
Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging


 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice and Tissue Preparation
2.2. Mouse Tissue Samples, Histology, and Immunohistology
2.3. RNA Isolation, Reverse Transcription, and Quantitative PCR
2.4. Endothelial Cell Magnetic-Activated Cell Sorting (MACS)
2.5. Protein Isolation, Quantification, and Western Blot
2.6. Statistics
3. Results
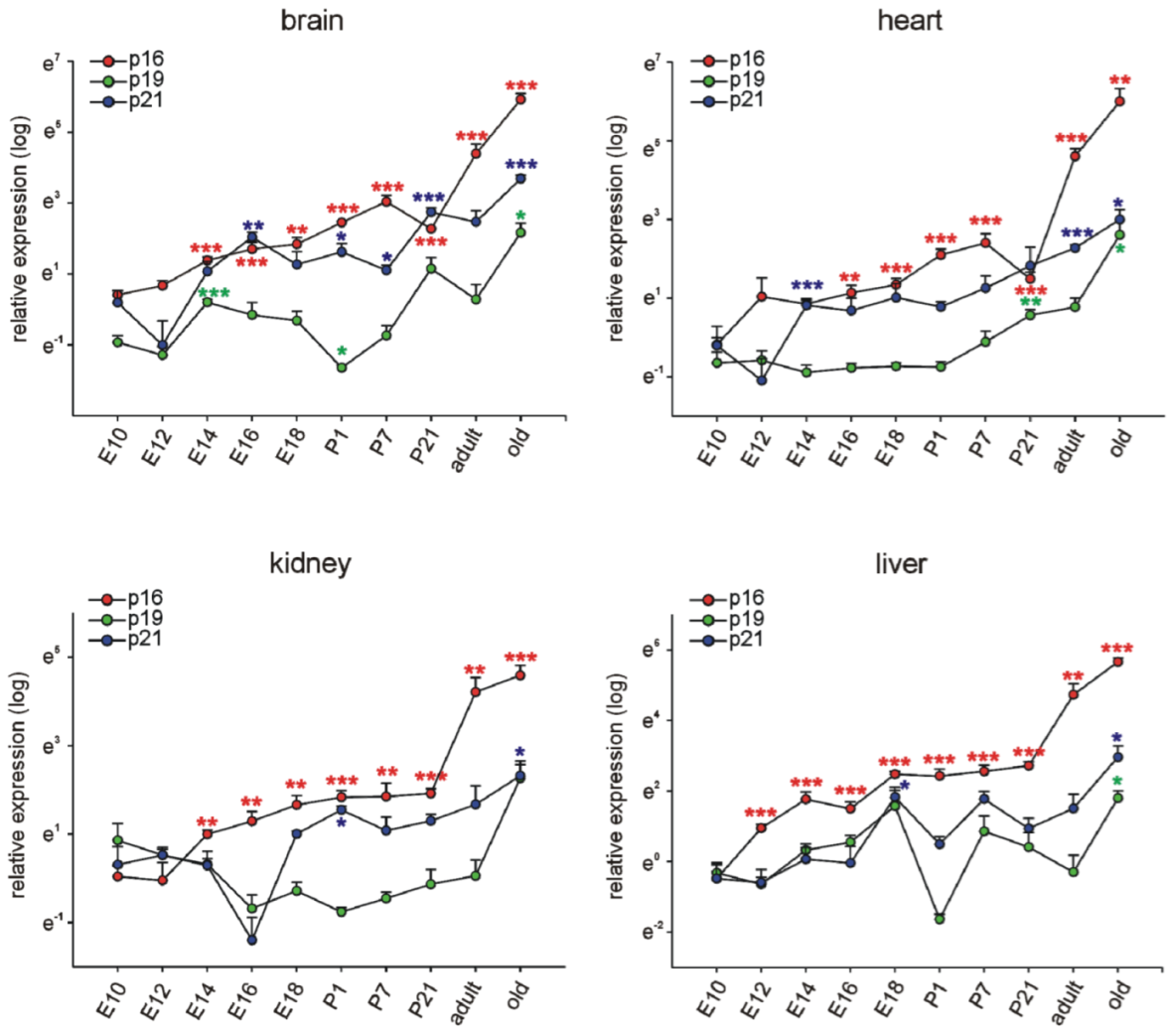
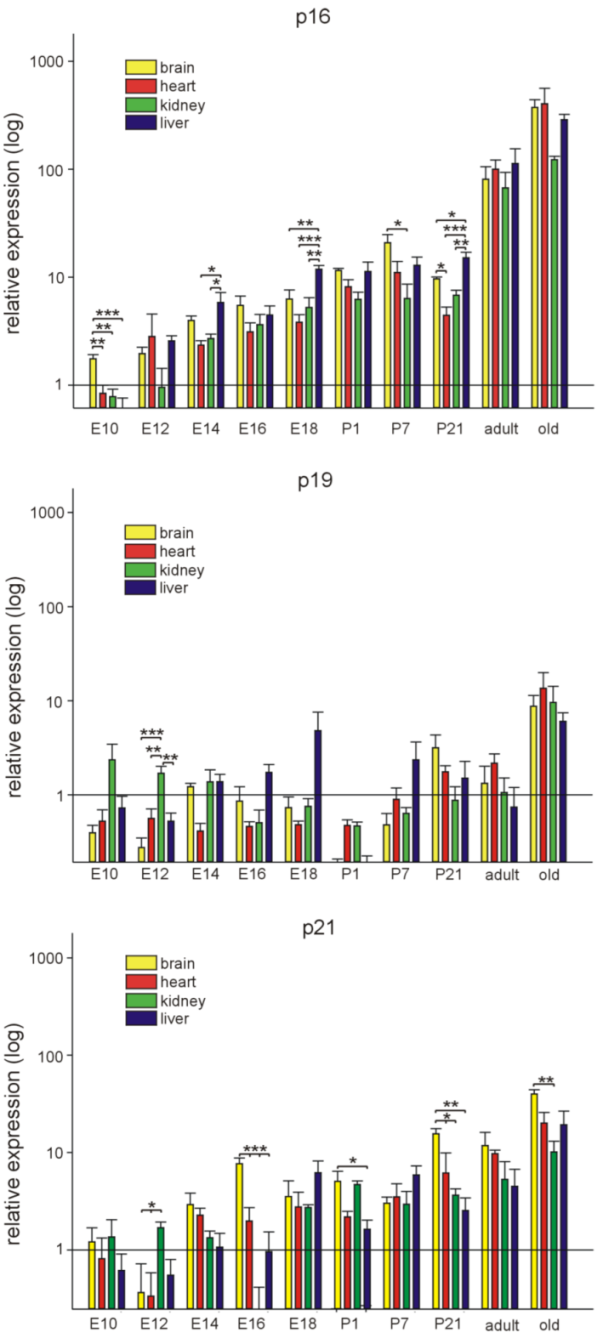
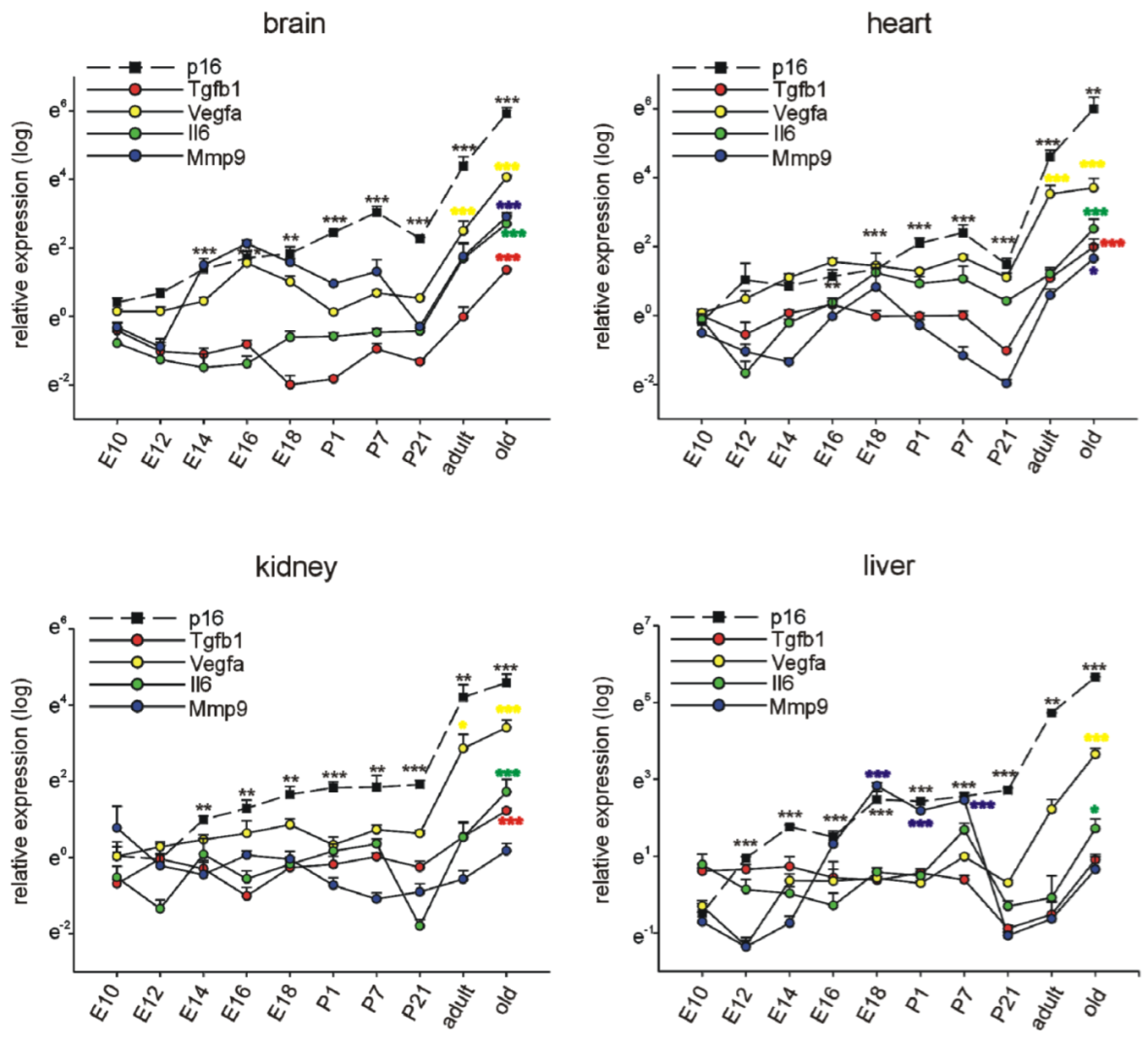
3.1. p16Ink4a, p19, and p21 mRNA Expression during Embryonic Development and Postnatal Stages in Different Organs
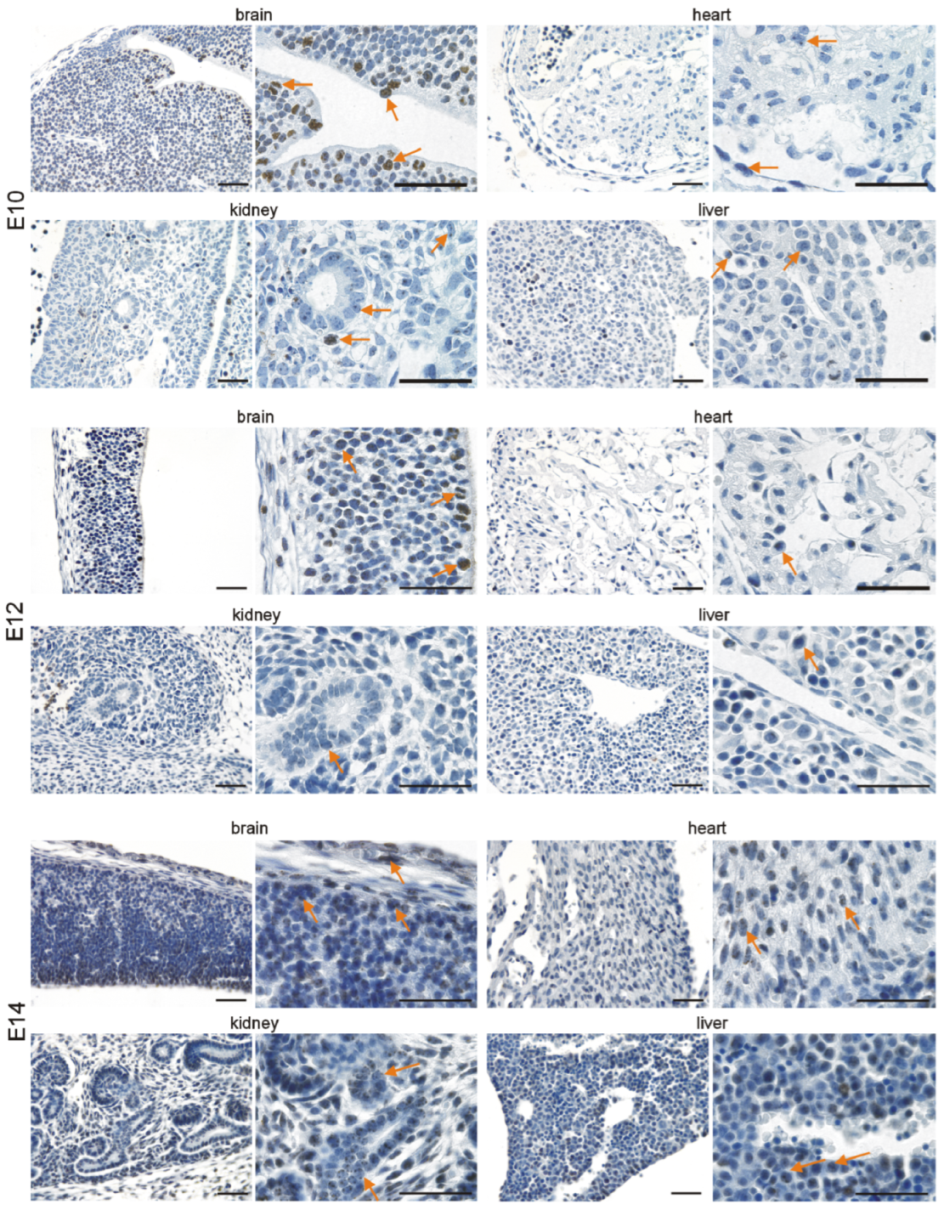
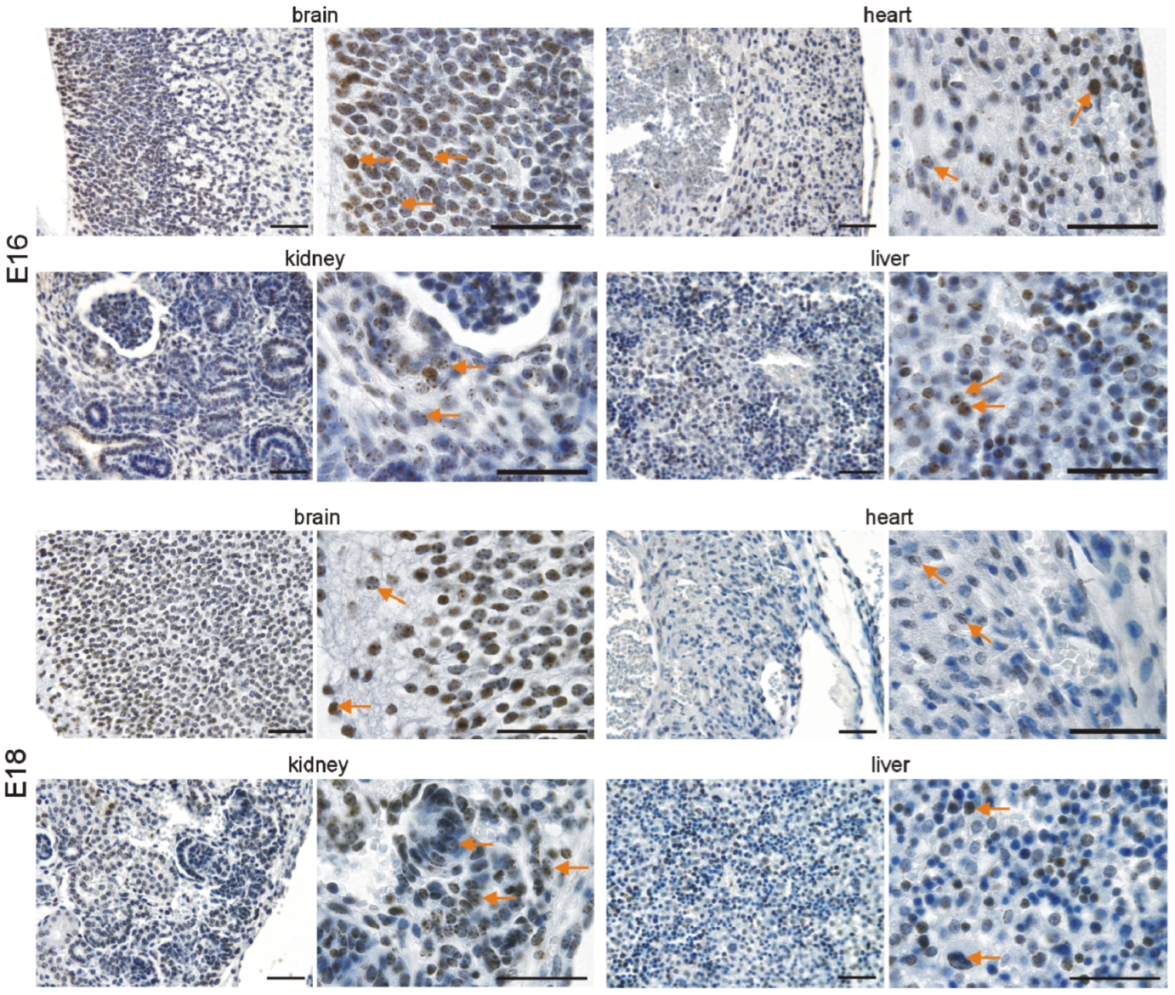
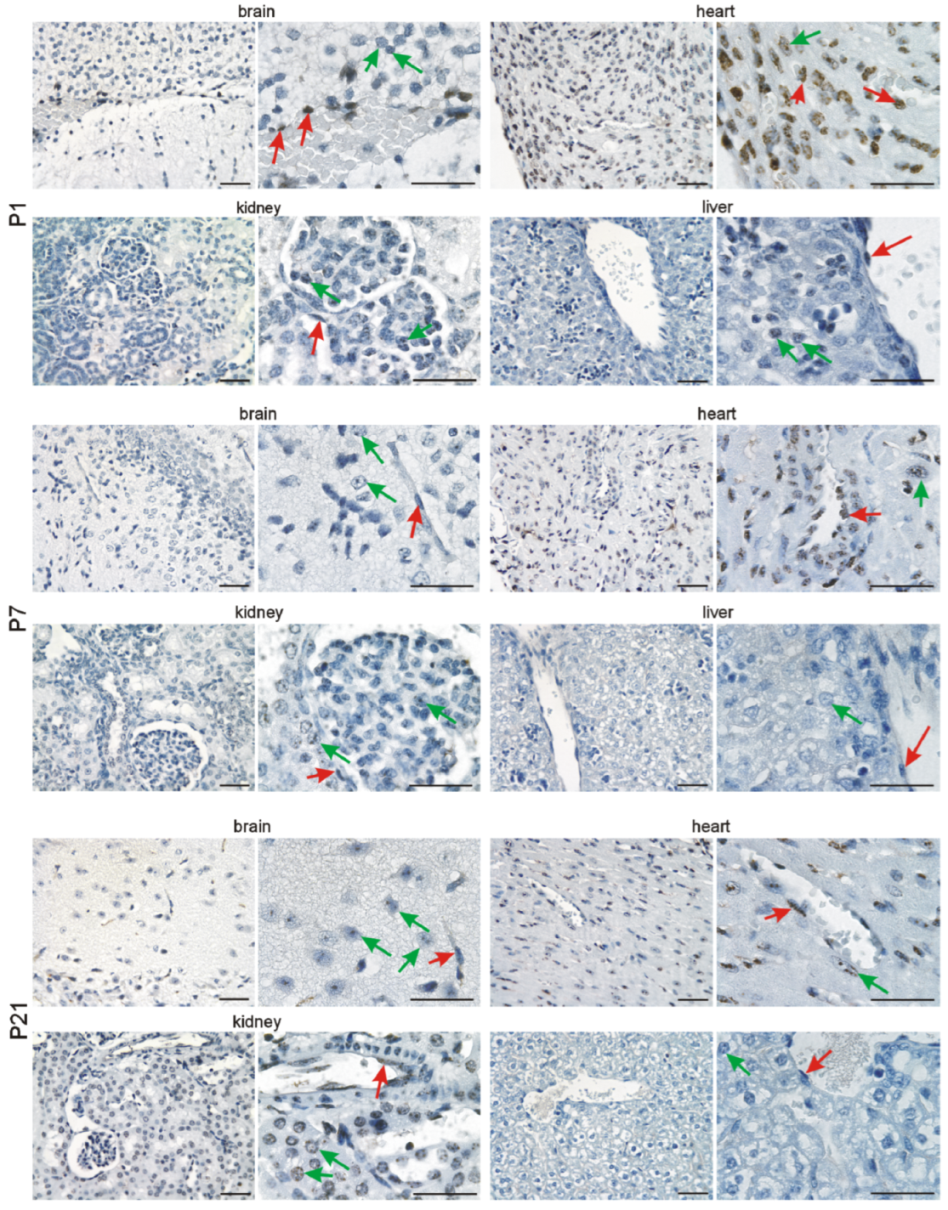
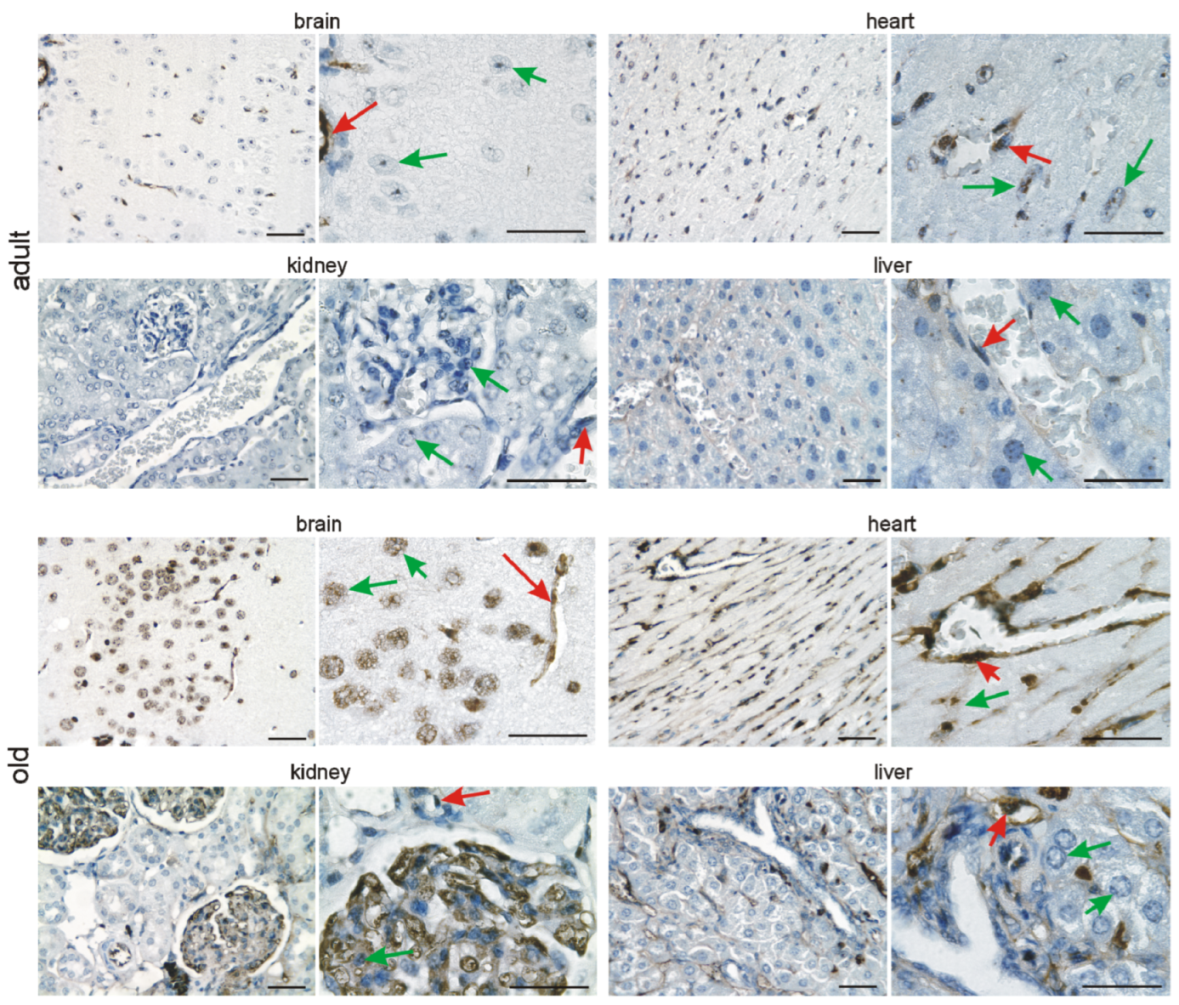
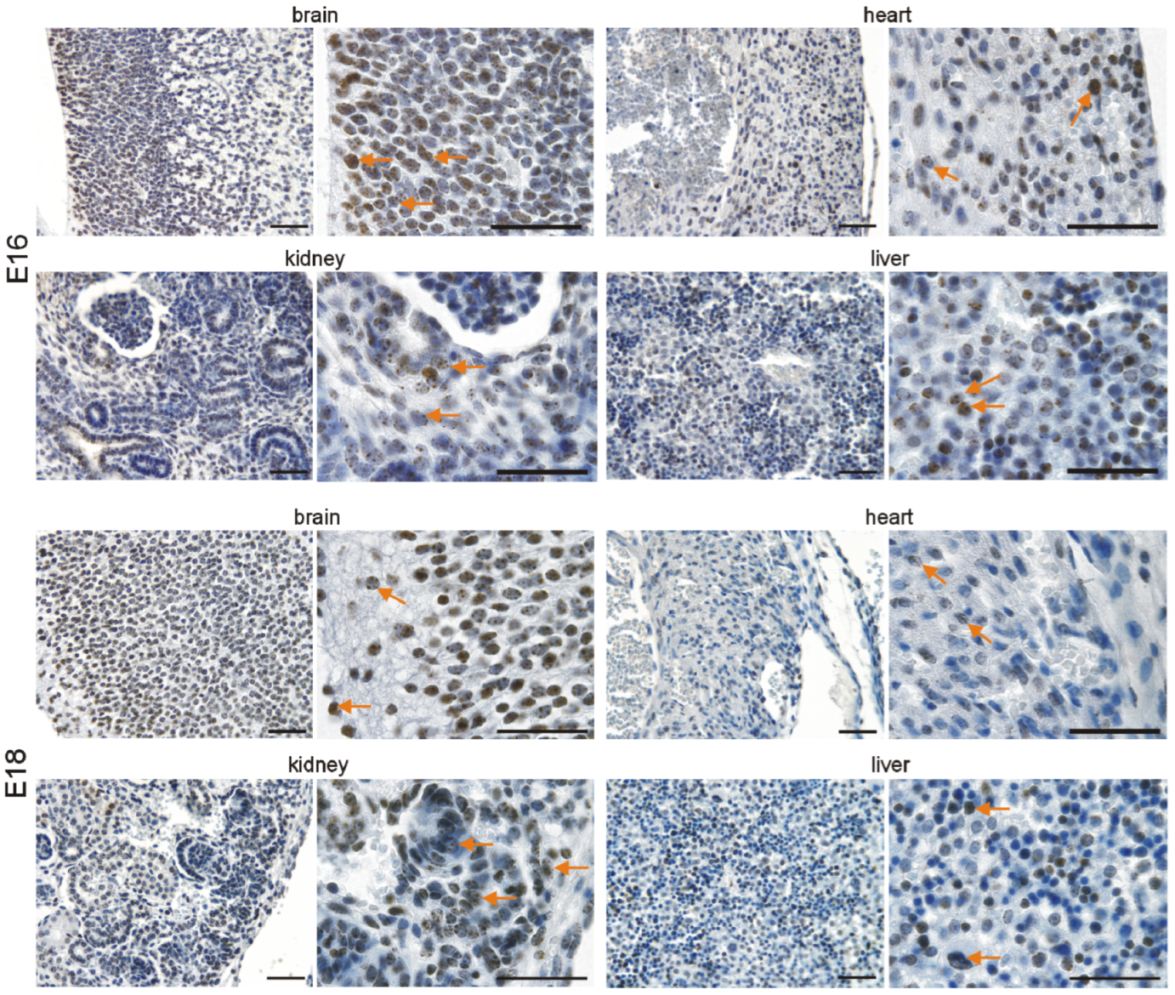
3.2. Immunohistochemical Investigation of p16 Expression
3.3. Selected SASP Factor Expression
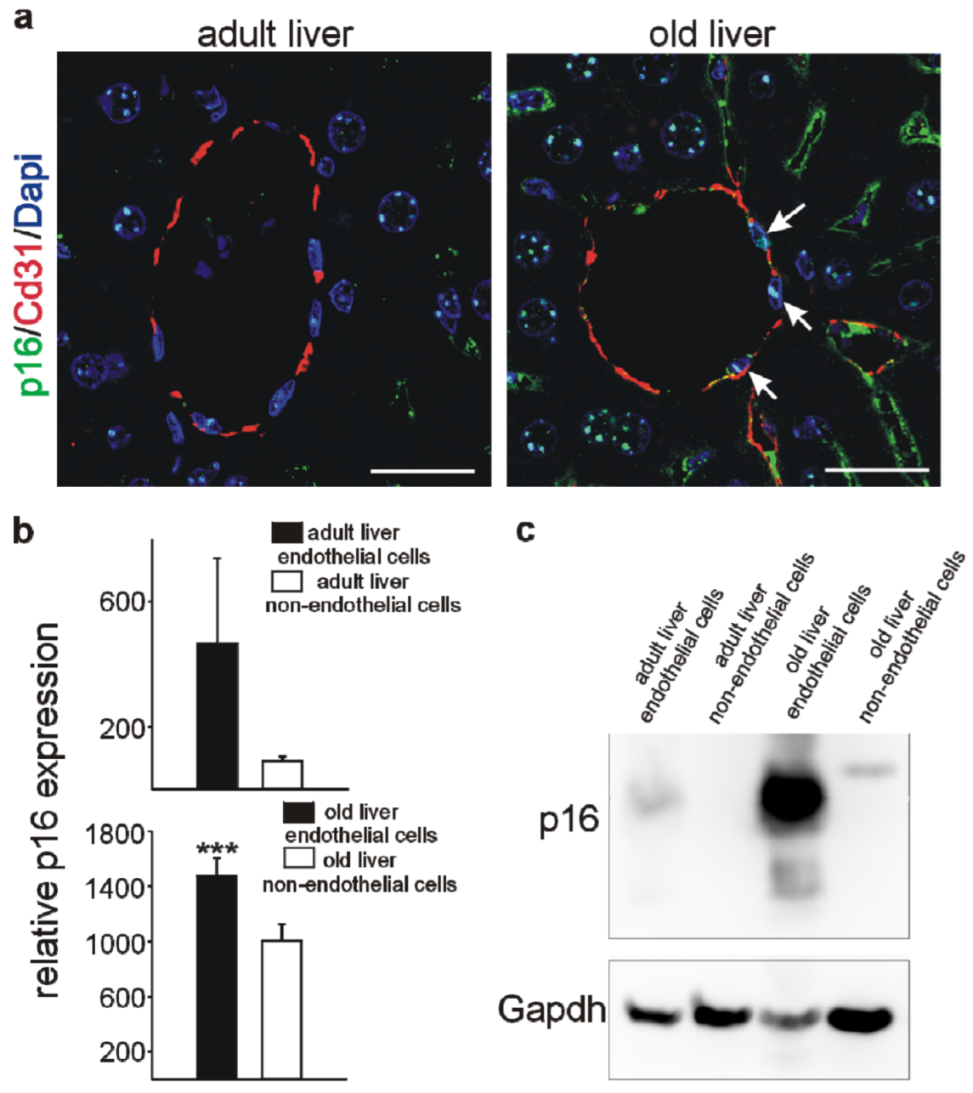
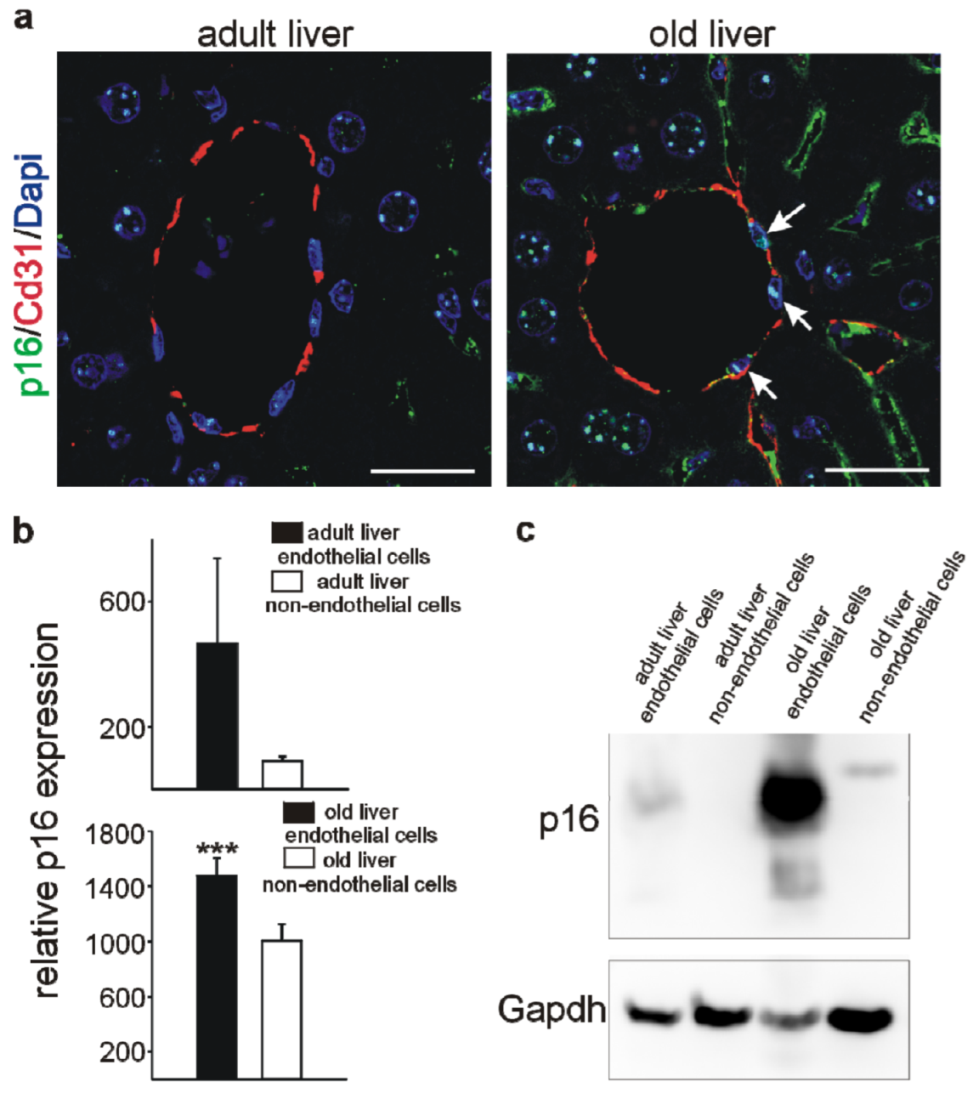
3.4. Higher p16 Expression in Endothelial versus Non-Endothelial Cells in the Liver
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [Green Version]
- Rose, M.R.; Charlesworth, B. A test of evolutionary theories of senescence. Nature 1980, 287, 141–142. [Google Scholar] [CrossRef]
- Tchkonia, T.; Kirkland, J.L. Aging, Cell Senescence, and Chronic Disease. J. Am. Med. Assoc. 2018, 320, 1319–1320. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- North, B.J.; Sinclair, D.A. The Intersection Between Aging and Cardiovascular Disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef]
- Ramly, E.; Kaafarani, H.M.; Velmahos, G.C. The Effect of Aging on Pulmonary Function. Surg. Clin. N. Am. 2015, 95, 53–69. [Google Scholar] [CrossRef]
- Meyer, B.R. Renal Function in Aging. J. Am. Geriatr. Soc. 1989, 37, 791–800. [Google Scholar] [CrossRef]
- Tung, S.; Iqbal, J. Evolution, Aging, and Osteoporosis. Ann. N. Y. Acad. Sci. 2007, 1116, 499–506. [Google Scholar] [CrossRef]
- Misra, D.; Seo, P.H.; Cohen, H.J. Aging and cancer. Clin. Adv. Hematol. Oncol. 2004, 2, 457–465. [Google Scholar]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; DELLA-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory Cytokines, Aging, and Age-Related Diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef]
- Burhans, W.C.; Weinberger, M. DNA replication stress, genome instability and aging. Nucleic Acids Res. 2007, 35, 7545–7556. [Google Scholar] [CrossRef]
