
Molecular Mechanisms of Lipid-Based Metabolic Adaptation Strategies in Response to Cold


{kind=link}
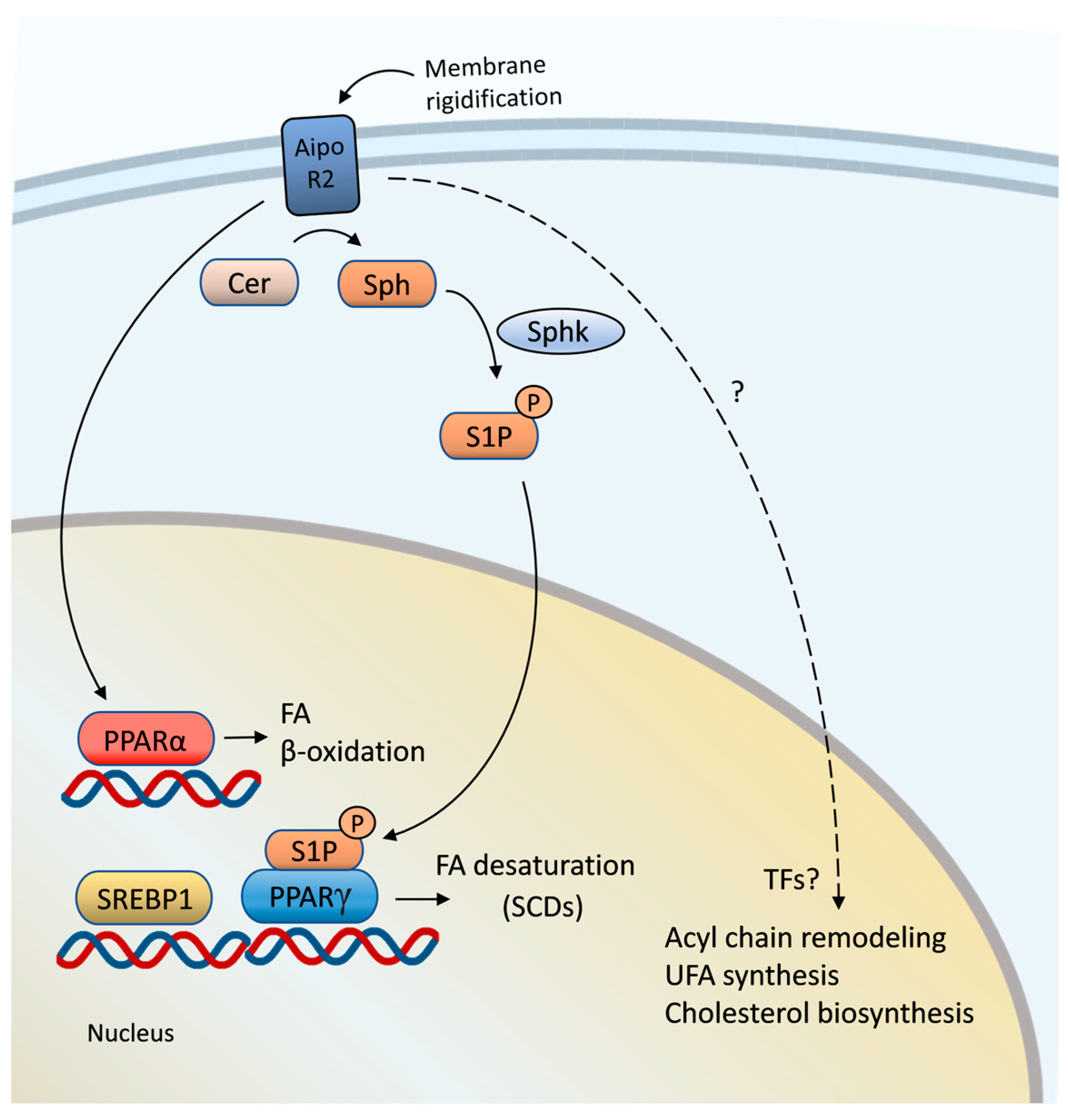{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Regulation of Membrane Fluidity in Poikilothermic and Cold-Adapted Organisms
3. Sensing Membrane Rigidification Is Essential for Membrane Fluidity Maintenance in Cold Adaptation

4. Mammalian Adiponectin Receptors Signal to Downstream Lipid Regulators
5. Lipid Bilayer Stress in the Endoplasmic Reticulum Induces the Unfolded Protein Response
6. PPARα Regulates Lipid and Ketone Metabolism in Heterothermic Hibernating Animals
7. Mitochondrial Function in Thermogenesis
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, D.G. Mitochondrial proton leaks and uncoupling proteins. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148428. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.J.; Squire, T.L.; Andrews, M.T. Coordinate expression of the PDK4 gene: A means of regulating fuel selection in a hibernating mammal. Physiol. Genom. 2002, 8, 5–13. [Google Scholar] [CrossRef]
- Wijenayake, S.; Tessier, S.N.; Storey, K.B. Regulation of pyruvate dehydrogenase (PDH) in the hibernating ground squirrel, (Ictidomys tridecemlineatus). J. Therm. Biol. 2017, 69, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Guschina, I.A.; Harwood, J.L. Mechanisms of temperature adaptation in poikilotherms. FEBS Lett. 2006, 580, 5477–5483. [Google Scholar] [CrossRef]
- Hayward, S.A.; Murray, P.A.; Gracey, A.Y.; Cossins, A.R. Beyond the lipid hypothesis: Mechanisms underlying phenotypic plasticity in inducible cold tolerance. Adv. Exp. Med. Biol. 2007, 594, 132–142. [Google Scholar] [CrossRef]
- Hayward, S.A.; Manso, B.; Cossins, A.R. Molecular basis of chill resistance adaptations in poikilothermic animals. J. Exp. Biol. 2014, 217, 6–15. [Google Scholar] [CrossRef]
- Sinensky, M. Homeoviscous adaptation—A homeostatic process that regulates the viscosity of membrane lipids in Escherichia coli. Proc. Natl. Acad. Sci. USA 1974, 71, 522–525. [Google Scholar] [CrossRef]
- Kostal, V. Cell structural modifications in insects at low temperatures. In Low Temperature Biology of Insects; Denlinger, D., Lee, R., Jr., Eds.; Cambridge University Press: Cambridge, UK, 2010; pp. 116–140. [Google Scholar]
- Chintalapati, S.; Kiran, M.D.; Shivaji, S. Role of membrane lipid fatty acids in cold adaptation. Cell Mol. Biol. 2004, 50, 631–642. [Google Scholar] [PubMed]
- Zhang, Y.M.; Rock, C.O. Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. [Google Scholar] [CrossRef]
- Cronan, J.E., Jr.; Gelmann, E.P. Physical properties of membrane lipids: Biological relevance and regulation. Bacteriol. Rev. 1975, 39, 232–256. [Google Scholar] [CrossRef] [PubMed]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Szekely, P.; Dvir, T.; Asor, R.; Resh, R.; Steiner, A.; Szekely, O.; Ginsburg, A.; Mosenkis, J.; Guralnick, V.; Dan, Y.; et al. Effect of temperature on the structure of charged membranes. J. Phys. Chem. B 2011, 115, 14501–14506. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Tristram-Nagle, S.; Kucerka, N.; Nagle, J.F. Temperature dependence of structure, bending rigidity, and bilayer interactions of dioleoylphosphatidylcholine bilayers. Biophys. J. 2008, 94, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Leonenko, Z.V.; Finot, E.; Ma, H.; Dahms, T.E.; Cramb, D.T. Investigation of temperature-induced phase transitions in DOPC and DPPC phospholipid bilayers using temperature-controlled scanning force microscopy. Biophys. J. 2004, 86, 3783–3793. [Google Scholar] [CrossRef]
- Nakamura, M.T.; Nara, T.Y. Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturases. Annu. Rev. Nutr. 2004, 24, 345–376. [Google Scholar] [CrossRef]
- Morita, R.Y. Psychrophilic bacteria. Bacteriol. Rev. 1975, 39, 144–167. [Google Scholar] [CrossRef]
- De Maayer, P.; Anderson, D.; Cary, C.; Cowan, D.A. Some like it cold: Understanding the survival strategies of psychrophiles. EMBO Rep. 2014, 15, 508–517. [Google Scholar] [CrossRef]
- Macdonald, P.M.; McDonough, B.; Sykes, B.D.; McElhaney, R.N. Fluorine-19 nuclear magnetic resonance studies of lipid fatty acyl chain order and dynamics in Acholeplasma laidlawii B membranes. Effects of methyl-branch substitution and of trans unsaturation upon membrane acyl-chain orientational order. Biochemistry 1983, 22, 5103–5111. [Google Scholar] [CrossRef] [PubMed]
- Roach, C.; Feller, S.E.; Ward, J.A.; Shaikh, S.R.; Zerouga, M.; Stillwell, W. Comparison of cis and trans fatty acid containing phosphatidylcholines on membrane properties. Biochemistry 2004, 43, 6344–6351. [Google Scholar] [CrossRef]
- Holtwick, R.; Meinhardt, F.; Keweloh, H. cis-trans isomerization of unsaturated fatty acids: Cloning and sequencing of the cti gene from Pseudomonas putida P8. Appl. Environ. Microbiol. 1997, 63, 4292–4297. [Google Scholar] [CrossRef] [PubMed]
