
Novel Insights into the Therapeutic Potential of Lung-Targeted Gene Transfer in the Most Common Respiratory Diseases

 ,
, 

 and
and 
Abstract
1. Introduction
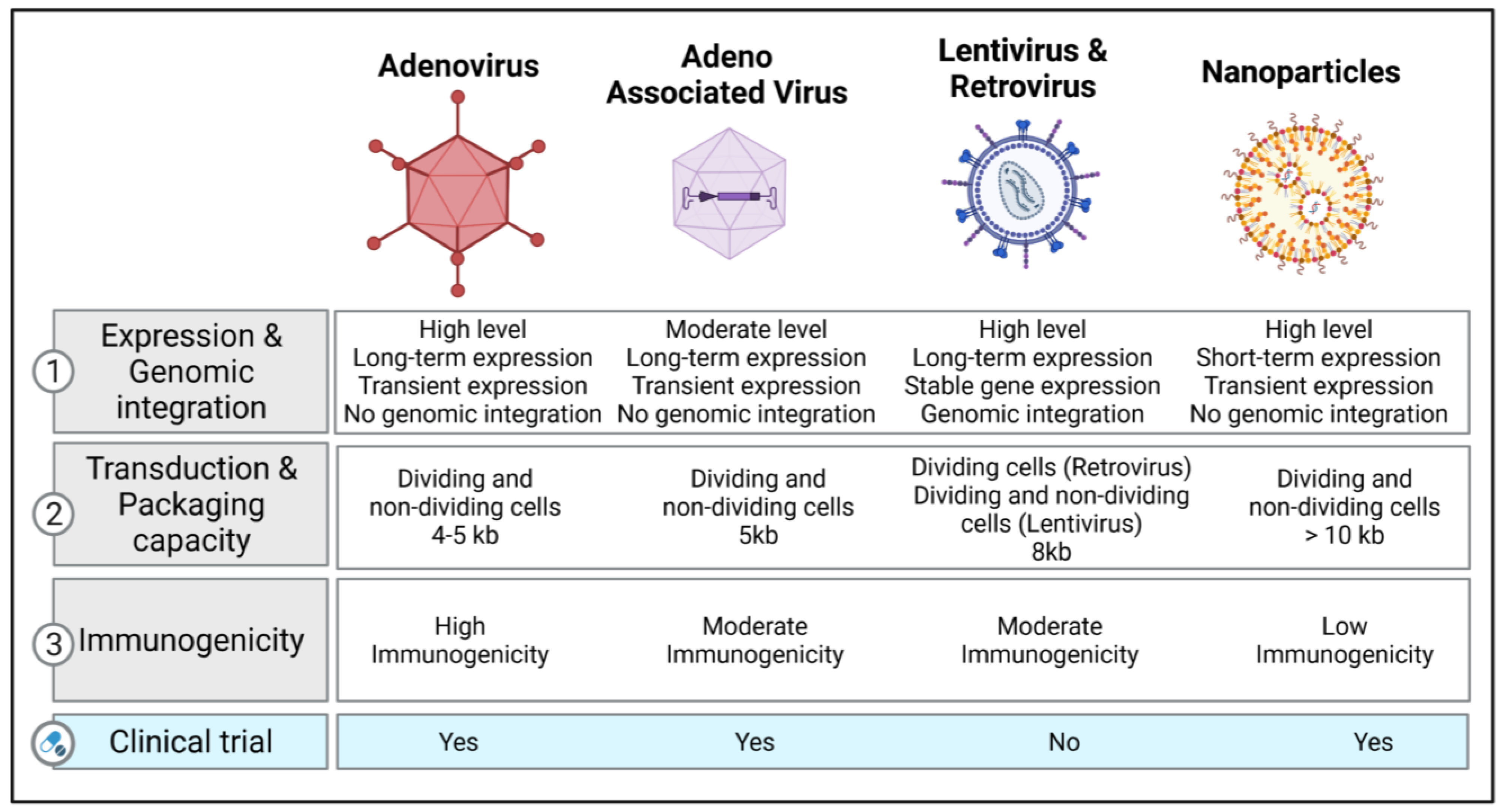
2. Vectors of Gene Therapy in Respiratory Diseases
2.1. Viral Vectors
2.1.1. Retroviral and Lentiviral Vectors
2.1.2. Adenovirus Vectors
2.1.3. Adeno-Associated Viruses (AAV) and Their Vectors
2.1.4. Other Viral Vectors
2.2. Non-Viral Vectors
Nanoparticle-Based Therapeutics
3. Gene-Editing Strategies
3.1. CRISPR-Cas9 as a Genome-Editing Tool
3.2. Base- and Prime-Editing Technologies
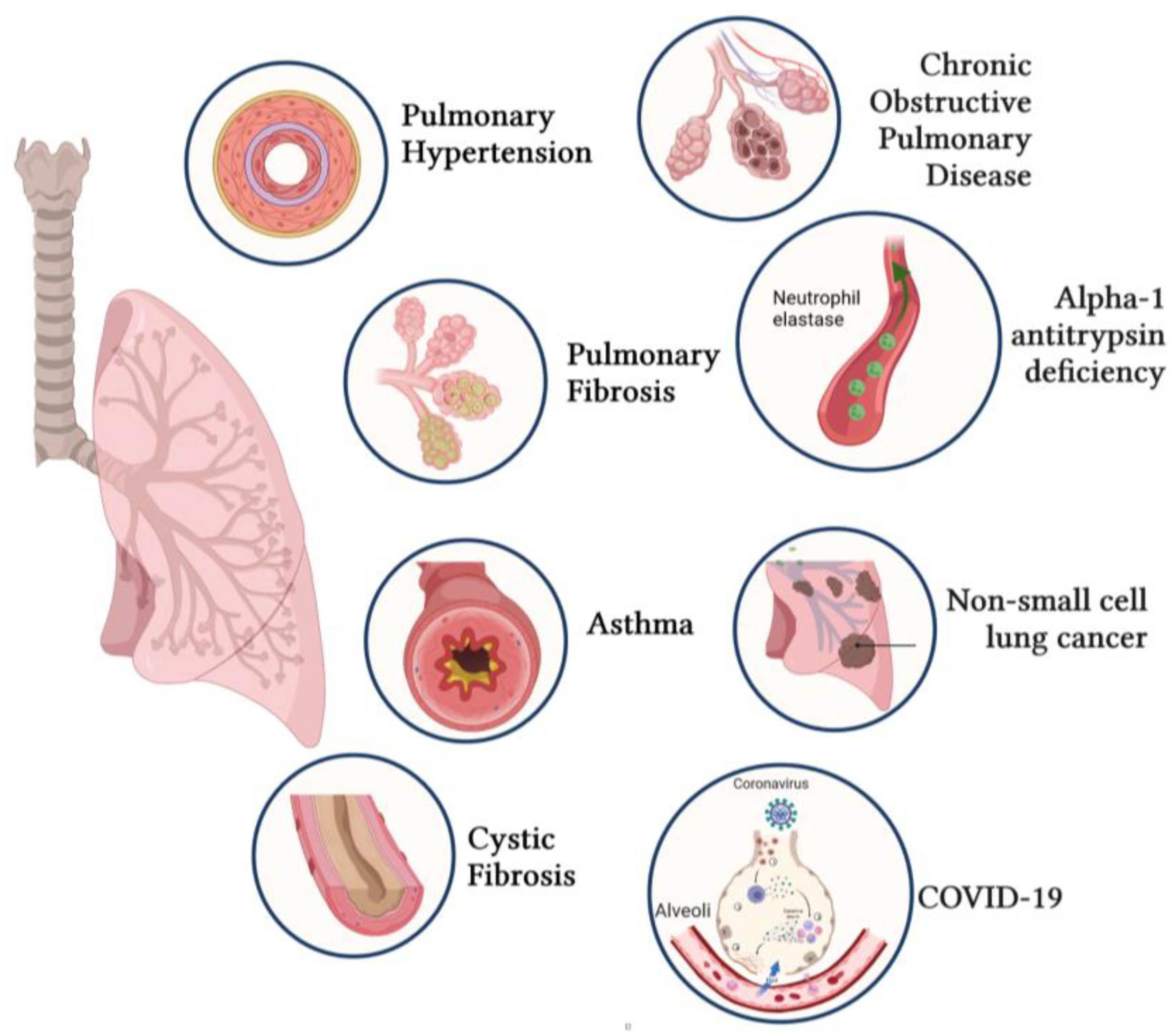
4. Applications of Gene Transfer for Respiratory Diseases
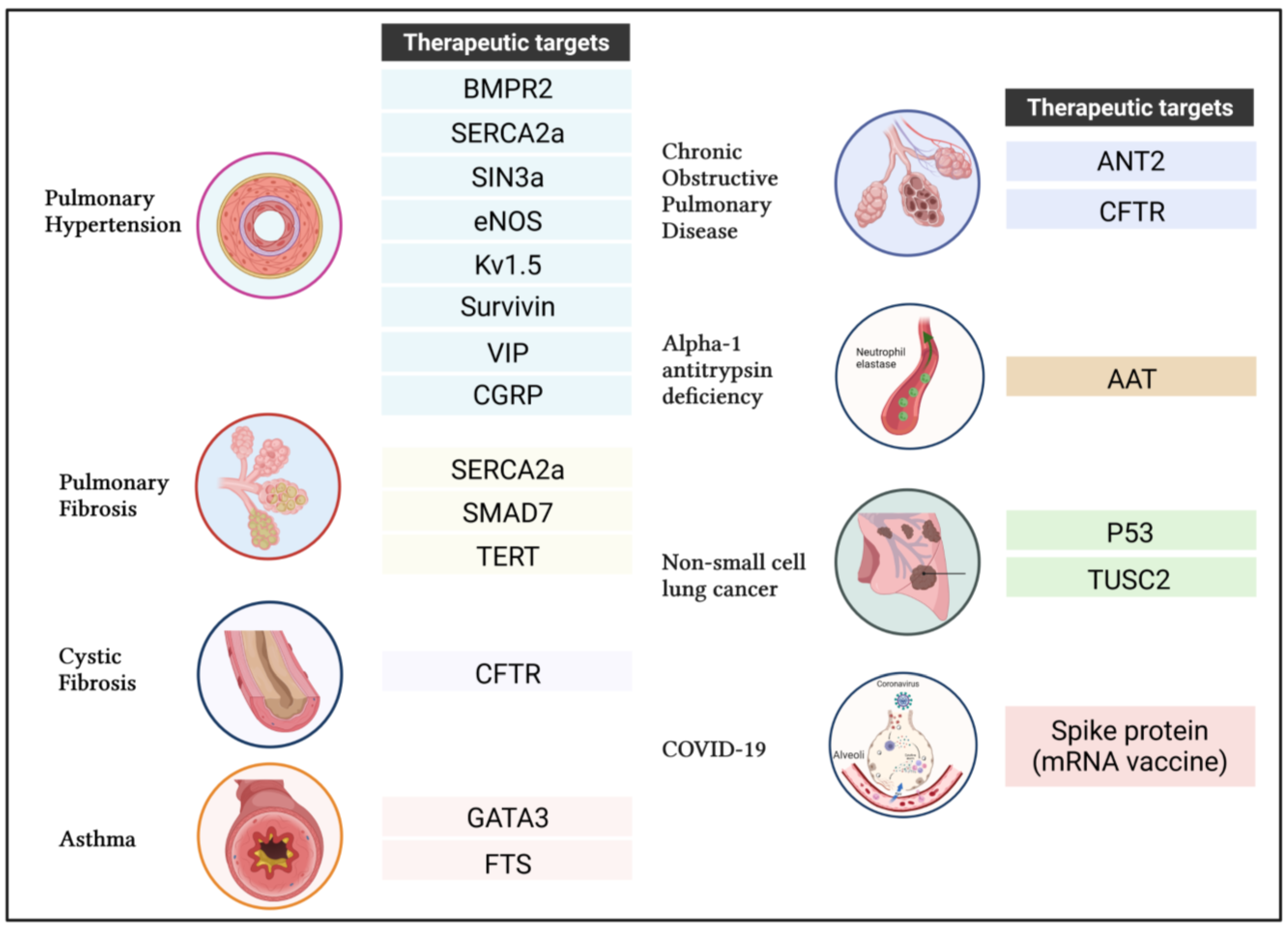
4.1. Pulmonary Arterial Hypertension (PAH)
4.1.1. Bone Morphogenetic Protein Type 2 Receptor (BMPR2)
4.1.2. Sarco-Endoplasmic Reticulum Calcium-ATPase 2a (SERCA2a)
4.1.3. SIN3 Transcription Regulator Family Member A
4.1.4. Endothelial Nitric Oxide Synthase (ENOS)
4.1.5. Voltage-Gated Potassium Channel Kv1.5
4.1.6. Survivin
4.1.7. Vasoactive Intestinal Peptide (VIP)
4.1.8. Calcitonin Gene-Related Peptide (CGRP)
4.2. Idiopathic Pulmonary Fibrosis (IPF)
4.2.1. SERCA2a
4.2.2. Caveolin-1
4.2.3. SMAD Family Member 7 (SMAD7)
4.2.4. Telomerase Reverse Transcriptase (TERT)
4.3. Cystic Fibrosis
Cystic Fibrosis Transmembrane Conductance Regulator (CFTR)
4.4. Asthma
4.4.1. GATA-Binding Protein 3 (GATA3)
4.4.2. Thymulin (Formerly Called “Serum Thymus Factor” or FTS)
4.5. Chronic Obstructive Pulmonary Disease (COPD)
4.6. Alpha-1 Antitrypsin Deficiency (AATD)
4.7. Non-Small-Cell Lung Cancer (NSCLC)
4.7.1. Tumor Suppressor p53
4.7.2. Tumor Suppressor Candidate 2 Gene (TUSC2)
4.8. Coronavirus Disease (COVID-19)
5. Limitations of Gene Therapy
6. Future Prospects
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Soriano, J.B.; Kendrick, P.J.; Paulson, K.R.; Gupta, V.; Abrams, E.M.; Adedoyin, R.A.; Adhikari, T.B.; Advani, S.M.; Agrawal, A.; Ahmadian, E.; et al. Prevalence and attributable health burden of chronic respiratory diseases, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Respir. Med. 2020, 8, 585–596. [Google Scholar] [CrossRef]
- Banuls, L.; Pellicer, D.; Castillo, S.; Navarro-Garcia, M.M.; Magallon, M.; Gonzalez, C.; Dasi, F. Gene Therapy in Rare Respiratory Diseases: What Have We Learned So Far? J. Clin. Med. 2020, 9, 2577. [Google Scholar] [CrossRef] [PubMed]
- Friedmann, T.; Roblin, R. Gene therapy for human genetic disease? Science 1972, 175, 949–955. [Google Scholar] [CrossRef] [PubMed]
- International, P.P.H.C.; Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A., 3rd; Loyd, J.E.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Stoltz, D.A.; Meyerholz, D.K.; Welsh, M.J. Origins of cystic fibrosis lung disease. N. Engl. J. Med. 2015, 372, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; McCray, P.B., Jr.; Sinn, P.L. Cystic Fibrosis Gene Therapy: Looking Back, Looking Forward. Genes 2018, 9, 538. [Google Scholar] [CrossRef] [PubMed]
- Driskell, R.A.; Engelhardt, J.F. Current status of gene therapy for inherited lung diseases. Annu. Rev. Physiol. 2003, 65, 585–612. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bishop, E.S.; Zhang, R.; Yu, X.; Farina, E.M.; Yan, S.; Zhao, C.; Zheng, Z.; Shu, Y.; Wu, X.; et al. Adenovirus-Mediated Gene Delivery: Potential Applications for Gene and Cell-Based Therapies in the New Era of Personalized Medicine. Genes Dis. 2017, 4, 43–63. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Corrigan-Curay, J.; Cohen-Haguenauer, O.; O’Reilly, M.; Ross, S.R.; Fan, H.; Rosenberg, N.; Somia, N.; King, N.; Friedmann, T.; Dunbar, C.; et al. Challenges in vector and trial design using retroviral vectors for long-term gene correction in hematopoietic stem cell gene therapy. Mol. Ther. 2012, 20, 1084–1094. [Google Scholar] [CrossRef] [PubMed]
- Wall, D.A.; Krueger, J. Chimeric antigen receptor T cell therapy comes to clinical practice. Curr. Oncol. 2020, 27, S115–S123. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.; Beekman, J.M.; Boyd, A.C.; Brand, J.; Carlon, M.S.; Connolly, M.M.; Chan, M.; Conlon, S.; Davidson, H.E.; Davies, J.C.; et al. Preparation for a first-in-man lentivirus trial in patients with cystic fibrosis. Thorax 2017, 72, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Mitomo, K.; Griesenbach, U.; Inoue, M.; Somerton, L.; Meng, C.; Akiba, E.; Tabata, T.; Ueda, Y.; Frankel, G.M.; Farley, R.; et al. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Mol. Ther. 2010, 18, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Aneja, M.K.; Geiger, J.P.; Himmel, A.; Rudolph, C. Targeted gene delivery to the lung. Expert. Opin. Drug Deliv. 2009, 6, 567–583. [Google Scholar] [CrossRef]
- Sinn, P.L.; Cooney, A.L.; Oakland, M.; Dylla, D.E.; Wallen, T.J.; Pezzulo, A.A.; Chang, E.H.; McCray, P.B., Jr. Lentiviral vector gene transfer to porcine airways. Mol. Ther. Nucleic Acids. 2012, 1, e56. [Google Scholar] [CrossRef] [PubMed]
- Sinn, P.L.; Burnight, E.R.; Hickey, M.A.; Blissard, G.W.; McCray, P.B., Jr. Persistent gene expression in mouse nasal epithelia following feline immunodeficiency virus-based vector gene transfer. J. Virol. 2005, 79, 12818–12827. [Google Scholar] [CrossRef]
- Ferrari, S.; Griesenbach, U.; Iida, A.; Farley, R.; Wright, A.M.; Zhu, J.; Munkonge, F.M.; Smith, S.N.; You, J.; Ban, H.; et al. Sendai virus-mediated CFTR gene transfer to the airway epithelium. Gene Ther. 2007, 14, 1371–1379. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Anson, D.S. The use of retroviral vectors for gene therapy-what are the risks? A review of retroviral pathogenesis and its relevance to retroviral vector-mediated gene delivery. Genet. Vaccines Ther. 2004, 2, 9. [Google Scholar] [CrossRef]
- Kay, M.A.; Glorioso, J.C.; Naldini, L. Viral vectors for gene therapy: The art of turning infectious agents into vehicles of therapeutics. Nat. Med. 2001, 7, 33–40. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Q.; Ertl, H.C.; Wilson, J.M. Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J. Virol. 1995, 69, 2004–2015. [Google Scholar] [CrossRef]
