
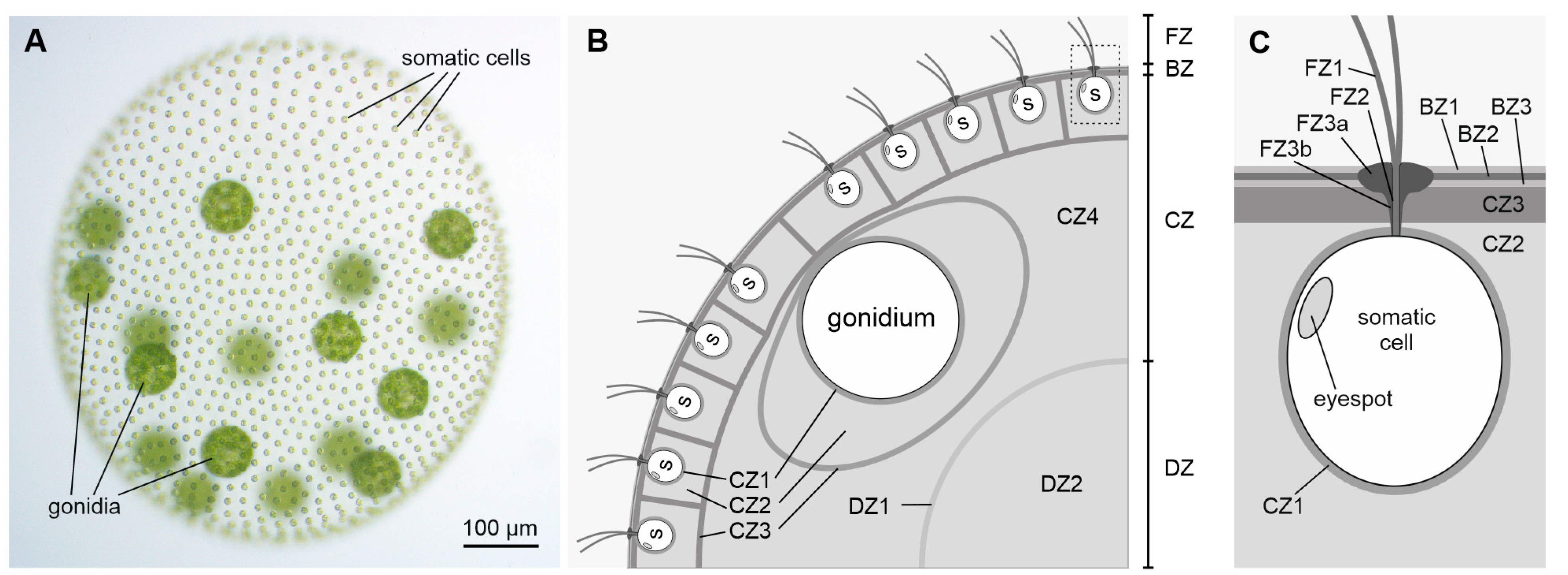
Cell Type-Specific Pherophorins of Volvox carteri Reveal Interplay of Both Cell Types in ECM Biosynthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sources of Sequences, Sequence Processing and Expression Analysis
2.2. Phylogenetic Analysis
2.3. Strains and Culture Conditions
2.4. Isolation of Genomic DNA
2.5. Genomic PCR
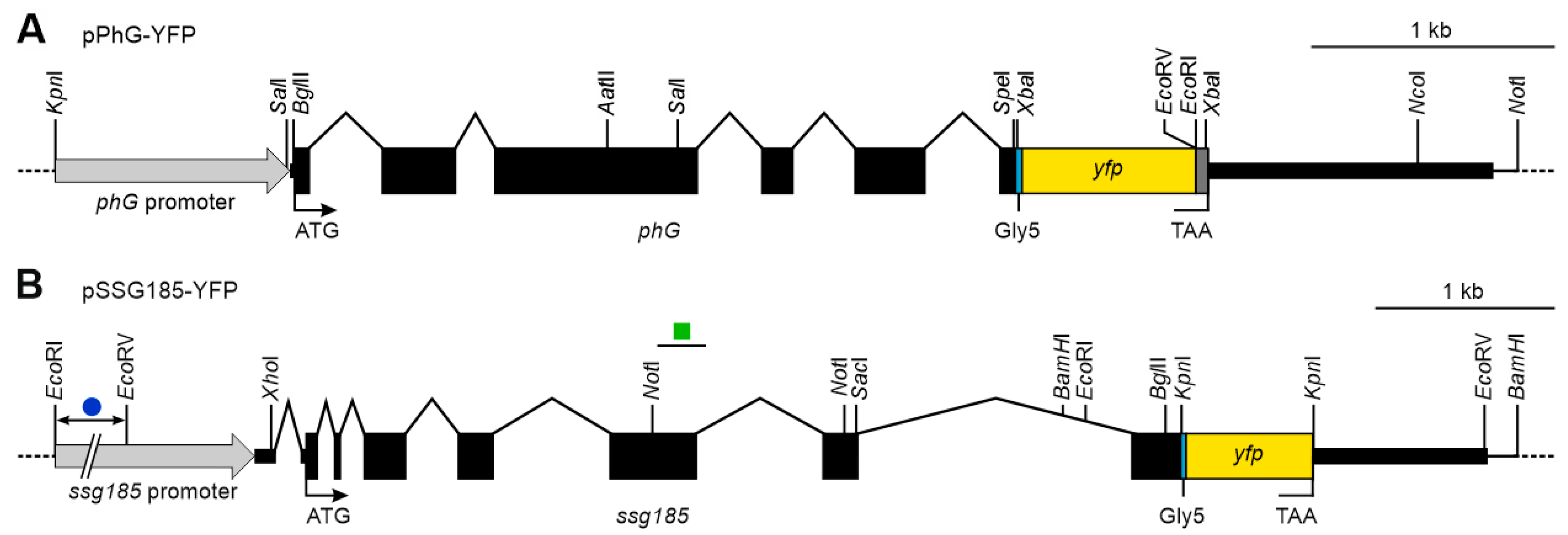
2.6. Construction of Vectors for Expression of Fusion Proteins in V. carteri
2.7. Stable Nuclear Transformation of V. carteri by Particle Bombardment
2.8. Confocal Laser Scanning Microscopy
3. Results
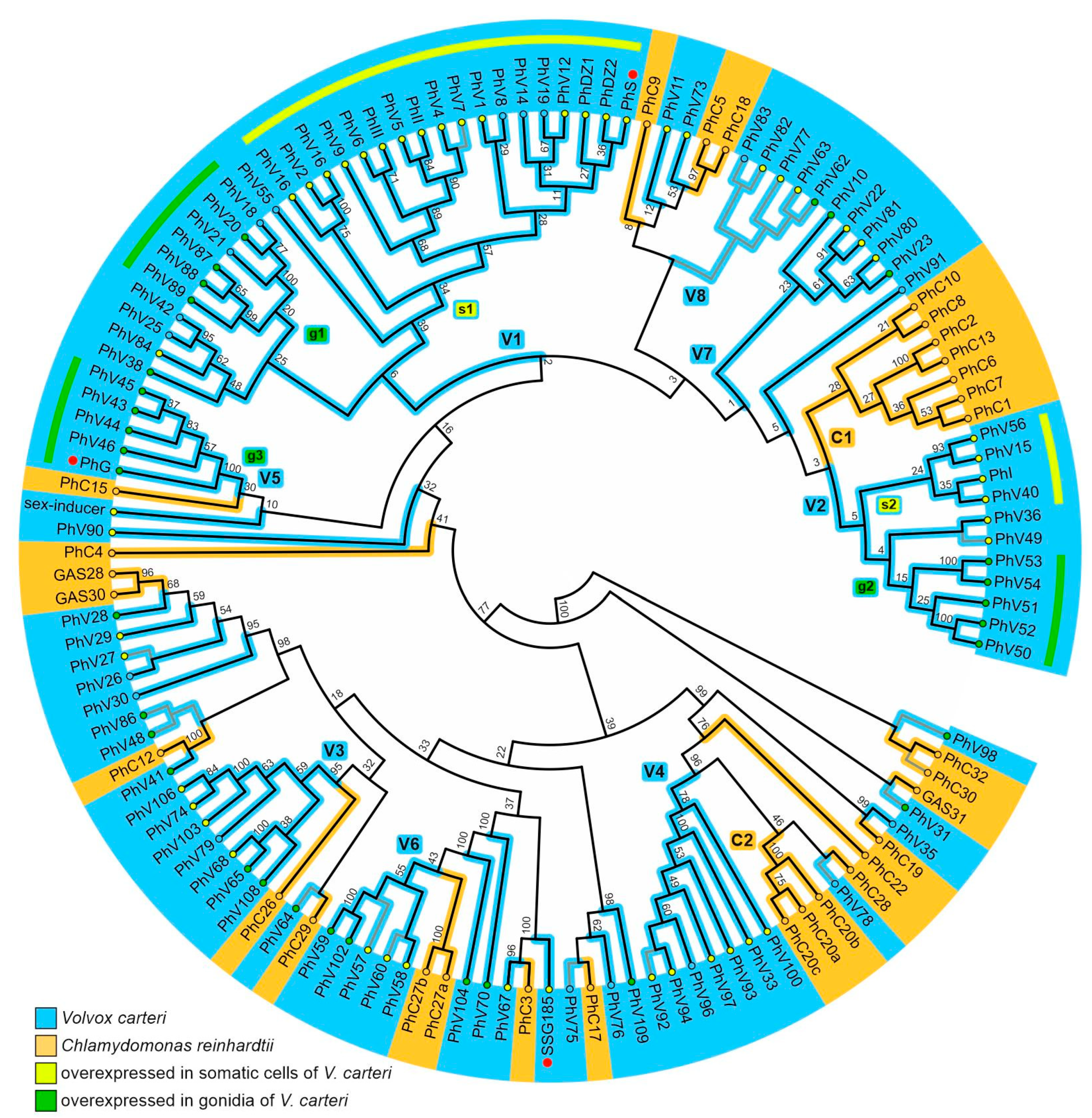
3.1. Phylogenetic Analysis of V. carteri and C. reinhardtii Pherophorins
3.2. The HR Domain of Pherophorins
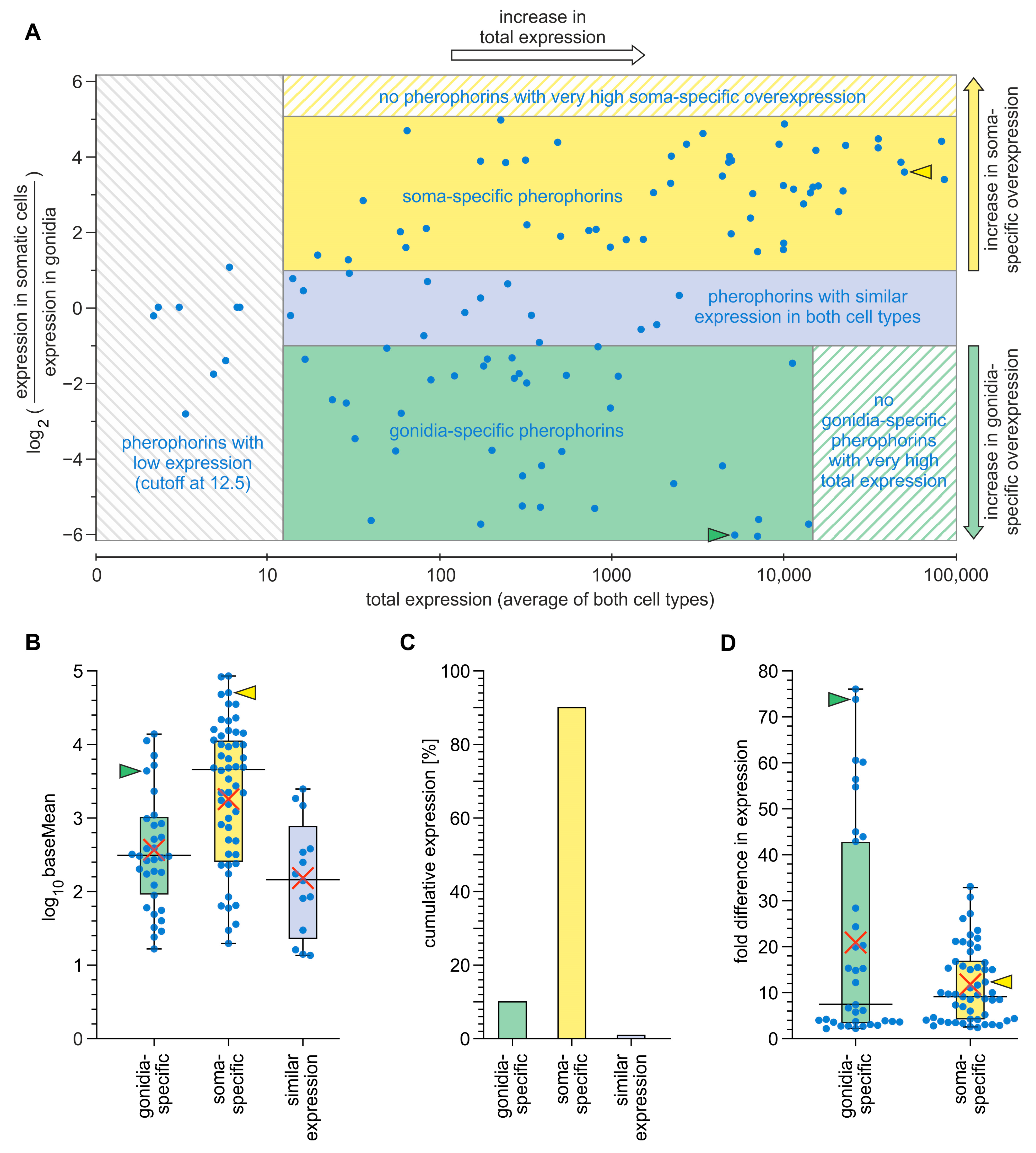
3.3. Expression Analysis of Pherophorin Genes of V. carteri
3.4. Generation of V. carteri Transformants Expressing Fluorescence-Tagged Pherophorins
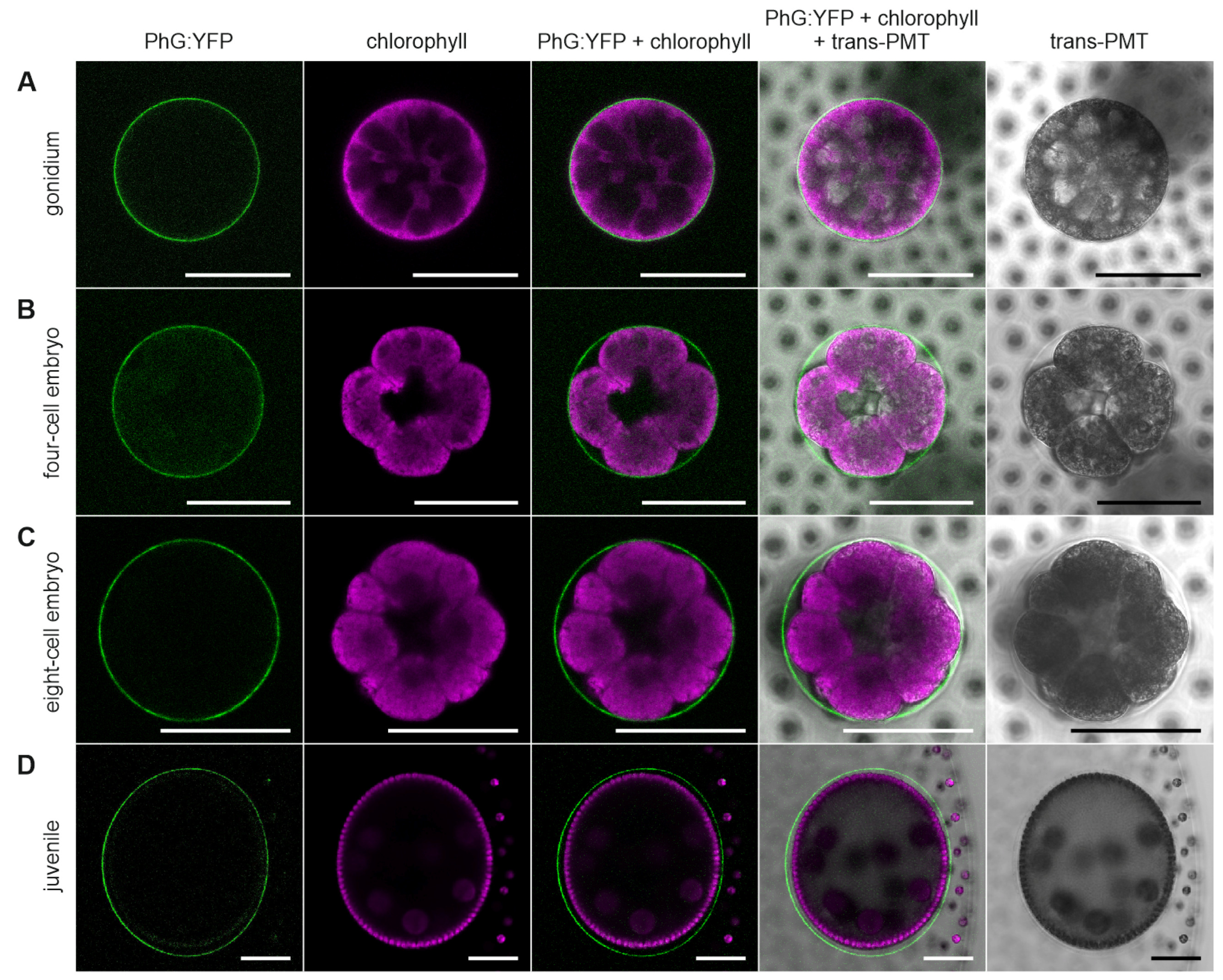
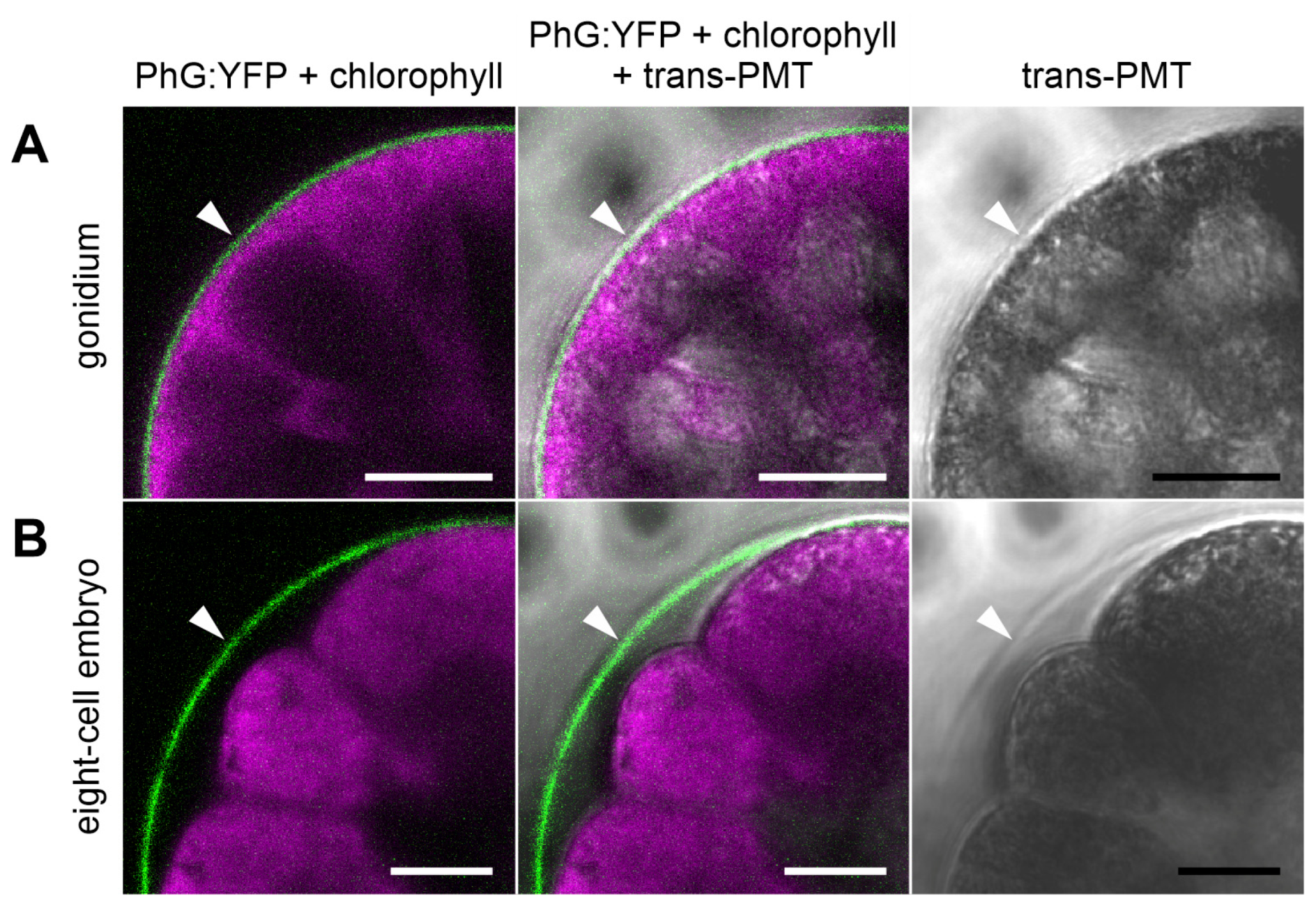
3.5. Development-Dependent In-Vivo Localization of the Gonidia-Specific Pherophorin PhG
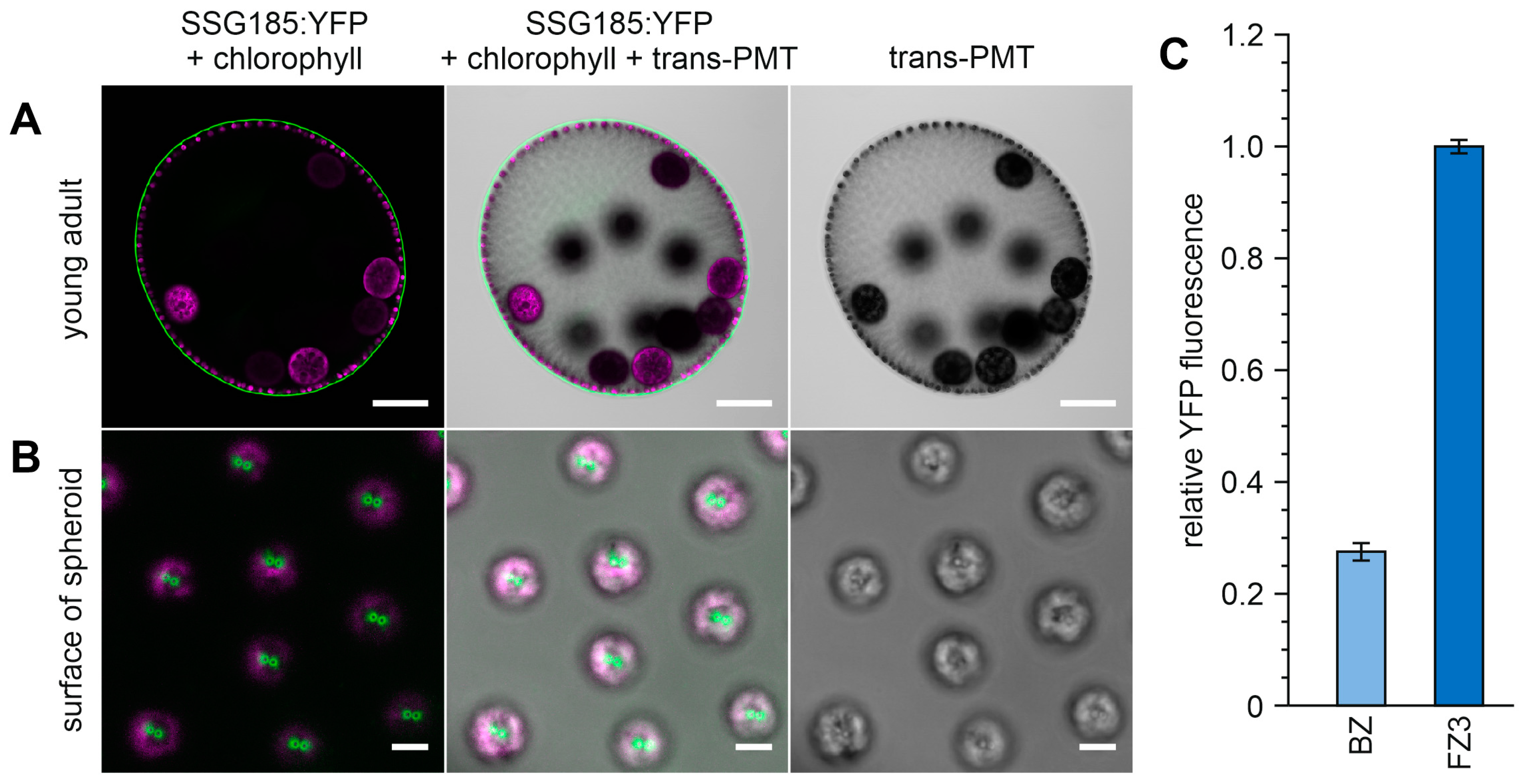
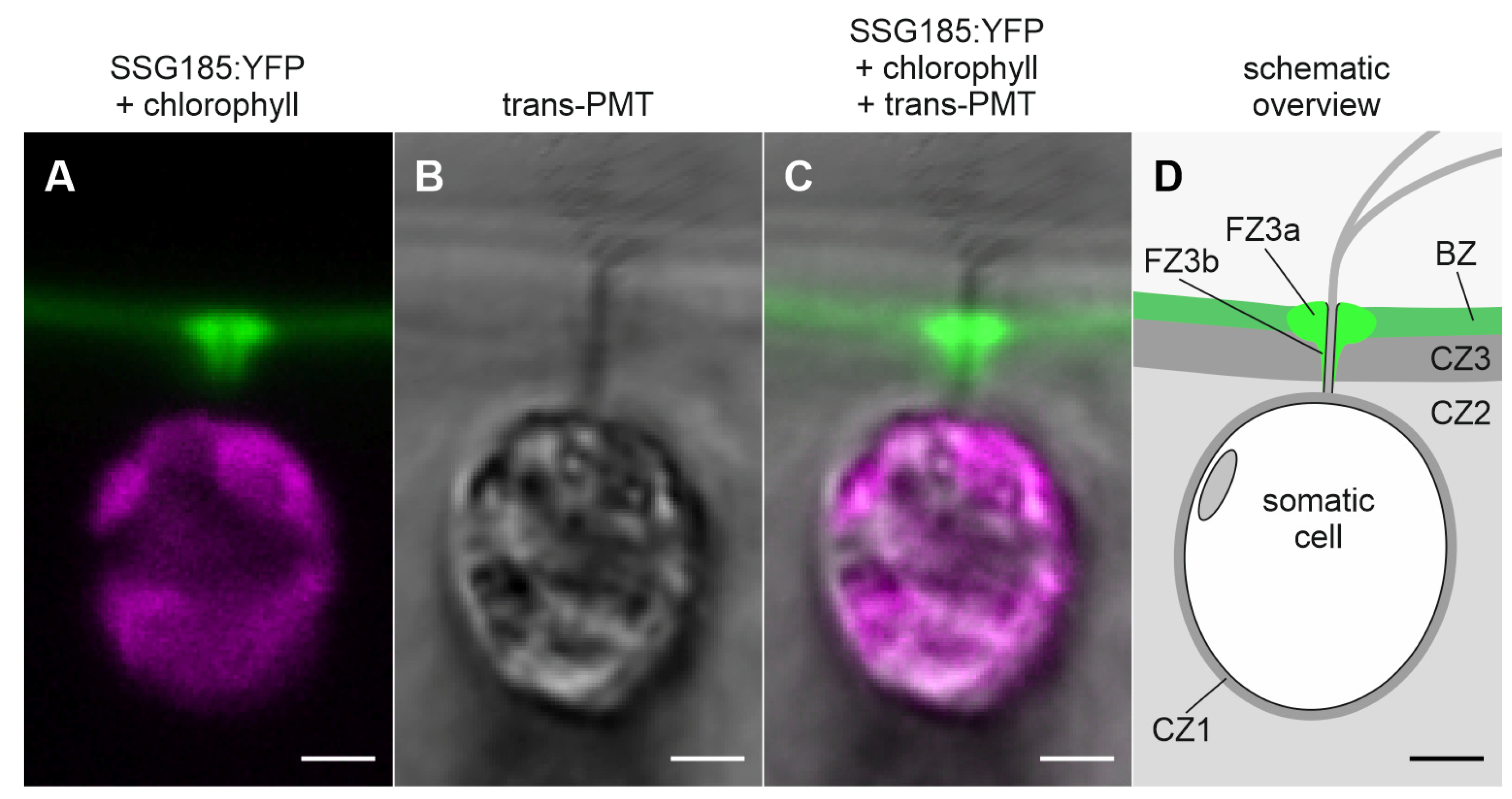
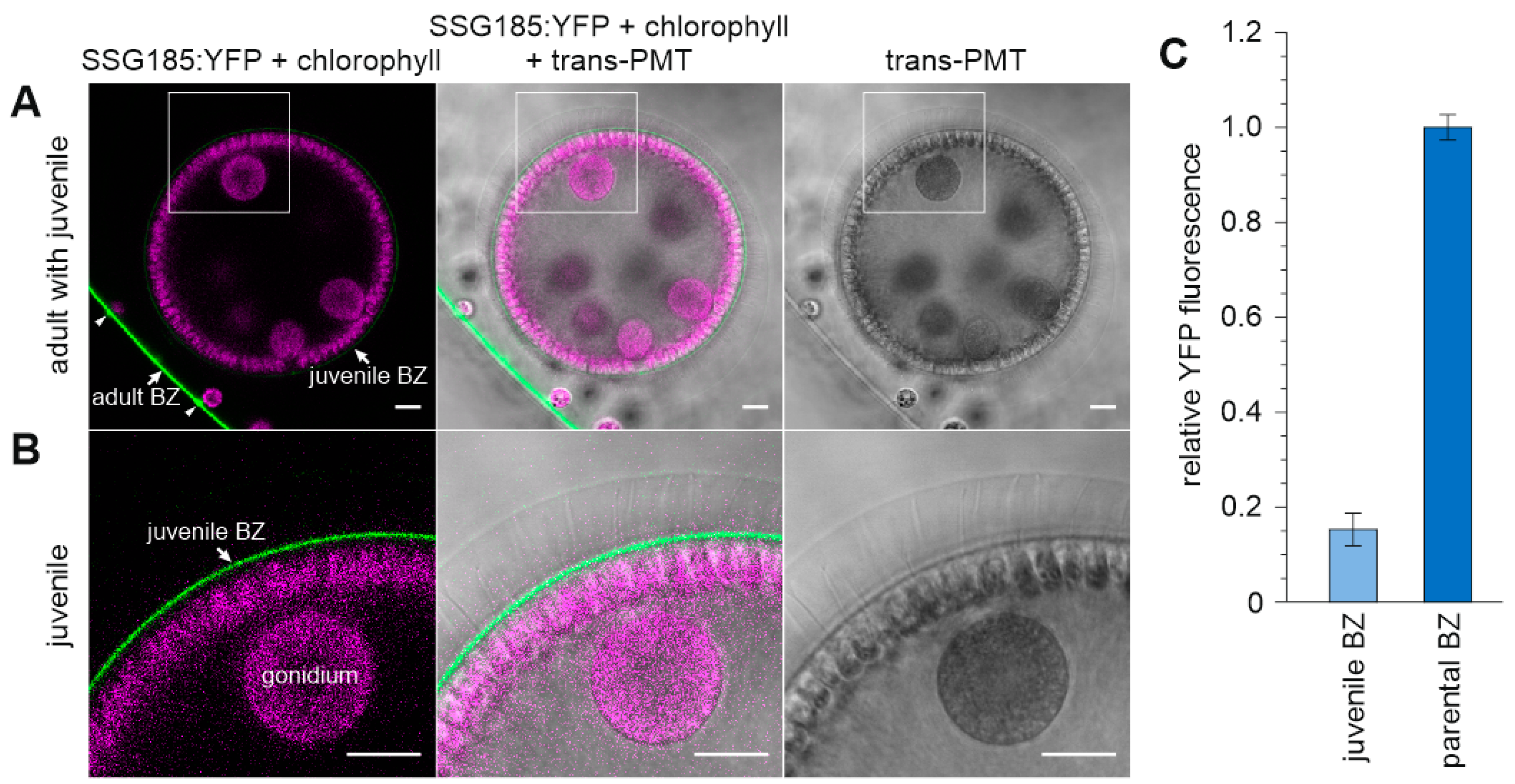
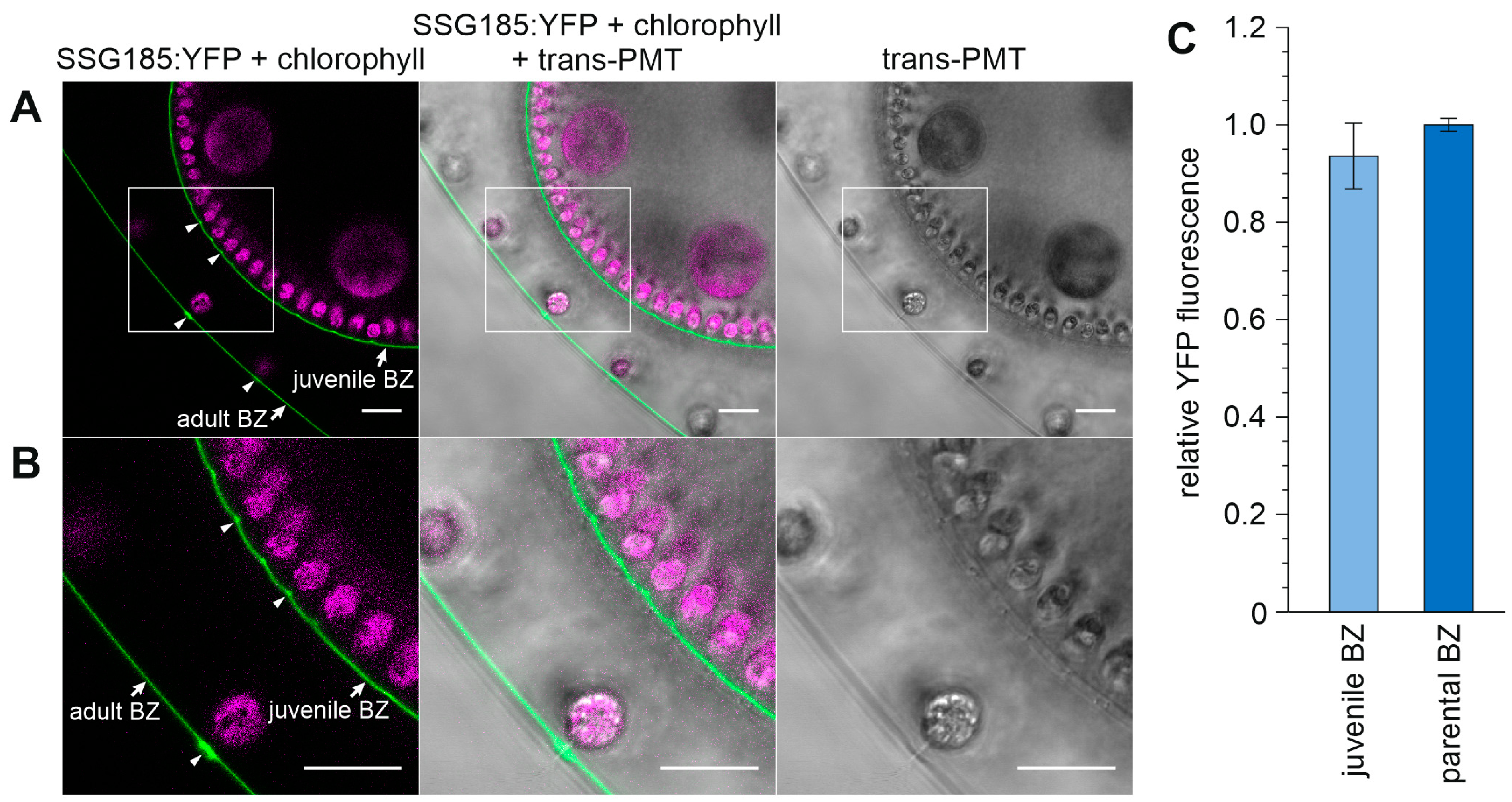
3.6. Development-Dependent In Vivo Localization of the Soma-Specific Pherophorin SSG185
4. Discussion
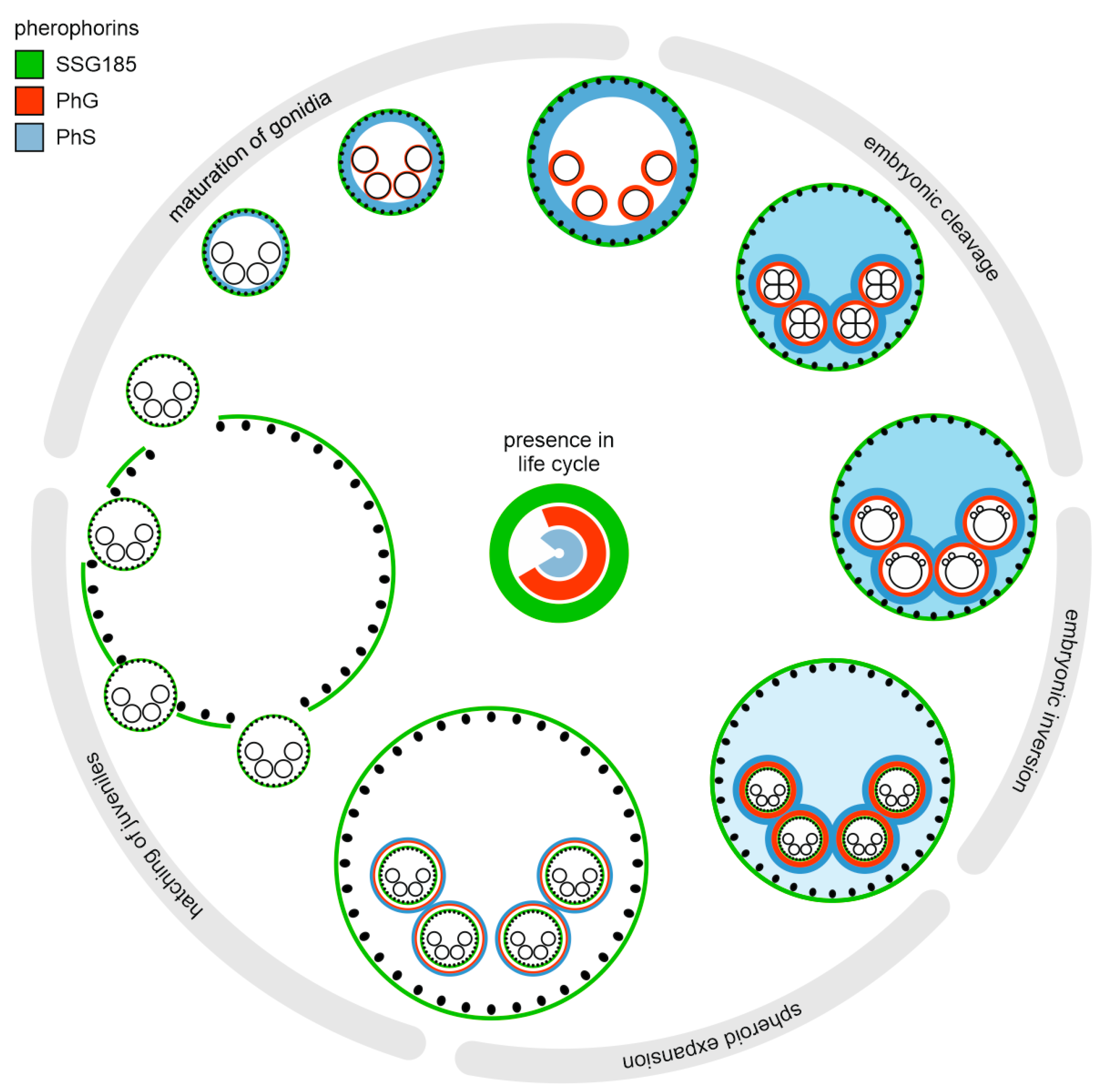
4.1. Not Only Somatic Cells but Also Gonidia Synthesize Pherophorins
4.2. PhG Is a Building Block of the Gonidial Vesicle
4.3. SSG185 Is Located in the Boundary Zone and FZ3 Where It Appears Soon after Embryonic Inversion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BZ | boundary zone |
| cDNA | complementary deoxyribonucleic acid |
| CLSM | confocal laser scanning microscope |
| CZ | cellular zone |
| DZ | deep zone |
| ECM | extracellular matrix |
| FZ | flagellar zone |
| HR | hydroxyproline-rich |
| HRGP | hydroxyproline-rich glycoprotein |
| IQR | inter quartile range |
| MBS | main beam splitter |
| mRNA | messenger ribonucleic acid |
| PhG | pherophorin-G |
| RT-PCR | reverse transcription polymerase chain reaction |
| SSG185 | sulfated surface glycoprotein with a mass of 185 kDa |
| trans-PMT | transmission-photomultiplier tube detector |
| UTR | untranslated region |
| UV | ultraviolet |
| YFP | yellow fluorescent protein (mVenus) |
References