- Eberlein, C.; Baumgarten, T.; Starke, S.; Heipieper, H.J. Immediate response mechanisms of Gram-negative solvent-tolerant bacteria to cope with environmental stress: Cis-trans isomerization of unsaturated fatty acids and outer membrane vesicle secretion. Appl. Microbiol. Biotechnol. 2018, 102, 2583–2593. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Shibahara, A.; Yamamoto, K.; Shinkai, K.; Kajimoto, G.; Okuyama, H. Evidence for cis-trans isomerization of a double bond in the fatty acids of the psychrophilic bacterium Vibrio sp. strain ABE-1. J. Bacteriol. 1993, 175, 916–918. [Google Scholar] [CrossRef]
- Erimban, S.; Daschakraborty, S. Cryostabilization of the Cell Membrane of a Psychrotolerant Bacteria via Homeoviscous Adaptation. J. Phys. Chem. Lett. 2020, 11, 7709–7716. [Google Scholar] [CrossRef] [PubMed]
- Klose, C.; Surma, M.A.; Gerl, M.J.; Meyenhofer, F.; Shevchenko, A.; Simons, K. Flexibility of a eukaryotic lipidome—Insights from yeast lipidomics. PLoS ONE 2012, 7, e350632012. [Google Scholar] [CrossRef] [PubMed]
- Chwastek, G.; Surma, M.A.; Rizk, S.; Grosser, D.; Lavrynenko, O.; Rucinska, M.; Jambor, H.; Saenz, J. Principles of Membrane Adaptation Revealed through Environmentally Induced Bacterial Lipidome Remodeling. Cell Rep. 2020, 32, 108165. [Google Scholar] [CrossRef]
- Bogdanov, M.; Pyrshev, K.; Yesylevskyy, S.; Ryabichko, S.; Boiko, V.; Ivanchenko, P.; Kiyamova, R.; Guan, Z.; Ramseyer, C.; Dowhan, W. Phospholipid distribution in the cytoplasmic membrane of Gram-negative bacteria is highly asymmetric, dynamic, and cell shape-dependent. Sci. Adv. 2020, 6, eaaz6333. [Google Scholar] [CrossRef] [PubMed]
- Vardhan Reddy, P.V.; Shiva Nageswara Rao, S.S.; Pratibha, M.S.; Sailaja, B.; Kavya, B.; Manorama, R.R.; Singh, S.M.; Radha Srinivas, T.N.; Shivaji, S. Bacterial diversity and bioprospecting for cold-active enzymes from culturable bacteria associated with sediment from a melt water stream of Midtre Lovenbreen glacier, an Arctic glacier. Res. Microbiol. 2009, 160, 538–546. [Google Scholar] [CrossRef]
- Czajka, J.J.; Abernathy, M.H.; Benites, V.T.; Baidoo, E.E.K.; Deming, J.W.; Tang, Y.J. Model metabolic strategy for heterotrophic bacteria in the cold ocean based on Colwellia psychrerythraea 34H. Proc. Natl. Acad. Sci. USA 2018, 115, 12507–12512. [Google Scholar] [CrossRef]
- Francais, M.; Bott, R.; Dargaignaratz, C.; Ginies, C.; Carlin, F.; Broussolle, V.; Nguyen-The, C. Short-Chain and Unsaturated Fatty Acids Increase Sequentially From the Lag Phase During Cold Growth of Bacillus cereus. Front. Microbiol. 2021, 12, 694757. [Google Scholar] [CrossRef]
- Denich, T.J.; Beaudette, L.A.; Lee, H.; Trevors, J.T. Effect of selected environmental and physico-chemical factors on bacterial cytoplasmic membranes. J. Microbiol. Methods 2003, 52, 149–182. [Google Scholar] [CrossRef] [PubMed]
- Ernst, R.; Ejsing, C.S.; Antonny, B. Homeoviscous Adaptation and the Regulation of Membrane Lipids. J. Mol. Biol. 2016, 428, 4776–4791. [Google Scholar] [CrossRef] [PubMed]
- Suutari, M.; Laakso, S. Microbial fatty acids and thermal adaptation. Crit. Rev. Microbiol. 1994, 20, 285–328. [Google Scholar] [CrossRef]
- Annous, B.A.; Becker, L.A.; Bayles, D.O.; Labeda, D.P.; Wilkinson, B.J. Critical role of anteiso-C15:0 fatty acid in the growth of Listeria monocytogenes at low temperatures. Appl. Environ. Microbiol. 1997, 63, 3887–3894. [Google Scholar] [CrossRef]
- Zhu, K.; Bayles, D.O.; Xiong, A.; Jayaswal, R.K.; Wilkinson, B.J. Precursor and temperature modulation of fatty acid composition and growth of Listeria monocytogenes cold-sensitive mutants with transposon-interrupted branched-chain alpha-keto acid dehydrogenase. Microbiology 2005, 151, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Saunders, L.P.; Sen, S.; Wilkinson, B.J.; Gatto, C. Insights into the Mechanism of Homeoviscous Adaptation to Low Temperature in Branched-Chain Fatty Acid-Containing Bacteria through Modeling FabH Kinetics from the Foodborne Pathogen Listeria monocytogenes. Front. Microbiol. 2016, 7, 1386. [Google Scholar] [CrossRef] [PubMed]
- Siliakus, M.F.; van der Oost, J.; Kengen, S.W.M. Adaptations of archaeal and bacterial membranes to variations in temperature, pH and pressure. Extremophiles 2017, 21, 651–670. [Google Scholar] [CrossRef]
- Dalluge, J.J.; Connell, L.B. On the potential of mass spectrometry-based metabolite profiling approaches to the study of biochemical adaptation in psychrophilic yeast. Extremophiles 2013, 17, 953–961. [Google Scholar] [CrossRef]
- Lands, W.E. Metabolism of glycerolipides; a comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [CrossRef]