- Murata, T.; Hori, M.; Lee, S.; Nakamura, A.; Kohama, K.; Karaki, H.; Ozaki, H. Vascular endothelium has a local anti-adenovirus vector system and glucocorticoid optimizes its gene transduction. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1796–1803. [Google Scholar] [CrossRef] [PubMed]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccin. Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Cots, D.; Bosch, A.; Chillon, M. Helper dependent adenovirus vectors: Progress and future prospects. Curr. Gene. Ther. 2013, 13, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Ouyang, H.; Grasemann, H.; Bartlett, C.; Du, K.; Duan, R.; Shi, F.; Estrada, M.; Seigel, K.E.; Coates, A.L.; et al. Transducing Airway Basal Cells with a Helper-Dependent Adenoviral Vector for Lung Gene Therapy. Hum. Gene Ther. 2018, 29, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Sondhi, D.; Stiles, K.M.; De, B.P.; Crystal, R.G. Genetic Modification of the Lung Directed Toward Treatment of Human Disease. Hum. Gene Ther. 2017, 28, 3–84. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; Thornell, I.M.; Singh, B.K.; Shah, V.S.; Stoltz, D.A.; McCray, P.B., Jr.; Zabner, J.; Sinn, P.L. A Novel AAV-mediated Gene Delivery System Corrects CFTR Function in Pigs. Am. J. Respir. Cell Mol. Biol. 2019, 61, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Allen, J.M.; Miller, A.D. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat. Biotechnol. 2002, 20, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Fakhiri, J.; Schneider, M.A.; Puschhof, J.; Stanifer, M.; Schildgen, V.; Holderbach, S.; Voss, Y.; El Andari, J.; Schildgen, O.; Boulant, S.; et al. Novel Chimeric Gene Therapy Vectors Based on Adeno-Associated Virus and Four Different Mammalian Bocaviruses. Mol. Ther. Methods Clin. Dev. 2019, 12, 202–222. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Feng, Z.; Sun, X.; Zhang, Y.; Zou, W.; Wang, Z.; Jensen-Cody, C.; Liang, B.; Park, S.Y.; Qiu, J.; et al. Human Bocavirus Type-1 Capsid Facilitates the Transduction of Ferret Airways by Adeno-Associated Virus Genomes. Hum. Gene Ther. 2017, 28, 612–625. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Keiser, N.W.; Song, Y.; Deng, X.; Cheng, F.; Qiu, J.; Engelhardt, J.F. A novel chimeric adenoassociated virus 2/human bocavirus 1 parvovirus vector efficiently transduces human airway epithelia. Mol. Ther. 2013, 21, 2181–2194. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, D.V.; Maheshri, N. Directed evolution of AAV mutants for enhanced gene delivery. Conf. Proc. IEEE Eng. Med. Biol Soc. 2004, 2004, 3520–3523. [Google Scholar] [CrossRef]
- Zhong, L.; Li, B.; Mah, C.S.; Govindasamy, L.; Agbandje-McKenna, M.; Cooper, M.; Herzog, R.W.; Zolotukhin, I.; Warrington, K.H., Jr.; Weigel-Van Aken, K.A.; et al. Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. USA 2008, 105, 7827–7832. [Google Scholar] [CrossRef]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol. Ther. 2006, 14, 316–327. [Google Scholar] [CrossRef]
- Lompre, A.M.; Hadri, L.; Merlet, E.; Keuylian, Z.; Mougenot, N.; Karakikes, I.; Chen, J.; Atassi, F.; Marchand, A.; Blaise, R.; et al. Efficient transduction of vascular smooth muscle cells with a translational AAV2.5 vector: A new perspective for in-stent restenosis gene therapy. Gene Ther. 2013, 20, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Markusic, D.M.; Herzog, R.W.; Aslanidi, G.V.; Hoffman, B.E.; Li, B.; Li, M.; Jayandharan, G.R.; Ling, C.; Zolotukhin, I.; Ma, W.; et al. High-efficiency transduction and correction of murine hemophilia B using AAV2 vectors devoid of multiple surface-exposed tyrosines. Mol. Ther. 2010, 18, 2048–2056. [Google Scholar] [CrossRef]
- Petrs-Silva, H.; Dinculescu, A.; Li, Q.; Min, S.H.; Chiodo, V.; Pang, J.J.; Zhong, L.; Zolotukhin, S.; Srivastava, A.; Lewin, A.S.; et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol. Ther. 2009, 17, 463–471. [Google Scholar] [CrossRef]
- Martini, S.V.; da Silva, A.L.; Ferreira, D.; Gomes, K.; Ornellas, F.M.; Lopes-Pacheco, M.; Zin, E.; Petrs-Silva, H.; Rocco, P.R.; Morales, M.M. Single tyrosine mutation in AAV8 vector capsid enhances gene lung delivery and does not alter lung morphofunction in mice. Cell. Physiol. Biochem. 2014, 34, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Pacheco, M.; Kitoko, J.Z.; Morales, M.M.; Petrs-Silva, H.; Rocco, P.R.M. Self-complementary and tyrosine-mutant rAAV vectors enhance transduction in cystic fibrosis bronchial epithelial cells. Exp. Cell Res. 2018, 372, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Grieger, J.C.; Samulski, R.J. Packaging capacity of adeno-associated virus serotypes: Impact of larger genomes on infectivity and postentry steps. J. Virol. 2005, 79, 9933–9944. [Google Scholar] [CrossRef]
- Gregory, S.M.; Nazir, S.A.; Metcalf, J.P. Implications of the innate immune response to adenovirus and adenoviral vectors. Future Virol. 2011, 6, 357–374. [Google Scholar] [CrossRef]
- Felberbaum, R.S. The baculovirus expression vector system: A commercial manufacturing platform for viral vaccines and gene therapy vectors. Biotechnol. J. 2015, 10, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Tokusumi, Y.; Ban, H.; Shirakura, M.; Kanaya, T.; Yoshizaki, M.; Hironaka, T.; Nagai, Y.; Iida, A.; Hasegawa, M. Recombinant Sendai virus vectors deleted in both the matrix and the fusion genes: Efficient gene transfer with preferable properties. J. Gene Med. 2004, 6, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Durymanov, M.; Reineke, J. Non-viral Delivery of Nucleic Acids: Insight Into Mechanisms of Overcoming Intracellular Barriers. Front. Pharmacol. 2018, 9, 971. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.K.; Vermerris, W. Recent Advances in Nanomaterials for Gene Delivery-A Review. Nanomaterials 2017, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Ball, R.L.; Bajaj, P.; Whitehead, K.A. Achieving long-term stability of lipid nanoparticles: Examining the effect of pH, temperature, and lyophilization. Int. J. Nanomedicine 2017, 12, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, I.C.; Eltoukhy, A.A.; Anderson, D.G.; Costa, K.D. Lipidoid mRNA Nanoparticles for Myocardial Delivery in Rodents. Methods Mol. Biol. 2017, 1521, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, I.C.; Eltoukhy, A.A.; Fish, K.M.; Nonnenmacher, M.; Ishikawa, K.; Chen, J.; Hajjar, R.J.; Anderson, D.G.; Costa, K.D. Myocardial Delivery of Lipidoid Nanoparticle Carrying modRNA Induces Rapid and Transient Expression. Mol. Ther. 2016, 24, 66–75. [Google Scholar] [CrossRef] [PubMed]
- McKiernan, P.J.; Cunningham, O.; Greene, C.M.; Cryan, S.A. Targeting miRNA-based medicines to cystic fibrosis airway epithelial cells using nanotechnology. Int. J. Nanomedicine 2013, 8, 3907–3915. [Google Scholar] [CrossRef] [PubMed]
