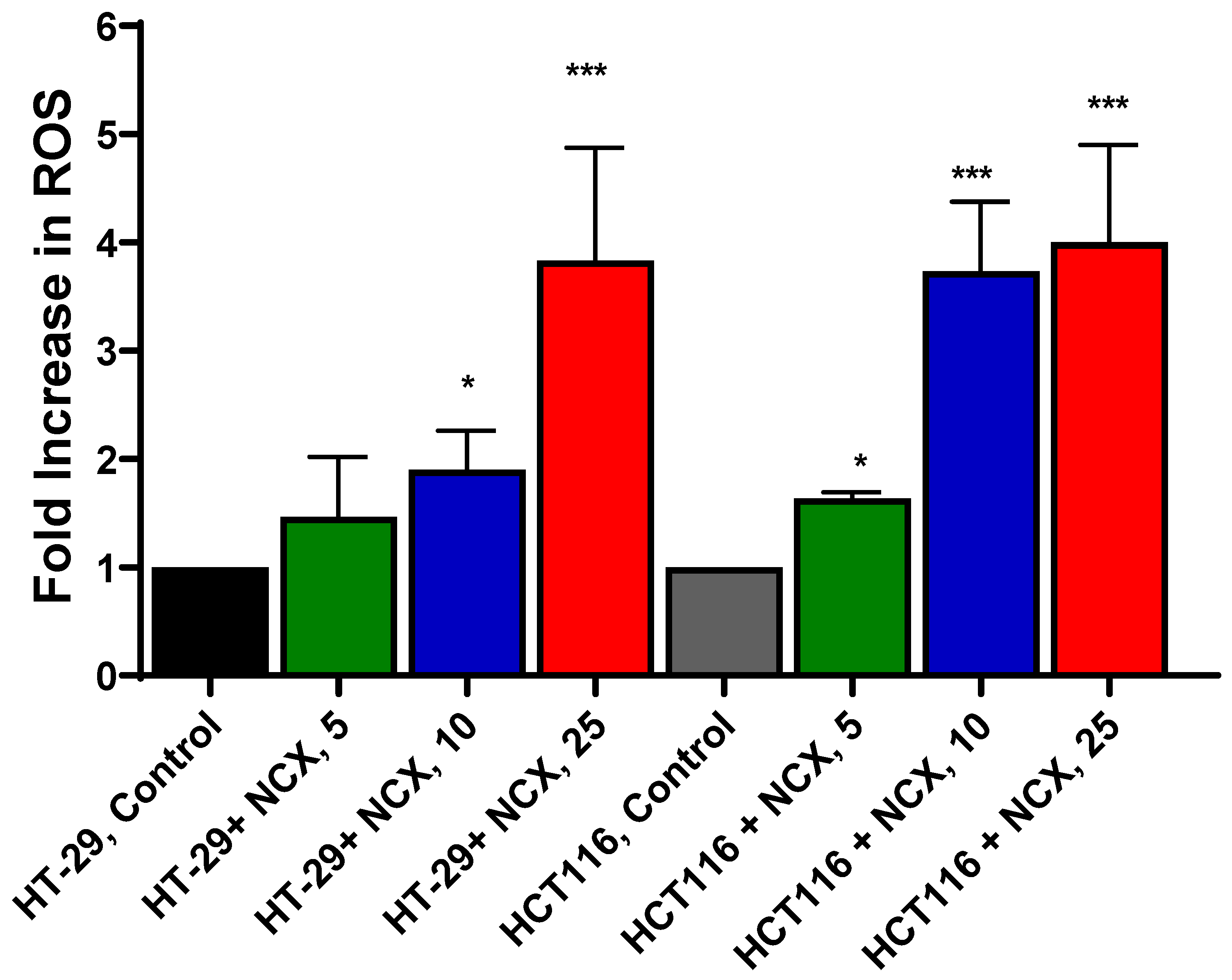
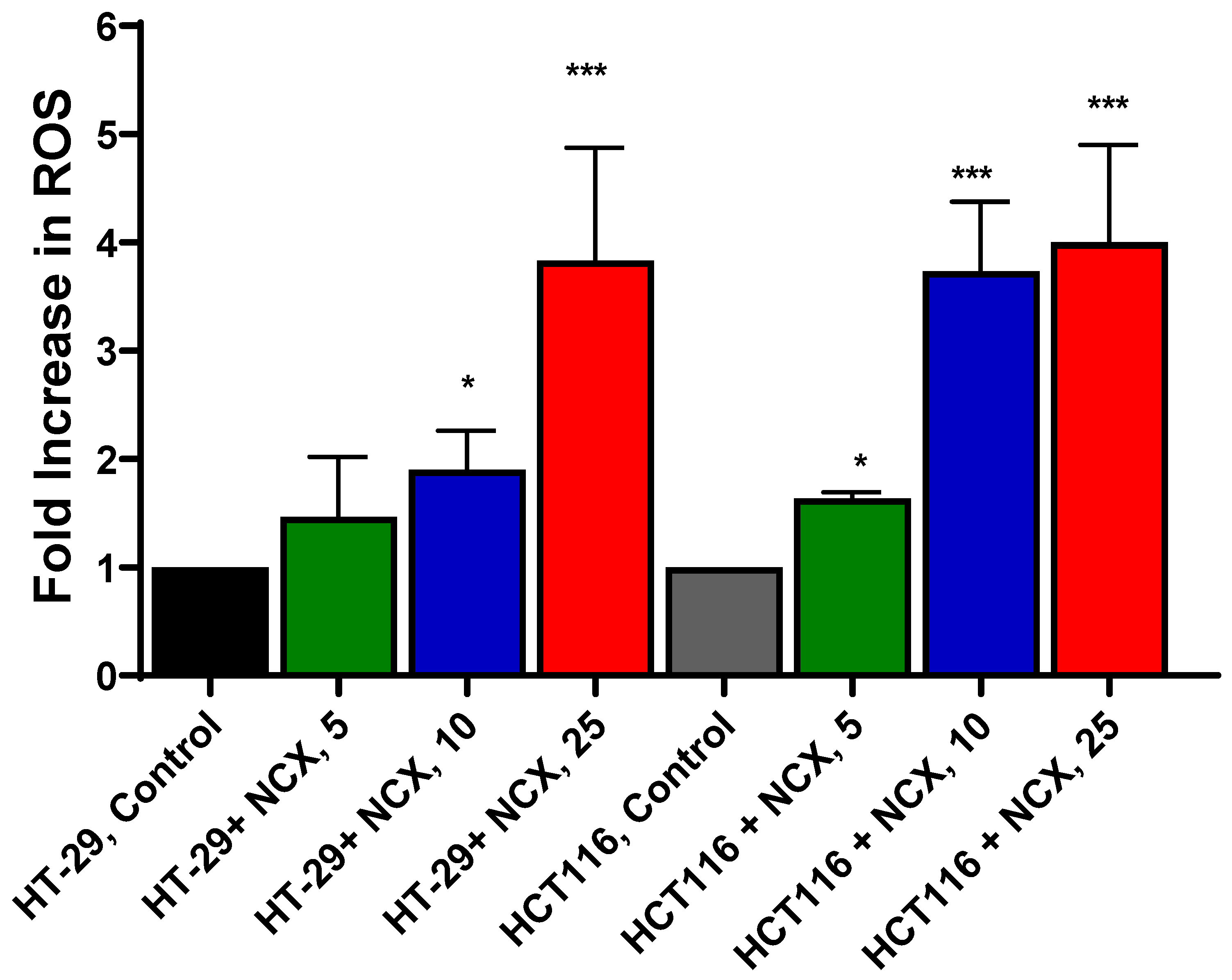
3.2. Measurements of Mitochondrial ROS
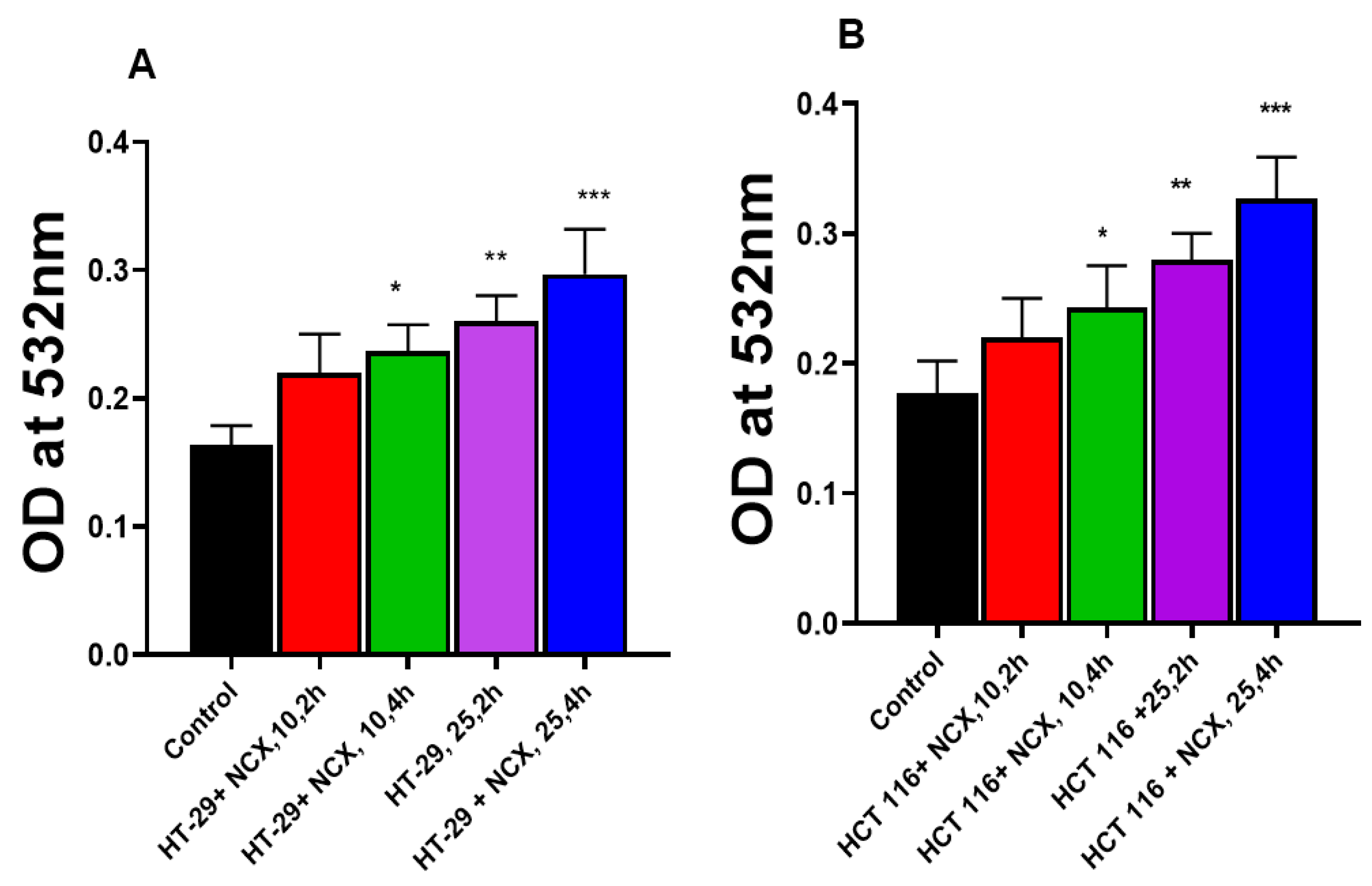
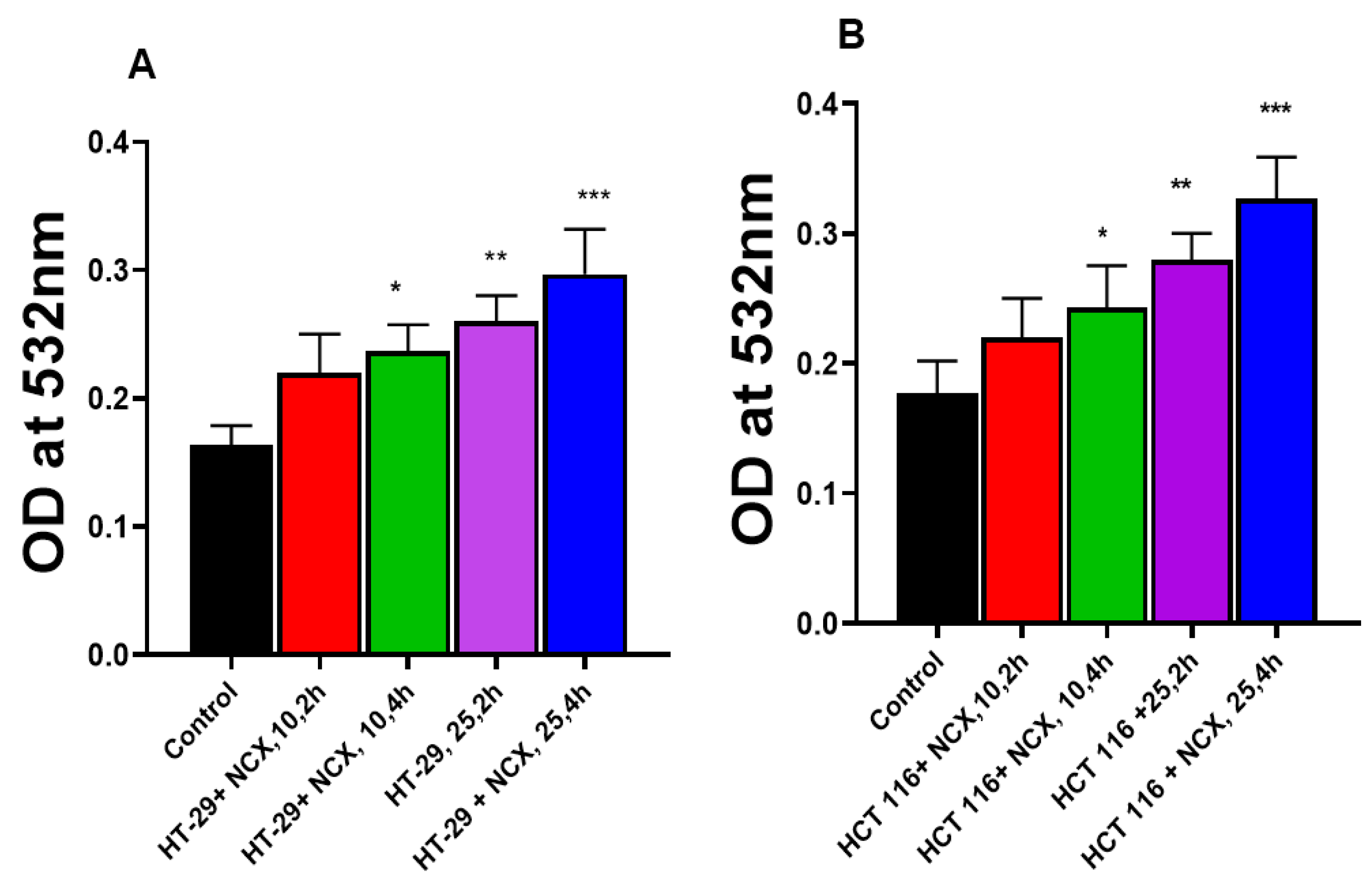
3.3. Lipid Peroxidation in HT-29 and HCT 116 Cells
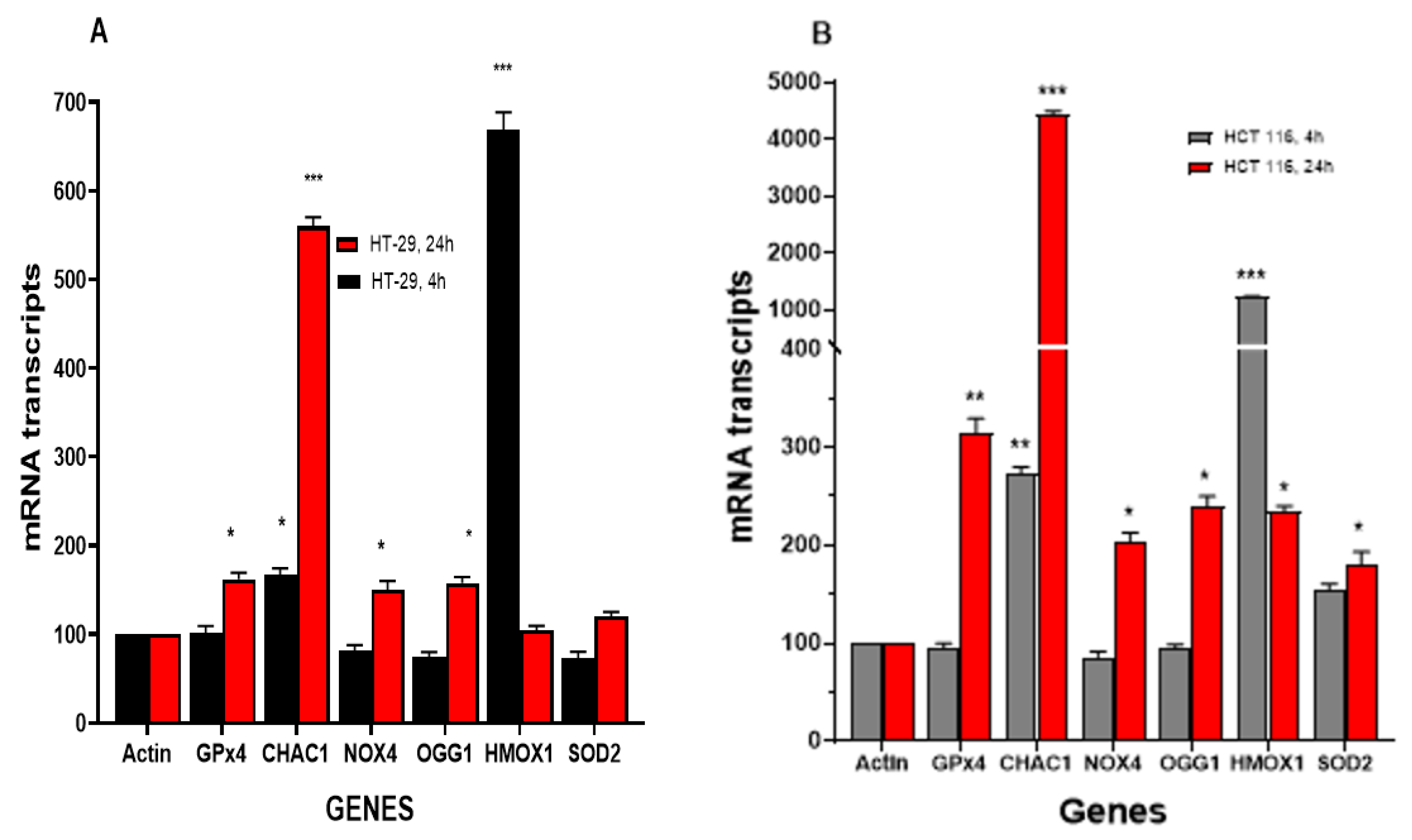
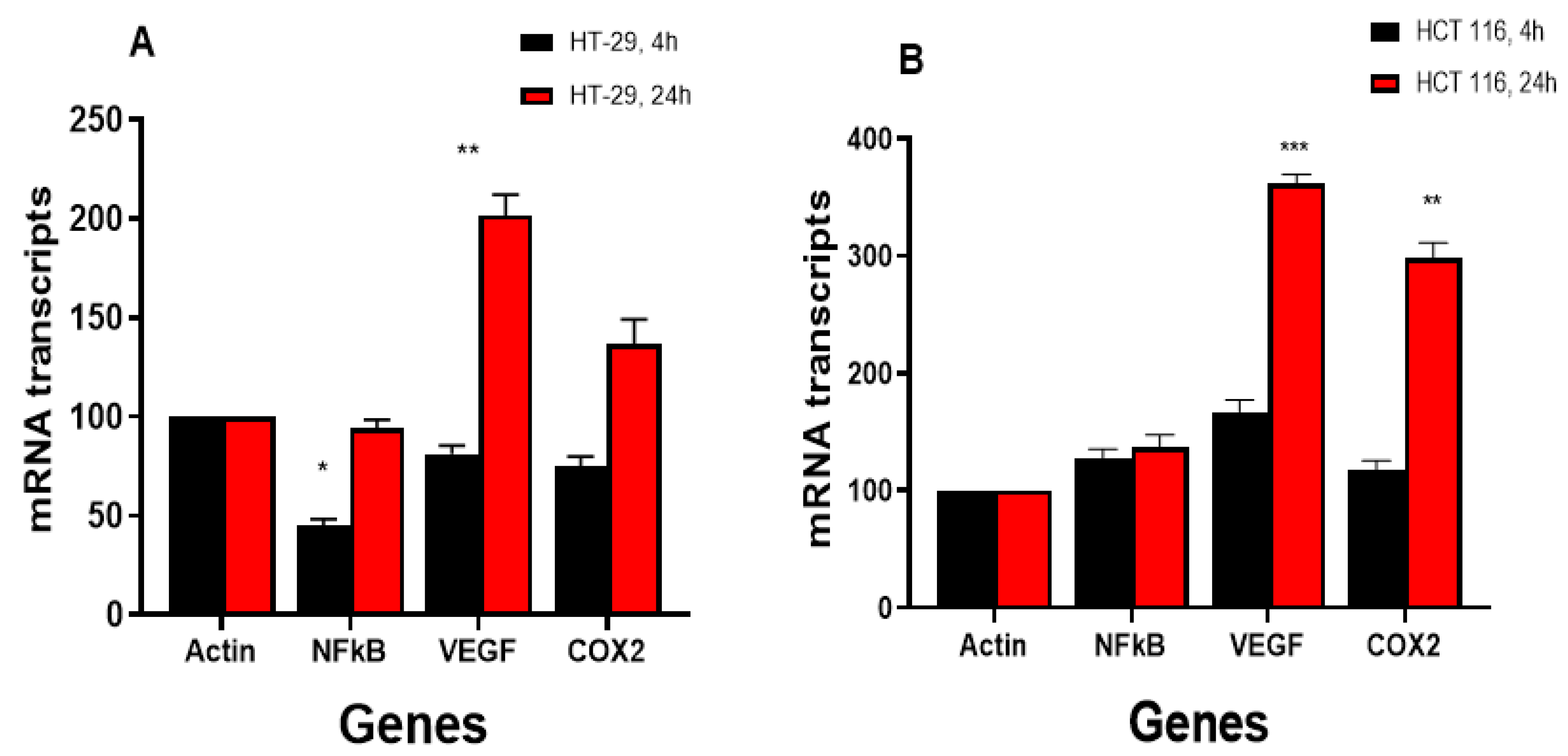
3.4. RT-PCR Studies
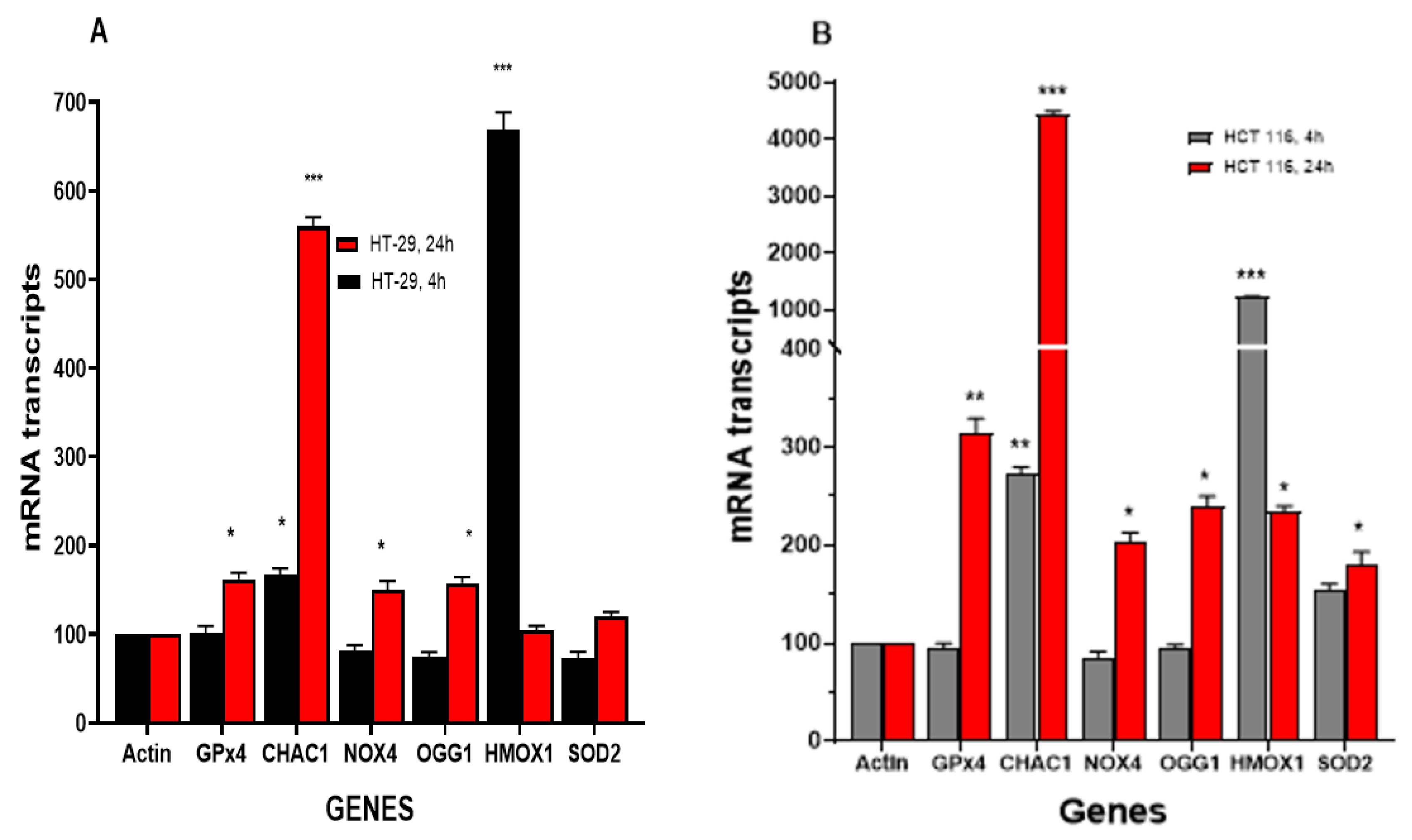
3.4.1. NCX4040 Induces Oxidative Stress Genes in HT-29 and HCT 116 Cells
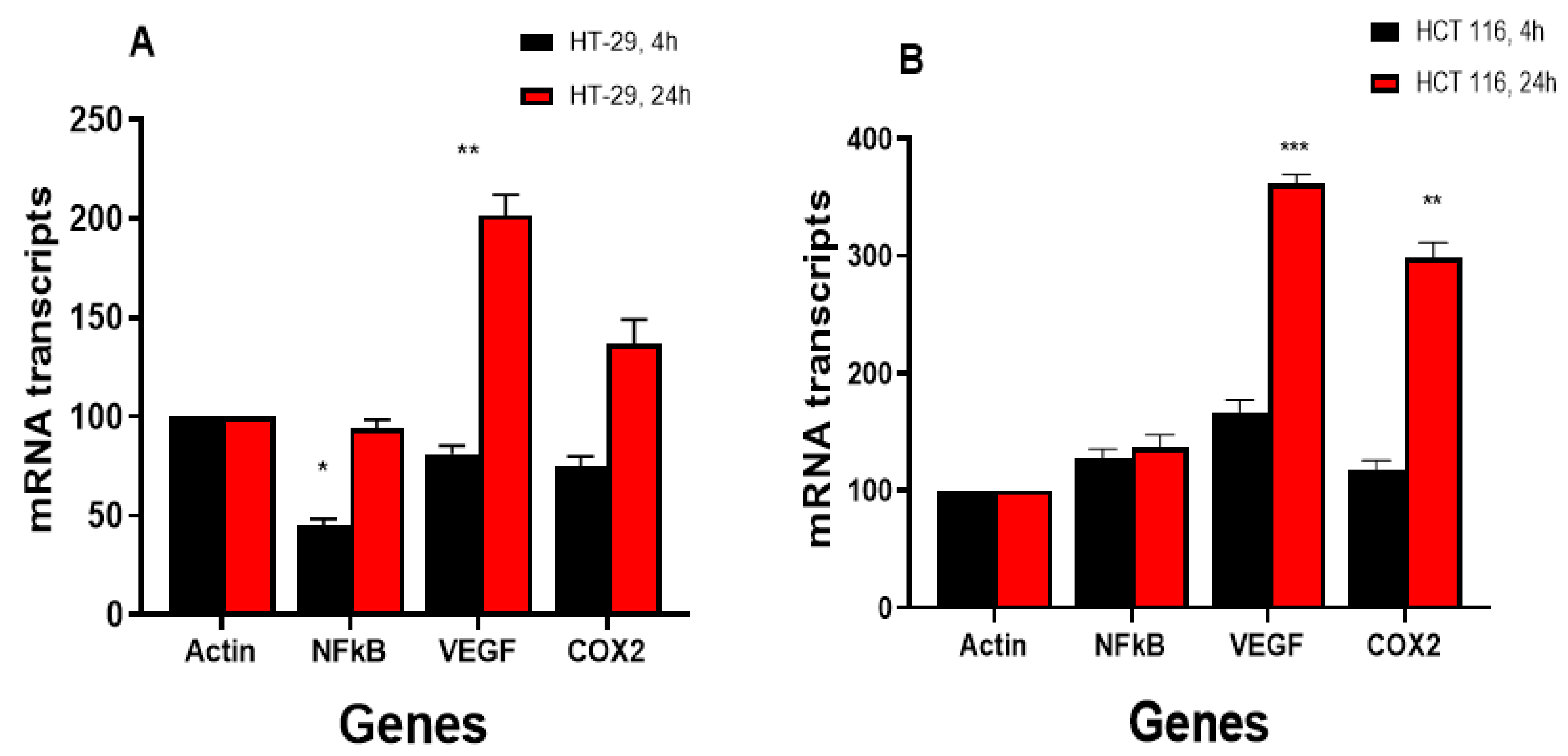
3.4.2. NCX4040 Induces Inflammatory Response Genes in HT-29 and HCT 116 Cells
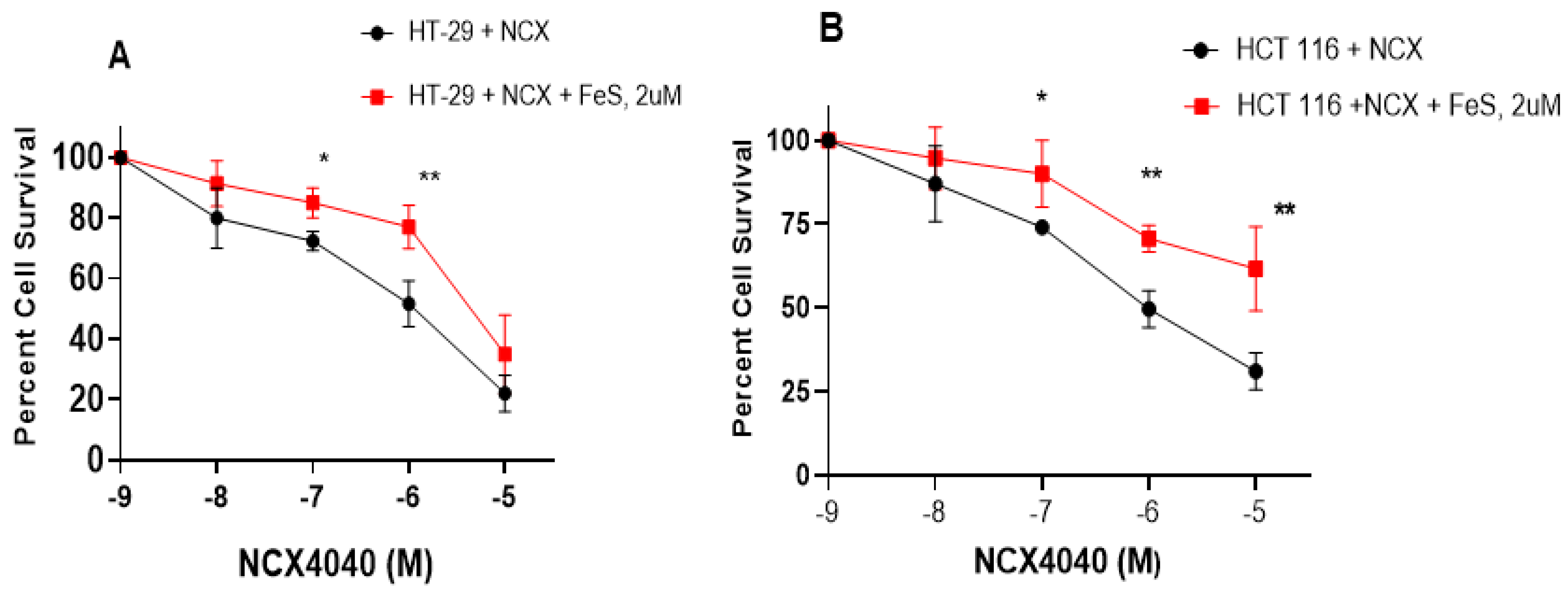
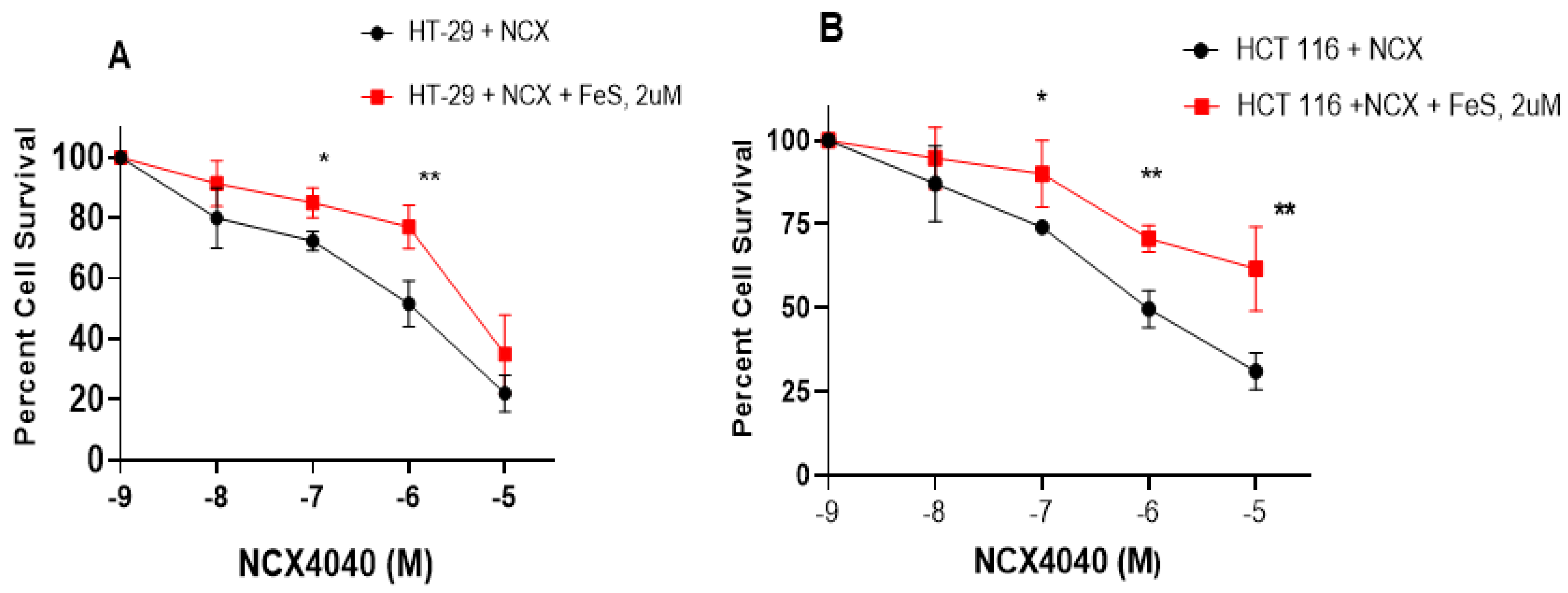
3.5. Ferrostatin-1 Inhibits NCX4040 Cytotoxicity in HT-29 and HCT 116 Cells
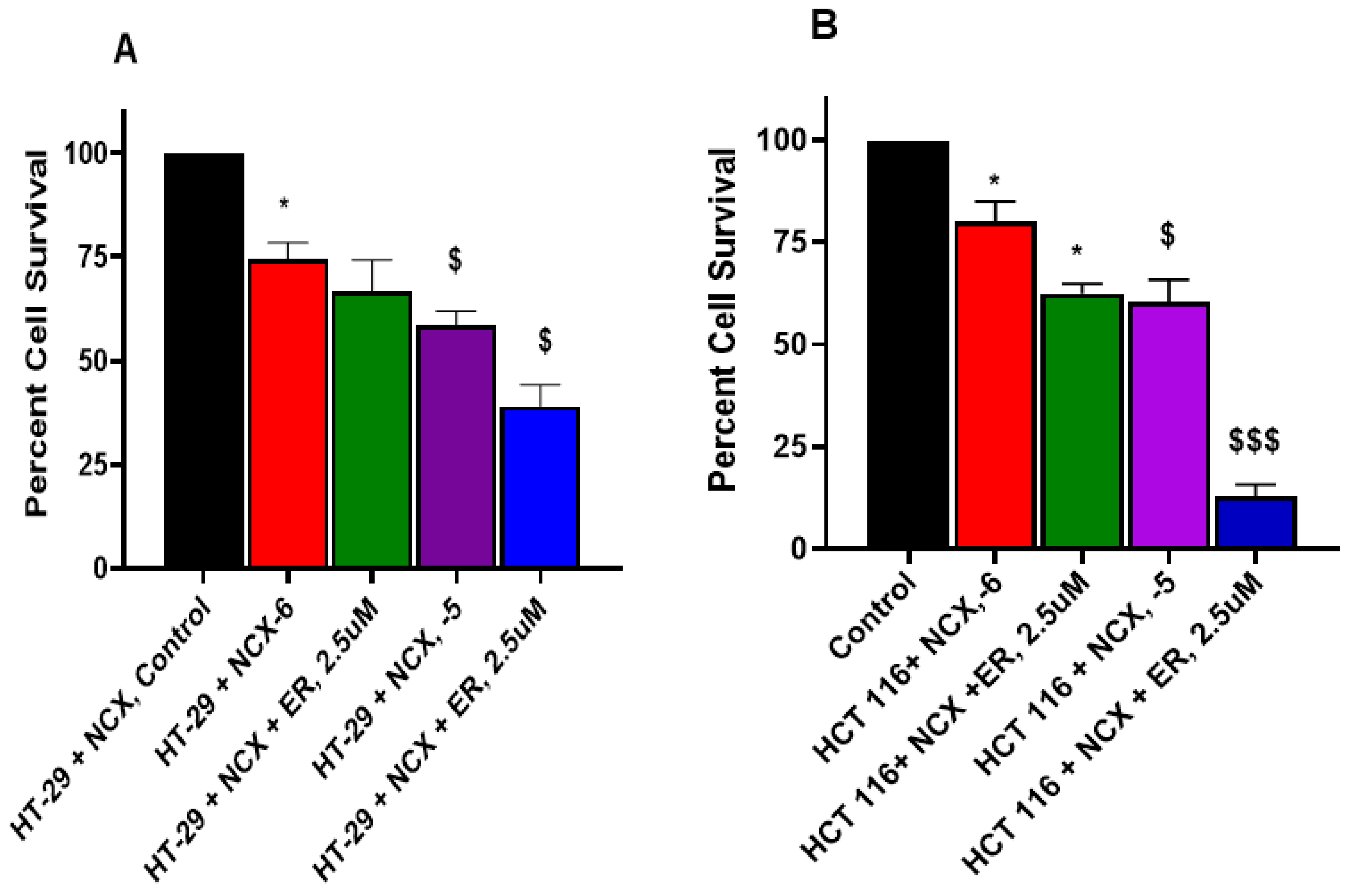
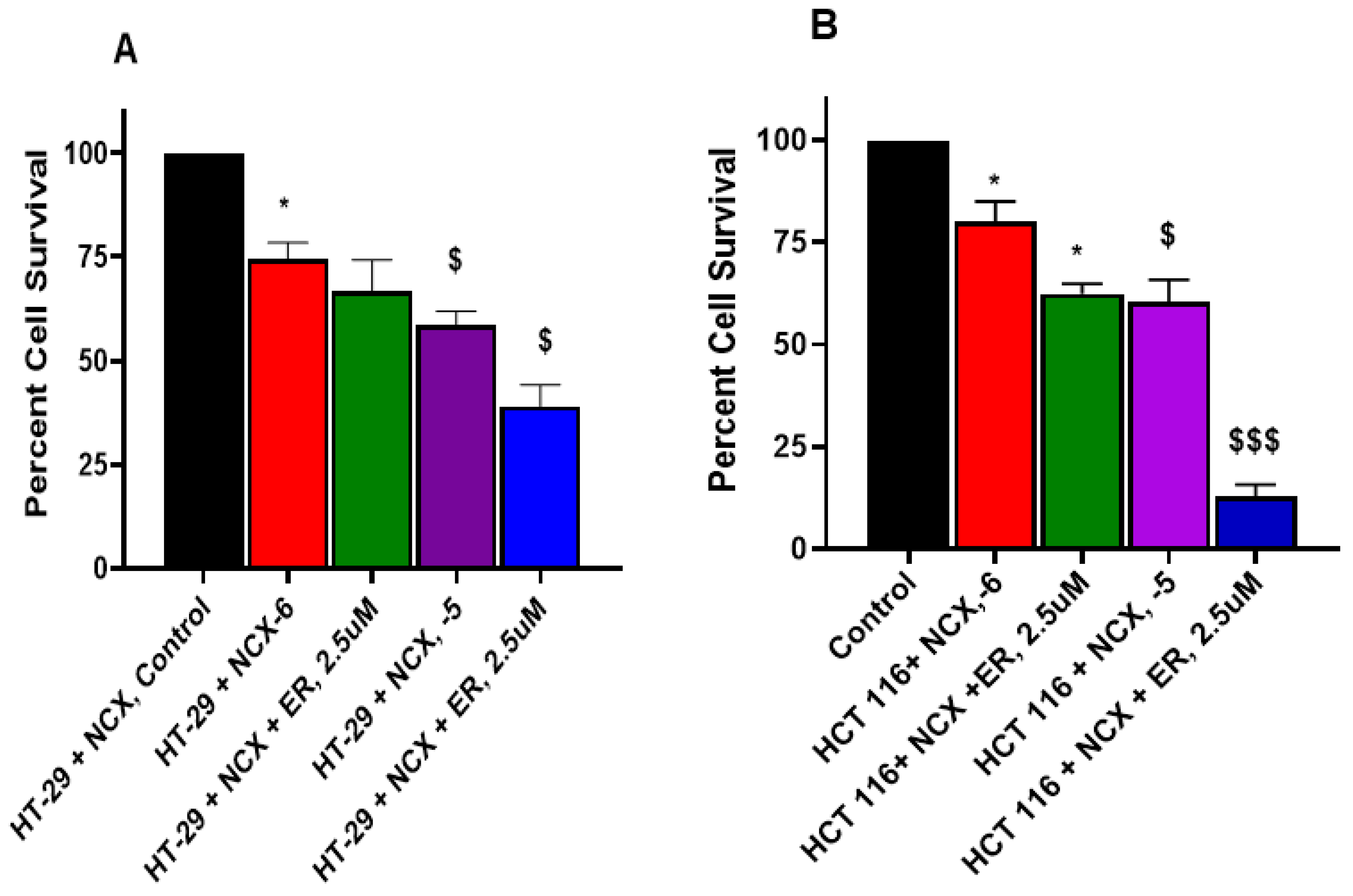
3.6. Erastin Enhances NCX4040 Cytotoxicity in HT-29 and HCT 116 Cells
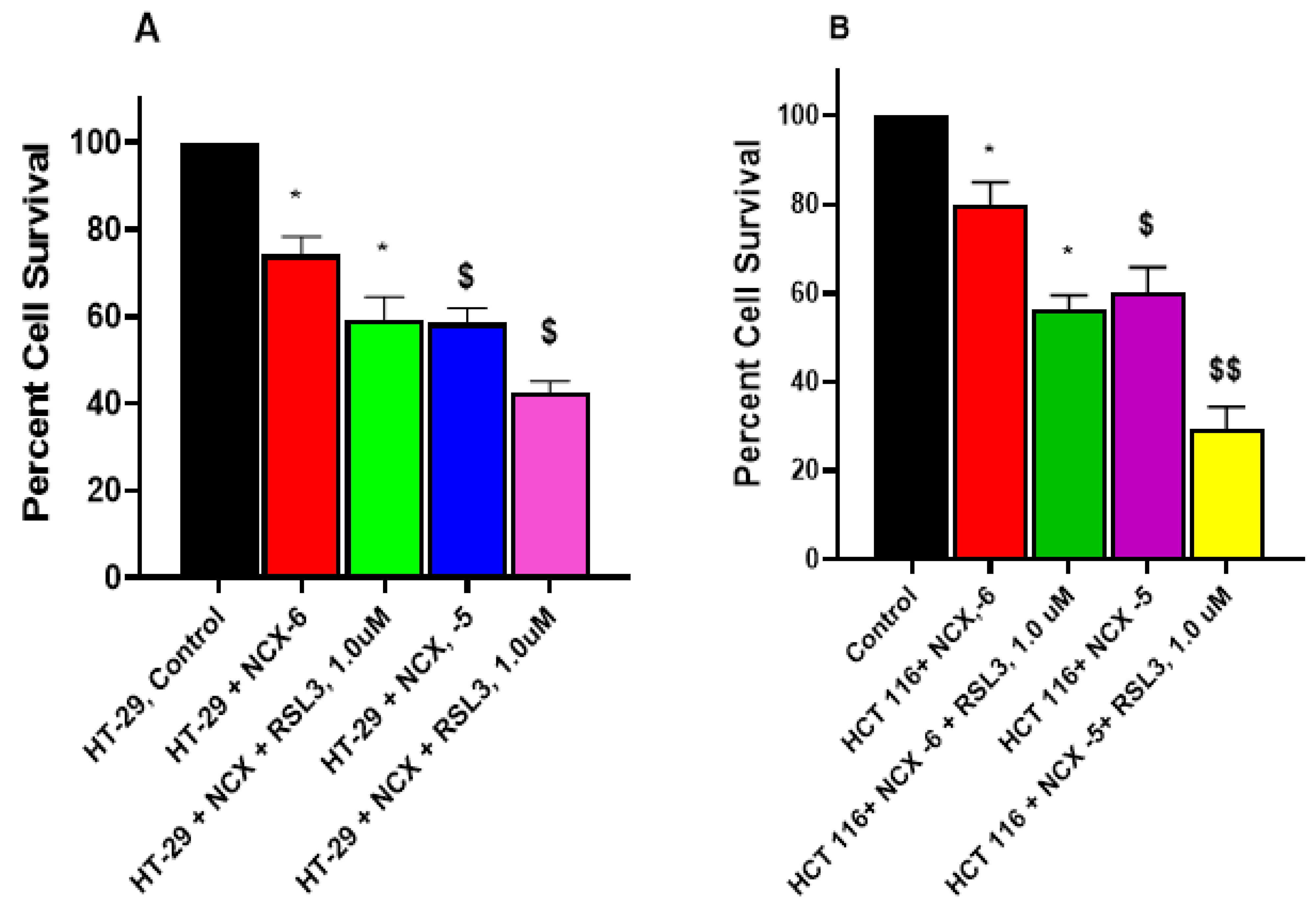
3.7. RSL3 Enhances NCX4040 Cytotoxicity in HT-29 and HCT 116 Cells
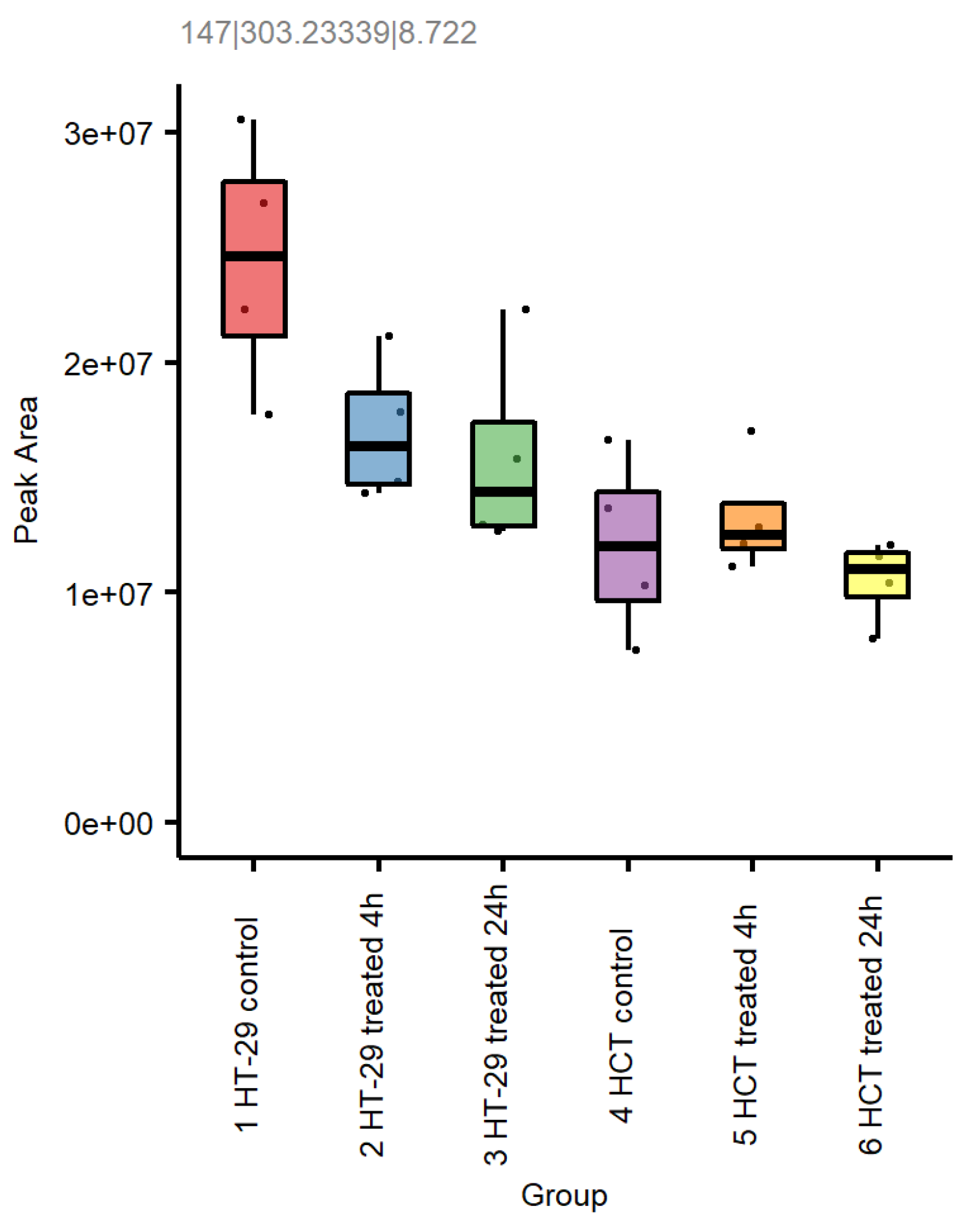
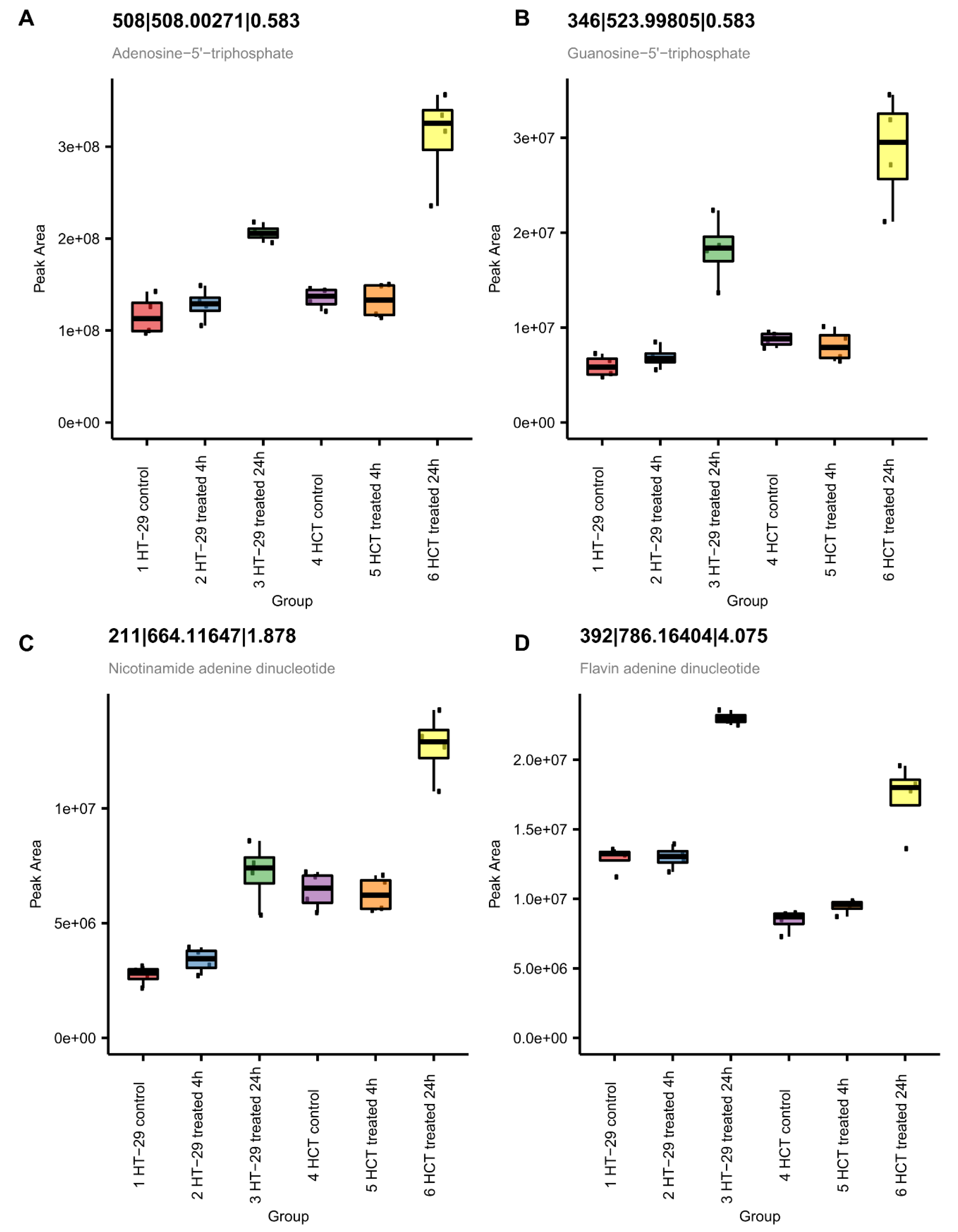
3.8. Metabolomic Studies in HT-29 and HCT 116 Cells
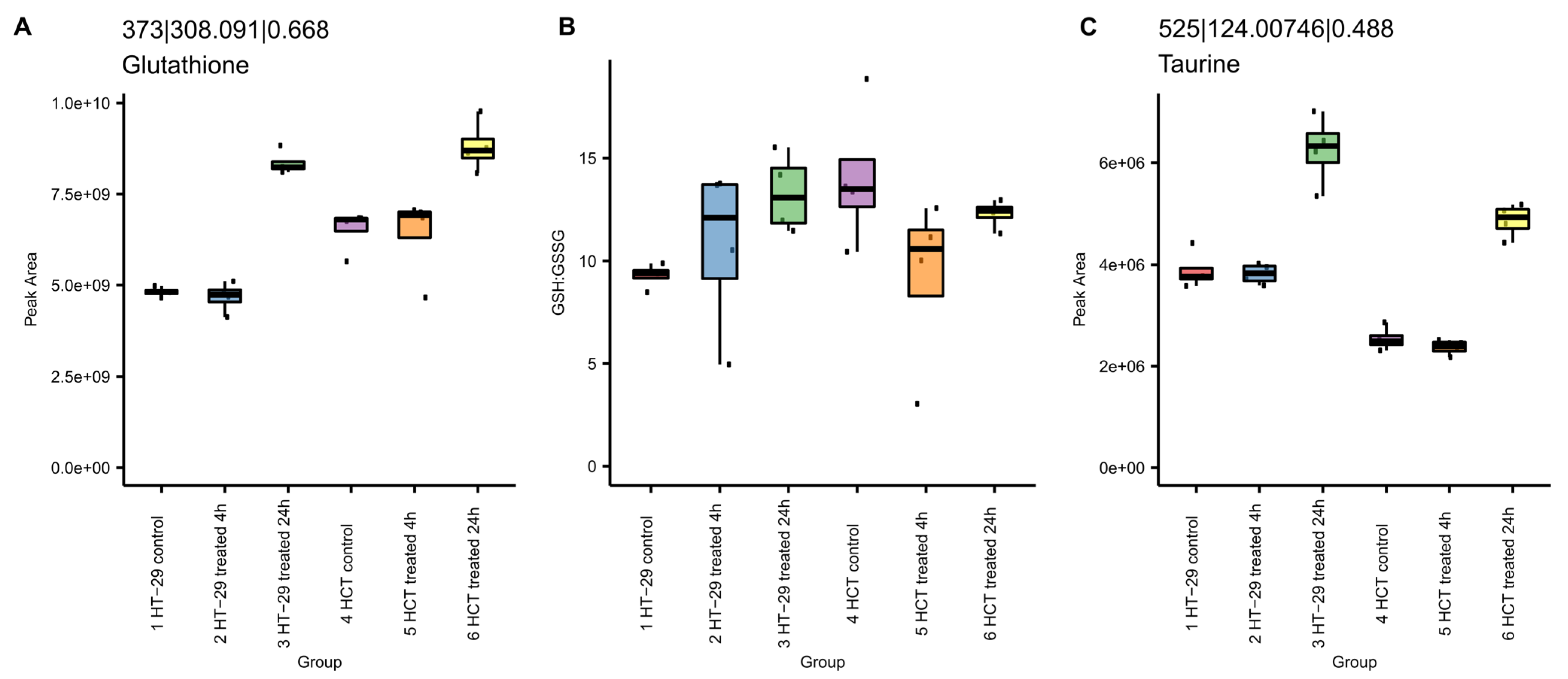
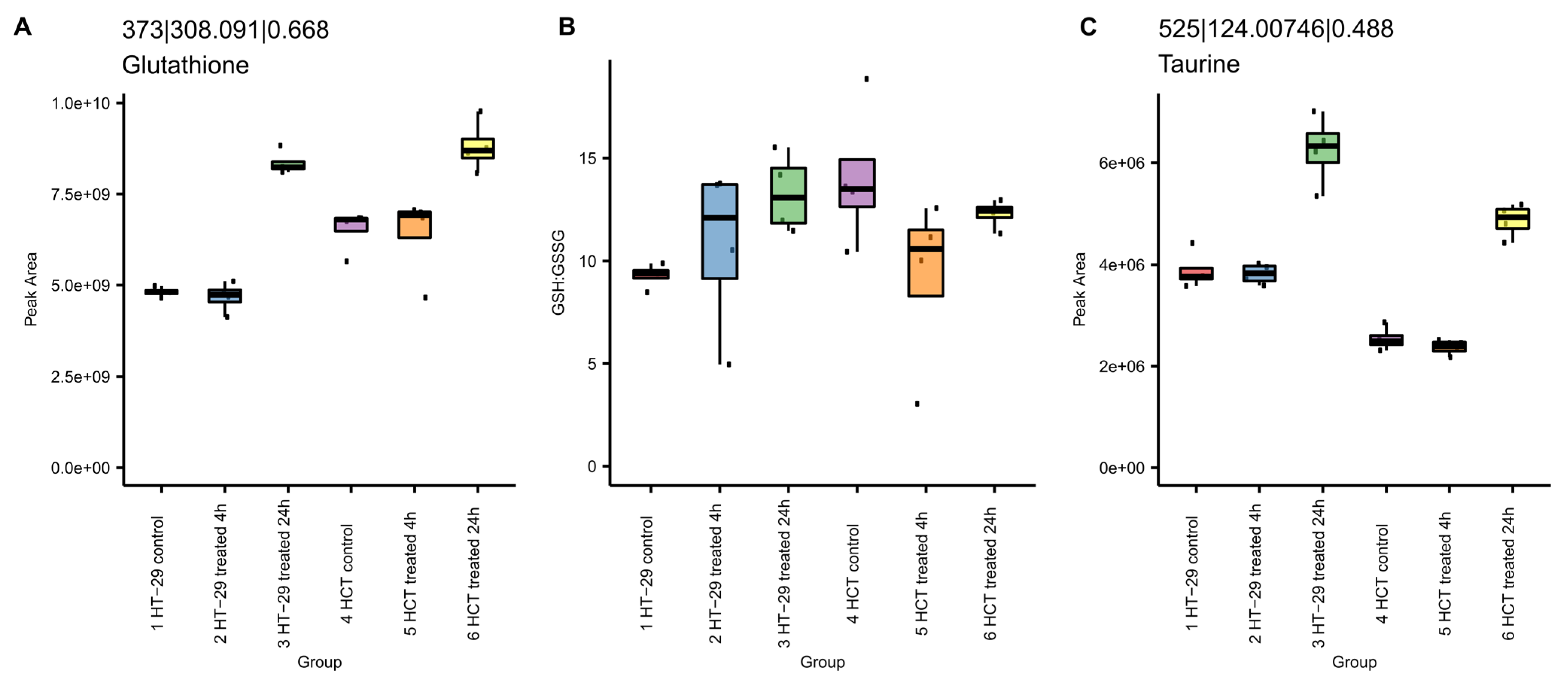
3.8.1. NCX4040 Increases Glutathione in HT-29 and HCT 116 Cells
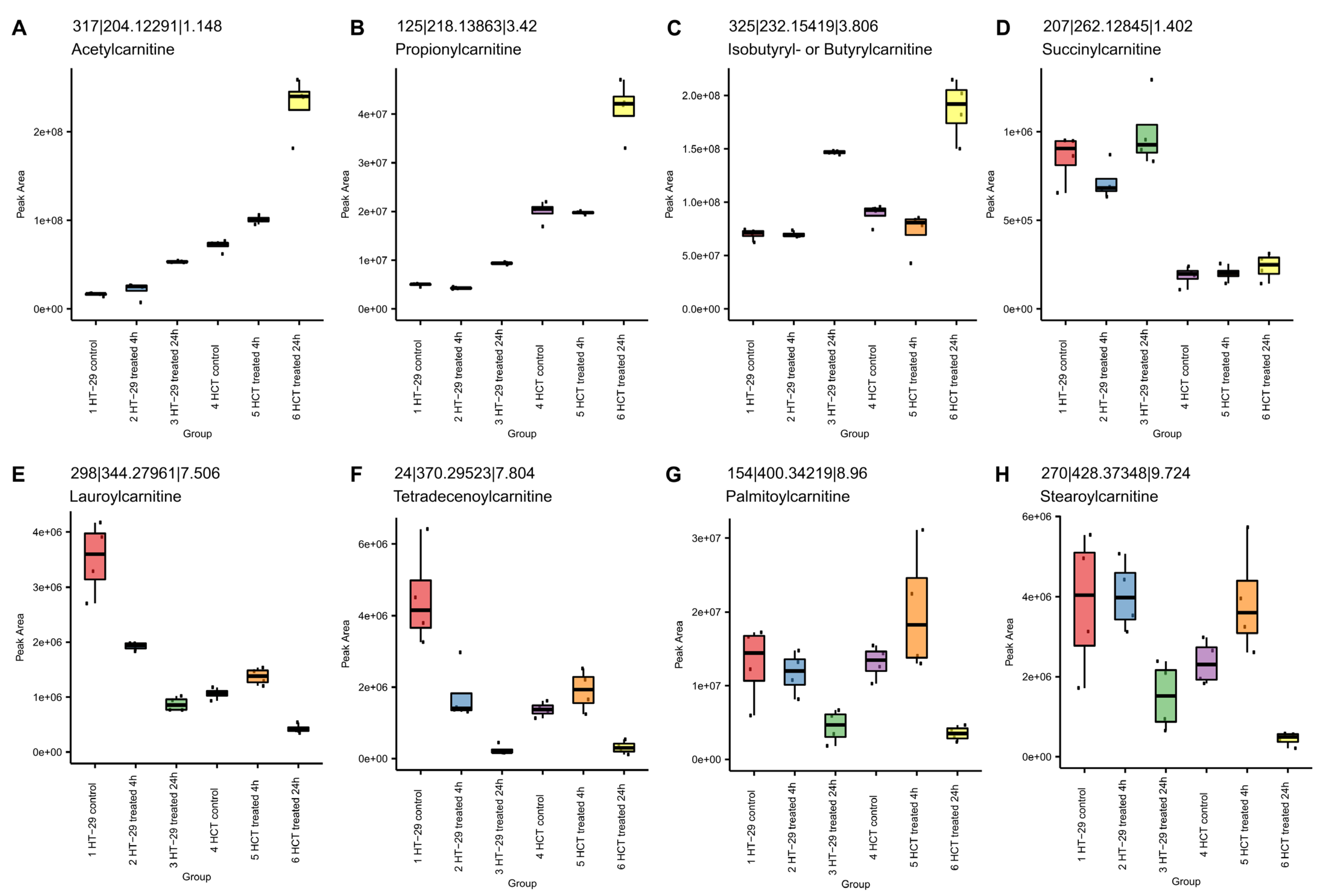
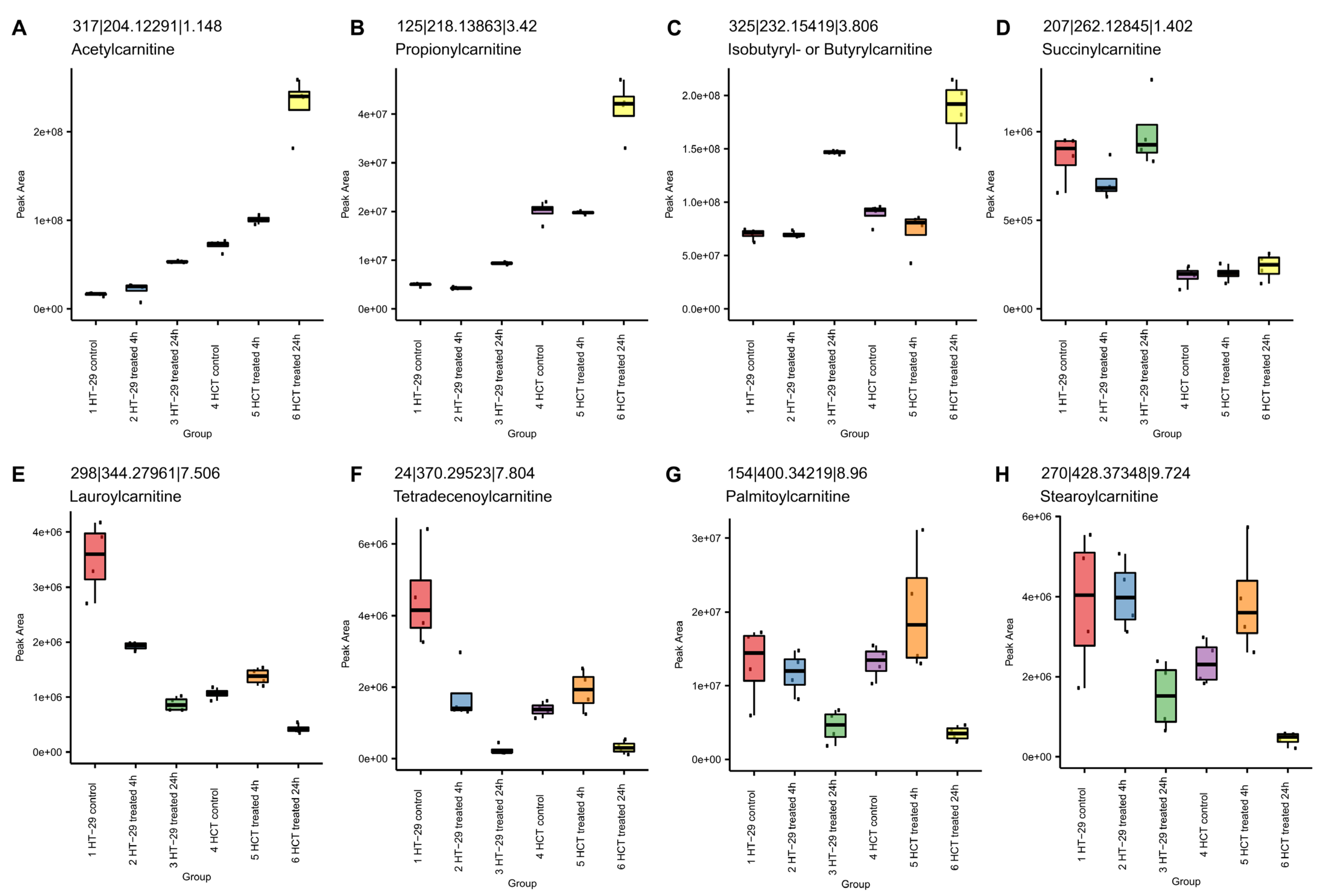
3.8.2. NCX4040 Enhances Lipid Metabolism in HT-29 and HCT116 Cells
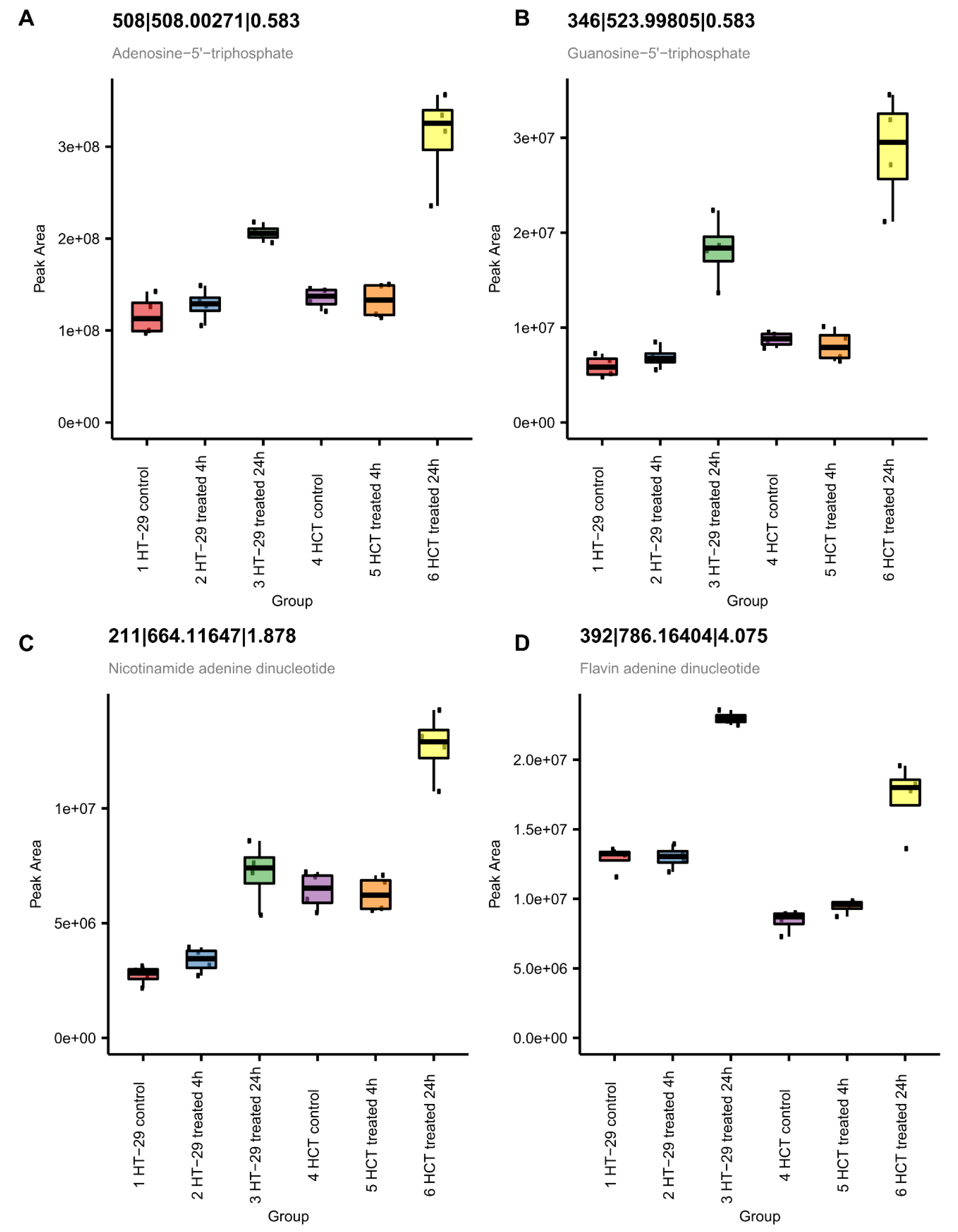
3.8.3. NCX4040 Enhances Energy Metabolism in HT-29 and HCT 116 Cells
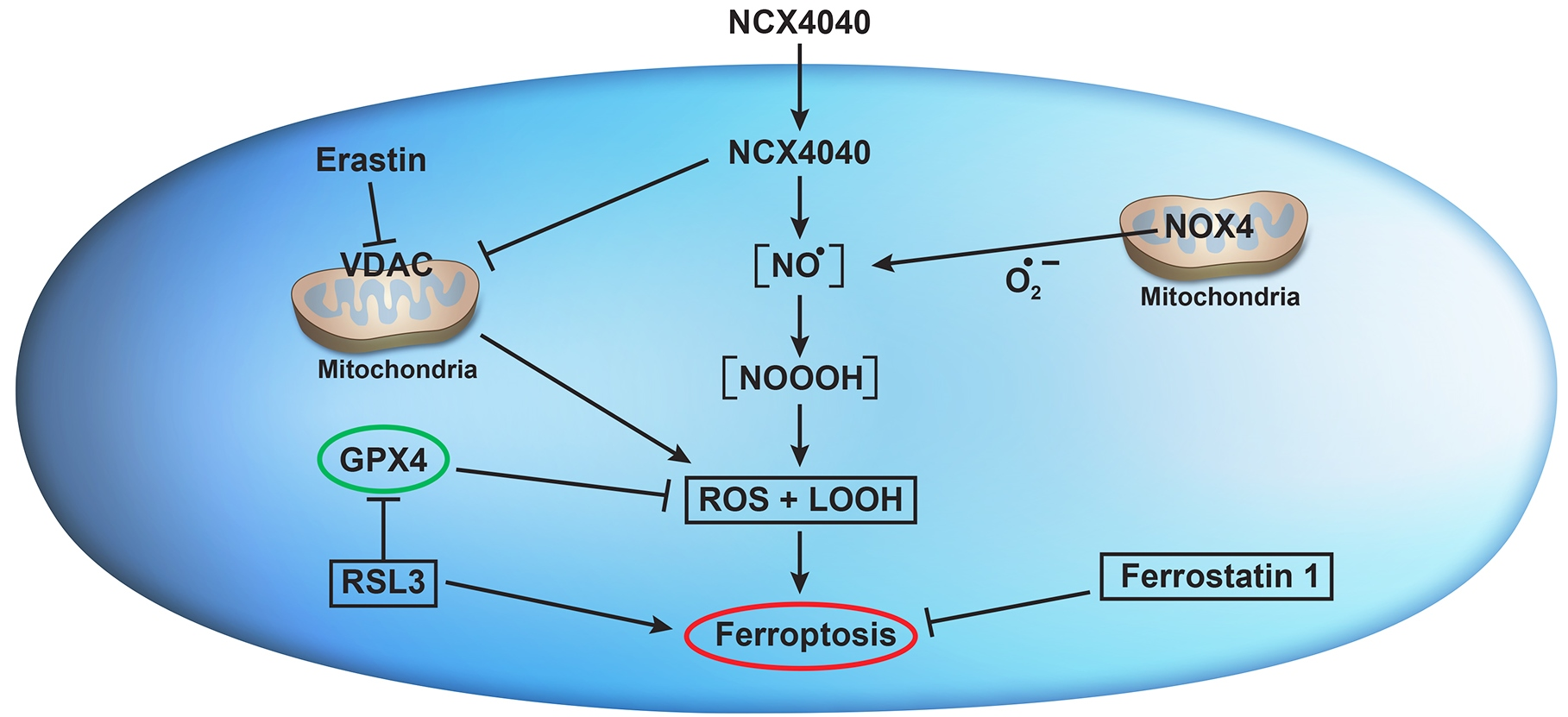