- Colinet, H.; Renault, D.; Javal, M.; Berkova, P.; Simek, P.; Kostal, V. Uncovering the benefits of fluctuating thermal regimes on cold tolerance of drosophila flies by combined metabolomic and lipidomic approach. Biochim. Biophys. Acta 2016, 1861, 1736–1745. [Google Scholar] [CrossRef]
- Kostal, V.; Urban, T.; Rimnacova, L.; Berkova, P.; Simek, P. Seasonal changes in minor membrane phospholipid classes, sterols and tocopherols in overwintering insect, Pyrrhocoris apterus. J. Insect Physiol. 2013, 59, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Enriquez, T.; Colinet, H. Cold acclimation triggers lipidomic and metabolic adjustments in the spotted wing drosophila Drosophila suzukii (Matsumara). Am. J. Physiol. Regul. Integr. Comp. Physiol. 2019, 316, R751–R763. [Google Scholar] [CrossRef]
- Suzuki, I.; Los, D.A.; Kanesaki, Y.; Mikami, K.; Murata, N. The pathway for perception and transduction of low-temperature signals in Synechocystis. EMBO J. 2000, 19, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Mansilla, M.C.; Aguilar, P.S.; Albanesi, D.; Cybulski, L.E.; Altabe, S.; de Mendoza, D. Regulation of fatty acid desaturation in Bacillus subtilis. Prostaglandins Leukot. Essent. Fat. Acids 2003, 68, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Sinetova, M.A.; Los, D.A. New insights in cyanobacterial cold stress responses: Genes, sensors, and molecular triggers. Biochim. Biophys. Acta 2016, 1860, 2391–2403. [Google Scholar] [CrossRef] [PubMed]
- de Mendoza, D. Temperature sensing by membranes. Annu. Rev. Microbiol. 2014, 68, 101–116. [Google Scholar] [CrossRef] [PubMed]
- Saita, E.; Albanesi, D.; de Mendoza, D. Sensing membrane thickness: Lessons learned from cold stress. Biochim. Biophys. Acta 2016, 1861, 837–846. [Google Scholar] [CrossRef]
- Cybulski, L.E.; del Solar, G.; Craig, P.O.; Espinosa, M.; de Mendoza, D. Bacillus subtilis DesR functions as a phosphorylation-activated switch to control membrane lipid fluidity. J. Biol. Chem. 2004, 279, 39340–39347. [Google Scholar] [CrossRef]
- Albanesi, D.; Mansilla, M.C.; de Mendoza, D. The membrane fluidity sensor DesK of Bacillus subtilis controls the signal decay of its cognate response regulator. J. Bacteriol. 2004, 186, 2655–2663. [Google Scholar] [CrossRef]
- Aguilar, P.S.; Cronan, J.E., Jr.; de Mendoza, D. A Bacillus subtilis gene induced by cold shock encodes a membrane phospholipid desaturase. J. Bacteriol. 1998, 180, 2194–2200. [Google Scholar] [CrossRef]
- Cybulski, L.E.; Martin, M.; Mansilla, M.C.; Fernandez, A.; de Mendoza, D. Membrane thickness cue for cold sensing in a bacterium. Curr. Biol. 2010, 20, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; de Mendoza, D. Regulation of Bacillus subtilis DesK thermosensor by lipids. Biochem. J. 2013, 451, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Porrini, L.; Cybulski, L.E.; Altabe, S.G.; Mansilla, M.C.; de Mendoza, D. Cerulenin inhibits unsaturated fatty acids synthesis in Bacillus subtilis by modifying the input signal of DesK thermosensor. Microbiologyopen 2014, 3, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Abriata, L.A.; Albanesi, D.; Dal Peraro, M.; de Mendoza, D. Signal Sensing and Transduction by Histidine Kinases as Unveiled through Studies on a Temperature Sensor. Acc. Chem. Res. 2017, 50, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Olsen, L.; Morck, C.; Brackmann, C.; Enejder, A.; Faergeman, N.J.; Pilon, M. The adiponectin receptor homologs in C. elegans promote energy utilization and homeostasis. PLoS ONE 2011, 6, e21343. [Google Scholar] [CrossRef] [PubMed]
- Svensk, E.; Stahlman, M.; Andersson, C.H.; Johansson, M.; Boren, J.; Pilon, M. PAQR-2 regulates fatty acid desaturation during cold adaptation in C. elegans. PLoS Genet. 2013, 9, e1003801. [Google Scholar] [CrossRef]
- Taubert, S.; Van Gilst, M.R.; Hansen, M.; Yamamoto, K.R. A Mediator subunit, MDT-15, integrates regulation of fatty acid metabolism by NHR-49-dependent and -independent pathways in C. elegans. Genes Dev. 2006, 20, 1137–1149. [Google Scholar] [CrossRef]
- Yang, F.; Vought, B.W.; Satterlee, J.S.; Walker, A.K.; Jim Sun, Z.Y.; Watts, J.L.; DeBeaumont, R.; Saito, R.M.; Hyberts, S.G.; Yang, S.; et al. An ARC/Mediator subunit required for SREBP control of cholesterol and lipid homeostasis. Nature 2006, 442, 700–704. [Google Scholar] [CrossRef]
- Walker, A.K.; Jacobs, R.L.; Watts, J.L.; Rottiers, V.; Jiang, K.; Finnegan, D.M.; Shioda, T.; Hansen, M.; Yang, F.; Niebergall, L.J.; et al. A conserved SREBP-1/phosphatidylcholine feedback circuit regulates lipogenesis in metazoans. Cell 2011, 147, 840–852. [Google Scholar] [CrossRef]
- Svensk, E.; Devkota, R.; Stahlman, M.; Ranji, P.; Rauthan, M.; Magnusson, F.; Hammarsten, S.; Johansson, M.; Boren, J.; Pilon, M. Caenorhabditis elegans PAQR-2 and IGLR-2 Protect against Glucose Toxicity by Modulating Membrane Lipid Composition. PLoS Genet. 2016, 12, e1005982. [Google Scholar] [CrossRef]