- Best, B.P. Nuclear DNA Damage as a Direct Cause of Aging. Rejuvenation Res. 2009, 12, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Freitas, A.; de Magalhaes, J.P. A review and appraisal of the DNA damage theory of ageing. Mutat. Res. Mutat. Res. 2011, 728, 12–22. [Google Scholar] [CrossRef]
- Ohtani, N.; Mann, D.J.; Hara, E. Cellular senescence: Its role in tumor suppression and aging. Cancer Sci. 2009, 100, 792–797. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; Van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef]
- Rhinn, M.; Ritschka, B.; Keyes, W.M. Cellular senescence in development, regeneration and disease. Development 2019, 146, dev151837. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Levy, M.Z.; Allsopp, R.C.; Futcher, A.; Greider, C.; Harley, C.B. Telomere end-replication problem and cell aging. J. Mol. Biol. 1992, 225, 951–960. [Google Scholar] [CrossRef]
- Allsopp, R.C.; Chang, E.; Kashefi-Aazam, M.; Rogaev, E.I.; Piatyszek, M.A.; Shay, J.W.; Harley, C.B. Telomere Shortening Is Associated with Cell Division in Vitro and in Vivo. Exp. Cell Res. 1995, 220, 194–200. [Google Scholar] [CrossRef]
- McEachern, M.J.; Krauskopf, A.; Blackburn, E.H. TELOMERES AND THEIR CONTROL. Annu. Rev. Genet. 2000, 34, 331–358. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras Provokes Premature Cell Senescence Associated with Accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, A.J.; Chiang, Y.J.; Hathcock, K.S.; Horikawa, I.; A Sedelnikova, O.; Hodes, R.J.; Bonner, W.M. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin 2008, 1, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Narita, M.; Nuñez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D. Healing and Hurting: Molecular Mechanisms, Functions, and Pathologies of Cellular Senescence. Mol. Cell 2009, 36, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munro, J.; I Barr, N.; Ireland, H.; Morrison, V.; Parkinson, E. Histone deacetylase inhibitors induce a senescence-like state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp. Cell Res. 2004, 295, 525–538. [Google Scholar] [CrossRef]
- Collins, C.J.; Sedivy, J.M. Involvement of the INK4a/Arf gene locus in senescence. Aging Cell 2003, 2, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Takahashi, A.; Motoi, N.; Yoshimoto, S.; Tajima, T.; Yamakoshi, K.; Hirao, A.; Yanagi, S.; Fukami, K.; Ishikawa, Y.; et al. Intrinsic Cooperation between p16INK4a and p21Waf1/Cip1 in the Onset of Cellular Senescence and Tumor Suppression In vivo. Cancer Res. 2010, 70, 9381–9390. [Google Scholar] [CrossRef] [Green Version]
