
Synergistic Anti-Ageing through Senescent Cells Specific Reprogramming


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- To propose synergistic anti-ageing in order to clarify the idea of combined anti-ageing with multiple rejuvenating factors and to serve the next research on new key pathways that can be applied in combination with known anti-ageing pathways.
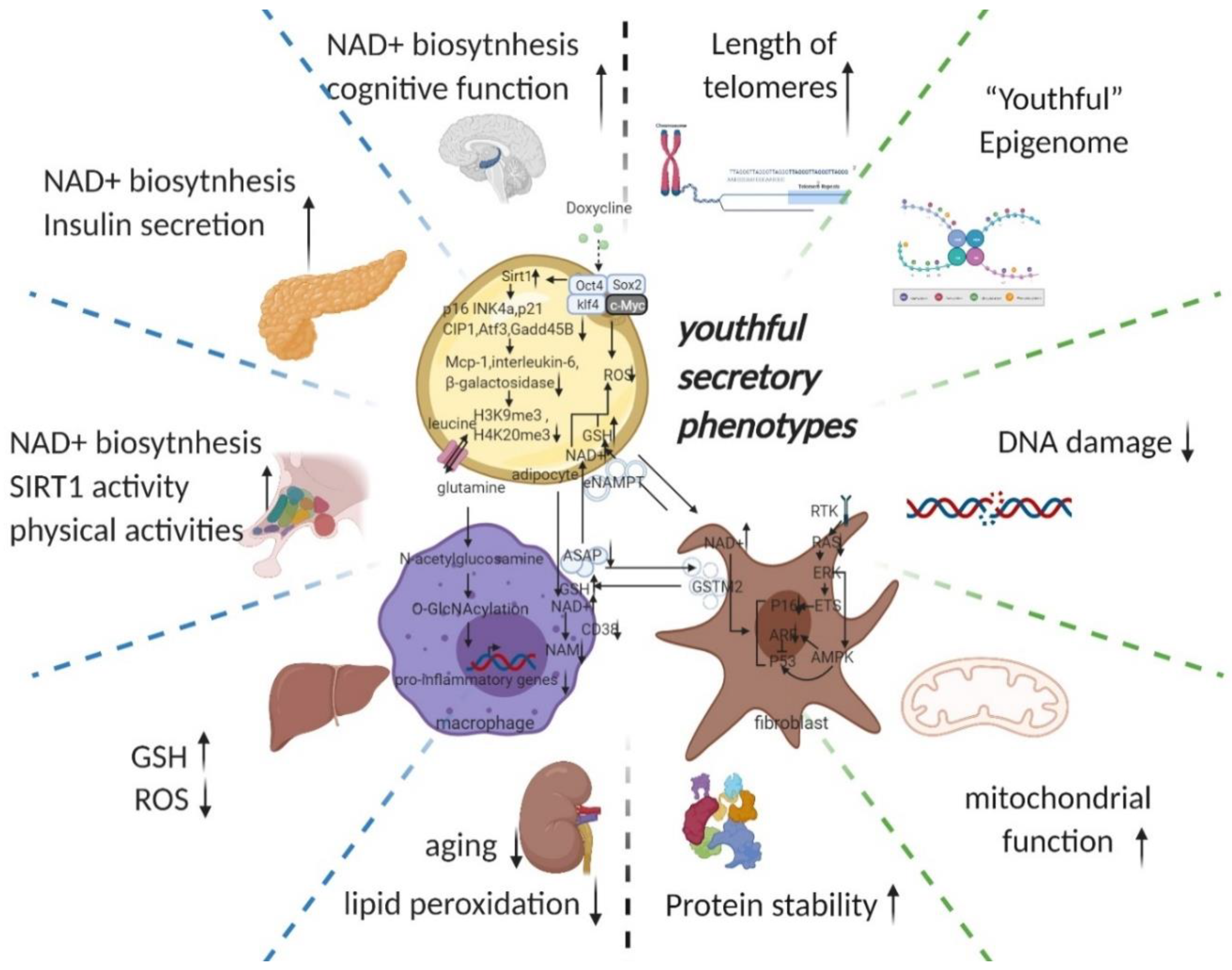
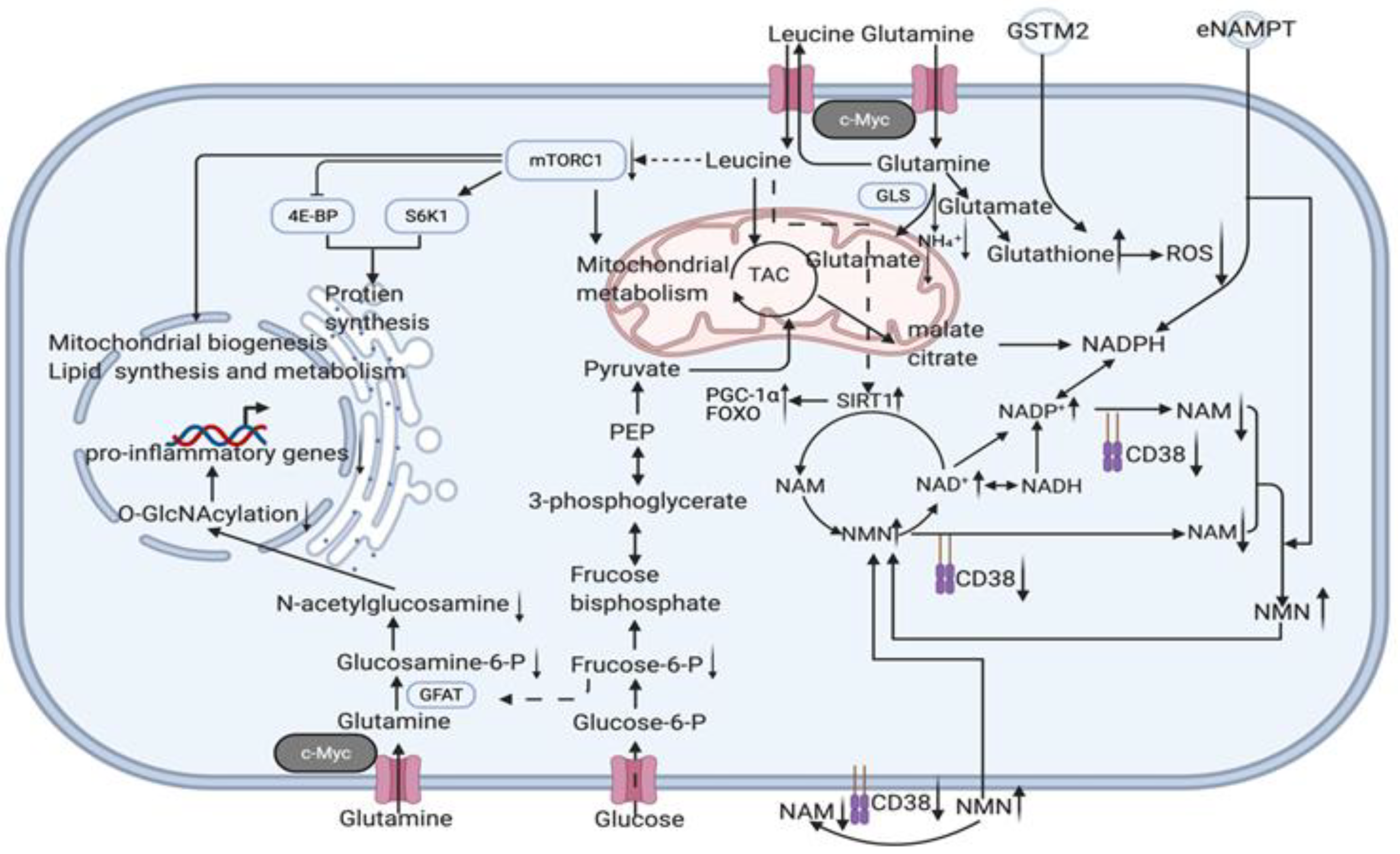
- The hypothesis of a youthful secretory phenotype is proposed to generalise the anti-ageing factors (particularly, NAD+, eNAMPT, GSTM2, etc. are involved in whole body anti-ageing by regulating the circulating NAD+/NADH balance) found in the secretome of young blood and young cells and to serve for future clinical translation and co-application.
- To propose the hypothesis that controlled reprogramming (defined as the induction of Yamanaka factors expression to reverse the ageing phenotype of cells but without iPSCs-induced pluripotent stem cell formation) may synergistically anti-age by a youthful secretory phenotype.
2. The Characteristics of Ageing and their Potential for Translation
2.1. “Asynchronous Effect” in Ageing
2.2. “Synergistic Effect” in Anti-Ageing
3. Strategies for Reversing Senescence and the Potential Underlying Mechanisms
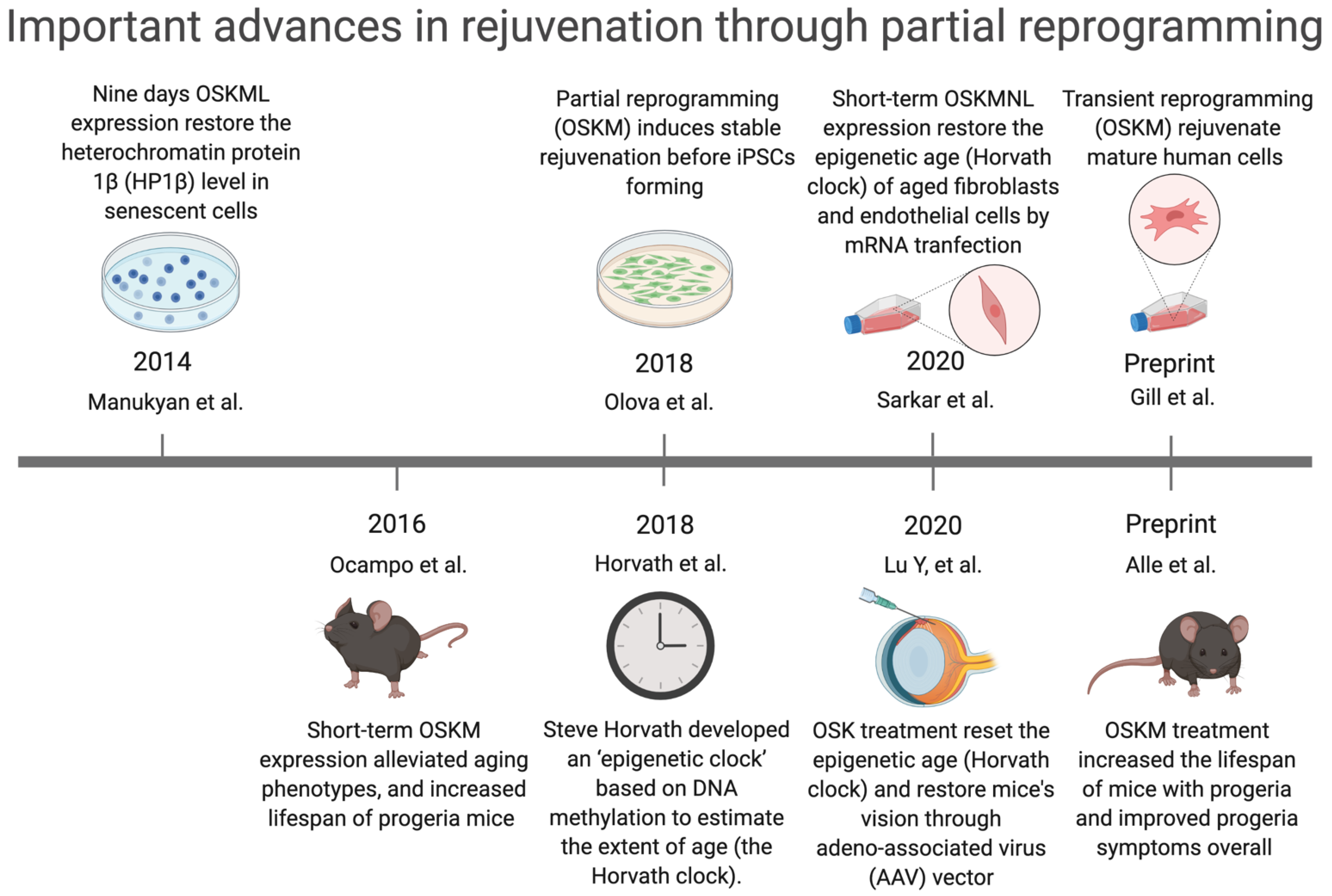
3.1. Reprogramming-Based Therapies to Reverse Senescence
3.2. Potential Key Mechanisms Related to Reprogramming-Based Therapies
3.2.1. Cyclin-Dependent Kinase Inhibitors (p16INK4A)
3.2.2. Senescence-Associated Secretory Phenotype (SASP)
3.2.3. DNA Methylation Level (Epigenetic Clock)
3.2.4. Telomeres
3.2.5. Youthful Secretory Phenotype (YSP)
4. Potential Candidates of Clinical Translation Scheme to Achieve Senescent Cells Specific Reprogramming
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Consortium, T.T.M. A single-cell transcriptomic atlas characterizes ageing tissues in the mouse. Nature 2020, 583, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Schaum, N.; Lehallier, B.; Hahn, O.; Pálovics, R.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature 2020, 583, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Correia-Melo, C.; Marques, F.D.; Anderson, R.; Hewitt, G.; Hewitt, R.; Cole, J.; Carroll, B.M.; Miwa, S.; Birch, J.; Merz, A.; et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016, 35, 724–742. [Google Scholar] [CrossRef]
- Vizioli, M.G.; Liu, T.; Miller, K.N.; Robertson, N.A.; Gilroy, K.; Lagnado, A.B.; Perez-Garcia, A.; Kiourtis, C.; Dasgupta, N.; Lei, X.; et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 2020, 34, 428–445. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Chiche, A.; Le Roux, I.; von Joest, M.; Sakai, H.; Aguín, S.B.; Cazin, C.; Salam, R.; Fiette, L.; Alegria, O.; Flamant, P.; et al. Injury-Induced Senescence Enables In Vivo Reprogramming in Skeletal Muscle. Cell Stem Cell 2017, 20, 407–414. [Google Scholar] [CrossRef]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Sagiv, A.; Burton, D.G.; Moshayev, Z.; Vadai, E.; Wensveen, F.; Ben-Dor, S.; Golani, O.; Polic, B.; Krizhanovsky, V. NKG2D ligands mediate immunosurveillance of senescent cells. Aging (Albany NY) 2016, 8, 328–344. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarragó, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020, 2, 1284–1304. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Mahmoudi, S.; Mancini, E.; Xu, L.; Moore, A.; Jahanbani, F.; Hebestreit, K.; Srinivasan, R.; Li, X.; Devarajan, K.; Prélot, L.; et al. Heterogeneity in old fibroblasts is linked to variability in reprogramming and wound healing. Nature 2019, 574, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, S.; Xu, L.; Brunet, A. Turning back time with emerging rejuvenation strategies. Nat. Cell Biol. 2019, 21, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Unnikrishnan, A.; Deepa, S.S.; Liu, Y.; Li, Y.; Ikeno, Y.; Sosnowska, D.; Van Remmen, H.; Richardson, A. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1(-/)(-) mice is correlated to increased cellular senescence. Redox Biol. 2017, 11, 30–37. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Wilkinson, J.E.; Hughes, B.; Gadela, N.; Ladiges, W.C.; Vo, N.; Niedernhofer, L.J.; Huffman, D.M.; Robbins, P.D. Heterochronic parabiosis regulates the extent of cellular senescence in multiple tissues. Geroscience 2020, 42, 951–961. [Google Scholar] [CrossRef]
- Castellano, J.M. Blood-Based Therapies to Combat Aging. Gerontology 2019, 65, 84–89. [Google Scholar] [CrossRef]
- Katsimpardi, L.; Litterman, N.K.; Schein, P.A.; Miller, C.M.; Loffredo, F.S.; Wojtkiewicz, G.R.; Chen, J.W.; Lee, R.T.; Wagers, A.J.; Rubin, L.L. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 2014, 344, 630–634. [Google Scholar] [CrossRef]
- Horowitz, A.M.; Fan, X.; Bieri, G.; Smith, L.K.; Sanchez-Diaz, C.I.; Schroer, A.B.; Gontier, G.; Casaletto, K.B.; Kramer, J.H.; Williams, K.E.; et al. Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 2020, 369, 167–173. [Google Scholar] [CrossRef] [PubMed]
- De Miguel, Z.; Khoury, N.; Betley, M.J.; Lehallier, B.; Willoughby, D.; Olsson, N.; Yang, A.C.; Hahn, O.; Lu, N.; Vest, R.T.; et al. Exercise plasma boosts memory and dampens brain inflammation via clusterin. Nature 2021, 600, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Sahu, A.; Clemens, Z.J.; Shinde, S.N.; Sivakumar, S.; Pius, A.; Bhatia, A.; Picciolini, S.; Carlomagno, C.; Gualerzi, A.; Bedoni, M.; et al. Regulation of aged skeletal muscle regeneration by circulating extracellular vesicles. Nat. Aging 2021, 1, 1148–1161. [Google Scholar] [CrossRef]
- Lombard, D.B.; Chua, K.F.; Mostoslavsky, R.; Franco, S.; Gostissa, M.; Alt, F.W. DNA repair, genome stability, and aging. Cell 2005, 120, 497–512. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Kirkland, K.T.; Fields, S.; Kennedy, B.K. Sir2-independent life span extension by calorie restriction in yeast. PLoS Biol. 2004, 2, 296. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, Y.; Paxman, J.; O’Laughlin, R.; Klepin, S.; Zhu, Y.; Pillus, L.; Tsimring, L.S.; Hasty, J.; Hao, N. A programmable fate decision landscape underlies single-cell aging in yeast. Science 2020, 369, 325–329. [Google Scholar] [CrossRef]
