
Molecular Mechanisms of Alveolar Epithelial Stem Cell Senescence and Senescence-Associated Differentiation Disorders in Pulmonary Fibrosis



Abstract
1. Introduction
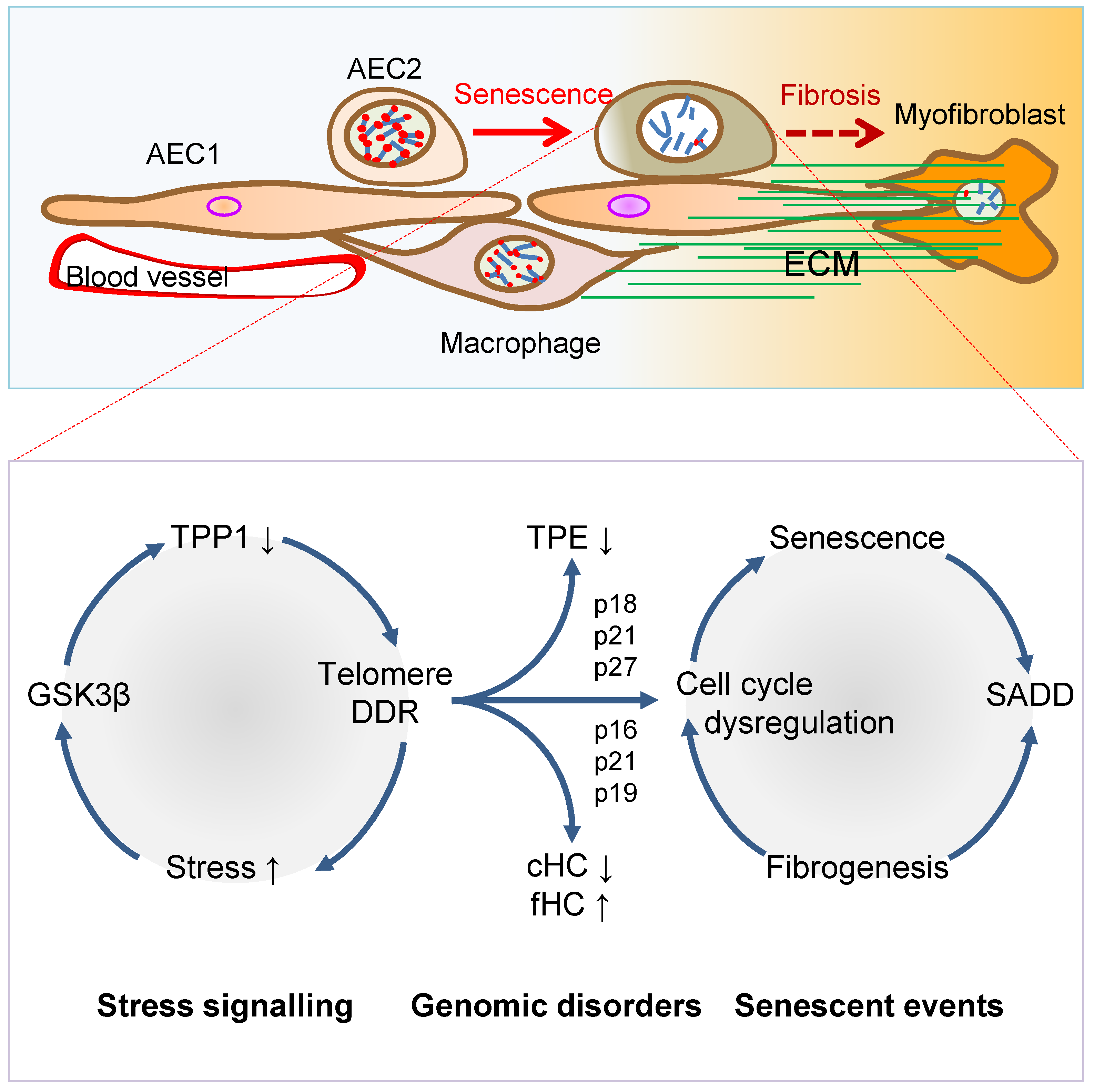
2. Senescence-Associated Differentiation Disorder (SADD)
3. Telomere Dysfunction Mediates Pulmonary Senescence and Fibrosis
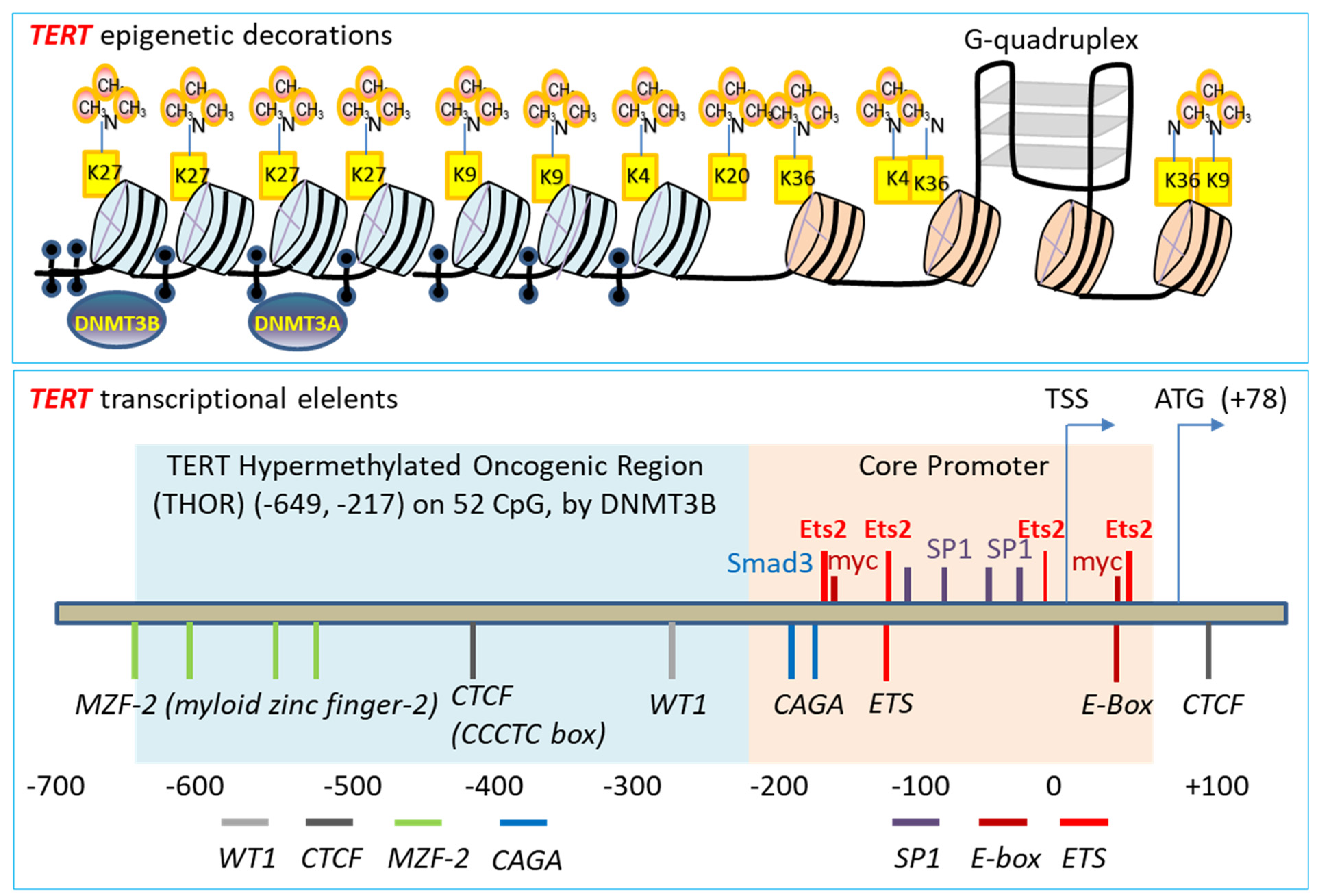
3.1. Telomerase Gene Deficiency in Ageing-Related Disorders
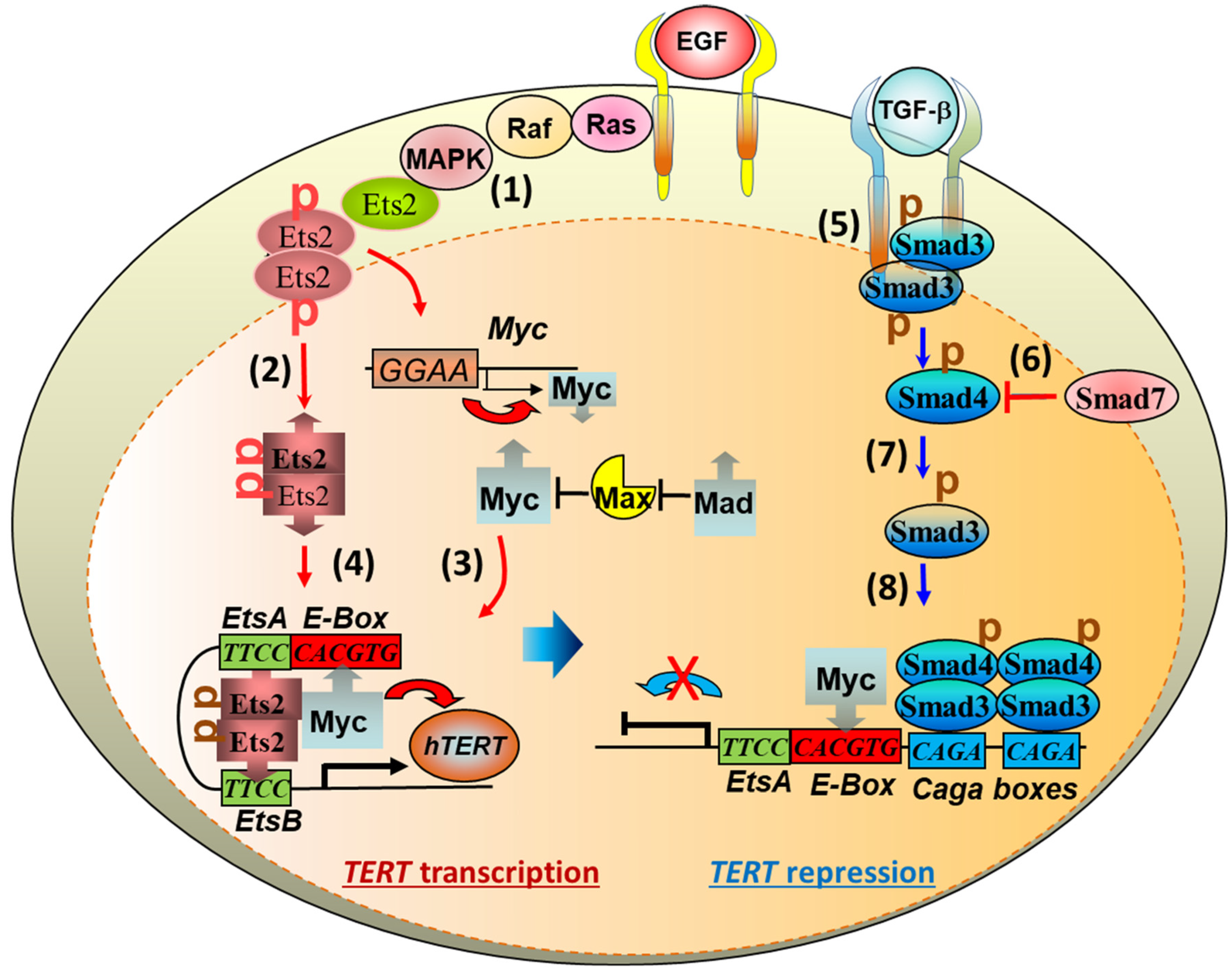
3.2. TGF-β Signaling to Telomerase TERT Gene Repression
3.3. Stress-Induced TPP1 Degradation and Telomere Uncapping
4. Targeted Intervention of Cellular Senescence and Tissue Fibrosis
4.1. Targeting Anti-Apoptotic Gene Bcl-2 to Clear Senescent Cells
4.2. Targeting TP53 and p16INK4A Tumor Suppressor Genes to Clear Senescent Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | Senolytic Agent | Mechanism of Action | Reference |
|---|---|---|---|
| BCL-2 Family Inhibitors | Dasatinib + Quercetin ABT-737 ABT-263 A-1331852 A-1155463 | PI3K/Akt inhibition/DNA intercalation Inhibiting BCL-2, BCL-XL, BCL-W Inhibiting BCL-2, BCL-XL, BCL-W Inhibiting BCL-XL Inhibiting BCL-XL | [3,217,218,220,224] |
| Targeting p53 | FOXO4-DRI UBX0101 RG7112 (RO5045337) P5091 P22077 NOX1 and NOX2 dual inhibitor PAI-1 inhibitor (TM5275) | Disrupting FOXO4-p53 interaction Disrupting MDM2-p53 interaction Disrupting MDM2-p53 interaction USP7 inhibitor USP7 inhibitor Activating p53 and apoptosis Activating p53 and apoptosis | [222,223,225,226] |
| HSP90 Inhibitors | 17-DMAG (alvespimycin) Geldanamycin 17-AAG (tanespimycin) Ganetespib | Disrupting HSP90-AKT interaction Attenuating HSP90 Attenuating HSP90 Attenuating HSP90 | [227,228] |
| Natural Products and their Analogues | Fisetin Curcumin O-Vanillin (curcumin metabolite) EF-24 (curcumin analogue) Piperlongumine and its analogues (compounds 47–49) GL-V9 | BCL-2, PI3K/AKT, p53, NF-κB and more Down-regulating Nrf2 and NF-κB pathways Down-regulating Nrf2 and NF-κB pathways Attenuating the BCL-2 family Unclear, OXR1, NF-κB - Unclear, increasing ROS, alkalizing lysosome | [224,229,230,231,232] |
| Cardiac Glycosides | Proscillaridin A Ouabain Ouabagenin Digoxin Bufalin K-Stropanthin Strophanthidin | Inhibiting Na+/K+-ATPase Inhibiting Na+/K+-ATPase, increasing BCL2-Family protein NOXA Inhibiting Na+/K+-ATPase Inhibiting Na+/K+-ATPase, increasing BCL2-Family protein NOXA Inhibiting Na+/K+-ATPase Inhibiting Na+/K+-ATPase Inhibiting Na+/K+-ATPase | [233,234] |
| Galactose Modified Prodrugs | SSK1 Pro-drug A (JHB75B) Nav-Gal 5FURGa | Targeting SA-β-galactosidase Targeting SA-β-galactosidase Targeting SA-β-galactosidase Targeting SA-β-galactosidase | [235,236,237] |
| PROTACs | PZ15227 ARV825 | Degrading BCL-XL Degrading BRD4 | [238,239] |
| Miscellaneous | Fenofibrate Azithromycin Roxithromycin Tamatinib (R406) MitoTam Panobinostat AT-406 Rapamycin Metformin | PPARα agonist Inducing autophagy and glycolysis NOX4 Syk inhibitor, FAK and p38MAPK Reducing mitochondrial membrane potential, Inhibiting OXPHOS Histone deacetylase inhibitor Inhibitor of c-IAP1, c-IAP2 and XIAP Inhibiting mTOR, p16 and p21 Inhibiting NF-κB pathways/AKT | [240,241,242,243,244,245,246,247] |
4.3. Preventing Telomere Dysfunction and Pulmonary Fibrosis by TELODIN
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cookson, W.O.; Moffatt, M.F. Bedside to gene and back in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2013, 368, 2228–2230. [Google Scholar] [CrossRef] [PubMed]
- Armanios, M.Y. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1370–1372. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-induced senescence is driver and therapeutic target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Yao, X.; Ma, S.; Ping, Y.; Fan, Y.; Sun, S.; He, Z.; Shi, Y.; Sun, L.; Xiao, S.; et al. A single-cell transcriptomic landscape of the lungs of patients with COVID-19. Nat. Cell Biol. 2021, 23, 1314–1328. [Google Scholar] [CrossRef]
- Nie, X.; Qian, L.; Sun, R.; Huang, B.; Dong, X.; Xiao, Q.; Zhang, Q.; Lu, T.; Yue, L.; Chen, S.; et al. Multi-organ proteomic landscape of COVID-19 autopsies. Cell 2021, 184, 775–791.e14. [Google Scholar] [CrossRef]