- A Weinberg, R. The Cat and Mouse Games That Genes, Viruses, and Cells Play. Cell 1997, 88, 573–575. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Hannon, G.J.; Beach, D.C. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Parry, D.; Bates, S.; Mann, D.J.; Peters, G. Lack of cyclin D-Cdk complexes in Rb-negative cells correlates with high levels of p16INK4/MTS1 tumour suppressor gene product. EMBO J. 1995, 14, 503–511. [Google Scholar] [CrossRef]
- Shamloo, B.; Usluer, S. p21 in Cancer Research. Cancers 2019, 11, 1178. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, H.; Huang, X.; Tang, J.; Zhang, S.; Li, Y.; Liu, X.; He, L.; Ju, Z.; Lui, K.O.; et al. Embryonic senescent cells re-enter cell cycle and contribute to tissues after birth. Cell Res. 2018, 28, 775–778. [Google Scholar] [CrossRef]
- Hosako, H.; Francisco, L.E.; Martin, G.S.; Mirkes, P.E. The roles of p53 and p21 in normal development and hyperthermia-induced malformations. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2009, 86, 40–47. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, À.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef] [Green Version]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; López, G.G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [Green Version]
- Zindy, F.; Soares, H.; Herzog, K.H.; Morgan, J.; Sherr, C.J.; Roussel, M.F. Expression of INK4 inhibitors of cyclin D-dependent kinases during mouse brain development. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1997, 8, 1139–1150. [Google Scholar]
- Zindy, F.; Quelle, D.; Roussel, M.F.; Sherr, C.J. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene 1997, 15, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Ouelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.-D.; Cherfils-Vicini, J.; Hosen, N.; Hohenstein, P.; Gilson, E.; Hastie, N.D.; Michiels, J.-F.; Wagner, N. The Wilms’ tumour suppressor Wt1 is a major regulator of tumour angiogenesis and progression. Nat. Commun. 2014, 5, 5852. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.; Ninkov, M.; Vukolic, A.; Deniz, G.C.; Rassoulzadegan, M.; Michiels, J.-F.; Wagner, K.-D. Implications of the Wilms’ Tumor Suppressor Wt1 in Cardiomyocyte Differentiation. Int. J. Mol. Sci. 2021, 22, 4346. [Google Scholar] [CrossRef]