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Averboukh, F.; Ziv, Y.; Kariv, Y.; Zmora, O.; Dotan, I.; Klausner, J.M.; Rabau, M.; Tulchinsky, H. Colorectal carcinoma in inflammatory bowel disease: A comparison between Crohn’s and ulcerative colitis. Color. Dis. 2011, 13, 1230–1235. [Google Scholar] [CrossRef]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhang, Z.; Sun, W.; Zhang, J.; Xu, Q.; Zhou, X.; Mao, L. Ferroptosis in colorectal cancer: Potential mechanisms and effective therapeutic targets. Biomed. Pharm. 2022, 153, 113524. [Google Scholar] [CrossRef]
- Rigas, B.; Kashfi, K. Nitric-oxide-donating NSAIDs as agents for cancer prevention. Trends Mol. Med. 2004, 10, 324–330. [Google Scholar] [CrossRef]
- Sinha, B.K.; Tokar, E.J.; Bortner, C.D. Molecular Mechanisms of Cytotoxicity of NCX4040, the Non-Steroidal Anti-Inflammatory NO-Donor, in Human Ovarian Cancer Cells. Int. J. Mol. Sci. 2022, 23, 8611. [Google Scholar] [CrossRef]
- Gao, J.; Liu, X.; Rigas, B. Nitric oxide-donating aspirin induces apoptosis in human colon cancer cells through induction of oxidative stress. Proc. Natl. Acad. Sci. USA 2005, 102, 17207–17212. [Google Scholar] [CrossRef] [Green Version]
- Bratasz, A.; Selvendiran, K.; Wasowicz, T.; Bobko, A.; Khramtsov, V.V.; Ignarro, L.J.; Kuppusamy, P. NCX-4040, a nitric oxide-releasing aspirin, sensitizes drug-resistant human ovarian xenograft tumors to cisplatin by depletion of cellular thiols. J. Transl. Med. 2008, 6, 9. [Google Scholar] [CrossRef] [Green Version]
- Dunlap, T.; Chandrasena, R.E.; Wang, Z.; Sinha, V.; Wang, Z.; Thatcher, G.R. Quinone formation as a chemoprevention strategy for hybrid drugs: Balancing cytotoxicity and cytoprotection. Chem. Res. Toxicol. 2007, 20, 1903–1912. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, N.; Chen, C.; Xu, X.; Luo, A.; Yan, Y.; Lu, Y.; Liu, J.; Ou, X.; Tan, Y.; et al. Sevoflurane Induces Ferroptosis of Glioma Cells Through Activating the ATF4-CHAC1 Pathway. Front. Oncol. 