- Devkota, R.; Henricsson, M.; Boren, J.; Pilon, M. The C. elegans PAQR-2 and IGLR-2 membrane homeostasis proteins are uniquely essential for tolerating dietary saturated fats. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158883. [Google Scholar] [CrossRef] [PubMed]
- Busayavalasa, K.; Ruiz, M.; Devkota, R.; Stahlman, M.; Bodhicharla, R.; Svensk, E.; Hermansson, N.O.; Boren, J.; Pilon, M. Leveraging a gain-of-function allele of Caenorhabditis elegans paqr-1 to elucidate membrane homeostasis by PAQR proteins. PLoS Genet. 2020, 16, e1008975. [Google Scholar] [CrossRef] [PubMed]
- Tanabe, H.; Fujii, Y.; Okada-Iwabu, M.; Iwabu, M.; Nakamura, Y.; Hosaka, T.; Motoyama, K.; Ikeda, M.; Wakiyama, M.; Terada, T.; et al. Crystal structures of the human adiponectin receptors. Nature 2015, 520, 312–316. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef] [PubMed]
- Villa, N.Y.; Kupchak, B.R.; Garitaonandia, I.; Smith, J.L.; Alonso, E.; Alford, C.; Cowart, L.A.; Hannun, Y.A.; Lyons, T.J. Sphingolipids function as downstream effectors of a fungal PAQR. Mol. Pharmacol. 2009, 75, 866–875. [Google Scholar] [CrossRef]
- Devkota, R.; Svensk, E.; Ruiz, M.; Stahlman, M.; Boren, J.; Pilon, M. The adiponectin receptor AdipoR2 and its Caenorhabditis elegans homolog PAQR-2 prevent membrane rigidification by exogenous saturated fatty acids. PLoS Genet. 2017, 13, e1007004. [Google Scholar] [CrossRef]
- Jeong, J.H.; Han, J.S.; Jung, Y.; Lee, S.M.; Park, S.H.; Park, M.; Shin, M.G.; Kim, N.; Kang, M.S.; Kim, S.; et al. A new AMPK isoform mediates glucose-restriction induced longevity non-cell autonomously by promoting membrane fluidity. Nat. Commun. 2023, 14, 288. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Ito, Y.; Tsuchida, A.; Yokomizo, T.; Kita, S.; Sugiyama, T.; Miyagishi, M.; Hara, K.; Tsunoda, M.; et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003, 423, 762–769. [Google Scholar] [CrossRef]
- Scherer, P.E.; Williams, S.; Fogliano, M.; Baldini, G.; Lodish, H.F. A novel serum protein similar to C1q, produced exclusively in adipocytes. J. Biol. Chem. 1995, 270, 26746–26749. [Google Scholar] [CrossRef]
- Hu, E.; Liang, P.; Spiegelman, B.M. AdipoQ is a novel adipose-specific gene dysregulated in obesity. J. Biol. Chem. 1996, 271, 10697–10703. [Google Scholar] [CrossRef]
- Fruebis, J.; Tsao, T.S.; Javorschi, S.; Ebbets-Reed, D.; Erickson, M.R.; Yen, F.T.; Bihain, B.E.; Lodish, H.F. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc. Natl. Acad. Sci. USA 2001, 98, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Combs, T.P.; Du, X.; Brownlee, M.; Scherer, P.E. The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat. Med. 2001, 7, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Combs, T.P.; Berg, A.H.; Obici, S.; Scherer, P.E.; Rossetti, L. Endogenous glucose production is inhibited by the adipose-derived protein Acrp30. J. Clin. Investig. 2001, 108, 1875–1881. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa, Y. The metabolic syndrome and adipocytokines. FEBS Lett. 2006, 580, 2917–2921. [Google Scholar] [CrossRef] [PubMed]
- Iwabu, M.; Okada-Iwabu, M.; Yamauchi, T.; Kadowaki, T. Adiponectin/AdipoR Research and Its Implications for Lifestyle-Related Diseases. Front. Cardiovasc. Med. 2019, 6, 116. [Google Scholar] [CrossRef]
- Imbeault, P.; Depault, I.; Haman, F. Cold exposure increases adiponectin levels in men. Metabolism 2009, 58, 552–559. [Google Scholar] [CrossRef]
- Mengel, L.A.; Seidl, H.; Brandl, B.; Skurk, T.; Holzapfel, C.; Stecher, L.; Claussnitzer, M.; Hauner, H. Gender Differences in the Response to Short-term Cold Exposure in Young Adults. J. Clin. Endocrinol. Metab. 2020, 105. [Google Scholar] [CrossRef]
- Hui, X.; Gu, P.; Zhang, J.; Nie, T.; Pan, Y.; Wu, D.; Feng, T.; Zhong, C.; Wang, Y.; Lam, K.S.; et al. Adiponectin Enhances Cold-Induced Browning of Subcutaneous Adipose Tissue via Promoting M2 Macrophage Proliferation. Cell Metab. 2015, 22, 279–290. [Google Scholar] [CrossRef]
- Wei, Q.; Lee, J.H.; Wang, H.; Bongmba, O.Y.N.; Wu, C.S.; Pradhan, G.; Sun, Z.; Chew, L.; Bajaj, M.; Chan, L.; et al. Adiponectin is required for maintaining normal body temperature in a cold environment. BMC Physiol. 2017, 17, 8. [Google Scholar] [CrossRef]
- Qiao, L.; Yoo, H.; Bosco, C.; Lee, B.; Feng, G.S.; Schaack, J.; Chi, N.W.; Shao, J. Adiponectin reduces thermogenesis by inhibiting brown adipose tissue activation in mice. Diabetologia 2014, 57, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Nio, Y.; Maki, T.; Kobayashi, M.; Takazawa, T.; Iwabu, M.; Okada-Iwabu, M.; Kawamoto, S.; Kubota, N.; Kubota, T.; et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat. Med. 2007, 13, 332–339. [Google Scholar] [CrossRef]