- Wagner, N.; Michiels, J.F.; Schedl, A.; Wagner, K.-D. The Wilms’ tumour suppressor WT1 is involved in endothelial cell proliferation and migration: Expression in tumour vessels in vivo. Oncogene 2008, 27, 3662–3672. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.-D.; Ying, Y.; Leong, W.; Jiang, J.; Hu, X.; Chen, Y.; Michiels, J.-F.; Lu, Y.; Gilson, E.; Wagner, N.; et al. The differential spatiotemporal expression pattern of shelterin genes throughout lifespan. Aging 2017, 9, 1219–1232. [Google Scholar] [CrossRef] [Green Version]
- Silver, N.; Best, S.; Jiang, J.; Thein, S.L. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol. Biol. 2006, 7, 33. [Google Scholar] [CrossRef] [Green Version]
- Keber, R.; Motaln, H.; Wagner, K.D.; Debeljak, N.; Rassoulzadegan, M.; Ačimovič, J.; Rozman, D.; Horvat, S. Mouse Knockout of the Cholesterogenic Cytochrome P450 Lanosterol 14α-Demethylase (Cyp51) Resembles Antley-Bixler Syndrome. J. Biol. Chem. 2011, 286, 29086–29097. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, A.; Lynam, E.; Purcell, R.; Jones, C.; Lopez, C.; Board, M.; Wagner, K.-D.; Wagner, N.; Carr, C.; Wheeler-Jones, C. Context-dependent regulation of endothelial cell metabolism: Differential effects of the PPARβ/δ agonist GW0742 and VEGF-A. Sci. Rep. 2020, 10, 7849. [Google Scholar] [CrossRef]
- Wagner, K.D.; Du, S.; Martin, L.; Leccia, N.; Michiels, J.-F.; Wagner, N. Vascular PPARβ/δ Promotes Tumor Angiogenesis and Progression. Cells 2019, 8, 1623. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.-D.; El Maï, M.; Ladomery, M.; Belali, T.; Leccia, N.; Michiels, J.-F.; Wagner, N. Altered VEGF Splicing Isoform Balance in Tumor Endothelium Involves Activation of Splicing Factors Srpk1 and Srsf1 by the Wilms’ Tumor Suppressor Wt1. Cells 2019, 8, 41. [Google Scholar] [CrossRef] [Green Version]
- Wagner, K.-D.; Vukolic, A.; Baudouy, D.; Michiels, J.-F.; Wagner, N. Inducible Conditional Vascular-Specific Overexpression of Peroxisome Proliferator-Activated Receptor Beta/Delta Leads to Rapid Cardiac Hypertrophy. PPAR Res. 2016, 2016, 7631085. [Google Scholar] [CrossRef] [Green Version]
- EL Maï, M.; Wagner, K.-D.; Michiels, J.-F.; Ambrosetti, D.; Borderie, A.; Destree, S.; Renault, V.; Djerbi, N.; Giraud-Panis, M.-J.; Gilson, E.; et al. The Telomeric Protein TRF2 Regulates Angiogenesis by Binding and Activating the PDGFRβ Promoter. Cell Rep. 2014, 9, 1047–1060. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.; Morrison, H.; Pagnotta, S.; Michiels, J.