- Armanios, M.Y.; Chen, J.J.; Cogan, J.D.; Alder, J.K.; Ingersoll, R.G.; Markin, C.; Lawson, W.E.; Xie, M.; Vulto, I.; Phillips, J.A., 3rd; et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2007, 356, 1317–1326. [Google Scholar] [CrossRef]
- Ting, C.; Aspal, M.; Vaishampayan, N.; Huang, S.K.; Wang, F.; Farver, C.; Zemans, R.L. Ineffectual AEC1 differentiation from KRT8 (hi) transitional state without fibrosis is associated with fatal COVID-19 ARDS. bioRxiv 2021. [Google Scholar] [CrossRef]
- Drake, T.M.; Docherty, A.B.; Harrison, E.M.; Quint, J.K.; Adamali, H.; Agnew, S.; Babu, S.; Barber, C.M.; Barratt, S.; Bendstrup, E.; et al. Outcome of Hospitalization for COVID-19 in Patients with Interstitial Lung Disease. An International Multicenter Study. Am. J. Respir. Crit. Care Med. 2020, 202, 1656–1665. [Google Scholar] [CrossRef]
- Jansing, N.L.; McClendon, J.; Henson, P.M.; Tuder, R.M.; Hyde, D.M.; Zemans, R.L. Unbiased Quantitation of Alveolar Type II to Alveolar Type I Cell Transdifferentiation during Repair after Lung Injury in Mice. Am. J. Respir. Cell Mol. Biol. 2017, 57, 519–526. [Google Scholar] [CrossRef]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Kathiriya, J.J.; Brumwell, A.N.; Jackson, J.R.; Tang, X.; Chapman, H.A. Distinct Airway Epithelial Stem Cells Hide among Club Cells but Mobilize to Promote Alveolar Regeneration. Cell Stem Cell 2020, 26, 346–358.e4. [Google Scholar] [CrossRef] [PubMed]
- Rao, W.; Wang, S.; Duleba, M.; Niroula, S.; Goller, K.; Xie, J.; Mahalingam, R.; Neupane, R.; Liew, A.A.; Vincent, M.; et al. Regenerative Metaplastic Clones in COPD Lung Drive Inflammation and Fibrosis. Cell 2020, 181, 848–864.e18. [Google Scholar] [CrossRef]
- Lee, J.H.; Bhang, D.H.; Beede, A.; Huang, T.L.; Stripp, B.R.; Bloch, K.D.; Wagers, A.J.; Tseng, Y.H.; Ryeom, S.; Kim, C.F. Lung stem cell differentiation in mice directed by endothelial cells via a BMP4-NFATc1-thrombospondin-1 axis. Cell 2014, 156, 440–455. [Google Scholar] [CrossRef]
- Chen, Q.; Suresh Kumar, V.; Finn, J.; Jiang, D.; Liang, J.; Zhao, Y.Y.; Liu, Y. CD44(high) alveolar type II cells show stem cell properties during steady-state alveolar homeostasis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L41–L51. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.; Buckley, S.; Doerken, M.; Barsky, L.; Weinberg, K.; Anderson, K.D.; Warburton, D.; Driscoll, B. Isolation of a putative progenitor subpopulation of alveolar epithelial type 2 cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Salahudeen, A.A.; Choi, S.S.; Rustagi, A.; Zhu, J.; van Unen, V.; de la O, S.M.; Flynn, R.A.; Margalef-Catala, M.; Santos, A.J.M.; Ju, J.; et al. Progenitor identification and SARS-CoV-2 infection in human distal lung organoids. Nature 2020, 588, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef]
- Choi, J.; Park, J.E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.K.; Han, N.; Lee, J.H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382.e7. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tata, A.; Konkimalla, A.; Katsura, H.; Lee, R.F.; Ou, J.; Banovich, N.E.; Kropski, J.A.; Tata, P.R. Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat. Cell Biol. 2020, 22, 934–946. [Google Scholar] [CrossRef]
- Strunz, M.; Simon, L.M.; Ansari, M.; Kathiriya, J.J.; Angelidis, I.; Mayr, C.H.; Tsidiridis, G.; Lange, M.; Mattner, L.F.; Yee, M.; et al. Alveolar regeneration through a Krt8+ transitional stem cell state that persists in human lung fibrosis. Nat. Commun. 2020, 11, 3559. [Google Scholar] [CrossRef]
- Jiang, P.; Gil de Rubio, R.; Hrycaj, S.M.; Gurczynski, S.J.; Riemondy, K.A.; Moore, B.B.; Omary, M.B.; Ridge, K.M.; Zemans, R.L. Ineffectual Type 2-to-Type 1 Alveolar Epithelial Cell Differentiation in Idiopathic Pulmonary Fibrosis: Persistence of the KRT8(hi) Transitional State. Am. J. Respir. Crit. Care Med. 2020, 201, 1443–1447. [Google Scholar] [CrossRef] [PubMed]
- Riemondy, K.A.; Jansing, N.L.; Jiang, P.; Redente, E.F.; Gillen, A.E.; Fu, R.; Miller, A.J.; Spence, J.R.; Gerber, A.N.; Hesselberth, J.R.; et al. Single cell RNA sequencing identifies TGFbeta as a key regenerative cue following LPS-induced lung injury. JCI Insight 2019, 4, e123637. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yu, Y.; Huang, H.; Hu, Y.; Fu, S.; Wang, Z.; Shi, M.; Zhao, X.; Yuan, J.; Li, J.; et al. Progressive Pulmonary Fibrosis Is Caused by Elevated Mechanical Tension on Alveolar Stem Cells. Cell 2020, 180, 107–121.e4. [Google Scholar] [CrossRef] [PubMed]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular senescence and its effector programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef]