2022, 12, 859621. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Patel, D.N.; Welsch, M.; Skouta, R.; Lee, E.D.; Hayano, M.; Thomas, A.G.; Gleason, C.E.; Tatonetti, N.P.; Slusher, B.S.; et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. eLife 2014, 3, e02523. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galaris, D.; Barbouti, A.; Pantopoulos, K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118535. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Zhang, R.; Liu, S.; Duan, T.; Zhai, L.; Zhang, M.; Han, X.; Xiang, Y.; Huang, X.; Lin, H.; et al. RSL3 Drives Ferroptosis Through GPX4 Inactivation and ROS Production in Colorectal Cancer. Front. Pharm. 2018, 9, 1371. [Google Scholar] [CrossRef] [Green Version]
- Tesei, A.; Zoli, W.; Fabbri, F.; Leonetti, C.; Rosetti, M.; Bolla, M.; Amadori, D.; Silvestrini, R. NCX 4040, an NO-donating acetylsalicylic acid derivative: Efficacy and mechanisms of action in cancer cells. Nitric Oxide 2008, 19, 225–236. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Delvaux, M.; Hague, P.; Craciun, L.; Wozniak, A.; Demetter, P.; Schoffski, P.; Erneux, C.; Vanderwinden, J.M. Ferroptosis Induction and YAP Inhibition as New Therapeutic Targets in Gastrointestinal Stromal Tumors (GISTs). Cancers 2022, 14, 5050. [Google Scholar] [CrossRef]
- Wang, D.; Tang, L.; Zhang, Y.; Ge, G.; Jiang, X.; Mo, Y.; Wu, P.; Deng, X.; Li, L.; Zuo, S.; et al. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis. 2022, 13, 544. [Google Scholar] [CrossRef] [PubMed]
- Mimnaugh, E.G.; Trush, M.A.; Gram, T.E. Stimulation by adriamycin of rat heart and liver microsomal NADPH-dependent lipid peroxidation. Biochem. Pharmacol. 1981, 30, 2797–2804. [Google Scholar] [CrossRef] [PubMed]