- Lan, J.; Rollins, J.A.; Zang, X.; Wu, D.; Zou, L.; Wang, Z.; Ye, C.; Wu, Z.; Kapahi, P.; Rogers, A.N.; et al. Translational Regulation of Non-autonomous Mitochondrial Stress Response Promotes Longevity. Cell Rep. 2019, 28, 1050–1062.e1056. [Google Scholar] [CrossRef]
- Chen, D.; Li, P.W.; Goldstein, B.A.; Cai, W.; Thomas, E.L.; Chen, F.; Hubbard, A.E.; Melov, S.; Kapahi, P. Germline signaling mediates the synergistically prolonged longevity produced by double mutations in daf-2 and rsks-1 in C. elegans. Cell Rep. 2013, 5, 1600–1610. [Google Scholar] [CrossRef]
- Zid, B.M.; Rogers, A.N.; Katewa, S.D.; Vargas, M.A.; Kolipinski, M.C.; Lu, T.A.; Benzer, S.; Kapahi, P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 2009, 139, 149–160. [Google Scholar] [CrossRef]
- Tanaka, T.; Biancotto, A.; Moaddel, R.; Moore, A.Z.; Gonzalez-Freire, M.; Aon, M.A.; Candia, J.; Zhang, P.; Cheung, F.; Fantoni, G.; et al. Plasma proteomic signature of age in healthy humans. Aging Cell 2018, 17, 12799. [Google Scholar] [CrossRef]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e1712. [Google Scholar] [CrossRef]
- Manukyan, M.; Singh, P.B. Epigenome rejuvenation: HP1β mobility as a measure of pluripotent and senescent chromatin ground states. Sci. Rep. 2014, 4, 4789. [Google Scholar] [CrossRef]
- Göbel, C.; Goetzke, R.; Eggermann, T.; Wagner, W. Interrupted reprogramming into induced pluripotent stem cells does not rejuvenate human mesenchymal stromal cells. Sci. Rep. 2018, 8, 11676. [Google Scholar] [CrossRef]
- Olova, N.; Simpson, D.J.; Marioni, R.E.; Chandra, T. Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 2019, 18, 12877. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Gill, D.; Parry, A.; Santos, F.; Hernando-Herraez, I.; Stubbs, T.M.; Milagre, I.; Reik, W. Multi-omic rejuvenation of human cells by maturation phase transient reprogramming. bioRxiv 2021, 426786. [Google Scholar] [CrossRef]
- Alle, Q.; Le Borgne, E.; Bensadoun, P.; Lemey, C.; Béchir, N.; Gabanou, M.; Estermann, F.; Bertrand-Gaday, C.; Pessemesse, L.; Toupet, K.; et al. A single short reprogramming early in life improves fitness and increases lifespan in old age. bioRxiv 2021, 443979. [Google Scholar] [CrossRef]
- Tomás-Loba, A.; Flores, I.; Fernández-Marcos, P.J.; Cayuela, M.L.; Maraver, A.; Tejera, A.; Borrás, C.; Matheu, A.; Klatt, P.; Flores, J.M.; et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell 2008, 135, 609–622. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef]
- Caso, G.; McNurlan, M.A.; Mileva, I.; Zemlyak, A.; Mynarcik, D.C.; Gelato, M.C. Peripheral fat loss and decline in adipogenesis in older humans. Metabolism 2013, 62, 337–340. [Google Scholar] [CrossRef]
- Khanh, V.C.; Zulkifli, A.F.; Tokunaga, C.; Yamashita, T.; Hiramatsu, Y.; Ohneda, O. Aging impairs beige adipocyte differentiation of mesenchymal stem cells via the reduced expression of Sirtuin 1. Biochem. Biophys. Res. Commun. 2018, 500, 682–690. [Google Scholar] [CrossRef]
- Berry, D.C.; Jiang, Y.; Arpke, R.W.; Close, E.L.; Uchida, A.; Reading, D.; Berglund, E.D.; Kyba, M.; Graff, J.M. Cellular Aging Contributes to Failure of Cold-Induced Beige Adipocyte Formation in Old Mice and Humans. Cell Metab. 2017, 25, 481. [Google Scholar] [CrossRef]