- Hayflick, L.; Moorhead, P.S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol 2014, 15, 482–496. [Google Scholar] [CrossRef]
- Maciel-Baron, L.A.; Morales-Rosales, S.L.; Aquino-Cruz, A.A.; Triana-Martinez, F.; Galvan-Arzate, S.; Luna-Lopez, A.; Gonzalez-Puertos, V.Y.; Lopez-Diazguerrero, N.E.; Torres, C.; Konigsberg, M. Senescence associated secretory phenotype profile from primary lung mice fibroblasts depends on the senescence induction stimuli. Age 2016, 38, 26. [Google Scholar] [CrossRef]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef]
- Rivera, T.; Haggblom, C.; Cosconati, S.; Karlseder, J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat. Struct. Mol. Biol. 2017, 24, 30–39. [Google Scholar] [CrossRef]
- Paluvai, H.; Di Giorgio, E.; Brancolini, C. The Histone Code of Senescence. Cells 2020, 9, 466. [Google Scholar] [CrossRef]
- Sati, S.; Bonev, B.; Szabo, Q.; Jost, D.; Bensadoun, P.; Serra, F.; Loubiere, V.; Papadopoulos, G.L.; Rivera-Mulia, J.C.; Fritsch, L.; et al. 4D Genome Rewiring during Oncogene-Induced and Replicative Senescence. Mol. Cell 2020, 78, 522–538.e9. [Google Scholar] [CrossRef] [PubMed]
- Campaner, S.; Doni, M.; Hydbring, P.; Verrecchia, A.; Bianchi, L.; Sardella, D.; Schleker, T.; Perna, D.; Tronnersjo, S.; Murga, M.; et al. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat. Cell Biol. 2010, 12, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef]
- Wang, L.; Chen, R.; Li, G.; Wang, Z.; Liu, J.; Liang, Y.; Liu, J.P. FBW7 Mediates Senescence and Pulmonary Fibrosis through Telomere Uncapping. Cell Metab. 2020, 32, 860–877.e9. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of In Vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Hoare, M.; Narita, M. Spatial and Temporal Control of Senescence. Trends Cell Biol. 2017, 27, 820–832. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Guan, X.; Carraro, G.; Parimon, T.; Liu, X.; Huang, G.; Mulay, A.; Soukiasian, H.J.; David, G.; Weigt, S.S.; et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2021, 203, 707–717. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, C.; Liu, Y.; Chen, H.; Liu, J.P. Cellular senescence occurred widespread to multiple selective sites in the fetal tissues and organs of mice. Clin. Exp. Pharmacol. Physiol. 2014, 41, 965–975. [Google Scholar] [CrossRef]
- Dorr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Dabritz, J.H.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic lethal metabolic targeting of cellular senescence in cancer therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of hTERT gene expression promotes escape from oncogene-induced cellular senescence. Proc. Natl. Acad. Sci. USA 2016, 113, 5024–5033. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.; Chen, J.Y.; Stewart-Ornstein, J.; Karhohs, K.W.; Mock, C.S.; Lahav, G. Fluctuations in p53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 2018, 71, 581–591.e5. [Google Scholar] [CrossRef] [PubMed]
- Baryshev, M.; Inashkina, I.; Salmina, K.; Huna, A.; Jackson, T.R.; Erenpreisa, J. DNA methylation of the Oct4A enhancers in embryonal carcinoma cells after etoposide treatment is associated with alternative splicing and altered pluripotency in reversibly senescent cells. Cell Cycle 2018, 17, 362–366. [Google Scholar] [CrossRef]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue damage and senescence provide critical signals for cellular reprogramming In Vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. CB 2017, 27, 2652–2660.e4. [Google Scholar] [CrossRef]
- Takasugi, M.; Okada, R.; Takahashi, A.; Virya Chen, D.; Watanabe, S.; Hara, E. Small extracellular vesicles secreted from senescent cells promote cancer cell proliferation through EphA2. Nat. Commun. 2017, 8, 15729. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Okada, R.; Nagao, K.; Kawamata, Y.; Hanyu, A.; Yoshimoto, S.; Takasugi, M.; Watanabe, S.; Kanemaki, M.T.; Obuse, C.; et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat. Commun. 2017, 8, 15287. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-associated exosome release from human prostate cancer cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef] [PubMed]