- Pilon, M. Paradigm shift: The primary function of the “Adiponectin Receptors” is to regulate cell membrane composition. Lipids Health Dis. 2021, 20, 43. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Palmgren, H.; Henricsson, M.; Devkota, R.; Jaiswal, H.; Maresca, M.; Bohlooly, Y.M.; Peng, X.R.; Boren, J.; Pilon, M. Extensive transcription mis-regulation and membrane defects in AdipoR2-deficient cells challenged with saturated fatty acids. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158884. [Google Scholar] [CrossRef]
- Ruiz, M.; Stahlman, M.; Boren, J.; Pilon, M. AdipoR1 and AdipoR2 maintain membrane fluidity in most human cell types and independently of adiponectin. J. Lipid Res. 2019, 60, 995–1004. [Google Scholar] [CrossRef] [PubMed]
- Yoda, M.; Nakano, Y.; Tobe, T.; Shioda, S.; Choi-Miura, N.H.; Tomita, M. Characterization of mouse GBP28 and its induction by exposure to cold. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 75–83. [Google Scholar] [CrossRef]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef]
- Kersten, S. Integrated physiology and systems biology of PPARalpha. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef]
- Keshvari, S.; Henstridge, D.C.; Ng, C.; Febbraio, M.A.; Whitehead, J.P. Muscle-specific overexpression of AdipoR1 or AdipoR2 gives rise to common and discrete local effects whilst AdipoR2 promotes additional systemic effects. Sci. Rep. 2017, 7, 41792. [Google Scholar] [CrossRef]
- Vasiliauskaite-Brooks, I.; Sounier, R.; Rochaix, P.; Bellot, G.; Fortier, M.; Hoh, F.; De Colibus, L.; Bechara, C.; Saied, E.M.; Arenz, C.; et al. Structural insights into adiponectin receptors suggest ceramidase activity. Nature 2017, 544, 120–123. [Google Scholar] [CrossRef]
- Green, C.D.; Maceyka, M.; Cowart, L.A.; Spiegel, S. Sphingolipids in metabolic disease: The good, the bad, and the unknown. Cell Metab. 2021, 33, 1293–1306. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, M.; Devkota, R.; Panagaki, D.; Bergh, P.O.; Kaper, D.; Henricsson, M.; Nik, A.; Petkevicius, K.; Hoog, J.L.; Bohlooly, Y.M.; et al. Sphingosine 1-phosphate mediates adiponectin receptor signaling essential for lipid homeostasis and embryogenesis. Nat. Commun. 2022, 13, 7162. [Google Scholar] [CrossRef] [PubMed]
- Fun, X.H.; Thibault, G. Lipid bilayer stress and proteotoxic stress-induced unfolded protein response deploy divergent transcriptional and non-transcriptional programmes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158449. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Gething, M.J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef]
- Thibault, G.; Shui, G.; Kim, W.; McAlister, G.C.; Ismail, N.; Gygi, S.P.; Wenk, M.R.; Ng, D.T. The membrane stress response buffers lethal effects of lipid disequilibrium by reprogramming the protein homeostasis network. Mol. Cell 2012, 48, 16–27. [Google Scholar] [CrossRef]
- Promlek, T.; Ishiwata-Kimata, Y.; Shido, M.; Sakuramoto, M.; Kohno, K.; Kimata, Y. Membrane aberrancy and unfolded proteins activate the endoplasmic reticulum stress sensor Ire1 in different ways. Mol. Biol. Cell 2011, 22, 3520–3532. [Google Scholar] [CrossRef]
- Pineau, L.; Colas, J.; Dupont, S.; Beney, L.; Fleurat-Lessard, P.; Berjeaud, J.M.; Berges, T.; Ferreira, T. Lipid-induced ER stress: Synergistic effects of sterols and saturated fatty acids. Traffic 2009, 10, 673–690. [Google Scholar] [CrossRef]
- Halbleib, K.; Pesek, K.; Covino, R.; Hofbauer, H.F.; Wunnicke, D.; Hanelt, I.; Hummer, G.; Ernst, R. Activation of the Unfolded Protein Response by Lipid Bilayer Stress. Mol. Cell 2017, 67, 673–684.e8. [Google Scholar] [CrossRef] [PubMed]
- Dudkevich, R.; Koh, J.H.; Beaudoin-Chabot, C.; Celik, C.; Lebenthal-Loinger, I.; Karako-Lampert, S.; Ahmad-Albukhari, S.; Thibault, G.; Henis-Korenblit, S. Neuronal IRE-1 coordinates an organism-wide cold stress response by regulating fat metabolism. Cell Rep. 2022, 41, 111739. [Google Scholar] [CrossRef] [PubMed]
- Toien, O.; Blake, J.; Edgar, D.M.; Grahn, D.A.; Heller, H.C.; Barnes, B.M. Hibernation in black bears: Independence of metabolic suppression from body temperature. Science 2011, 331, 906–909. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Storey, K.B. Life in the cold: Links between mammalian hibernation and longevity. Biomol. Concepts 2016, 7, 41–52. [Google Scholar] [CrossRef]
- Andrews, M.T. Molecular interactions underpinning the phenotype of hibernation in mammals. J. Exp. Biol. 2019, 222. [Google Scholar] [CrossRef]
- Nelson, C.J.; Otis, J.P.; Carey, H.V. A role for nuclear receptors in mammalian hibernation. J. Physiol. 2009, 587, 1863–1870. [Google Scholar] [CrossRef]
- Cahill, G.F., Jr. Fuel metabolism in starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef]
- Wu, P.; Inskeep, K.; Bowker-Kinley, M.M.; Popov, K.M.; Harris, R.A. Mechanism responsible for inactivation of skeletal muscle pyruvate dehydrogenase complex in starvation and diabetes. Diabetes 1999, 48, 1593–1599. [Google Scholar] [CrossRef]