- Brown, N.H. Extracellular matrix in development: Insights from mechanisms conserved between invertebrates and vertebrates. Cold Spring Harb. Perspect. Biol. 2011, 3, a005082. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.R.; Cho, S.; Discher, D.E. Stem cell differentiation is regulated by extracellular matrix mechanics. Physiology (Bethesda) 2018, 33, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Bacete, L.; Melida, H.; Miedes, E.; Molina, A. Plant cell wall-mediated immunity: Cell wall changes trigger disease resistance responses. Plant J. 2018, 93, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Plant cell wall extensibility: Connecting plant cell growth with cell wall structure, mechanics, and the action of wall-modifying enzymes. J. Exp. Bot. 2016, 67, 463–476. [Google Scholar] [CrossRef]
- Kirk, D.L. Volvox: Molecular-Genetic Origins of Multicellularity and Cellular Differentiation, 1st ed.; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar]
- Kirk, D.L. Germ-soma differentiation in Volvox. Dev. Biol. 2001, 238, 213–223. [Google Scholar] [CrossRef]
- Nozaki, H. Origin and evolution of the genera Pleodorina and Volvox (Volvocales). Biologia 2003, 58, 425–431. [Google Scholar]
- Hallmann, A. Extracellular matrix and sex-inducing pheromone in Volvox. Int. Rev. Cytol. 2003, 227, 131–182. [Google Scholar]
- Schmitt, R. Differentiation of germinal and somatic cells in Volvox carteri. Curr. Opin. Microbiol. 2003, 6, 608–613. [Google Scholar] [CrossRef]
- Kirk, D.L. Seeking the ultimate and proximate causes of Volvox multicellularity and cellular differentiation. Integr. Comp. Biol. 2003, 43, 247–253. [Google Scholar] [CrossRef]
- Kirk, M.M.; Kirk, D.L. Exploring germ-soma differentiation in Volvox. J. Biosci. 2004, 29, 143–152. [Google Scholar] [CrossRef]
- Kirk, D.L. A twelve-step program for evolving multicellularity and a division of labor. BioEssays 2005, 27, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Michod, R.E.; Viossat, Y.; Solari, C.A.; Hurand, M.; Nedelcu, A.M. Life-history evolution and the origin of multicellularity. J. Theor. Biol. 2006, 239, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Kirk, D.L.; Birchem, R.; King, N. The extracellular matrix of Volvox: A comparative study and proposed system of nomenclature. J. Cell Sci. 1986, 80, 207–231. [Google Scholar] [CrossRef]