- McLendon, J.M.; Joshi, S.R.; Sparks, J.; Matar, M.; Fewell, J.G.; Abe, K.; Oka, M.; McMurtry, I.F.; Gerthoffer, W.T. Lipid nanoparticle delivery of a microRNA-145 inhibitor improves experimental pulmonary hypertension. J. Control. Release 2015, 210, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Satija, S.; Paudel, K.R.; Malyla, V.; Kannaujiya, V.K.; Chellappan, D.K.; Bebawy, M.; Hansbro, P.M.; Wich, P.R.; Dua, K. Targeting respiratory diseases using miRNA inhibitor based nanotherapeutics: Current status and future perspectives. Nanomedicine 2021, 31, 102303. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.; Paudel, K.R.; Shukla, S.D.; Allam, V.; Kannaujiya, V.K.; Panth, N.; Das, A.; Parihar, V.K.; Chakraborty, A.; Ali, M.K.; et al. Recent trends of NFkappaB decoy oligodeoxynucleotide-based nanotherapeutics in lung diseases. J. Control. Release 2021, 337, 629–644. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, D.; Ungaro, F.; Giovino, C.; Polimeno, A.; Quaglia, F.; Carnuccio, R. Sustained inhibition of IL-6 and IL-8 expression by decoy ODN to NF-kappaB delivered through respirable large porous particles in LPS-stimulated cystic fibrosis bronchial cells. J. Gene Med. 2011, 13, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Wardwell, P.R.; Bader, R.A. Immunomodulation of cystic fibrosis epithelial cells via NF-kappaB decoy oligonucleotide-coated polysaccharide nanoparticles. J. Biomed. Mater. Res. A 2015, 103, 1622–1631. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Egashira, K.; Chen, L.; Nakano, K.; Iwata, E.; Miyagawa, M.; Tsujimoto, H.; Hara, K.; Morishita, R.; Sueishi, K.; et al. Nanoparticle-mediated delivery of nuclear factor kappaB decoy into lungs ameliorates monocrotaline-induced pulmonary arterial hypertension. Hypertension 2009, 53, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.W.; Boyd, A.C.; Porteous, D.J.; Davies, G.; Davies, J.C.; Griesenbach, U.; Higgins, T.E.; Gill, D.R.; Hyde, S.C.; Innes, J.A.; et al. A Phase I/IIa Safety and Efficacy Study of Nebulized Liposome-mediated Gene Therapy for Cystic Fibrosis Supports a Multidose Trial. Am. J. Respir. Crit. Care Med. 2015, 192, 1389–1392. [Google Scholar] [CrossRef] [PubMed]
- Alton, E.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691. [Google Scholar] [CrossRef]
- Konstan, M.W.; Davis, P.B.; Wagener, J.S.; Hilliard, K.A.; Stern, R.C.; Milgram, L.J.; Kowalczyk, T.H.; Hyatt, S.L.; Fink, T.L.; Gedeon, C.R.; et al. Compacted DNA nanoparticles administered to the nasal mucosa of cystic fibrosis subjects are safe and demonstrate partial to complete cystic fibrosis transmembrane regulator reconstitution. Hum. Gene Ther. 2004, 15, 1255–1269. [Google Scholar] [CrossRef] [PubMed]
- Alapati, D.; Morrisey, E.E. Gene Editing and Genetic Lung Disease. Basic Research Meets Therapeutic Application. Am. J. Respir. Cell Mol. Biol 2017, 56, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wen, Y.; Guo, X. CRISPR/Cas9 for genome editing: Progress, implications and challenges. Hum. Mol. Genet. 2014, 23, R40–R46. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Komor, A.C.; Rees, H.A.; Packer, M.S.; Badran, A.H.; Bryson, D.I.; Liu, D.R. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Alapati, D.; Zacharias, W.J.; Hartman, H.A.; Rossidis, A.C.; Stratigis, J.D.; Ahn, N.J.; Coons, B.; Zhou, S.; Li, H.; Singh, K.; et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med. 2019, 11, 8375. [Google Scholar] [CrossRef]
- Maule, G.; Arosio, D.; Cereseto, A. Gene Therapy for Cystic Fibrosis: Progress and Challenges of Genome Editing. Int. J. Mol. Sci 2020, 21, 3903. [Google Scholar] [CrossRef] [PubMed]
- Mohammadzadeh, I.; Qujeq, D.; Yousefi, T.; Ferns, G.A.; Maniati, M.; Vaghari-Tabari, M. CRISPR/Cas9 gene editing: A new therapeutic approach in the treatment of infection and autoimmunity. IUBMB Life 2020, 72, 1603–1621. [Google Scholar] [CrossRef]
- Nair, J.; Nair, A.; Veerappan, S.; Sen, D. Translatable gene therapy for lung cancer using Crispr CAS9-an exploratory review. Cancer Gene Ther. 2020, 27, 116–124. [Google Scholar] [CrossRef]
- Ferdosi, S.R.; Ewaisha, R.; Moghadam, F.; Krishna, S.; Park, J.G.; Ebrahimkhani, M.R.; Kiani, S.; Anderson, K.S. Multifunctional CRISPR-Cas9 with engineered immunosilenced human T cell epitopes. Nat. Commun. 2019, 10, 1842. [Google Scholar] [CrossRef]
- Cyranoski, D. CRISPR gene-editing tested in a person for the first time. Nature 2016, 539, 479. [Google Scholar] [CrossRef]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Ooi, K.H.; Liu, M.M.; Tay, J.W.D.; Teo, S.Y.; Kaewsapsak, P.; Jin, S.; Lee, C.K.; Hou, J.; Maurer-Stroh, S.; Lin, W.; et al. An engineered CRISPR-Cas12a variant and DNA-RNA hybrid guides enable robust and rapid COVID-19 testing. Nat. Commun. 2021, 12, 1739. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Wang, X.; Li, S.; Luo, W.; Zhang, X.; Wang, C.; Chen, Q.; Yu, S.; Tai, J.; Wang, Y. Rapid, Ultrasensitive, and Highly Specific Diagnosis of COVID-19 by CRISPR-Based Detection. ACS Sens. 2021, 6, 881–888. [Google Scholar] [CrossRef] [PubMed]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef] [PubMed]
- Kantor, A.; McClements, M.E.; MacLaren, R.E. CRISPR-Cas9 DNA Base-Editing and Prime-Editing. Int. J. Mol. Sci. 2020, 21, 6240. [Google Scholar] [CrossRef] [PubMed]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, I.G. Prime Editing: Genome Editing for Rare Genetic Diseases Without Double-Strand Breaks or Donor DNA. Front. Genet. 2020, 11, 528. [Google Scholar] [CrossRef] [PubMed]
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. 2013, 22, 543–551. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.C.; Potoka, K.C.; Champion, H.C.; Mora, A.L.; Gladwin, M.T. Pulmonary arterial hypertension: The clinical syndrome. Circ. Res. 2014, 115, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Janostiak, R.; Lezoualc’h, F.; Hadri, L. Targeting epigenetic mechanisms as an emerging therapeutic strategy in pulmonary hypertension disease. Vasc Biol 2020, 2, R17–R34. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Pradhan, N.; Hadri, L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev. Cardiovasc Med. 2020, 21, 163–179. [Google Scholar] [CrossRef]
- Duarte, J.D.; Hanson, R.L.; Machado, R.F. Pharmacologic treatments for pulmonary hypertension: Exploring pharmacogenomics. Future Cardiol. 2013, 9, 335–349. [Google Scholar] [CrossRef] [PubMed]