- Konopka, K.E.; Nguyen, T.; Jentzen, J.M.; Rayes, O.; Schmidt, C.J.; Wilson, A.M.; Farver, C.F.; Myers, J.L. Diffuse alveolar damage (DAD) resulting from coronavirus disease 2019 Infection is Morphologically Indistinguishable from Other Causes of DAD. Histopathology 2020, 77, 570–578. [Google Scholar] [CrossRef]
- Carsana, L.; Sonzogni, A.; Nasr, A.; Rossi, R.S.; Pellegrinelli, A.; Zerbi, P.; Rech, R.; Colombo, R.; Antinori, S.; Corbellino, M.; et al. Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: A two-centre descriptive study. Lancet Infect. Dis. 2020, 20, 1135–1140. [Google Scholar] [CrossRef]
- Adams, T.S.; Schupp, J.C.; Poli, S.; Ayaub, E.A.; Neumark, N.; Ahangari, F.; Chu, S.G.; Raby, B.A.; DeIuliis, G.; Januszyk, M.; et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1983. [Google Scholar] [CrossRef] [PubMed]
- Habermann, A.C.; Gutierrez, A.J.; Bui, L.T.; Yahn, S.L.; Winters, N.I.; Calvi, C.L.; Peter, L.; Chung, M.I.; Taylor, C.J.; Jetter, C.; et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci. Adv. 2020, 6, eaba1972. [Google Scholar] [CrossRef]
- Alder, J.K.; Barkauskas, C.E.; Limjunyawong, N.; Stanley, S.E.; Kembou, F.; Tuder, R.M.; Hogan, B.L.; Mitzner, W.; Armanios, M. Telomere dysfunction causes alveolar stem cell failure. Proc. Natl. Acad. Sci. USA 2015, 112, 5099–5104. [Google Scholar] [CrossRef]
- Razdan, N.; Vasilopoulos, T.; Herbig, U. Telomere dysfunction promotes transdifferentiation of human fibroblasts into myofibroblasts. Aging Cell 2018, 17, e12838. [Google Scholar] [CrossRef]
- Lee, J.; Reddy, R.; Barsky, L.; Scholes, J.; Chen, H.; Shi, W.; Driscoll, B. Lung alveolar integrity is compromised by telomere shortening in telomerase-null mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, 57–70. [Google Scholar] [CrossRef]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Marmai, C.; Sutherland, R.E.; Kim, K.K.; Dolganov, G.M.; Fang, X.; Kim, S.S.; Jiang, S.; Golden, J.A.; Hoopes, C.W.; Matthay, M.A.; et al. Alveolar epithelial cells express mesenchymal proteins in patients with idiopathic pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops In Vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef] [PubMed]
- Bharat, A.; Querrey, M.; Markov, N.S.; Kim, S.; Kurihara, C.; Garza-Castillon, R.; Manerikar, A.; Shilatifard, A.; Tomic, R.; Politanska, Y.; et al. Lung transplantation for patients with severe COVID-19. Sci. Transl. Med. 2020, 12, eabe4282. [Google Scholar] [CrossRef]
- Chen, J.; Wu, H.; Yu, Y.; Tang, N. Pulmonary alveolar regeneration in adult COVID-19 patients. Cell Res. 2020, 30, 708–710. [Google Scholar] [CrossRef]
- Kulkarni, T.; de Andrade, J.; Zhou, Y.; Luckhardt, T.; Thannickal, V.J. Alveolar epithelial disintegrity in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, 185–191. [Google Scholar] [CrossRef]
- Chung, M.I.; Bujnis, M.; Barkauskas, C.E.; Kobayashi, Y.; Hogan, B.L.M. Niche-mediated BMP/SMAD signaling regulates lung alveolar stem cell proliferation and differentiation. Development 2018, 145, dev163014. [Google Scholar] [CrossRef]
- Schruf, E.; Schroeder, V.; Le, H.Q.; Schonberger, T.; Raedel, D.; Stewart, E.L.; Fundel-Clemens, K.; Bluhmki, T.; Weigle, S.; Schuler, M.; et al. Recapitulating idiopathic pulmonary fibrosis related alveolar epithelial dysfunction in a human iPSC-derived air-liquid interface model. FASEB J. 2020, 34, 7825–7846. [Google Scholar] [CrossRef]
- Gokey, J.J.; Snowball, J.; Green, J.; Waltamath, M.; Spinney, J.J.; Black, K.E.; Hariri, L.P.; Xu, Y.; Perl, A.K. Pretreatment of aged mice with retinoic acid supports alveolar regeneration via upregulation of reciprocal PDGFA signalling. Thorax 2021, 76, 456–467. [Google Scholar] [CrossRef]
- Zheng, X.; Qi, C.; Zhang, S.; Fang, Y.; Ning, W. TGF-beta1 induces Fstl1 via the Smad3-c-Jun pathway in lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, 240–251. [Google Scholar] [CrossRef]