- Fukada, K.; Inoue, T.; Shiraishi, H. A posttranslationally regulated protease, VheA, is involved in the liberation of juveniles from parental spheroids in Volvox carteri. Plant Cell 2006, 18, 2554–2566. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, M.; Nagashio, R.; Sato, Y.; Hasegawa, T. Late Somatic Gene 2 disrupts parental spheroids cooperatively with Volvox hatching enzyme A in Volvox. Planta 2017, 245, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Godl, K.; Hallmann, A.; Wenzl, S.; Sumper, M. Differential targeting of closely related ECM glycoproteins: The pherophorin family from Volvox. EMBO J. 1997, 16, 25–34. [Google Scholar] [CrossRef]
- Sumper, M.; Hallmann, A. Biochemistry of the extracellular matrix of Volvox. Int. Rev. Cytol. 1998, 180, 51–85. [Google Scholar]
- von der Heyde, B.; Hallmann, A. Targeted migration of pherophorin-S indicates extensive extracellular matrix dynamics in Volvox carteri. Plant J. 2020, 103, 2301–2317. [Google Scholar] [CrossRef]
- Miller, D.H.; Mellman, I.S.; Lamport, D.T.A.; Miller, M. The chemical composition of the cell wall of Chlamydomonas gymnogama and the concept of a plant cell wall protein. J. Cell Biol. 1974, 63, 420–429. [Google Scholar] [CrossRef]
- Miller, D.H.; Lamport, D.T.A.; Miller, M. Hydroxyproline heterooligosaccharides in Chlamydomonas. Science 1972, 176, 918–920. [Google Scholar] [CrossRef]
- Sommer-Knudsen, J.; Bacic, A.; Clarke, A.E. Hydroxyproline-rich plant glycoproteins. Phytochemistry 1998, 47, 483–497. [Google Scholar] [CrossRef]
- Showalter, A.M.; Basu, D. Extensin and arabinogalactan-protein biosynthesis: Glycosyltransferases, research challenges, and biosensors. Front. Plant Sci. 2016, 7, 814. [Google Scholar] [CrossRef] [PubMed]
- Woessner, J.P.; Goodenough, U.W. Volvocine cell walls and their constituent glycoproteins: An evolutionary perspective. Protoplasma 1994, 181, 245–258. [Google Scholar] [CrossRef]
- Woessner, J.P.; Goodenough, U.W. Zygote and vegetative cell wall proteins in Chlamydomonas reinhardtii share a common epitope, (SerPro)x. Plant Sci. 1992, 83, 65–76. [Google Scholar] [CrossRef]
- Adair, W.S.; Snell, W.J. The Chlamydomonas reinhardtii cell wall: Structure, biochemistry, and molecular biology. In Organization and Assembly of Plant and Animal Extracellular Matrix; Adair, W.S., Mecham, R.P., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 15–84. [Google Scholar]