- Meloche, J.; Paulin, R.; Courboulin, A.; Lambert, C.; Barrier, M.; Bonnet, P.; Bisserier, M.; Roy, M.; Sussman, M.A.; Agharazii, M.; et al. RAGE-dependent activation of the oncoprotein Pim1 plays a critical role in systemic vascular remodeling processes. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2114–2124. [Google Scholar] [CrossRef] [PubMed]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone inhibits the Src/STAT3 constitutive activation in pulmonary arterial hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef]
- Courboulin, A.; Tremblay, V.L.; Barrier, M.; Meloche, J.; Jacob, M.H.; Chapolard, M.; Bisserier, M.; Paulin, R.; Lambert, C.; Provencher, S.; et al. Kruppel-like factor 5 contributes to pulmonary artery smooth muscle proliferation and resistance to apoptosis in human pulmonary arterial hypertension. Respir. Res. 2011, 12, 128. [Google Scholar] [CrossRef]
- Meloche, J.; Courchesne, A.; Barrier, M.; Carter, S.; Bisserier, M.; Paulin, R.; Lauzon-Joset, J.F.; Breuils-Bonnet, S.; Tremblay, E.; Biardel, S.; et al. Critical role for the advanced glycation end-products receptor in pulmonary arterial hypertension etiology. J. Am. Heart Assoc. 2013, 2, e005157. [Google Scholar] [CrossRef]
- Jones, C.; Bisserier, M.; Bueno-Beti, C.; Bonnet, G.; Neves-Zaph, S.; Lee, S.Y.; Milara, J.; Dorfmuller, P.; Humbert, M.; Leopold, J.A.; et al. A novel secreted-cAMP pathway inhibits pulmonary hypertension via a feed-forward mechanism. Cardiovasc. Res. 2020, 116, 1500–1513. [Google Scholar] [CrossRef] [PubMed]
- Fazal, S.; Bisserier, M.; Hadri, L. Molecular and Genetic Profiling for Precision Medicines in Pulmonary Arterial Hypertension. Cells 2021, 10, 638. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.D.; Southgate, L.; Eichstaedt, C.A.; Aldred, M.A.; Austin, E.D.; Best, D.H.; Chung, W.K.; Benjamin, N.; Elliott, C.G.; Eyries, M.; et al. Pulmonary Arterial Hypertension: A Current Perspective on Established and Emerging Molecular Genetic Defects. Hum. Mutat. 2015, 36, 1113–1127. [Google Scholar] [CrossRef]
- Andruska, A.; Spiekerkoetter, E. Consequences of BMPR2 Deficiency in the Pulmonary Vasculature and Beyond: Contributions to Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2499. [Google Scholar] [CrossRef] [PubMed]
- Feng, F.; Harper, R.L.; Reynolds, P.N. BMPR2 gene delivery reduces mutation-related PAH and counteracts TGF-beta-mediated pulmonary cell signalling. Respirology 2016, 21, 526–532. [Google Scholar] [CrossRef]
- Reynolds, A.M.; Xia, W.; Holmes, M.D.; Hodge, S.J.; Danilov, S.; Curiel, D.T.; Morrell, N.W.; Reynolds, P.N. Bone morphogenetic protein type 2 receptor gene therapy attenuates hypoxic pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1182–L1192. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Moudgil, R.; Hashimoto, K.; Bonnet, S.; Michelakis, E.D.; Archer, S.L. Overexpression of human bone morphogenetic protein receptor 2 does not ameliorate monocrotaline pulmonary arterial hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L872–L878. [Google Scholar] [CrossRef] [PubMed]
- Harper, R.L.; Reynolds, A.M.; Bonder, C.S.; Reynolds, P.N. BMPR2 gene therapy for PAH acts via Smad and non-Smad signalling. Respirology 2016, 21, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Orriols, M.; Gomez-Puerto, M.C.; Ten Dijke, P. BMP type II receptor as a therapeutic target in pulmonary arterial hypertension. Cell Mol. Life Sci. 2017, 74, 2979–2995. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Katz, M.G.; Bueno-Beti, C.; Brojakowska, A.; Zhang, S.; Gubara, S.; Kohlbrenner, E.; Fazal, S.; Fargnoli, A.; Dorfmuller, P.; et al. Combination Therapy with STAT3 Inhibitor Enhances SERCA2a-Induced BMPR2 Expression and Inhibits Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2021, 22, 9105. [Google Scholar] [CrossRef] [PubMed]
- Harvey, L.D.; Chan, S.Y. Emerging Metabolic Therapies in Pulmonary Arterial Hypertension. J. Clin. Med. 2017, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium. 2018, 69, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Lipskaia, L.; del Monte, F.; Capiod, T.; Yacoubi, S.; Hadri, L.; Hours, M.; Hajjar, R.J.; Lompre, A.M. Sarco/endoplasmic reticulum Ca2+-ATPase gene transfer reduces vascular smooth muscle cell proliferation and neointima formation in the rat. Circ. Res. 2005, 97, 488–495. [Google Scholar] [CrossRef]
- Hadri, L.; Kratlian, R.G.; Benard, L.; Maron, B.A.; Dorfmuller, P.; Ladage, D.; Guignabert, C.; Ishikawa, K.; Aguero, J.; Ibanez, B.; et al. Therapeutic efficacy of AAV1.SERCA2a in monocrotaline-induced pulmonary arterial hypertension. Circulation 2013, 128, 512–523. [Google Scholar] [CrossRef] [PubMed]
- Strauss, B.; Sassi, Y.; Bueno-Beti, C.; Ilkan, Z.; Raad, N.; Cacheux, M.; Bisserier, M.; Turnbull, I.C.; Kohlbrenner, E.; Hajjar, R.J.; et al. Intra-tracheal gene delivery of aerosolized SERCA2a to the lung suppresses ventricular arrhythmias in a model of pulmonary arterial hypertension. J. Mol. Cell Cardiol. 2019, 127, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Strauss, B.; Bisserier, M.; Obus, E.; Katz, M.G.; Fargnoli, A.; Cacheux, M.; Akar, J.G.; Hummel, J.P.; Hadri, L.; Sassi, Y.; et al. Right predominant electrical remodeling in a pure model of pulmonary hypertension promotes reentrant arrhythmias. Heart Rhythm. 2022, 19, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Ishikawa, K.; Plataki, M.; Bikou, O.; Kohlbrenner, E.; Aguero, J.; Hadri, L.; Zarragoikoetxea, I.; Fish, K.; Leopold, J.A.; et al. Safety and long-term efficacy of AAV1.SERCA2a using nebulizer delivery in a pig model of pulmonary hypertension. Pulm. Circ. 2018, 8, 2045894018799738. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.G.; Fargnoli, A.S.; Gubara, S.M.; Bisserier, M.; Sassi, Y.; Bridges, C.R.; Hajjar, R.J.; Hadri, L. The Left Pneumonectomy Combined with Monocrotaline or Sugen as a Model of Pulmonary Hypertension in Rats. J. Vis. Exp. 2019, 10, 59050. [Google Scholar] [CrossRef] [PubMed]
- Grzenda, A.; Lomberk, G.; Zhang, J.S.; Urrutia, R. Sin3: Master scaffold and transcriptional corepressor. Biochim. Biophys. Acta 2009, 1789, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Mathiyalagan, P.; Zhang, S.; Elmastour, F.; Dorfmuller, P.; Humbert, M.; David, G.; Tarzami, S.; Weber, T.; Perros, F.; et al. Regulation of the Methylation and Expression Levels of the BMPR2 Gene by SIN3a as a Novel Therapeutic Mechanism in Pulmonary Arterial Hypertension. Circulation 2021, 144, 52–73. [Google Scholar] [CrossRef] [PubMed]