- Kim, K.K.; Wei, Y.; Szekeres, C.; Kugler, M.C.; Wolters, P.J.; Hill, M.L.; Frank, J.A.; Brumwell, A.N.; Wheeler, S.E.; Kreidberg, J.A.; et al. Epithelial cell alpha3beta1 integrin links beta-catenin and Smad signaling to promote myofibroblast formation and pulmonary fibrosis. J. Clin. Investig. 2009, 119, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Namba, T.; Tanaka, K.I.; Ito, Y.; Hoshino, T.; Matoyama, M.; Yamakawa, N.; Isohama, Y.; Azuma, A.; Mizushima, T. Induction of EMT-like phenotypes by an active metabolite of leflunomide and its contribution to pulmonary fibrosis. Cell Death Differ. 2010, 17, 1882–1895. [Google Scholar] [CrossRef] [PubMed]
- Schneider, D.J.; Wu, M.; Le, T.T.; Cho, S.H.; Brenner, M.B.; Blackburn, M.R.; Agarwal, S.K. Cadherin-11 contributes to pulmonary fibrosis: Potential role in TGF-beta production and epithelial to mesenchymal transition. FASEB J. 2012, 26, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Tanjore, H.; Cheng, D.S.; Degryse, A.L.; Zoz, D.F.; Abdolrasulnia, R.; Lawson, W.E.; Blackwell, T.S. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J. Biol. Chem. 2011, 286, 30972–30980. [Google Scholar] [CrossRef]
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; Blackwell, T.S.; et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 657–665. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, Y.; Kahn, M.; Ann, D.K.; Han, A.; Wang, H.; Nguyen, C.; Flodby, P.; Zhong, Q.; Krishnaveni, M.S.; et al. Interactions between β-catenin and transforming growth factor-β signaling pathways mediate epithelial-mesenchymal transition and are dependent on the transcriptional co-activator cAMP-response element-binding protein (CREB)-binding protein (CBP). J. Biol. Chem. 2012, 287, 7026–7038. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, L.; Hong, X.; Chen, H.; Shi, Y.; Liu, Y.; Liu, J.; Liu, J.P. Pulmonary Alveolar Stem Cell Senescence, Apoptosis, and Differentiation by p53-Dependent and -Independent Mechanisms in Telomerase-Deficient Mice. Cells 2021, 10, 2892. [Google Scholar] [CrossRef]
- Chen, R.; Zhang, K.; Chen, H.; Zhao, X.; Wang, J.; Li, L.; Cong, Y.; Ju, Z.; Xu, D.; Williams, B.R.; et al. Telomerase deficiency causes alveolar stem cell senescence-associated low-grade inflammation in lungs. J. Biol. Chem. 2015, 290, 30813–30829. [Google Scholar] [CrossRef]
- Wang, L.; Yu, X.; Liu, J.P. Telomere Damage Response and Low-Grade Inflammation. Adv. Exp. Med. Biol 2017, 1024, 213–224. [Google Scholar] [CrossRef]
- Birnhuber, A.; Egemnazarov, B.; Biasin, V.; Bonyadi Rad, E.; Wygrecka, M.; Olschewski, H.; Kwapiszewska, G.; Marsh, L.M. CDK4/6 inhibition enhances pulmonary inflammatory infiltration in bleomycin-induced lung fibrosis. Respir. Res. 2020, 21, 167. [Google Scholar] [CrossRef]
- Sala, M.A.; Balderas-Martinez, Y.I.; Buendia-Roldan, I.; Abdala-Valencia, H.; Nam, K.; Jain, M.; Bhorade, S.; Bharat, A.; Reyfman, P.A.; Ridge, K.M.; et al. Inflammatory pathways are upregulated in the nasal epithelium in patients with idiopathic pulmonary fibrosis. Respir. Res. 2018, 19, 233. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Krishnaveni, M.S.; Li, C.; Zhou, B.; Xing, Y.; Banfalvi, A.; Li, A.; Lombardi, V.; Akbari, O.; Borok, Z.; et al. Epithelium-specific deletion of TGF-beta receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 2011, 121, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Aumiller, V.; Balsara, N.; Wilhelm, J.; Gunther, A.; Konigshoff, M. WNT/beta-catenin signaling induces IL-1beta expression by alveolar epithelial cells in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Tzouvelekis, A.; Wang, R.; Herazo-Maya, J.D.; Ibarra, G.H.; Srivastava, A.; de Castro, J.P.W.; DeIuliis, G.; Ahangari, F.; Woolard, T.; et al. Thyroid hormone inhibits lung fibrosis in mice by improving epithelial mitochondrial function. Nat. Med. 2018, 24, 39–49. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Otoupalova, E.; Smith, S.; Cheng, G.; Thannickal, V.J. Oxidative Stress in Pulmonary Fibrosis. Compr. Physiol. 2020, 10, 509–547. [Google Scholar] [CrossRef]
- Korthagen, N.M.; van Moorsel, C.H.; Barlo, N.P.; Kazemier, K.M.; Ruven, H.J.; Grutters, J.C. Association between variations in cell cycle genes and idiopathic pulmonary fibrosis. PLoS ONE 2012, 7, e30442. [Google Scholar] [CrossRef]