-F.; Schwab, Y.; Tryggvason, K.; Schedl, A.; Wagner, K.-D. The podocyte protein nephrin is required for cardiac vessel formation. Hum. Mol. Genet. 2011, 20, 2182–2194. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Kawamata, Y.; Takahashi, A.; Imai, Y.; Hanyu, A.; Okuma, A.; Takasugi, M.; Yamakoshi, K.; Sorimachi, H.; Kanda, H.; et al. Ablation of the p16INK4a tumour suppressor reverses ageing phenotypes of klotho mice. Nat. Commun. 2015, 6, 7035. [Google Scholar] [CrossRef] [Green Version]
- Bruggeman, S.W.; Valk-Lingbeek, M.E.; van der Stoop, P.P.; Jacobs, J.J.; Kieboom, K.; Tanger, E.; Hulsman, D.; Leung, C.; Arsenijevic, Y.; Marino, S.; et al. Ink4a and Arf differentially affect cell proliferation and neural stem cell self-renewal in Bmi1-deficient mice. Genes Dev. 2005, 19, 1438–1443. [Google Scholar] [CrossRef] [Green Version]
- Sladky, V.C.; Knapp, K.; Soratroi, C.; Heppke, J.; Eichin, F.; Rocamora-Reverte, L.; Szabó, T.G.; Bongiovanni, L.; Westendorp, B.; Moreno, E.; et al. E2F-Family Members Engage the PIDDosome to Limit Hepatocyte Ploidy in Liver Development and Regeneration. Dev. Cell 2020, 52, 335–349.e7. [Google Scholar] [CrossRef]
- Pulido, T.; Velarde, M.C.; Alimirah, F. The senescence-associated secretory phenotype: Fueling a wound that never heals. Mech. Ageing Dev. 2021, 199, 111561. [Google Scholar] [CrossRef]
- Chambers, C.R.; Ritchie, S.; Pereira, B.A.; Timpson, P. Overcoming the senescence-associated secretory phenotype (SASP): A complex mechanism of resistance in the treatment of cancer. Mol. Oncol. 2021. [Google Scholar] [CrossRef]
- Grosse, L.; Wagner, N.; Emelyanov, A.; Molina, C.; Lacas-Gervais, S.; Wagner, K.-D.; Bulavin, D.V. Defined p16High Senescent Cell Types Are Indispensable for Mouse Healthspan. Cell Metab. 2020, 32, 87–99.e6. [Google Scholar] [CrossRef]
- Costantini, F.; Kopan, R. Patterning a Complex Organ: Branching Morphogenesis and Nephron Segmentation in Kidney Development. Dev. Cell 2010, 18, 698–712. [Google Scholar] [CrossRef] [Green Version]
- Wagner, N.; Wagner, K.-D. Every Beat You Take—The Wilms′ Tumor Suppressor WT1 and the Heart. Int. J. Mol. Sci. 2021, 22, 7675. [Google Scholar] [CrossRef]
- Henry, A.M.; Hohmann, J.G. High-resolution gene expression atlases for adult and developing mouse brain and spinal cord. Mamm. Genome 2012, 23, 539–549. [Google Scholar] [CrossRef]
- Zhao, R.; Duncan, S.A. Embryonic development of the liver. Hepatology 2005, 41, 956–967. [Google Scholar] [CrossRef]