- Heitzer, M.; Hallmann, A. An extracellular matrix-localized metalloproteinase with an exceptional QEXXH metal binding site prefers copper for catalytic activity. J. Biol. Chem. 2002, 277, 28280–28286. [Google Scholar] [CrossRef]
- Hallmann, A. The pherophorins: Common, versatile building blocks in the evolution of extracellular matrix architecture in Volvocales. Plant J. 2006, 45, 292–307. [Google Scholar] [CrossRef]
- Hallmann, A. VCRPs, small cysteine-rich proteins, might be involved in extracellular signaling in the green alga Volvox. Plant Signal. Behav. 2008, 3, 124–127. [Google Scholar] [CrossRef][Green Version]
- Harris, E.H.; Stern, D.B.; Witman, G.B. The Chlamydomonas Sourcebook, 2nd ed.; Academic Press: San Diego, CA, USA, 2009. [Google Scholar]
- Harris, E.H. The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use; Academic Press: San Diego, CA, USA, 1989. [Google Scholar]
- Wenzl, S.; Sumper, M. The occurrence of different sulphated cell surface glycoproteins correlates with defined developmental events in Volvox. FEBS Lett. 1982, 143, 311–315. [Google Scholar] [CrossRef]
- Ertl, H.; Mengele, R.; Wenzl, S.; Engel, J.; Sumper, M. The extracellular matrix of Volvox carteri: Molecular structure of the cellular compartment. J. Cell Biol. 1989, 109, 3493–3501. [Google Scholar] [CrossRef]
- Sumper, M.; Berg, E.; Wenzl, S.; Godl, K. How a sex pheromone might act at a concentration below 10−16 M. EMBO J. 1993, 12, 831–836. [Google Scholar] [CrossRef]
- Godl, K.; Hallmann, A.; Rappel, A.; Sumper, M. Pherophorins: A family of extracellular matrix glycoproteins from Volvox structurally related to the sex-inducing pheromone. Planta 1995, 196, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Ender, F.; Godl, K.; Wenzl, S.; Sumper, M. Evidence for autocatalytic cross-linking of hydroxyproline-rich glycoproteins during extracellular matrix assembly in Volvox. Plant Cell 2002, 14, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Ertl, H.; Hallmann, A.; Wenzl, S.; Sumper, M. A novel extensin that may organize extracellular matrix biogenesis in Volvox carteri. EMBO J. 1992, 11, 2055–2062. [Google Scholar] [CrossRef]
- Hallmann, A.; Kirk, D.L. The developmentally regulated ECM glycoprotein ISG plays an essential role in organizing the ECM and orienting the cells of Volvox. J. Cell Sci. 2000, 113, 4605–4617. [Google Scholar] [CrossRef] [PubMed]