- Pearce, C.G.; Najjar, S.F.; Kapadia, M.R.; Murar, J.; Eng, J.; Lyle, B.; Aalami, O.O.; Jiang, Q.; Hrabie, J.A.; Saavedra, J.E.; et al. Beneficial effect of a short-acting NO donor for the prevention of neointimal hyperplasia. Free Radic. Biol. Med. 2008, 44, 73–81. [Google Scholar] [CrossRef]
- Janssens, S.P.; Bloch, K.D.; Nong, Z.; Gerard, R.D.; Zoldhelyi, P.; Collen, D. Adenoviral-mediated transfer of the human endothelial nitric oxide synthase gene reduces acute hypoxic pulmonary vasoconstriction in rats. J. Clin. Investig. 1996, 98, 317–324. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Budts, W.; Pokreisz, P.; Nong, Z.; Van Pelt, N.; Gillijns, H.; Gerard, R.; Lyons, R.; Collen, D.; Bloch, K.D.; Janssens, S. Aerosol gene transfer with inducible nitric oxide synthase reduces hypoxic pulmonary hypertension and pulmonary vascular remodeling in rats. Circulation 2000, 102, 2880–2885. [Google Scholar] [CrossRef] [PubMed]
- Pu, X.; Du, L.; Hu, Y.; Fan, Y.; Xu, Q. Stem/Progenitor Cells and Pulmonary Arterial Hypertension. Arterioscler Thromb. Vasc. Biol. 2021, 41, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Moudgil, R.; Michelakis, E.D.; Archer, S.L. The role of k+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: Implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 2006, 13, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.L.; Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Powell, F.; Haddad, G.H.; Yuan, J.X. Hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Ann. N. Y. Acad. Sci. 2009, 1177, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Platoshyn, O.; Brevnova, E.E.; Burg, E.D.; Yu, Y.; Remillard, C.V.; Yuan, J.X. Acute hypoxia selectively inhibits KCNA5 channels in pulmonary artery smooth muscle cells. Am. J. Physiol. Cell Physiol. 2006, 290, C907–C916. [Google Scholar] [CrossRef] [PubMed]
- Brevnova, E.E.; Platoshyn, O.; Zhang, S.; Yuan, J.X. Overexpression of human KCNA5 increases IK V and enhances apoptosis. Am. J. Physiol. Cell Physiol. 2004, 287, C715–C722. [Google Scholar] [CrossRef] [PubMed]
- Pozeg, Z.I.; Michelakis, E.D.; McMurtry, M.S.; Thebaud, B.; Wu, X.C.; Dyck, J.R.; Hashimoto, K.; Wang, S.; Moudgil, R.; Harry, G.; et al. In vivo gene transfer of the O2-sensitive potassium channel Kv1.5 reduces pulmonary hypertension and restores hypoxic pulmonary vasoconstriction in chronically hypoxic rats. Circulation 2003, 107, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- McMurtry, M.S.; Archer, S.L.; Altieri, D.C.; Bonnet, S.; Haromy, A.; Harry, G.; Bonnet, S.; Puttagunta, L.; Michelakis, E.D. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J. Clin. Investig. 2005, 115, 1479–1491. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, Y.; Cao, L.; Han, H.; Wang, J.; Yang, B.; Nattel, S.; Wang, Z. HERG K+ channel, a regulator of tumor cell apoptosis and proliferation. Cancer Res. 2002, 62, 4843–4848. [Google Scholar]
- Petkov, V.; Mosgoeller, W.; Ziesche, R.; Raderer, M.; Stiebellehner, L.; Vonbank, K.; Funk, G.C.; Hamilton, G.; Novotny, C.; Burian, B.; et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J. Clin. Investig. 2003, 111, 1339–1346. [Google Scholar] [CrossRef]
- Haberl, I.; Frei, K.; Ramsebner, R.; Doberer, D.; Petkov, V.; Albinni, S.; Lang, I.; Lucas, T.; Mosgoeller, W. Vasoactive intestinal peptide gene alterations in patients with idiopathic pulmonary arterial hypertension. Eur. J. Hum. Genet. 2007, 15, 18–22. [Google Scholar] [CrossRef]
- Said, S.I.; Hamidi, S.A.; Dickman, K.G.; Szema, A.M.; Lyubsky, S.; Lin, R.Z.; Jiang, Y.P.; Chen, J.J.; Waschek, J.A.; Kort, S. Moderate pulmonary arterial hypertension in male mice lacking the vasoactive intestinal peptide gene. Circulation 2007, 115, 1260–1268. [Google Scholar] [CrossRef]
- St Hilaire, R.C.; Kadowitz, P.J.; Jeter, J.R., Jr. Adenoviral transfer of vasoactive intestinal peptide (VIP) gene inhibits rat aortic and pulmonary artery smooth muscle cell proliferation. Peptides 2009, 30, 2323–2329. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tjen, A.L.S.; Kraiczi, H.; Ekman, R.; Keith, I.M. Sensory CGRP depletion by capsaicin exacerbates hypoxia-induced pulmonary hypertension in rats. Regul. Pept. 1998, 74, 1–10. [Google Scholar] [CrossRef]
- Chattergoon, N.N.; D’Souza, F.M.; Deng, W.; Chen, H.; Hyman, A.L.; Kadowitz, P.J.; Jeter, J.R., Jr. Antiproliferative effects of calcitonin gene-related peptide in aortic and pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L202–L211. [Google Scholar] [CrossRef] [PubMed]
- Champion, H.C.; Bivalacqua, T.J.; Toyoda, K.; Heistad, D.D.; Hyman, A.L.; Kadowitz, P.J. In vivo gene transfer of prepro-calcitonin gene-related peptide to the lung attenuates chronic hypoxia-induced pulmonary hypertension in the mouse. Circulation 2000, 101, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Bivalacqua, T.J.; Hyman, A.L.; Kadowitz, P.J.; Paolocci, N.; Kass, D.A.; Champion, H.C. Role of calcitonin gene-related peptide (CGRP) in chronic hypoxia-induced pulmonary hypertension in the mouse. Influence of gene transfer in vivo. Regul. Pept. 2002, 108, 129–133. [Google Scholar] [CrossRef]
- Zhao, Q.; Liu, Z.; Wang, Z.; Yang, C.; Liu, J.; Lu, J. Effect of prepro-calcitonin gene-related peptide-expressing endothelial progenitor cells on pulmonary hypertension. Ann. Thorac. Surg. 2007, 84, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Gunther, A.; Korfei, M.; Mahavadi, P.; von der Beck, D.; Ruppert, C.; Markart, P. Unravelling the progressive pathophysiology of idiopathic pulmonary fibrosis. Eur. Respir. Rev. 2012, 21, 152–160. [Google Scholar] [CrossRef]
- Wolters, P.J.; Collard, H.R.; Jones, K.D. Pathogenesis of idiopathic pulmonary fibrosis. Annu. Rev. Pathol. 2014, 9, 157–179. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Milara, J.; Abdeldjebbar, Y.; Gubara, S.; Jones, C.; Bueno-Beti, C.; Chepurko, E.; Kohlbrenner, E.; Katz, M.G.; Tarzami, S.; et al. AAV1.SERCA2a Gene Therapy Reverses Pulmonary Fibrosis by Blocking the STAT3/FOXM1 Pathway and Promoting the SNON/SKI Axis. Mol. Ther. 2020, 28, 394–410. [Google Scholar] [CrossRef] [PubMed]