- Mimnaugh, E.G.; Kennedy, K.A.; Trush, M.A.; Sinha, B.K. Adriamycin-enhanced membrane lipid peroxidation in isolated rat nuclei. Cancer Res. 1985, 45, 3296–3304. [Google Scholar] [PubMed]
- Zielonka, J.; Kalyanaraman, B. Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: Another inconvenient truth. Free Radic. Biol. Med. 2010, 48, 983–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffman, M.E.; Kauffman, M.K.; Traore, K.; Zhu, H.; Trush, M.A.; Jia, Z.; Li, Y.R. MitoSOX-Based Flow Cytometry for Detecting Mitochondrial ROS. React. Oxyg. Species 2016, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Sinha, B.K.; Tokar, E.J.; Li, J.; Bushel, P.R. Gene Expression Profiling Elucidates Cellular Responses to NCX4040 in Human Ovarian Tumor Cells: Implications in the Mechanisms of Action of NCX4040. Cancers 2022, 15, 285. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, Y.; Zheng, T.; Yang, G.; Zhang, X.; Sun, Z.; Shi, C.; Zhao, S. Inhibition of heme oxygenase-1 enhances anti-cancer effects of arsenic trioxide on glioma cells. J. Neurooncol. 2011, 104, 449–458. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015, 2, e1054549. [Google Scholar] [CrossRef] [Green Version]
- Crawford, R.R.; Prescott, E.T.; Sylvester, C.F.; Higdon, A.N.; Shan, J.; Kilberg, M.S.; Mungrue, I.N. Human CHAC1 Protein Degrades Glutathione, and mRNA Induction Is Regulated by the Transcription Factors ATF4 and ATF3 and a Bipartite ATF/CRE Regulatory Element. J. Biol. Chem. 2015, 290, 15878–15891. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.; Liu, C.X.; Song, R.; Li, Q.L. Ferrostatin-1 protects HT-22 cells from oxidative toxicity. Neural Regen. Res. 2020, 15, 528–536. [Google Scholar] [CrossRef]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vuckovic, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef] [Green Version]