- Ender, F.; Hallmann, A.; Amon, P.; Sumper, M. Response to the sexual pheromone and wounding in the green alga Volvox: Induction of an extracellular glycoprotein consisting almost exclusively of hydroxyproline. J. Biol. Chem. 1999, 274, 35023–35028. [Google Scholar] [CrossRef]
- Hallmann, A. A small cysteine-rich extracellular protein, VCRP, is inducible by the sex-inducer of Volvox carteri and by wounding. Planta 2007, 226, 719–727. [Google Scholar] [CrossRef]
- Hallmann, A.; Godl, K.; Wenzl, S.; Sumper, M. The highly efficient sex-inducing pheromone system of Volvox. Trends Microbiol. 1998, 6, 185–189. [Google Scholar] [CrossRef]
- Hallmann, A.; Amon, P.; Godl, K.; Heitzer, M.; Sumper, M. Transcriptional activation by the sexual pheromone and wounding: A new gene family from Volvox encoding modular proteins with (hydroxy)proline-rich and metalloproteinase homology domains. Plant J. 2001, 26, 583–593. [Google Scholar] [CrossRef]
- Hallmann, A.; Sumper, M. An inducible arylsulfatase of Volvox carteri with properties suitable for a reporter-gene system. Purification, characterization and molecular cloning. Eur. J. Biochem. 1994, 221, 143–150. [Google Scholar] [CrossRef]
- Hallmann, A. Enzymes in the extracellular matrix of Volvox: An inducible, calcium-dependent phosphatase with a modular composition. J. Biol. Chem. 1999, 274, 1691–1697. [Google Scholar] [CrossRef]
- Amon, P.; Haas, E.; Sumper, M. The sex-inducing pheromone and wounding trigger the same set of genes in the multicellular green alga Volvox. Plant Cell 1998, 10, 781–789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prochnik, S.E.; Umen, J.; Nedelcu, A.M.; Hallmann, A.; Miller, S.M.; Nishii, I.; Ferris, P.; Kuo, A.; Mitros, T.; Fritz-Laylin, L.K.; et al. Genomic analysis of organismal complexity in the multicellular green alga Volvox carteri. Science 2010, 329, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Marechal-Drouard, L.; et al. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Hanschen, E.R.; Marriage, T.N.; Ferris, P.J.; Hamaji, T.; Toyoda, A.; Fujiyama, A.; Neme, R.; Noguchi, H.; Minakuchi, Y.; Suzuki, M.; et al. The Gonium pectorale genome demonstrates co-option of cell cycle regulation during the evolution of multicellularity. Nat. Commun. 2016, 7, 11370. [Google Scholar] [CrossRef]
- Wenzl, S.; Sumper, M. Sulfation of a cell surface glycoprotein correlates with the developmental program during embryogenesis of Volvox carteri. Proc. Natl. Acad. Sci. USA 1981, 78, 3716–3720. [Google Scholar] [CrossRef]