- Dressen, A.; Abbas, A.R.; Cabanski, C.; Reeder, J.; Ramalingam, T.R.; Neighbors, M.; Bhangale, T.R.; Brauer, M.J.; Hunkapiller, J.; Reeder, J.; et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: A candidate gene sequencing study. Lancet Respir. Med. 2018, 6, 603–614. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Tsakiri, K.D.; Cronkhite, J.T.; Kuan, P.J.; Xing, C.; Raghu, G.; Weissler, J.C.; Rosenblatt, R.L.; Shay, J.W.; Garcia, C.K. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc. Natl. Acad. Sci. USA 2007, 104, 7552–7557. [Google Scholar] [CrossRef]
- Stuart, B.D.; Choi, J.; Zaidi, S.; Xing, C.; Holohan, B.; Chen, R.; Choi, M.; Dharwadkar, P.; Torres, F.; Girod, C.E.; et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 2015, 47, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.R.; Caetano Mota, P.; Melo, N.; Bastos, H.N.; Padrao, E.; Pereira, J.M.; Cunha, R.; Souto Moura, C.; Guimaraes, S.; Morais, A. Heterozygous TERT gene mutation associated with familial idiopathic pulmonary fibrosis. Respir. Med. Case Rep. 2019, 26, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Chen, J.J.; Lancaster, L.; Danoff, S.; Su, S.C.; Cogan, J.D.; Vulto, I.; Xie, M.; Qi, X.; Tuder, R.M.; et al. Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13051–13056. [Google Scholar] [CrossRef] [PubMed]
- Alder, J.K.; Cogan, J.D.; Brown, A.F.; Anderson, C.J.; Lawson, W.E.; Lansdorp, P.M.; Phillips, J.A., 3rd; Loyd, J.E.; Chen, J.J.; Armanios, M. Ancestral mutation in telomerase causes defects in repeat addition processivity and manifests as familial pulmonary fibrosis. PLoS Genet. 2011, 7, e1001352. [Google Scholar] [CrossRef]
- Armanios, M. Syndromes of Telomere Shortening. Annu Rev. Genom. Hum. Genet. 2009, 62, 6405–6409. [Google Scholar] [CrossRef]
- Petrovski, S.; Todd, J.L.; Durheim, M.T.; Wang, Q.; Chien, J.W.; Kelly, F.L.; Frankel, C.; Mebane, C.M.; Ren, Z.; Bridgers, J.; et al. An Exome Sequencing Study to Assess the Role of Rare Genetic Variation in Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2017, 196, 82–93. [Google Scholar] [CrossRef]
- Chilosi, M.; Poletti, V.; Rossi, A. The pathogenesis of COPD and IPF: Distinct horns of the same devil? Respir. Res. 2012, 13, 3. [Google Scholar] [CrossRef]
- Nogee, L.M.; Dunbar, A.E., 3rd; Wert, S.E.; Askin, F.; Hamvas, A.; Whitsett, J.A. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 2001, 344, 573–579. [Google Scholar] [CrossRef]
- Cottin, V.; Reix, P.; Khouatra, C.; Thivolet-Bejui, F.; Feldmann, D.; Cordier, J.F. Combined pulmonary fibrosis and emphysema syndrome associated with familial SFTPC mutation. Thorax 2011, 66, 918–919. [Google Scholar] [CrossRef]
- Nathan, N.; Giraud, V.; Picard, C.; Nunes, H.; Dastot-Le Moal, F.; Copin, B.; Galeron, L.; De Ligniville, A.; Kuziner, N.; Reynaud-Gaubert, M.; et al. Germline SFTPA1 mutation in familial idiopathic interstitial pneumonia and lung cancer. Hum. Mol. Genet. 2016, 25, 1457–1467. [Google Scholar] [CrossRef]
- Wu, J.; McKeague, M.; Sturla, S.J. Nucleotide-Resolution Genome-Wide Mapping of Oxidative DNA Damage by Click-Code-Seq. J. Am. Chem Soc. 2018, 140, 9783–9787. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Kumar, N.; Roginskaya, V.; Fouquerel, E.; Opresko, P.L.; Shiva, S.; Watkins, S.C.; Kolodieznyi, D.; Bruchez, M.P.; Van Houten, B. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 18435–18444. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, G.; Jurk, D.; Marques, F.D.; Correia-Melo, C.; Hardy, T.; Gackowska, A.; Anderson, R.; Taschuk, M.; Mann, J.; Passos, J.F. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012, 3, 708. [Google Scholar] [CrossRef] [PubMed]
- Degryse, A.L.; Xu, X.C.; Newman, J.L.; Mitchell, D.B.; Tanjore, H.; Polosukhin, V.V.; Jones, B.R.; McMahon, F.B.; Gleaves, L.A.; Phillips, J.A., 3rd; et al. Telomerase deficiency does not alter bleomycin-induced fibrosis in mice. Exp. Lung Res. 2012, 38, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, M.; Rossiello, F.; Clerici, M.; Barozzi, S.; Cittaro, D.; Kaplunov, J.M.; Bucci, G.; Dobreva, M.; Matti, V.; Beausejour, C.M.; et al. Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 2012, 14, 355–365. [Google Scholar] [CrossRef]