- Bisserier, M.; Hadri, L. Lung-targeted SERCA2a Gene Therapy: From Discovery to Therapeutic Application in Bleomycin-Induced Pulmonary Fibrosis. J. Cell Immunol. 2020, 2, 149–156. [Google Scholar] [PubMed]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G. Caveolin, a protein component of caveolae membrane coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Bucci, M.; Gratton, J.P.; Rudic, R.D.; Acevedo, L.; Roviezzo, F.; Cirino, G.; Sessa, W.C. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 2000, 6, 1362–1367. [Google Scholar] [CrossRef]
- Wang, X.M.; Zhang, Y.; Kim, H.P.; Zhou, Z.; Feghali-Bostwick, C.A.; Liu, F.; Ifedigbo, E.; Xu, X.; Oury, T.D.; Kaminski, N.; et al. Caveolin-1: A critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J. Exp. Med. 2006, 203, 2895–2906. [Google Scholar] [CrossRef]
- Nakao, A.; Fujii, M.; Matsumura, R.; Kumano, K.; Saito, Y.; Miyazono, K.; Iwamoto, I. Transient gene transfer and expression of Smad7 prevents bleomycin-induced lung fibrosis in mice. J. Clin. Investig. 1999, 104, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Colebatch, A.J.; Dobrovic, A.; Cooper, W.A. TERT gene: Its function and dysregulation in cancer. J. Clin. Pathol. 2019, 72, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, A.; Gomez-Lopez, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. Elife 2018, 7, 31299. [Google Scholar] [CrossRef]
- Brown, S.D.; White, R.; Tobin, P. Keep them breathing: Cystic fibrosis pathophysiology, diagnosis, and treatment. JAAPA 2017, 30, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, C.; Davies, J.C. Current and future treatment options for cystic fibrosis lung disease: Latest evidence and clinical implications. Ther. Adv. Chronic. Dis. 2016, 7, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Caplan, A.I. Adult mesenchymal stem cells: An innovative therapeutic for lung diseases. Discov. Med. 2010, 9, 337–345. [Google Scholar]
- Cambraia, A.; Junior, M.C.; Zembrzuski, V.M.; Junqueira, R.M.; Cabello, P.H.; de Cabello, G.M.K. Next-Generation Sequencing for Molecular Diagnosis of Cystic Fibrosis in a Brazilian Cohort. Dis. Markers 2021, 2021, 9812074. [Google Scholar] [CrossRef] [PubMed]
- Donnelley, M.; Parsons, D.W. Gene Therapy for Cystic Fibrosis Lung Disease: Overcoming the Barriers to Translation to the Clinic. Front. Pharmacol. 2018, 9, 1381. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Couture, L.A.; Gregory, R.J.; Graham, S.M.; Smith, A.E.; Welsh, M.J. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell 1993, 75, 207–216. [Google Scholar] [CrossRef]
- Knowles, M.R.; Hohneker, K.W.; Zhou, Z.; Olsen, J.C.; Noah, T.L.; Hu, P.C.; Leigh, M.W.; Engelhardt, J.F.; Edwards, L.J.; Jones, K.R.; et al. A controlled study of adenoviral-vector-mediated gene transfer in the nasal epithelium of patients with cystic fibrosis. N. Engl. J. Med. 1995, 333, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.G.; McElvaney, N.G.; Herena, J.; Crystal, R.G. Modification of nasal epithelial potential differences of individuals with cystic fibrosis consequent to local administration of a normal CFTR cDNA adenovirus gene transfer vector. Hum. Gene Ther. 1995, 6, 1487–1496. [Google Scholar] [CrossRef] [PubMed]
- Zabner, J.; Ramsey, B.W.; Meeker, D.P.; Aitken, M.L.; Balfour, R.P.; Gibson, R.L.; Launspach, J.; Moscicki, R.A.; Richards, S.M.; Standaert, T.A.; et al. Repeat administration of an adenovirus vector encoding cystic fibrosis transmembrane conductance regulator to the nasal epithelium of patients with cystic fibrosis. J. Clin. Investig. 1996, 97, 1504–1511. [Google Scholar] [CrossRef] [PubMed]
- Cooney, A.L.; Singh, B.K.; Loza, L.M.; Thornell, I.M.; Hippee, C.E.; Powers, L.S.; Ostedgaard, L.S.; Meyerholz, D.K.; Wohlford-Lenane, C.; Stoltz, D.A.; et al. Widespread airway distribution and short-term phenotypic correction of cystic fibrosis pigs following aerosol delivery of piggyBac/adenovirus. Nucleic Acids Res. 2018, 46, 9591–9600. [Google Scholar] [CrossRef] [PubMed]
- Guggino, W.B.; Cebotaru, L. Adeno-Associated Virus (AAV) gene therapy for cystic fibrosis: Current barriers and recent developments. Expert. Opin. Biol. Ther. 2017, 17, 1265–1273. [Google Scholar] [CrossRef] [PubMed]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, S.; Salahudeen, A.A.; Sellers, Z.M.; Bravo, D.T.; Choi, S.S.; Batish, A.; Le, W.; Baik, R.; de la, O.S.; Kaushik, M.P.; et al. High-Efficiency, Selection-free Gene Repair in Airway Stem Cells from Cystic Fibrosis Patients Rescues CFTR Function in Differentiated Epithelia. Cell Stem Cell 2020, 26, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Santos, L.; Mention, K.; Cavusoglu-Doran, K.; Sanz, D.J.; Bacalhau, M.; Lopes-Pacheco, M.; Harrison, P.T.; Farinha, C.M. Comparison of Cas9 and Cas12a CRISPR editing methods to correct the W1282X-CFTR mutation. J. Cyst. Fibros. 2022, 21, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Oren, Y.S.; Avizur-Barchad, O.; Ozeri-Galai, E.; Elgrabli, R.; Schirelman, M.R.; Blinder, T.; Stampfer, C.D.; Ordan, M.; Laselva, O.; Cohen-Cymberknoh, M.; et al. Antisense oligonucleotide splicing modulation as a novel Cystic Fibrosis therapeutic approach for the W1282X nonsense mutation. J. Cyst. Fibros. 2021, 16, 12. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Sivetz, N.; Layne, J.; Voss, D.M.; Yang, L.; Zhang, Q.; Krainer, A.R. Exon-skipping antisense oligonucleotides for cystic fibrosis therapy. Proc. Natl. Acad. Sci. USA 2022, 119, 118. [Google Scholar] [CrossRef] [PubMed]
- Limb, S.L.; Brown, K.C.; Wood, R.A.; Wise, R.A.; Eggleston, P.A.; Tonascia, J.; Adkinson, N.F., Jr. Irreversible lung function deficits in young adults with a history of childhood asthma. J. Allergy Clin. Immunol. 2005, 116, 1213–1219. [Google Scholar] [CrossRef] [PubMed]
- Horak, F.; Doberer, D.; Eber, E.; Horak, E.; Pohl, W.; Riedler, J.; Szepfalusi, Z.; Wantke, F.; Zacharasiewicz, A.; Studnicka, M. Diagnosis and management of asthma—Statement on the 2015 GINA Guidelines. Wien. Klin. Wochenschr. 2016, 128, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Ghaffar, O.; Olivensstein, R.; Taha, R.A.; Soussi-Gounni, A.; Zhang, D.H.; Ray, A.; Hamid, Q. Gene expression of the GATA-3 transcription factor is increased in atopic asthma. J. Allergy Clin. Immunol. 1999, 103, 215–222. [Google Scholar] [CrossRef]