- Holst, O.; Christoffel, V.; Fründ, R.; Moll, H.; Sumper, M. A phosphodiester bridge between two arabinose residues as a structural element of an extracellular glycoprotein of Volvox carteri. Eur. J. Biochem. 1989, 181, 345–350. [Google Scholar] [CrossRef]
- Wenzl, S.; Thym, D.; Sumper, M. Development-dependent modification of the extracellular matrix by a sulphated glycoprotein in Volvox carteri. EMBO J. 1984, 3, 739–744. [Google Scholar] [CrossRef]
- Klein, B.; Wibberg, D.; Hallmann, A. Whole transcriptome RNA-Seq analysis reveals extensive cell type-specific compartmentalization in Volvox carteri. BMC Biol. 2017, 15, 111. [Google Scholar] [CrossRef]
- Mengele, R.; Sumper, M. Gulose as a constituent of a glycoprotein. FEBS Lett. 1992, 298, 14–16. [Google Scholar] [CrossRef]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2012, 40, D1178–D1186. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Hilker, R.; Stadermann, K.B.; Doppmeier, D.; Kalinowski, J.; Stoye, J.; Straube, J.; Winnebald, J.; Goesmann, A. ReadXplorer—Visualization and analysis of mapped sequences. Bioinformatics 2014, 30, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef]
- Anders, S.; McCarthy, D.J.; Chen, Y.; Okoniewski, M.; Smyth, G.K.; Huber, W.; Robinson, M.D. Count-based differential expression analysis of RNA sequencing data using R and Bioconductor. Nat. Protoc. 2013, 8, 1765–1786. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016; Available online: https://www.R-project.org/ (accessed on 20 April 2022).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Nicholas, K.B.; Nicholas, H.B.; Deerfield, D.W. GeneDoc: Analysis and visualization of genetic variation. EMBnet News 1997, 4, 14. [Google Scholar]
- Felsenstein, J. Phylip—Phylogeny Inference Package (Version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Stöver, B.C.; Müller, K.F. TreeGraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef]
- Kianianmomeni, A.; Nematollahi, G.; Hallmann, A. A gender-specific retinoblastoma-related protein in Volvox carteri implies a role for the retinoblastoma protein family in sexual development. Plant Cell 2008, 20, 2399–2419. [Google Scholar] [CrossRef]
- Starr, R.C. Structure, reproduction and differentiation in Volvox carteri f. nagariensis Iyengar, strains HK 9 & 10. Arch. Protistenkd. 1969, 111, 204–222. [Google Scholar]
- Starr, R.C. Control of differentiation in Volvox. Dev. Biol. Suppl. 1970, 4, 59–100. [Google Scholar]