- Vasey, D.B.; Wolf, C.R.; Brown, K.; Whitelaw, C.B.A. Spatial p21 expression profile in the mid-term mouse embryo. Transgenic Res. 2010, 20, 23–28. [Google Scholar] [CrossRef]
- Parker, S.B.; Eichele, G.; Zhang, P.; Rawls, A.; Sands, A.T.; Bradley, A.; Olson, E.N.; Harper, J.W.; Elledge, S.J. p53-Independent Expression of p21 Cip1 in Muscle and Other Terminally Differentiating Cells. Science 1995, 267, 1024–1027. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.-D.; Scholz, H.; Kirschner, K.; Schedl, A. Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms’ tumor suppressor Wt1. Am. J. Physiol. Integr. Comp. Physiol. 2006, 291, R779–R787. [Google Scholar] [CrossRef]
- Krishnamurthy, J.; Torrice, C.; Ramsey, M.R.; Kovalev, G.I.; Al-Regaiey, K.; Su, L.; Sharpless, N.E. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 2004, 114, 1299–1307. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef]
- López-Domínguez, J.A.; Rodríguez-López, S.; Ahumada-Castro, U.; Desprez, P.-Y.; Konovalenko, M.; Laberge, R.-M.; Cárdenas, C.; Villalba, J.M.; Campisi, J. Cdkn1a transcript variant 2 is a marker of aging and cellular senescence. Aging 2021, 13, 13380–13392. [Google Scholar] [CrossRef]
- Idda, M.L.; McClusky, W.G.; Lodde, V.; Munk, R.; Abdelmohsen, K.; Rossi, M.; Gorospe, M. Survey of senescent cell markers with age in human tissues. Aging 2020, 12, 4052–4066. [Google Scholar] [CrossRef]
- Hudgins, A.D.; Tazearslan, C.; Tare, A.; Zhu, Y.; Huffman, D.; Suh, Y. Age- and Tissue-Specific Expression of Senescence Biomarkers in Mice. Front. Genet. 2018, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Yousefzadeh, M.J.; Zhao, J.; Bukata, C.; Wade, E.A.; McGowan, S.J.; Angelini, L.A.; Bank, M.P.; Gurkar, A.; McGuckian, C.A.; Calubag, M.F.; et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 2020, 19, e13094. [Google Scholar] [CrossRef]
- Wanner, E.; Thoppil, H.; Riabowol, K. Senescence and Apoptosis: Architects of Mammalian Development. Front. Cell Dev. Biol. 2021, 8, 620089. [Google Scholar] [CrossRef]
- Cheong, C.; Sung, Y.H.; Lee, J.; Choi, Y.S.; Song, J.; Kee, C.; Lee, H.-W. Role of INK4a locus in normal eye development and cataract genesis. Mech. Ageing Dev. 2006, 127, 633–638. [Google Scholar] [CrossRef]
- An, S.; Chen, Y.; Gao, C.; Qin, B.; Du, X.; Meng, F.; Qi, Y. Inactivation of INK4a and ARF induces myocardial proliferation and improves cardiac repair following ischemia-reperfusion. Mol. Med. Rep. 2015, 12, 5911–5916. [Google Scholar] [CrossRef] [Green Version]