- Palikaras, K.; Mari, M.; Petanidou, B.; Pasparaki, A.; Filippidis, G.; Tavernarakis, N. Ectopic fat deposition contributes to age-associated pathology in Caenorhabditis elegans. J. Lipid Res. 2017, 58, 72–80. [Google Scholar] [CrossRef]
- Tchkonia, T.; Pirtskhalava, T.; Thomou, T.; Cartwright, M.J.; Wise, B.; Karagiannides, I.; Shpilman, A.; Lash, T.L.; Becherer, J.D.; Kirkland, J.L. Increased TNFalpha and CCAAT/enhancer-binding protein homologous protein with aging predispose preadipocytes to resist adipogenesis. Am. J. Physiol. Endocrinol. Metab. 2007, 293, 1810–1819. [Google Scholar] [CrossRef]
- Graja, A.; Gohlke, S.; Schulz, T.J. Aging of Brown and Beige/Brite Adipose Tissue. Handb. Exp. Pharm. 2019, 251, 55–72. [Google Scholar] [CrossRef]
- Petrus, P.; Lecoutre, S.; Dollet, L.; Wiel, C.; Sulen, A.; Gao, H.; Tavira, B.; Laurencikiene, J.; Rooyackers, O.; Checa, A.; et al. Glutamine Links Obesity to Inflammation in Human White Adipose Tissue. Cell Metab. 2020, 31, 375–390. [Google Scholar] [CrossRef]
- Yoshida, M.; Satoh, A.; Lin, J.B.; Mills, K.F.; Sasaki, Y.; Rensing, N.; Wong, M.; Apte, R.S.; Imai, S.-I. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab. 2019, 30, 329–342. [Google Scholar] [CrossRef]
- Ma, C.; Pi, C.; Yang, Y.; Lin, L.; Shi, Y.; Li, Y.; Li, Y.; He, X. Nampt Expression Decreases Age-Related Senescence in Rat Bone Marrow Mesenchymal Stem Cells by Targeting Sirt1. PLoS ONE 2017, 12, e0170930. [Google Scholar] [CrossRef]
- Fafián-Labora, J.A.; Rodríguez-Navarro, J.A.; O’Loghlen, A. Small Extracellular Vesicles Have GST Activity and Ameliorate Senescence-Related Tissue Damage. Cell Metab. 2020, 32, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Biran, A.; Perelmutter, M.; Gal, H.; Burton, D.G.; Ovadya, Y.; Vadai, E.; Geiger, T.; Krizhanovsky, V. Senescent cells communicate via intercellular protein transfer. Genes Dev. 2015, 29, 791–802. [Google Scholar] [CrossRef]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [PubMed]
- Altshuler-Keylin, S.; Shinoda, K.; Hasegawa, Y.; Ikeda, K.; Hong, H.; Kang, Q.; Yang, Y.; Perera, R.M.; Debnath, J.; Kajimura, S. Beige Adipocyte Maintenance Is Regulated by Autophagy-Induced Mitochondrial Clearance. Cell Metab. 2016, 24, 402–419. [Google Scholar] [CrossRef]
- Vassallo, P.F.; Simoncini, S.; Ligi, I.; Chateau, A.L.; Bachelier, R.; Robert, S.; Morere, J.; Fernandez, S.; Guillet, B.; Marcelli, M.; et al. Accelerated senescence of cord blood endothelial progenitor cells in premature neonates is driven by SIRT1 decreased expression. Blood 2014, 123, 2116–2126. [Google Scholar] [CrossRef] [PubMed]
- Johmura, Y.; Yamanaka, T.; Omori, S.; Wang, T.W.; Sugiura, Y.; Matsumoto, M.; Suzuki, N.; Kumamoto, S.; Yamaguchi, K.; Hatakeyama, S.; et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 2021, 371, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Yoshida, M.; Johnson, S.; Takikawa, A.; Usui, I.; Tobe, K.; Nakagawa, T.; Yoshino, J.; Imai, S.-i. SIRT1-Mediated eNAMPT Secretion from Adipose Tissue Regulates Hypothalamic NAD+ and Function in Mice. Cell Metab. 2015, 21, 706–717. [Google Scholar] [CrossRef]