- Coluzzi, E.; Colamartino, M.; Cozzi, R.; Leone, S.; Meneghini, C.; O’Callaghan, N.; Sgura, A. Oxidative stress induces persistent telomeric DNA damage responsible for nuclear morphology change in mammalian cells. PLoS ONE 2014, 9, e110963. [Google Scholar] [CrossRef]
- Aitken, M.L.; Dugowson, C.; Schmidt, R.A.; Fer, M. Bleomycin-induced pulmonary fibrosis in a patient with rheumatoid arthritis. A possible synergistic effect? West. J. Med. 1989, 150, 344–346. [Google Scholar]
- Santrach, P.J.; Askin, F.B.; Wells, R.J.; Azizkhan, R.G.; Merten, D.F. Nodular form of bleomycin-related pulmonary injury in patients with osteogenic sarcoma. Cancer 1989, 64, 806–811. [Google Scholar] [CrossRef]
- Parfrey, H.; Babar, J.; Fiddler, C.A.; Chilvers, E.R. Idiopathic pulmonary fibrosis in a Christmas Island nuclear test veteran. BMJ Case Rep. 2010, 2010, bcr0620103102. [Google Scholar] [CrossRef]
- Desai, M.Y.; Karunakaravel, K.; Wu, W.; Agarwal, S.; Smedira, N.G.; Lytle, B.W.; Griffin, B.P. Pulmonary fibrosis on multidetector computed tomography and mortality in patients with radiation-associated cardiac disease undergoing cardiac surgery. J. Thorac. Cardiovasc. Surg. 2014, 148, 475–481.e3. [Google Scholar] [CrossRef]
- Gross, N.J. Pulmonary effects of radiation therapy. Ann. Intern. Med. 1977, 86, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.P.; Hsu, C.L.; Fan, L.C.; Huang, Z.; Bhatia, D.; Chen, Y.J.; Hisata, S.; Cho, S.J.; Nakahira, K.; Imamura, M.; et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat. Commun. 2019, 10, 3390. [Google Scholar] [CrossRef]
- Anathy, V.; Lahue, K.G.; Chapman, D.G.; Chia, S.B.; Casey, D.T.; Aboushousha, R.; van der Velden, J.L.J.; Elko, E.; Hoffman, S.M.; McMillan, D.H.; et al. Reducing protein oxidation reverses lung fibrosis. Nat. Med. 2018, 24, 1128–1135. [Google Scholar] [CrossRef] [PubMed]
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2005, 353, 2229–2242. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Buhl, R.; Grimes, G.J.; Bokser, A.D.; Hubbard, R.C.; Holroyd, K.J.; Roum, J.H.; Czerski, D.B.; Cantin, A.M.; Crystal, R.G. Effect of glutathione aerosol on oxidant-antioxidant imbalance in idiopathic pulmonary fibrosis. Lancet 1991, 338, 215–216. [Google Scholar] [CrossRef]
- Guan, L.L.; Kuwahara, J.; Sugiura, Y. Guanine-specific binding by bleomycin-nickel(III) complex and its reactivity for guanine-quartet telomeric DNA. Biochemistry 1993, 32, 6141–6145. [Google Scholar] [CrossRef] [PubMed]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef]
- Cornforth, M.N.; Meyne, J.; Littlefield, L.G.; Bailey, S.M.; Moyzis, R.K. Telomere staining of human chromosomes and the mechanism of radiation-induced dicentric formation. Radiat. Res. 1989, 120, 205–212. [Google Scholar] [CrossRef]
- Metcalfe, J.A.; Parkhill, J.; Campbell, L.; Stacey, M.; Biggs, P.; Byrd, P.J.; Taylor, A.M. Accelerated telomere shortening in ataxia telangiectasia. Nat. Genet. 1996, 13, 350–353. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Pereira-Smith, O.M. Expression of hTERT does not prevent stress-induced senescence in normal human fibroblasts, but protects the cells from stress-induced apoptosis and necrosis. J. Biol. Chem. 2002, 24, 24. [Google Scholar] [CrossRef]
- Petersen, S.; Saretzki, G.; von Zglinicki, T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp. Cell Res. 1998, 239, 152–160. [Google Scholar] [CrossRef] [PubMed]
- von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Halu, A.; Liu, S.; Baek, S.H.; Hobbs, B.D.; Hunninghake, G.M.; Cho, M.H.; Silverman, E.K.; Sharma, A. Exploring the cross-phenotype network region of disease modules reveals concordant and discordant pathways between chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Hum. Mol. Genet. 2019, 28, 2352–2364. [Google Scholar] [CrossRef]
- Kumar, M.; Seeger, W.; Voswinckel, R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2014, 51, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Huang, W.Y.; Wong, J.Y.; Chatterjee, N.; Reding, D.; Crawford, E.D.; De Vivo, I.; Hayes, R.B.; Savage, S.A. The association between leukocyte telomere length and cigarette smoking, dietary and physical variables, and risk of prostate cancer. Aging Cell 2009, 8, 405–413. [Google Scholar] [CrossRef]