- Yagoda, N.; von Rechenberg, M.; Zaganjor, E.; Bauer, A.J.; Yang, W.S.; Fridman, D.J.; Wolpaw, A.J.; Smukste, I.; Peltier, J.M.; Boniface, J.J.; et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007, 447, 864–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.S.; Stockwell, B.R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem. Biol. 2008, 15, 234–245. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. OncoTargets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef] [PubMed]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-kappaB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxid. Med. Cell Longev. 2021, 2021, 2915019. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct. Target Ther. 2020, 5, 108. [Google Scholar] [CrossRef]
- Lee, J.Y.; Nam, M.; Son, H.Y.; Hyun, K.; Jang, S.Y.; Kim, J.W.; Kim, M.W.; Jung, Y.; Jang, E.; Yoon, S.J.; et al. Polyunsaturated fatty acid biosynthesis pathway determines ferroptosis sensitivity in gastric cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 32433–32442. [Google Scholar] [CrossRef]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Kang, R.; Kroemer, G.; Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 2019, 133, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Zheng, Y.; Wang, C.; Liu, Y. Glutathione depletion induces ferroptosis, autophagy, and premature cell senescence in retinal pigment epithelial cells. Cell Death Dis. 2018, 9, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Mo, J.; Dai, J.; Ye, C.; Cen, W.; Zheng, X.; Jiang, L.; Ye, L. Cetuximab promotes RSL3-induced ferroptosis by suppressing the Nrf2/HO-1 signalling pathway in KRAS mutant colorectal cancer. Cell Death Dis. 2021, 12, 1079. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.Y.; Dixon, S.J. Mechanisms of ferroptosis. Cell Mol. Life Sci. 2016, 73, 2195–2209. [Google Scholar] [CrossRef] [Green Version]
- Szwarc, M.M.; Guarnieri, A.L.; Joshi, M.; Duc, H.N.; Laird, M.C.; Pandey, A.; Khanal, S.; Dohm, E.; Bui, A.K.; Sullivan, K.D.; et al. FAM193A is a positive regulator of p53 activity. Cell Rep. 2023, 42, 112230. [Google Scholar] [CrossRef]
- Chen, M.S.; Wang, S.F.; Hsu, C.Y.; Yin, P.H.; Yeh, T.S.; Lee, H.C.; Tseng, L.M. CHAC1 degradation of glutathione enhances cystine-starvation-induced necroptosis and ferroptosis in human triple negative breast cancer cells via the GCN2-eIF2alpha-ATF4 pathway. Oncotarget 2017, 8, 114588–114602. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Zeng, G.Z.; Yin, J.L.; Bian, Z.X. Artesunate activates the ATF4-CHOP-CHAC1 pathway and affects ferroptosis in Burkitt’s Lymphoma. Biochem. Biophys. Res. Commun. 2019, 519, 533–539. [Google Scholar] [CrossRef]
- Xiao, R.; Wang, S.; Guo, J.; Liu, S.; Ding, A.; Wang, G.; Li, W.; Zhang, Y.; Bian, X.; Zhao, S.; et al. Ferroptosis-related gene NOX4, CHAC1 and HIF1A are valid biomarkers for stomach adenocarcinoma. J. Cell Mol. Med. 2022, 26, 1183–1193. [Google Scholar] [CrossRef]
- Iwakiri, Y. S-nitrosylation of proteins: A new insight into endothelial cell function regulated by eNOS-derived NO. Nitric Oxide 2011, 25, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Ruiz, A.; Araujo, I.M.; Izquierdo-Alvarez, A.; Hernansanz-Agustin, P.; Lamas, S.; Serrador, J.M. Specificity in S-nitrosylation: A short-range mechanism for NO signaling? Antioxid. Redox Signal. 