- Baker, D.J.; Perez-Terzic, C.; Jin, F.; Pitel, K.S.; Pitel, K.; Niederländer, N.J.; Jeganathan, K.; Yamada, S.; Reyes, S.; Rowe, L.; et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat. Cell Biol. 2008, 10, 825–836. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Sato, S.; Kawamata, Y.; Takahashi, A.; Imai, Y.; Hanyu, A.; Okuma, A.; Takasugi, M.; Yamakoshi, K.; Sorimachi, H.; Kanda, H.; et al. Ablation of the p16(INK4a) tumour suppressor reverses ageing phenotypes of klotho mice. Nat. Commun. 2015, 6, 7035. [Google Scholar] [CrossRef] [PubMed]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e86. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T. Senolytic drugs: From discovery to translation. J. Intern. Med. 2020, 288, 518–536. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lu, T.; Sun, G.; Zheng, Y.; Yang, S.; Zhang, H.; Hao, S.; Liu, Y.; Ma, S.; Zhang, H.; et al. Targeting of apoptosis gene loci by reprogramming factors leads to selective eradication of leukemia cells. Nat. Commun. 2019, 10, 5594. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-Y.; Souroullas, G.P.; Diekman, B.O.; Krishnamurthy, J.; Hall, B.M.; Sorrentino, J.A.; Parker, J.S.; Sessions, G.A.; Gudkov, A.V.; Sharpless, N.E. Cells exhibiting strong promoter activation in vivo display features of senescence. Proc. Natl. Acad. Sci. USA 2019, 116, 2603–2611. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; de Martino, A.; Serrano, M. Senescence promotes in vivo reprogramming through p16 and IL-6. Aging Cell 2018, 17. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Borghesan, M.; Fafián-Labora, J.; Eleftheriadou, O.; Carpintero-Fernández, P.; Paez-Ribes, M.; Vizcay-Barrena, G.; Swisa, A.; Kolodkin-Gal, D.; Ximénez-Embún, P.; Lowe, R.; et al. Small Extracellular Vesicles Are Key Regulators of Non-cell Autonomous Intercellular Communication in Senescence via the Interferon Protein IFITM3. Cell Rep. 2019, 27, 3956–3971.e3956. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Müller, M.; Green, A.R.; Adams, P.D.; et al. Notch Signaling Mediates Secondary Senescence. Cell. Rep. 2019, 27, 997–1007. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef]
- Wang, W.; Zheng, Y.; Sun, S.; Li, W.; Song, M.; Ji, Q.; Wu, Z.; Liu, Z.; Fan, Y.; Liu, F.; et al. A genome-wide CRISPR-based screen identifies KAT7 as a driver of cellular senescence. Sci. Transl. Med. 2021, 13, eabd2655. [Google Scholar] [CrossRef]
- Lee, J.; Bignone, P.A.; Coles, L.S.; Liu, Y.; Snyder, E.; Larocca, D. Induced pluripotency and spontaneous reversal of cellular aging in supercentenarian donor cells. Biochem. Biophys. Res. Commun. 2020, 525, 563–569. [Google Scholar] [CrossRef]
- Chen, Z.; Chang, W.Y.; Etheridge, A.; Strickfaden, H.; Jin, Z.; Palidwor, G.; Cho, J.-H.; Wang, K.; Kwon, S.Y.; Doré, C.; et al. Reprogramming progeria fibroblasts re-establishes a normal epigenetic landscape. Aging Cell 2017, 16, 870–887. [Google Scholar] [CrossRef]
- Roake, C.M.; Artandi, S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef]
- Jaskelioff, M.; Muller, F.L.; Paik, J.-H.; Thomas, E.; Jiang, S.; Adams, A.C.; Sahin, E.; Kost-Alimova, M.; Protopopov, A.; Cadiñanos, J.; et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 2011, 469, 102–106. [Google Scholar] [CrossRef]