- Nacher, V.; Carretero, A.; Navarro, M.; Ayuso, E.; Ramos, D.; Luppo, M.; Rodríguez, A.; Mendes, L.; Herrero-Fresneda, I.; Ruberte, J. Endothelial Cell Transduction in Primary Cultures from Regressing Mesonephros. Cells Tissues Organs 2010, 191, 84–95. [Google Scholar] [CrossRef]
- Dominguez-Bautista, J.A.; Acevo-Rodríguez, P.S.; Castro-Obregón, S. Programmed Cell Senescence in the Mouse Developing Spinal Cord and Notochord. Front. Cell Dev. Biol. 2021, 9, 587096. [Google Scholar] [CrossRef]
- Frescas, D.; Hall, B.M.; Strom, E.; Virtuoso, L.P.; Gupta, M.; Gleiberman, A.S.; Rydkina, E.; Balan, V.; Vujcic, S.; Chernova, O.B.; et al. Murine mesenchymal cells that express elevated levels of the CDK inhibitor p16(Ink4a) in vivo are not necessarily senescent. Cell Cycle 2017, 16, 1526–1533. [Google Scholar] [CrossRef]
- Hall, B.M.; Balan, V.; Gleiberman, A.S.; Strom, E.; Krasnov, P.; Virtuoso, L.P.; Rydkina, E.; Vujcic, S.; Balan, K.; Gitlin, I.; et al. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 2016, 8, 1294–1315. [Google Scholar] [CrossRef] [Green Version]
- Minami, R.; Muta, K.; Umemura, T.; Motomura, S.; Abe, Y.; Nishimura, J.; Nawata, H. p16INK4a induces differentiation and apoptosis in erythroid lineage cells. Exp. Hematol. 2003, 31, 355–362. [Google Scholar] [CrossRef]
- Wang, X.; Yang, L.; Wang, Y.-C.; Xu, Z.-R.; Feng, Y.; Zhang, J.; Wang, Y.; Xu, C.-R. Comparative analysis of cell lineage differentiation during hepatogenesis in humans and mice at the single-cell transcriptome level. Cell Res. 2020, 30, 1109–1126. [Google Scholar] [CrossRef]
- Wu, T.; Liang, Z.; Zhang, Z.; Liu, C.; Zhang, L.; Gu, Y.; Peterson, K.L.; Evans, S.M.; Fu, X.-D.; Chen, J. PRDM16 Is a Compact Myocardium-Enriched Transcription Factor Required to Maintain Compact Myocardial Cardiomyocyte Identity in Left Ventricle. Circulation 2021. [Google Scholar] [CrossRef]
- Sedmera, D.; Pexieder, T.; Vuillemin, M.; Thompson, R.P.; Anderson, R.H. Developmental patterning of the myocardium. Anat. Rec. 2000, 258, 319–337. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2017, 553, 96–100. [Google Scholar] [CrossRef] [Green Version]