- Tian, Y.; Gao, S.; von der Heyde, E.L.; Hallmann, A.; Nagel, G. Two-component cyclase opsins of green algae are ATP-dependent and light-inhibited guanylyl cyclases. BMC Biol. 2018, 16, 144. [Google Scholar] [CrossRef]
- Provasoli, L.; Pintner, I.J. Artificial media for fresh-water algae: Problems and suggestions. In The Ecology of Algae, a Symposium Held at the Pymatuning Laboratory of Field Biology on June 18 and 19, 1959, 1st ed.; The Pymatuning Symposia in Ecology, Special Publication No. 2.; Tryon, C.A., Hartman, R.T., Eds.; University of Pittsburgh: Pittsburgh, PA, USA, 1959; pp. 84–96. [Google Scholar]
- Starr, R.C.; Jaenicke, L. Purification and characterization of the hormone initiating sexual morphogenesis in Volvox carteri f. nagariensis Iyengar. Proc. Natl. Acad. Sci. USA 1974, 71, 1050–1054. [Google Scholar] [CrossRef]
- Edwards, K.; Johnstone, C.; Thompson, C. A simple and rapid method for the preparation of plant genomic DNA for PCR analysis. Nucleic Acids Res. 1991, 19, 1349. [Google Scholar] [CrossRef]
- Lerche, K.; Hallmann, A. Stable nuclear transformation of Gonium pectorale. BMC Biotechnol. 2009, 9, 64. [Google Scholar] [CrossRef]
- Lerche, K.; Hallmann, A. Stable nuclear transformation of Eudorina elegans. BMC Biotechnol. 2013, 13, 11. [Google Scholar] [CrossRef] [PubMed]
- Lerche, K.; Hallmann, A. Stable nuclear transformation of Pandorina morum. BMC Biotechnol. 2014, 14, 65. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.; Messing, J. The pUC plasmids, an M13mp7-derived system for insertion mutagenesis and sequencing with synthetic universal primers. Gene 1982, 19, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Lauersen, K.J.; Kruse, O.; Mussgnug, J.H. Targeted expression of nuclear transgenes in Chlamydomonas reinhardtii with a versatile, modular vector toolkit. Appl. Microbiol. Biotechnol. 2015, 99, 3491–3503. [Google Scholar] [CrossRef]
- von der Heyde, E.L.; Hallmann, A. Babo1, formerly Vop1 and Cop1/2, is no eyespot photoreceptor but a basal body protein illuminating cell division in Volvox carteri. Plant J. 2020, 102, 276–298. [Google Scholar] [CrossRef]
- Schiedlmeier, B.; Schmitt, R.; Müller, W.; Kirk, M.M.; Gruber, H.; Mages, W.; Kirk, D.L. Nuclear transformation of Volvox carteri. Proc. Natl. Acad. Sci. USA 1994, 91, 5080–5084. [Google Scholar] [CrossRef]