- Bernardes de Jesus, B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef]
- Sun, L.; Chiang, J.Y.; Choi, J.Y.; Xiong, Z.-M.; Mao, X.; Collins, F.S.; Hodes, R.J.; Cao, K. Transient induction of telomerase expression mediates senescence and reduces tumorigenesis in primary fibroblasts. Proc. Natl. Acad. Sci. USA 2019, 116, 18983–18993. [Google Scholar] [CrossRef]
- Chakravarti, D.; Hu, B.; Mao, X.; Rashid, A.; Li, J.; Li, J.; Liao, W.-T.; Whitley, E.M.; Dey, P.; Hou, P.; et al. Telomere dysfunction activates YAP1 to drive tissue inflammation. Nat. Commun. 2020, 11, 4766. [Google Scholar] [CrossRef]
- Conboy, I.M.; Conboy, M.J.; Wagers, A.J.; Girma, E.R.; Weissman, I.L.; Rando, T.A. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 2005, 433, 760–764. [Google Scholar] [CrossRef]
- Lei, Q.; Gao, F.; Liu, T.; Ren, W.; Chen, L.; Cao, Y.; Chen, W.; Guo, S.; Zhang, Q.; Chen, W.; et al. Extracellular vesicles deposit PCNA to rejuvenate aged bone marrow-derived mesenchymal stem cells and slow age-related degeneration. Sci. Transl. Med. 2021, 13, eaaz8697. [Google Scholar] [CrossRef]
- Loffredo, F.S.; Steinhauser, M.L.; Jay, S.M.; Gannon, J.; Pancoast, J.R.; Yalamanchi, P.; Sinha, M.; Dall’Osso, C.; Khong, D.; Shadrach, J.L.; et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 2013, 153, 828–839. [Google Scholar] [CrossRef]
- Sinha, M.; Jang, Y.C.; Oh, J.; Khong, D.; Wu, E.Y.; Manohar, R.; Miller, C.; Regalado, S.G.; Loffredo, F.S.; Pancoast, J.R.; et al. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science 2014, 344, 649–652. [Google Scholar] [CrossRef]
- Harper, S.C.; Brack, A.; MacDonnell, S.; Franti, M.; Olwin, B.B.; Bailey, B.A.; Rudnicki, M.A.; Houser, S.R. Is Growth Differentiation Factor 11 a Realistic Therapeutic for Aging-Dependent Muscle Defects? Circ. Res. 2016, 118, 1143–1150. [Google Scholar] [CrossRef]
- Villeda, S.A.; Plambeck, K.E.; Middeldorp, J.; Castellano, J.M.; Mosher, K.I.; Luo, J.; Smith, L.K.; Bieri, G.; Lin, K.; Berdnik, D.; et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 2014, 20, 659–663. [Google Scholar] [CrossRef]
- Liu, A.; Guo, E.; Yang, J.; Yang, Y.; Liu, S.; Jiang, X.; Hu, Q.; Dirsch, O.; Dahmen, U.; Zhang, C.; et al. Young plasma reverses age-dependent alterations in hepatic function through the restoration of autophagy. Aging Cell 2018, 17. [Google Scholar] [CrossRef]
- Lee, B.-R.; Kim, J.-H.; Choi, E.-S.; Cho, J.-H.; Kim, E. Effect of young exosomes injected in aged mice. Int. J. Nanomed. 2018, 13, 5335–5345. [Google Scholar] [CrossRef]
- Ghosh, A.K.; O’Brien, M.; Mau, T.; Qi, N.; Yung, R. Adipose Tissue Senescence and Inflammation in Aging is Reversed by the Young Milieu. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1709–1715. [Google Scholar] [CrossRef] [PubMed]
- Grigorian-Shamagian, L.; Liu, W.; Fereydooni, S.; Middleton, R.C.; Valle, J.; Cho, J.H.; Marbán, E. Cardiac and systemic rejuvenation after cardiosphere-derived cell therapy in senescent rats. Eur. Heart J. 2017, 38, 2957–2967. [Google Scholar] [CrossRef] [PubMed]