- E Sharpless, N.; Ramsey, M.; Balasubramanian, P.; Castrillon, D.H.; DePinho, R. The differential impact of p16INK4a or p19ARF deficiency on cell growth and tumorigenesis. Oncogene 2004, 23, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Sharpless, N.; Bardeesy, N.; Lee, K.-H.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; DePinho, R. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Serrano, M.; Lee, H.-W.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R. Role of the INK4a Locus in Tumor Suppression and Cell Mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.; Li, N.; Kong, M.; Wu, J.; Fan, Z.; Miao, D.; Xu, Y.; Ye, Q.; Wang, Y. CDKN2a/p16 Antagonizes Hepatic Stellate Cell Activation and Liver Fibrosis by Modulating ROS Levels. Front. Cell Dev. Biol. 2020, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Wu, J.; Miao, D.; An, W.; Wang, Y. p16 deficiency promotes nonalcoholic steatohepatitis via regulation of hepatic oxidative stress. Biochem. Biophys. Res. Commun. 2017, 486, 264–269. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene of Interest | Oligonucleotide Sequences | References |
|---|---|---|
| p16ink4 | F: AGGGCCGTGTGCATGACGTG R: GCACCGGGCGGGAGAAGGTA | [59] |
| p19arf | F: CGCTCTGGCTTTCGTGAAC R: GTGCGGCCCTCTTCTCAA | [60] |
| p21 | F: AATTGGAGTCAGGCGCAGAT R: CATGAGCGCATCGCAATCAC | [61] |
| Tgf-b1 | F: AGCTGGTGAAACGGAAGCG R: GCGAGCCTTAGTTTGGACAGG | This study |
| Vegfa | F: CTCACCAAAGCCAGCACATA R: AATGCTTTCTCCGCTCTGAA | [54] |
| Il-6 | F: CACTTCACAAGTCGGAGGCT R: TGCCATTGCACAACTCTTTTCT | [54] |
| Mmp9 | F: CCATGCACTGGGCTTAGATCA B: GGCCTTGGGTCAGGCTTAGA | [54] |
| Gapdh | F: AGGTCGGTGTGAACGGATTTG R: TGTAGACCATGTAGTTGAGGTCA | [47,48,54,58] |
| β-actin | F: CTTCCTCCCTGGAGAAGAGC R: ATGCCACAGGATTCCATACC | [47,48,54,58] |
| Rplp0 | F: CACTGGTCTAGGACCCGAGAAG R: GGTGCCTCTGGAGATTTTCG | [47,48,54,58] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Safwan-Zaiter, H.; Wagner, N.; Michiels, J.-F.; Wagner, K.-D. Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging. Cells 2022, 11, 541. https://doi.org/10.3390/cells11030541
Safwan-Zaiter H, Wagner N, Michiels J-F, Wagner K-D. Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging. Cells. 2022; 11(3):541. https://doi.org/10.3390/cells11030541
Chicago/Turabian StyleSafwan-Zaiter, Hasan, Nicole Wagner, Jean-François Michiels, and Kay-Dietrich Wagner. 2022. "Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging" Cells 11, no. 3: 541. https://doi.org/10.3390/cells11030541
APA StyleSafwan-Zaiter, H., Wagner, N., Michiels, J.-F., & Wagner, K.-D. (2022). Dynamic Spatiotemporal Expression Pattern of the Senescence-Associated Factor p16Ink4a in Development and Aging. Cells, 11(3), 541. https://doi.org/10.3390/cells11030541