- Hallmann, A.; Wodniok, S. Swapped green algal promoters: aphVIII-based gene constructs with Chlamydomonas flanking sequences work as dominant selectable markers in Volvox and vice versa. Plant Cell Rep. 2006, 25, 582–591. [Google Scholar] [CrossRef]
- Gruber, H.; Kirzinger, S.H.; Schmitt, R. Expression of the Volvox gene encoding nitrate reductase: Mutation-dependent activation of cryptic splice sites and intron-enhanced gene expression from a cDNA. Plant Mol. Biol. 1996, 31, 1–12. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Society. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Solari, C.A.; Drescher, K.; Goldstein, R.E. The flagellar photoresponse in Volvox species (Volvocaceae, Chlorophyceae). J. Phycol. 2011, 47, 580–583. [Google Scholar] [CrossRef] [PubMed]
- Ueki, N.; Nishii, I. Controlled enlargement of the glycoprotein vesicle surrounding a Volvox embryo requires the InvB nucleotide-sugar transporter and is required for normal morphogenesis. Plant Cell 2009, 21, 1166–1181. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huskey, R.J. Mutants affecting vegetative cell orientation in Volvox carteri. Dev. Biol. 1979, 72, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Strenkert, D.; Schmollinger, S.; Gallaher, S.D.; Salome, P.A.; Purvine, S.O.; Nicora, C.D.; Mettler-Altmann, T.; Soubeyrand, E.; Weber, A.P.M.; Lipton, M.S.; et al. Multiomics resolution of molecular events during a day in the life of Chlamydomonas. Proc. Natl. Acad. Sci. USA 2019, 116, 2374–2383. [Google Scholar] [CrossRef] [PubMed]











Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von der Heyde, B.; Hallmann, A. Cell Type-Specific Pherophorins of Volvox carteri Reveal Interplay of Both Cell Types in ECM Biosynthesis. Cells 2023, 12, 134. https://doi.org/10.3390/cells12010134
von der Heyde B, Hallmann A. Cell Type-Specific Pherophorins of Volvox carteri Reveal Interplay of Both Cell Types in ECM Biosynthesis. Cells. 2023; 12(1):134. https://doi.org/10.3390/cells12010134
Chicago/Turabian Stylevon der Heyde, Benjamin, and Armin Hallmann. 2023. "Cell Type-Specific Pherophorins of Volvox carteri Reveal Interplay of Both Cell Types in ECM Biosynthesis" Cells 12, no. 1: 134. https://doi.org/10.3390/cells12010134
APA Stylevon der Heyde, B., & Hallmann, A. (2023). Cell Type-Specific Pherophorins of Volvox carteri Reveal Interplay of Both Cell Types in ECM Biosynthesis. Cells, 12(1), 134. https://doi.org/10.3390/cells12010134