- Makkar, R.R.; Kereiakes, D.J.; Aguirre, F.; Kowalchuk, G.; Chakravarty, T.; Malliaras, K.; Francis, G.S.; Povsic, T.J.; Schatz, R.; Traverse, J.H.; et al. Intracoronary ALLogeneic heart STem cells to Achieve myocardial Regeneration (ALLSTAR): A randomized, placebo-controlled, double-blinded trial. Eur. Heart J. 2020, 41, 3451–3458. [Google Scholar] [CrossRef]
- Li, J.; Li, S.-H.; Dong, J.; Alibhai, F.J.; Zhang, C.; Shao, Z.-B.; Song, H.-F.; He, S.; Yin, W.-J.; Wu, J.; et al. Long-term repopulation of aged bone marrow stem cells using young Sca-1 cells promotes aged heart rejuvenation. Aging Cell 2019, 18, e13026. [Google Scholar] [CrossRef]
- Saha, P.; Sharma, S.; Korutla, L.; Datla, S.R.; Shoja-Taheri, F.; Mishra, R.; Bigham, G.E.; Sarkar, M.; Morales, D.; Bittle, G.; et al. Circulating exosomes derived from transplanted progenitor cells aid the functional recovery of ischemic myocardium. Sci. Transl. Med. 2019, 11, eaau1168. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Bernier, M.; Aon, M.A.; Cortassa, S.; Kim, E.Y.; Fang, E.F.; Palacios, H.H.; Ali, A.; Navas-Enamorado, I.; Di Francesco, A.; et al. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 2018, 27, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhou, Q.; Gao, M.; Nowsheen, S.; Zhao, F.; Kim, W.; Zhu, Q.; Kojima, Y.; Yin, P.; Zhang, Y.; et al. Tandem Deubiquitination and Acetylation of SPRTN Promotes DNA-Protein Crosslink Repair and Protects against Aging. Mol. Cell 2020, 79, 824–835. [Google Scholar] [CrossRef]
- Kim, J.-H.; Lee, B.-R.; Choi, E.-S.; Lee, K.-M.; Choi, S.-K.; Cho, J.H.; Jeon, W.B.; Kim, E. Reverse Expression of Aging-Associated Molecules through Transfection of miRNAs to Aged Mice. Mol. Ther. Nucleic Acids 2017, 6, 106–115. [Google Scholar] [CrossRef]
- Bae, Y.-U.; Son, Y.; Kim, C.-H.; Kim, K.S.; Hyun, S.H.; Woo, H.G.; Jee, B.A.; Choi, J.-H.; Sung, H.-K.; Choi, H.-C.; et al. Embryonic Stem Cell-Derived mmu-miR-291a-3p Inhibits Cellular Senescence in Human Dermal Fibroblasts Through the TGF-β Receptor 2 Pathway. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1359–1367. [Google Scholar] [CrossRef]
- Su, T.; Xiao, Y.; Xiao, Y.; Guo, Q.; Li, C.; Huang, Y.; Deng, Q.; Wen, J.; Zhou, F.; Luo, X.-H. Bone Marrow Mesenchymal Stem Cells-Derived Exosomal MiR-29b-3p Regulates Aging-Associated Insulin Resistance. ACS Nano 2019, 13, 2450–2462. [Google Scholar] [CrossRef]
- Arif, A.; Terenzi, F.; Potdar, A.A.; Jia, J.; Sacks, J.; China, A.; Halawani, D.; Vasu, K.; Li, X.; Brown, J.M.; et al. EPRS is a critical mTORC1-S6K1 effector that influences adiposity in mice. Nature 2017, 542, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, F.; Guo, Q.; Duan, Y.; Wang, W.; Zhong, Y.; Yang, Y.; Yin, Y. Leucine Supplementation: A Novel Strategy for Modulating Lipid Metabolism and Energy Homeostasis. Nutrients 2020, 12, 1299. [Google Scholar] [CrossRef] [PubMed]
- Richardson, N.E.; Konon, E.N.; Schuster, H.S.; Mitchell, A.T.; Boyle, C.; Rodgers, A.C.; Finke, M.; Haider, L.R.; Yu, D.; Flores, V.; et al. Lifelong restriction of dietary branched-chain amino acids has sex-specific benefits for frailty and life span in mice. Nat. Aging 2021, 1, 73–86. [Google Scholar] [CrossRef]