- Sel, S.; Wegmann, M.; Dicke, T.; Sel, S.; Henke, W.; Yildirim, A.O.; Renz, H.; Garn, H. Effective prevention and therapy of experimental allergic asthma using a GATA-3-specific DNAzyme. J. Allergy Clin. Immunol. 2008, 121, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Homburg, U.; Renz, H.; Timmer, W.; Hohlfeld, J.M.; Seitz, F.; Luer, K.; Mayer, A.; Wacker, A.; Schmidt, O.; Kuhlmann, J.; et al. Safety and tolerability of a novel inhaled GATA3 mRNA targeting DNAzyme in patients with TH2-driven asthma. J. Allergy Clin. Immunol. 2015, 136, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Garn, H.; Renz, H. GATA-3-specific DNAzyme—A novel approach for stratified asthma therapy. Eur. J. Immunol. 2017, 47, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Yara, S.; Kawakami, K.; Kudeken, N.; Tohyama, M.; Teruya, K.; Chinen, T.; Awaya, A.; Saito, A. FTS reduces bleomycin-induced cytokine and chemokine production and inhibits pulmonary fibrosis in mice. Clin. Exp. Immunol. 2001, 124, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Savino, W.; Dardenne, M. Neuroendocrine control of thymus physiology. Endocr. Rev. 2000, 21, 412–443. [Google Scholar] [CrossRef] [PubMed]
- da Silva, A.L.; Martini, S.V.; Abreu, S.C.; Samary Cdos, S.; Diaz, B.L.; Fernezlian, S.; de Sa, V.K.; Capelozzi, V.L.; Boylan, N.J.; Goya, R.G.; et al. DNA nanoparticle-mediated thymulin gene therapy prevents airway remodeling in experimental allergic asthma. J. Control. Release 2014, 180, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Burney, P.G.; Silverman, E.K.; Celli, B.R.; Vestbo, J.; Wedzicha, J.A.; Wouters, E.F. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers 2015, 1, 15076. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.R.; Mannino, D.M.; Soriano, J.B.; Vermeire, P.A.; Buist, A.S.; Thun, M.J.; Connell, C.; Jemal, A.; Lee, T.A.; Miravitlles, M.; et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur. Respir. J. 2006, 27, 188–207. [Google Scholar] [CrossRef] [PubMed]
- Montuschi, P. Pharmacological treatment of chronic obstructive pulmonary disease. Int J. Chron. Obstruct. Pulmon. Dis. 2006, 1, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Phougat, N.; Ruhil, S.; Dhankhar, S.; Balhara, M.; Chhillar, A.K. Genomics of Chronic Obstructive Pulmonary Disease (COPD); Exploring the SNPs of Protease-Antiprotease Pathway. Curr. Genomics 2013, 14, 204–213. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rab, A.; Rowe, S.M.; Raju, S.V.; Bebok, Z.; Matalon, S.; Collawn, J.F. Cigarette smoke and CFTR: Implications in the pathogenesis of COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L530–L541. [Google Scholar] [CrossRef] [PubMed]
- Torres-Duran, M.; Lopez-Campos, J.L.; Barrecheguren, M.; Miravitlles, M.; Martinez-Delgado, B.; Castillo, S.; Escribano, A.; Baloira, A.; Navarro-Garcia, M.M.; Pellicer, D.; et al. Alpha-1 antitrypsin deficiency: Outstanding questions and future directions. Orphanet J. Rare Dis. 2018, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Dunlea, D.M.; Fee, L.T.; McEnery, T.; McElvaney, N.G.; Reeves, E.P. The impact of alpha-1 antitrypsin augmentation therapy on neutrophil-driven respiratory disease in deficient individuals. J. Inflamm Res. 2018, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Seixas, S.; Marques, P.I. Known Mutations at the Cause of Alpha-1 Antitrypsin Deficiency an Updated Overview of SERPINA1 Variation Spectrum. Appl. Clin. Genet. 2021, 14, 173–194. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.A.; Siegfried, W.; Yoshimura, K.; Yoneyama, K.; Fukayama, M.; Stier, L.E.; Paakko, P.K.; Gilardi, P.; Stratford-Perricaudet, L.D.; Perricaudet, M.; et al. Adenovirus-mediated transfer of a recombinant alpha 1-antitrypsin gene to the lung epithelium in vivo. Science 1991, 252, 431–434. [Google Scholar] [CrossRef] [PubMed]
- Brantly, M.L.; Chulay, J.D.; Wang, L.; Mueller, C.; Humphries, M.; Spencer, L.T.; Rouhani, F.; Conlon, T.J.; Calcedo, R.; Betts, M.R.; et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 16363–16368. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Choi, Y.K.; Campbell-Thompson, M.; Li, C.; Tang, Q.; Crawford, J.M.; Flotte, T.R.; Song, S. Therapeutic level of functional human alpha 1 antitrypsin (hAAT) secreted from murine muscle transduced by adeno-associated virus (rAAV1) vector. J. Gene Med. 2006, 8, 730–735. [Google Scholar] [CrossRef] [PubMed]
- Brantly, M.L.; Spencer, L.T.; Humphries, M.; Conlon, T.J.; Spencer, C.T.; Poirier, A.; Garlington, W.; Baker, D.; Song, S.; Berns, K.I.; et al. Phase I trial of intramuscular injection of a recombinant adeno-associated virus serotype 2 alphal-antitrypsin (AAT) vector in AAT-deficient adults. Hum. Gene Ther. 2006, 17, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Flotte, T.R.; Trapnell, B.C.; Humphries, M.; Carey, B.; Calcedo, R.; Rouhani, F.; Campbell-Thompson, M.; Yachnis, A.T.; Sandhaus, R.A.; McElvaney, N.G.; et al. Phase 2 clinical trial of a recombinant adeno-associated viral vector expressing alpha1-antitrypsin: Interim results. Hum. Gene Ther. 2011, 22, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Chiuchiolo, M.J.; Kaminsky, S.M.; Sondhi, D.; Mancenido, D.; Hollmann, C.; Crystal, R.G. Phase I/II study of intrapleural administration of a serotype rh.10 replication-deficient adeno-associated virus gene transfer vector expressing the human alpha1-antitrypsin cDNA to individuals with alpha1-antitrypsin deficiency. Hum. Gene Ther. Clin. Dev. 2014, 25, 112–133. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Lara-Guerra, H.; Roth, J.A. Gene Therapy for Lung Cancer. Crit. Rev. Oncog. 2016, 21, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Cai, D.W.; Georges, R.N.; Mukhopadhyay, T.; Grimm, E.A.; Roth, J.A. Therapeutic effect of a retroviral wild-type p53 expression vector in an orthotopic lung cancer model. J. Natl. Cancer Inst. 1994, 86, 1458–1462. [Google Scholar] [CrossRef]
- Zhang, W.W.; Fang, X.; Mazur, W.; French, B.A.; Georges, R.N.; Roth, J.A. High-efficiency gene transfer and high-level expression of wild-type p53 in human lung cancer cells mediated by recombinant adenovirus. Cancer Gene Ther. 1994, 1, 5–13. [Google Scholar] [PubMed]
- Bouvet, M.; Fang, B.; Ekmekcioglu, S.; Ji, L.; Bucana, C.D.; Hamada, K.; Grimm, E.A.; Roth, J.A. Suppression of the immune response to an adenovirus vector and enhancement of intratumoral transgene expression by low-dose etoposide. Gene Ther. 1998, 5, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Weill, D.; Mack, M.; Roth, J.; Swisher, S.; Proksch, S.; Merritt, J.; Nemunaitis, J. Adenoviral-mediated p53 gene transfer to non-small cell lung cancer through endobronchial injection. Chest 2000, 118, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.A.; Nguyen, D.; Lawrence, D.D.; Kemp, B.L.; Carrasco, C.H.; Ferson, D.Z.; Hong, W.K.; Komaki, R.; Lee, J.J.; Nesbitt, J.C.; et al. Retrovirus-mediated wild-type p53 gene transfer to tumors of patients with lung cancer. Nat. Med. 1996, 2, 985–991. [Google Scholar] [CrossRef]