2013, 19, 1220–1235. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.K.; Kumar, A.; Kumari, A.; Tokar, E.J.; Waalkes, M.P.; Bortner, C.D.; Williams, J.; Ehrenshaft, M.; Mason, R.P.; Sinha, B.K. Nitric Oxide Down-Regulates Topoisomerase I and Induces Camptothecin Resistance in Human Breast MCF-7 Tumor Cells. PLoS ONE 2015, 10, e0141897. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.; Chen, J.; Li, Y.; Li, Y.; Zhong, Y.; Li, G.; Liu, L.; Li, Y. Glabridin, a bioactive component of licorice, ameliorates diabetic nephropathy by regulating ferroptosis and the VEGF/Akt/ERK pathways. Mol. Med. 2022, 28, 58. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Xie, J.; Li, X.; Li, Y.; Thirupathi, A.; Zhang, J.; Yu, P.; Gao, G.; Chang, Y.; Shi, Z. Ferritinophagy-Mediated Ferroptosis Involved in Paraquat-Induced Neurotoxicity of Dopaminergic Neurons: Implication for Neurotoxicity in PD. Oxid. Med. Cell Longev. 2021, 2021, 9961628. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Gao, M.; Liang, J.; Chen, Y.; Wang, Y.; Wang, Y.; Xiao, Y.; Zhao, Z.; Wan, X.; Jiang, M.; et al. SLC7A11 Reduces Laser-Induced Choroidal Neovascularization by Inhibiting RPE Ferroptosis and VEGF Production. Front. Cell Dev. Biol. 2021, 9, 639851. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, Y.; Zhao, H. Identification of ferroptosis-related genes and predicted overall survival in patients with burns. Front. Surg. 2022, 9, 1060036. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, J.; Wang, Y.; Liu, Z.; Wu, Y. Ferroptosis drives photoreceptor degeneration in mice with defects in all-trans-retinal clearance. J. Biol. Chem. 2021, 296, 100187. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Krelin, Y.; Shteinfer-Kuzmine, A.; Arif, T. Voltage-Dependent Anion Channel 1 As an Emerging Drug Target for Novel Anti-Cancer Therapeutics. Front. Oncol. 2017, 7, 154. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Sedlic, F.; Pravdic, D.; Bosnjak, Z.J.; Kwok, W.M. Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channel. FEBS Lett. 2011, 585, 328–334. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, B.K.; Bortner, C.D.; Jarmusch, A.K.; Tokar, E.J.; Murphy, C.; Wu, X.; Winter, H.; Cannon, R.E. Ferroptosis-Mediated Cell Death Induced by NCX4040, The Non-Steroidal Nitric Oxide Donor, in Human Colorectal Cancer Cells: Implications in Therapy. Cells 2023, 12, 1626. https://doi.org/10.3390/cells12121626
Sinha BK, Bortner CD, Jarmusch AK, Tokar EJ, Murphy C, Wu X, Winter H, Cannon RE. Ferroptosis-Mediated Cell Death Induced by NCX4040, The Non-Steroidal Nitric Oxide Donor, in Human Colorectal Cancer Cells: Implications in Therapy. Cells. 2023; 12(12):1626. https://doi.org/10.3390/cells12121626
Chicago/Turabian StyleSinha, Birandra K., Carl D. Bortner, Alan K. Jarmusch, Erik J. Tokar, Carri Murphy, Xian Wu, Heather Winter, and Ronald E. Cannon. 2023. "Ferroptosis-Mediated Cell Death Induced by NCX4040, The Non-Steroidal Nitric Oxide Donor, in Human Colorectal Cancer Cells: Implications in Therapy" Cells 12, no. 12: 1626. https://doi.org/10.3390/cells12121626
APA StyleSinha, B. K., Bortner, C. D., Jarmusch, A. K., Tokar, E. J., Murphy, C., Wu, X., Winter, H., & Cannon, R. E. (2023). Ferroptosis-Mediated Cell Death Induced by NCX4040, The Non-Steroidal Nitric Oxide Donor, in Human Colorectal Cancer Cells: Implications in Therapy. Cells, 12(12), 1626. https://doi.org/10.3390/cells12121626