- Sugden, M.C.; Bulmer, K.; Gibbons, G.F.; Holness, M.J. Role of peroxisome proliferator-activated receptor-alpha in the mechanism underlying changes in renal pyruvate dehydrogenase kinase isoform 4 protein expression in starvation and after refeeding. Arch. Biochem. Biophys. 2001, 395, 246–252. [Google Scholar] [CrossRef]
- Zhang, S.; Hulver, M.W.; McMillan, R.P.; Cline, M.A.; Gilbert, E.R. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr. Metab. 2014, 11, 10. [Google Scholar] [CrossRef]
- Xu, Y.; Shao, C.; Fedorov, V.B.; Goropashnaya, A.V.; Barnes, B.M.; Yan, J. Molecular signatures of mammalian hibernation: Comparisons with alternative phenotypes. BMC Genom. 2013, 14, 567. [Google Scholar] [CrossRef] [PubMed]
- Kabine, M.; Clemencet, M.C.; Bride, J.; El Kebbaj, M.S.; Latruffe, N.; Cherkaoui-Malki, M. Changes of peroxisomal fatty acid metabolism during cold acclimatization in hibernating jerboa (Jaculus orientalis). Biochimie 2003, 85, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Epperson, L.E.; Li, W.; Hughes, M.A.; Taylor, R.; Rogers, J.; Martin, S.L.; Cossins, A.R.; Gracey, A.Y. Seasonally hibernating phenotype assessed through transcript screening. Physiol. Genom. 2005, 24, 13–22. [Google Scholar] [CrossRef]
- Yan, J.; Barnes, B.M.; Kohl, F.; Marr, T.G. Modulation of gene expression in hibernating arctic ground squirrels. Physiol. Genom. 2008, 32, 170–181. [Google Scholar] [CrossRef] [PubMed]
- El Kebbaj, Z.; Andreoletti, P.; Mountassif, D.; Kabine, M.; Schohn, H.; Dauca, M.; Latruffe, N.; El Kebbaj, M.S.; Cherkaoui-Malki, M. Differential regulation of peroxisome proliferator-activated receptor (PPAR)-alpha1 and truncated PPARalpha2 as an adaptive response to fasting in the control of hepatic peroxisomal fatty acid beta-oxidation in the hibernating mammal. Endocrinology 2009, 150, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Wu, Y.; Sun, K.; Wang, H.; Zhang, B.; Song, S.; Du, Z.; Jiang, T.; Shi, L.; Wang, L.; et al. Differential Expression of Hepatic Genes of the Greater Horseshoe Bat (Rhinolophus ferrumequinum) between the Summer Active and Winter Torpid States. PLoS ONE 2015, 10, e0145702. [Google Scholar] [CrossRef]
- Eddy, S.F.; Morin, P., Jr.; Storey, K.B. Cloning and expression of PPAR-gamma and PGC-1alpha from the hibernating ground squirrel, Spermophilus tridecemlineatus. Mol. Cell Biochem. 2005, 269, 175–182. [Google Scholar] [CrossRef]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Finck, B.N.; Kelly, D.P. PGC-1 coactivators: Inducible regulators of energy metabolism in health and disease. J. Clin. Investig. 2006, 116, 615–622. [Google Scholar] [CrossRef]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine regulation of the fasting response by PPARalpha-mediated induction of fibroblast growth factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef]
- Krilowicz, B.L. Ketone body metabolism in a ground squirrel during hibernation and fasting. Am. J. Physiol. 1985, 249, R462–R470. [Google Scholar] [CrossRef]
- Rauch, J.C.; Behrisch, H.W. Ketone bodies: A source of energy during hibernation. Can. J. Zool. 1981, 59, 754–760. [Google Scholar] [CrossRef]
- LeBlanc, P.J.; Obbard, M.; Battersby, B.J.; Felskie, A.K.; Brown, L.; Wright, P.A.; Ballantyne, J.S. Correlations of plasma lipid metabolites with hibernation and lactation in wild black bears Ursus americanus. J. Comp. Physiol. B 2001, 171, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Schonfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow. Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef]
- Epperson, L.E.; Dahl, T.A.; Martin, S.L. Quantitative analysis of liver protein expression during hibernation in the golden-mantled ground squirrel. Mol. Cell Proteom. 2004, 3, 920–933. [Google Scholar] [CrossRef]
- Andrews, M.T.; Russeth, K.P.; Drewes, L.R.; Henry, P.G. Adaptive mechanisms regulate preferred utilization of ketones in the heart and brain of a hibernating mammal during arousal from torpor. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 296, R383–R393. [Google Scholar] [CrossRef] [PubMed]
- Konig, B.; Koch, A.; Giggel, K.; Dordschbal, B.; Eder, K.; Stangl, G.I. Monocarboxylate transporter (MCT)-1 is up-regulated by PPARalpha. Biochim. Biophys. Acta 2008, 1780, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Owen, O.E. Ketone Bodies as a Fuel for the Brain during Starvation. Biochem. Mol. Biol. Educ. 2005, 33, 246–251. [Google Scholar] [CrossRef]
- Klein, A.H.; Wendroth, S.M.; Drewes, L.R.; Andrews, M.T. Small-volume d-beta-hydroxybutyrate solution infusion increases survivability of lethal hemorrhagic shock in rats. Shock 2010, 34, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Mulier, K.E.; Lexcen, D.R.; Luzcek, E.; Greenberg, J.J.; Beilman, G.J. Treatment with beta-hydroxybutyrate and melatonin is associated with improved survival in a porcine model of hemorrhagic shock. Resuscitation 2012, 83, 253–258. [Google Scholar] [CrossRef]