- Green, C.R.; Wallace, M.; Divakaruni, A.S.; Phillips, S.A.; Murphy, A.N.; Ciaraldi, T.P.; Metallo, C.M. Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol 2016, 12, 15–21. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Wilen, C.B.; Moley, J.R.; Li, Y.; Zou, W.; Malvin, N.P.; Rowen, M.N.; Saunders, B.T.; Ma, H.; Mack, M.R.; et al. Intercellular Mitochondria Transfer to Macrophages Regulates White Adipose Tissue Homeostasis and Is Impaired in Obesity. Cell Metab 2021, 33, 270–282.e278. [Google Scholar] [CrossRef]
- Baur, J.A.; Pearson, K.J.; Price, N.L.; Jamieson, H.A.; Lerin, C.; Kalra, A.; Prabhu, V.V.; Allard, J.S.; Lopez-Lluch, G.; Lewis, K.; et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006, 444, 337–342. [Google Scholar] [CrossRef]
- Zhao, W.; Kruse, J.P.; Tang, Y.; Jung, S.Y.; Qin, J.; Gu, W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature 2008, 451, 587–590. [Google Scholar] [CrossRef]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Cantó, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef]
- Yilmazer, A.; de Lázaro, I.; Bussy, C.; Kostarelos, K. In vivo cell reprogramming towards pluripotency by virus-free overexpression of defined factors. PLoS ONE 2013, 8, e54754. [Google Scholar] [CrossRef][Green Version]
- Caulier, B.; Berthoin, L.; Coradin, H.; Garban, F.; Dagher, M.C.; Polack, B.; Toussaint, B.; Lenormand, J.L.; Laurin, D. Targeted release of transcription factors for human cell reprogramming by ZEBRA cell-penetrating peptide. Int. J. Pharm. 2017, 529, 65–74. [Google Scholar] [CrossRef]
- Senís, E.; Mosteiro, L.; Wilkening, S.; Wiedtke, E.; Nowrouzi, A.; Afzal, S.; Fronza, R.; Landerer, H.; Abad, M.; Niopek, D.; et al. AAVvector-mediated in vivo reprogramming into pluripotency. Nat. Commun. 2018, 9, 2651. [Google Scholar] [CrossRef]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef]
- Roux, A.; Zhang, C.; Paw, J.; Zavala-Solorio, J.; Vijay, T.; Kolumam, G.; Kenyon, C.; Kimmel, J.C. Partial reprogramming restores youthful gene expression through transient suppression of cell identity. bioRxiv 2021, 444–556. [Google Scholar] [CrossRef]
- Park, I.H.; Zhao, R.; West, J.A.; Yabuuchi, A.; Huo, H.; Ince, T.A.; Lerou, P.H.; Lensch, M.W.; Daley, G.Q. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008, 451, 141–146. [Google Scholar] [CrossRef]
- Yen, K.; Mehta, H.H.; Kim, S.-J.; Lue, Y.; Hoang, J.; Guerrero, N.; Port, J.; Bi, Q.; Navarrete, G.; Brandhorst, S.; et al. The mitochondrial derived peptide humanin is a regulator of lifespan and healthspan. Aging (Albany NY) 2020, 12, 11185–11199. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.; Skutella, T. Synergistic Anti-Ageing through Senescent Cells Specific Reprogramming. Cells 2022, 11, 830. https://doi.org/10.3390/cells11050830
Chen R, Skutella T. Synergistic Anti-Ageing through Senescent Cells Specific Reprogramming. Cells. 2022; 11(5):830. https://doi.org/10.3390/cells11050830
Chicago/Turabian StyleChen, Rui, and Thomas Skutella. 2022. "Synergistic Anti-Ageing through Senescent Cells Specific Reprogramming" Cells 11, no. 5: 830. https://doi.org/10.3390/cells11050830
APA StyleChen, R., & Skutella, T. (2022). Synergistic Anti-Ageing through Senescent Cells Specific Reprogramming. Cells, 11(5), 830. https://doi.org/10.3390/cells11050830