- Swisher, S.G.; Roth, J.A.; Nemunaitis, J.; Lawrence, D.D.; Kemp, B.L.; Carrasco, C.H.; Connors, D.G.; El-Naggar, A.K.; Fossella, F.; Glisson, B.S.; et al. Adenovirus-mediated p53 gene transfer in advanced non-small-cell lung cancer. J. Natl. Cancer Inst. 1999, 91, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Saeki, T.; Mhashilkar, A.; Swanson, X.; Zou-Yang, X.H.; Sieger, K.; Kawabe, S.; Branch, C.D.; Zumstein, L.; Meyn, R.E.; Roth, J.A.; et al. Inhibition of human lung cancer growth following adenovirus-mediated mda-7 gene expression in vivo. Oncogene 2002, 21, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Lerman, M.I.; Minna, J.D. The 630-kb lung cancer homozygous deletion region on human chromosome 3p21.3: Identification and evaluation of the resident candidate tumor suppressor genes. The International Lung Cancer Chromosome 3p21.3 Tumor Suppressor Gene Consortium. Cancer Res. 2000, 60, 6116–6133. [Google Scholar]
- Ito, I.; Ji, L.; Tanaka, F.; Saito, Y.; Gopalan, B.; Branch, C.D.; Xu, K.; Atkinson, E.N.; Bekele, B.N.; Stephens, L.C.; et al. Liposomal vector mediated delivery of the 3p FUS1 gene demonstrates potent antitumor activity against human lung cancer in vivo. Cancer Gene Ther. 2004, 11, 733–739. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lu, C.; Stewart, D.J.; Lee, J.J.; Ji, L.; Ramesh, R.; Jayachandran, G.; Nunez, M.I.; Wistuba, I.; Erasmus, J.J.; Hicks, M.E.; et al. Phase I clinical trial of systemically administered TUSC2(FUS1)-nanoparticles mediating functional gene transfer in humans. PLoS ONE 2012, 7, e34833. [Google Scholar] [CrossRef]
- Hamouche, W.; Bisserier, M.; Brojakowska, A.; Eskandari, A.; Fish, K.; Goukassian, D.A.; Hadri, L. Pathophysiology and pharmacological management of pulmonary and cardiovascular features of COVID-19. J. Mol. Cell Cardiol. 2021, 153, 72–85. [Google Scholar] [CrossRef]
- Brojakowska, A.; Eskandari, A.; Bisserier, M.; Bander, J.; Garikipati, V.N.S.; Hadri, L.; Goukassian, D.A.; Fish, K.M. Comorbidities, sequelae, blood biomarkers and their associated clinical outcomes in the Mount Sinai Health System COVID-19 patients. PLoS ONE 2021, 16, e0253660. [Google Scholar] [CrossRef]
- Eskandari, A.; Brojakowska, A.; Bisserier, M.; Bander, J.; Garikipati, V.N.S.; Hadri, L.; Goukassian, D.; Fish, K. Retrospective analysis of demographic factors in COVID-19 patients entering the Mount Sinai Health System. PLoS ONE 2021, 16, e0254707. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.K.; Wang, S.K.; Chu, C.J.; Copland, D.A.; Letizia, A.J.; Costa Verdera, H.; Chiang, J.J.; Sethi, M.; Wang, M.K.; Neidermyer, W.J., Jr.; et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci. Transl. Med. 2021, 13, 3438. [Google Scholar] [CrossRef] [PubMed]
- Sultana, N.; Hadas, Y.; Sharkar, M.T.K.; Kaur, K.; Magadum, A.; Kurian, A.A.; Hossain, N.; Alburquerque, B.; Ahmed, S.; Chepurko, E.; et al. Optimization of 5’ Untranslated Region of Modified mRNA for Use in Cardiac or Hepatic Ischemic Injury. Mol. Ther. Methods Clin. Dev. 2020, 17, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Alagoz, M.; Kherad, N. Advance genome editing technologies in the treatment of human diseases: CRISPR therapy (Review). Int J. Mol. Med. 2020, 46, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Cardenes, N.; Sembrat, J.; Noda, K.; Lovelace, T.; Alvarez, D.; Bittar, H.E.T.; Philips, B.J.; Nouraie, M.; Benos, P.V.; Sanchez, P.G.; et al. Human ex vivo lung perfusion: A novel model to study human lung diseases. Sci. Rep. 2021, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Alysandratos, K.D.; Russo, S.J.; Petcherski, A.; Taddeo, E.P.; Acin-Perez, R.; Villacorta-Martin, C.; Jean, J.C.; Mulugeta, S.; Rodriguez, L.R.; Blum, B.C.; et al. Patient-specific iPSCs carrying an SFTPC mutation reveal the intrinsic alveolar epithelial dysfunction at the inception of interstitial lung disease. Cell Rep. 2021, 36, 109636. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene | PH Models | Vector | Delivery Methods | Results | References |
|---|---|---|---|---|---|
| BMPR2 | Chronic hypoxia Monocrotaline Sugen/hypoxia Pneumonectomy/monocrotaline | Adenovirus AAV1 | Intravenous Intratracheal | ↓ RVSP ↓ mPAP ↓ RV hypertrophy ↓ Vascular remodeling | [90,91,92,93,94,95] |
| SERCA2A | Sugen/hypoxia Monocrotaline Pneumonectomy/monocrotaline Pulmonary vein banding | AAV1 | Intratracheal | ↓ RVSP ↓ mPAP ↓ RV hypertrophy ↓ Vascular remodeling ↑ Cardiac function ↓ PVR | [95,99,100,101,102,103] |
| SIN3A | Sugen/hypoxia Monocrotaline | AAV1 | Intratracheal | ↓ RVSP ↓ mPAP ↓ RV hypertrophy ↓ Vascular remodeling | [105] |
| ENOS | Chronic hypoxia | Adenovirus | Intratracheal | ↓ mPAP ↓ RV hypertrophy ↓ PVR | [107] |
| KV1.5 | Chronic hypoxia | Adenovirus | Intratracheal | ↑ Cardiac function ↓ PVR ↓ RV hypertrophy ↓ Vasoconstriction | [114] |
| SURVIVIN | Monocrotaline | Adenovirus | Intratracheal | ↓Vascular remodeling ↓ PVR ↓ RV hypertrophy | [115] |
| CGRP | Chronic hypoxia | Adenovirus | Intratracheal | ↓RV hypertrophy ↓Vascular remodeling ↓ PVR | [123,124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bisserier, M.; Sun, X.-Q.; Fazal, S.; Turnbull, I.C.; Bonnet, S.; Hadri, L. Novel Insights into the Therapeutic Potential of Lung-Targeted Gene Transfer in the Most Common Respiratory Diseases. Cells 2022, 11, 984. https://doi.org/10.3390/cells11060984
Bisserier M, Sun X-Q, Fazal S, Turnbull IC, Bonnet S, Hadri L. Novel Insights into the Therapeutic Potential of Lung-Targeted Gene Transfer in the Most Common Respiratory Diseases. Cells. 2022; 11(6):984. https://doi.org/10.3390/cells11060984
Chicago/Turabian StyleBisserier, Malik, Xiao-Qing Sun, Shahood Fazal, Irene C. Turnbull, Sébastien Bonnet, and Lahouaria Hadri. 2022. "Novel Insights into the Therapeutic Potential of Lung-Targeted Gene Transfer in the Most Common Respiratory Diseases" Cells 11, no. 6: 984. https://doi.org/10.3390/cells11060984
APA StyleBisserier, M., Sun, X.-Q., Fazal, S., Turnbull, I. C., Bonnet, S., & Hadri, L. (2022). Novel Insights into the Therapeutic Potential of Lung-Targeted Gene Transfer in the Most Common Respiratory Diseases. Cells, 11(6), 984. https://doi.org/10.3390/cells11060984