- Perez de Lara Rodriguez, C.E.; Drewes, L.R.; Andrews, M.T. Hibernation-based blood loss therapy increases survivability of lethal hemorrhagic shock in rats. J. Comp. Physiol. B 2017, 187, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Mulier, K.E.; Muratore, S.L.; Beilman, G.J. D-beta-Hydroxybutyrate and melatonin for treatment of porcine hemorrhagic shock and injury: A melatonin dose-ranging study. BMC Res. Notes 2017, 10, 649. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.; Thakral, S.; Mulier, K.E.; Suryanarayanan, R.; Beilman, G.J. Evaluation of novel formulations of d-beta-hydroxybutyrate and melatonin in a rat model of hemorrhagic shock. Int. J. Pharm. 2018, 548, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Heinemann, M.; Howard, J.; Huber, G.; Iyer-Biswas, S.; Le Treut, G.; Lynch, M.; Montooth, K.L.; Needleman, D.J.; Pigolotti, S.; et al. Physical bioenergetics: Energy fluxes, budgets, and constraints in cells. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Thornton, W.M. The relation of oxygen to the heat of combustion of organic compounds. Philos. Mag. Ser. 1917, 33, 196–203. [Google Scholar] [CrossRef]
- Rodriguez, A.; Becerril, S.; Ezquerro, S.; Mendez-Gimenez, L.; Fruhbeck, G. Crosstalk between adipokines and myokines in fat browning. Acta Physiol. 2017, 219, 362–381. [Google Scholar] [CrossRef]
- Arhire, L.I.; Mihalache, L.; Covasa, M. Irisin: A Hope in Understanding and Managing Obesity and Metabolic Syndrome. Front. Endocrinol. 2019, 10, 524. [Google Scholar] [CrossRef]
- Rajakumari, S.; Wu, J.; Ishibashi, J.; Lim, H.W.; Giang, A.H.; Won, K.J.; Reed, R.R.; Seale, P. EBF2 determines and maintains brown adipocyte identity. Cell Metab. 2013, 17, 562–574. [Google Scholar] [CrossRef]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef]
- Tabuchi, C.; Sul, H.S. Signaling Pathways Regulating Thermogenesis. Front. Endocrinol. 2021, 12, 595020. [Google Scholar] [CrossRef]
- Bertholet, A.M.; Kirichok, Y. Mitochondrial H(+) Leak and Thermogenesis. Annu. Rev. Physiol. 2022, 84, 381–407. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S. Transdifferentiation properties of adipocytes in the adipose organ. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E977–E986. [Google Scholar] [CrossRef] [PubMed]
- Forner, F.; Kumar, C.; Luber, C.A.; Fromme, T.; Klingenspor, M.; Mann, M. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metab. 2009, 10, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Stefanidis, A.; Wiedmann, N.M.; Adler, E.S.; Oldfield, B.J. Hypothalamic control of adipose tissue. Best. Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Munzberg, H.; Floyd, E.; Chang, J.S. Sympathetic Innervation of White Adipose Tissue: To Beige or Not to Beige? Physiology 2021, 36, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Bartness, T.J.; Liu, Y.; Shrestha, Y.B.; Ryu, V. Neural innervation of white adipose tissue and the control of lipolysis. Front. Neuroendocrinol. 2014, 35, 473–493. [Google Scholar] [CrossRef]
- Bachman, E.S.; Dhillon, H.; Zhang, C.Y.; Cinti, S.; Bianco, A.C.; Kobilka, B.K.; Lowell, B.B. βAR signaling required for diet-induced thermogenesis and obesity resistance. Science 2002, 297, 843–845. [Google Scholar] [CrossRef]
- Ueta, C.B.; Fernandes, G.W.; Capelo, L.P.; Fonseca, T.L.; Maculan, F.D.; Gouveia, C.H.; Brum, P.C.; Christoffolete, M.A.; Aoki, M.S.; Lancellotti, C.L.; et al. β(1) Adrenergic receptor is key to cold- and diet-induced thermogenesis in mice. J. Endocrinol. 2012, 214, 359–365. [Google Scholar] [CrossRef]
- Yang, X.; Ruan, H.B. Neuronal Control of Adaptive Thermogenesis. Front. Endocrinol. 2015, 6, 149. [Google Scholar] [CrossRef]
- Tiraby, C.; Tavernier, G.; Lefort, C.; Larrouy, D.; Bouillaud, F.; Ricquier, D.; Langin, D. Acquirement of brown fat cell features by human white adipocytes. J. Biol. Chem. 2003, 278, 33370–33376. [Google Scholar] [CrossRef]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Medvedev, A.V.; Daniel, K.W.; Collins, S. beta-Adrenergic activation of p38 MAP kinase in adipocytes: cAMP induction of the uncoupling protein 1 (UCP1) gene requires p38 MAP kinase. J. Biol. Chem. 2001, 276, 27077–27082. [Google Scholar] [CrossRef] [PubMed]
- Hondares, E.; Mora, O.; Yubero, P.; Rodriguez de la Concepcion, M.; Iglesias, R.; Giralt, M.; Villarroya, F. Thiazolidinediones and rexinoids induce peroxisome proliferator-activated receptor-coactivator (PGC)-1alpha gene transcription: An autoregulatory loop controls PGC-1alpha expression in adipocytes via peroxisome proliferator-activated receptor-gamma coactivation. Endocrinology 2006, 147, 2829–2838. [Google Scholar] [CrossRef] [PubMed]
- Barbera, M.J.; Schluter, A.; Pedraza, N.; Iglesias, R.; Villarroya, F.; Giralt, M. Peroxisome proliferator-activated receptor alpha activates transcription of the brown fat uncoupling protein-1 gene. A link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J. Biol. Chem. 2001, 276, 1486–1493. [Google Scholar] [CrossRef] [PubMed]
- Fedorenko, A.; Lishko, P.V.; Kirichok, Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012, 151, 400–413. [Google Scholar] [CrossRef]
- Wikstrom, J.D.; Mahdaviani, K.; Liesa, M.; Sereda, S.B.; Si, Y.; Las, G.; Twig, G.; Petrovic, N.; Zingaretti, C.; Graham, A.; et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J. 2014, 33, 418–436. [Google Scholar] [CrossRef]
- Ahlabo, I.; Barnard, T. Observations on peroxisomes in brown adipose tissue of the rat. J. Histochem. Cytochem. 1971, 19, 670–675. [Google Scholar] [CrossRef]
- Bagattin, A.; Hugendubler, L.; Mueller, E. Transcriptional coactivator PGC-1alpha promotes peroxisomal remodeling and biogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 20376–20381. [Google Scholar] [CrossRef]
- Guardiola-Diaz, H.M.; Rehnmark, S.; Usuda, N.; Albrektsen, T.; Feltkamp, D.; Gustafsson, J.A.; Alexson, S.E. Rat peroxisome proliferator-activated receptors and brown adipose tissue function during cold acclimatization. J. Biol. Chem. 1999, 274, 23368–23377. [Google Scholar] [CrossRef]
- Park, H.; He, A.; Tan, M.; Johnson, J.M.; Dean, J.M.; Pietka, T.A.; Chen, Y.; Zhang, X.; Hsu, F.F.; Razani, B.; et al. Peroxisome-derived lipids regulate adipose thermogenesis by mediating cold-induced mitochondrial fission. J. Clin. Investig. 2019, 129, 694–711. [Google Scholar] [CrossRef]
- Broniec, A.; Klosinski, R.; Pawlak, A.; Wrona-Krol, M.; Thompson, D.; Sarna, T. Interactions of plasmalogens and their diacyl analogs with singlet oxygen in selected model systems. Free. Radic. Biol. Med. 2011, 50, 892–898. [Google Scholar] [CrossRef]
- Kroger, J.; Jacobs, S.; Jansen, E.H.; Fritsche, A.; Boeing, H.; Schulze, M.B. Erythrocyte membrane fatty acid fluidity and risk of type 2 diabetes in the EPIC-Potsdam study. Diabetologia 2015, 58, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Winocour, P.D.; Bryszewska, M.; Watala, C.; Rand, M.L.; Epand, R.M.; Kinlough-Rathbone, R.L.; Packham, M.A.; Mustard, J.F. Reduced membrane fluidity in platelets from diabetic patients. Diabetes 1990, 39, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Bianchetti, G.; Viti, L.; Scupola, A.; Di Leo, M.; Tartaglione, L.; Flex, A.; De Spirito, M.; Pitocco, D.; Maulucci, G. Erythrocyte membrane fluidity as a marker of diabetic retinopathy in type 1 diabetes mellitus. Eur. J. Clin. Investig. 2021, 51, e13455. [Google Scholar] [CrossRef] [PubMed]
- Szekely, M.; Szelenyi, Z.; Sumegi, I. Brown adipose tissue as a source of heat during pyrogen-induced fever. Acta Physiol. Acad. Sci. Hung. 1973, 43, 85–88. [Google Scholar]
- Blatteis, C.M. Effect of propranolol on endotoxin-induced pyrogenesis in newborn and adult guinea pigs. J. Appl. Physiol. 1976, 40, 35–39. [Google Scholar] [CrossRef]
- Liu, J.; Zeng, D.; Luo, J.; Wang, H.; Xiong, J.; Chen, X.; Chen, T.; Sun, J.; Xi, Q.; Zhang, Y. LPS-Induced Inhibition of miR-143 Expression in Brown Adipocytes Promotes Thermogenesis and Fever. Int. J. Mol. Sci. 2022, 23, 13805. [Google Scholar] [CrossRef]
- Szentirmai, E.; Kapas, L. Brown adipose tissue plays a central role in systemic inflammation-induced sleep responses. PLoS ONE 2018, 13, e0197409. [Google Scholar] [CrossRef]
- Eskilsson, A.; Shionoya, K.; Enerback, S.; Engblom, D.; Blomqvist, A. The generation of immune-induced fever and emotional stress-induced hyperthermia in mice does not involve brown adipose tissue thermogenesis. FASEB J. 2020, 34, 5863–5876. [Google Scholar] [CrossRef]
- Seki, T.; Yang, Y.; Sun, X.; Lim, S.; Xie, S.; Guo, Z.; Xiong, W.; Kuroda, M.; Sakaue, H.; Hosaka, K.; et al. Brown-fat-mediated tumour suppression by cold-altered global metabolism. Nature 2022, 608, 421–428. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, G.; Baumeister, R.; Heimbucher, T. Molecular Mechanisms of Lipid-Based Metabolic Adaptation Strategies in Response to Cold. Cells 2023, 12, 1353. https://doi.org/10.3390/cells12101353
Wu G, Baumeister R, Heimbucher T. Molecular Mechanisms of Lipid-Based Metabolic Adaptation Strategies in Response to Cold. Cells. 2023; 12(10):1353. https://doi.org/10.3390/cells12101353
Chicago/Turabian StyleWu, Gang, Ralf Baumeister, and Thomas Heimbucher. 2023. "Molecular Mechanisms of Lipid-Based Metabolic Adaptation Strategies in Response to Cold" Cells 12, no. 10: 1353. https://doi.org/10.3390/cells12101353
APA StyleWu, G., Baumeister, R., & Heimbucher, T. (2023). Molecular Mechanisms of Lipid-Based Metabolic Adaptation Strategies in Response to Cold. Cells, 12(10), 1353. https://doi.org/10.3390/cells12101353