- Stanley, S.E.; Chen, J.J.; Podlevsky, J.D.; Alder, J.K.; Hansel, N.N.; Mathias, R.A.; Qi, X.; Rafaels, N.M.; Wise, R.A.; Silverman, E.K.; et al. Telomerase mutations in smokers with severe emphysema. J. Clin. Investig. 2015, 125, 563–570. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Epel, E.S.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef]
- Aalbers, A.M.; Kajigaya, S.; van den Heuvel-Eibrink, M.M.; van der Velden, V.H.; Calado, R.T.; Young, N.S. Human telomere disease due to disruption of the CCAAT box of the TERC promoter. Blood 2012, 119, 3060–3063. [Google Scholar] [CrossRef][Green Version]
- Hao, L.Y.; Armanios, M.; Strong, M.A.; Karim, B.; Feldser, D.M.; Huso, D.; Greider, C.W. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 2005, 123, 1121–1131. [Google Scholar] [CrossRef]
- Fogarty, P.F.; Yamaguchi, H.; Wiestner, A.; Baerlocher, G.M.; Sloand, E.; Zeng, W.S.; Read, E.J.; Lansdorp, P.M.; Young, N.S. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. Lancet 2003, 362, 1628–1630. [Google Scholar] [CrossRef]
- Cawthon, R.M.; Smith, K.R.; O’Brien, E.; Sivatchenko, A.; Kerber, R.A. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet 2003, 361, 393–395. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Wood, E.; Collins, K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 1999, 402, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Kadota, T.; Yoshioka, Y.; Fujita, Y.; Araya, J.; Minagawa, S.; Hara, H.; Miyamoto, A.; Suzuki, S.; Fujimori, S.; Kohno, T.; et al. Extracellular Vesicles from Fibroblasts Induce Epithelial-Cell Senescence in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2020, 63, 623–636. [Google Scholar] [CrossRef] [PubMed]
- Khayrullin, A.; Krishnan, P.; Martinez-Nater, L.; Mendhe, B.; Fulzele, S.; Liu, Y.; Mattison, J.A.; Hamrick, M.W. Very Long-Chain C24:1 Ceramide Is Increased in Serum Extracellular Vesicles with Aging and Can Induce Senescence in Bone-Derived Mesenchymal Stem Cells. Cells 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed]
- Misawa, T.; Tanaka, Y.; Okada, R.; Takahashi, A. Biology of extracellular vesicles secreted from senescent cells as senescence-associated secretory phenotype factors. Geriatr. Gerontol. Int. 2020, 20, 539–546. [Google Scholar] [CrossRef]
- Pardo, A.; Selman, M. Lung Fibroblasts, Aging, and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac Soc. 2016, 13 (Suppl. 5), S417–S421. [Google Scholar] [CrossRef]
- Kelley, W.J.; Zemans, R.L.; Goldstein, D.R. Cellular senescence: Friend or foe to respiratory viral infections? Eur. Respir. J. 2020, 56, 2002708. [Google Scholar] [CrossRef]
- Ley, B.; Newton, C.A.; Arnould, I.; Elicker, B.M.; Henry, T.S.; Vittinghoff, E.; Golden, J.A.; Jones, K.D.; Batra, K.; Torrealba, J.; et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: An observational cohort-control study. Lancet Respir. Med. 2017, 5, 639–647. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Gall, J.G. A tandemly repeated sequence at the termini of the extrachromosomal ribosomal RNA genes in Tetrahymena. J. Mol. Biol. 1978, 120, 33–53. [Google Scholar] [CrossRef]
- Shampay, J.; Szostak, J.W.; Blackburn, E.H. DNA sequences of telomeres maintained in yeast. Nature 1984, 310, 154–157. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase enzyme with two kinds of primer specificity. Cell 1985, 51, 405–413. [Google Scholar] [CrossRef]
- de Lange, T.; Shiue, L.; Myers, R.M.; Cox, D.R.; Naylor, S.L.; Killery, A.M.; Varmus, H.E. Structure and variability of human chromosome ends. Mol. Cell. Biol. 1990, 10, 518–527. [Google Scholar] [PubMed]
- de Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Hastie, N.D.; Dempster, M.; Dunlop, M.G.; Thompson, A.M.; Green, D.K.; Allshire, R.C. Telomere reduction in human colorectal carcinoma and with ageing. Nature 1990, 346, 866–868. [Google Scholar] [CrossRef]
- Oexle, K.; Zwirner, A. Advanced telomere shortening in respiratory chain disorders. Hum. Mol. Genet. 1997, 6, 905–908. [Google Scholar] [CrossRef]
- Vaziri, H.; West, M.D.; Allsopp, R.C.; Davison, T.S.; Wu, Y.S.; Arrowsmith, C.H.; Poirier, G.G.; Benchimol, S. ATM-dependent telomere loss in aging human diploid fibroblasts and DNA damage lead to the post-translational activation of p53 protein involving poly(ADP-ribose) polymerase. EMBO J. 1997, 16, 6018–6033. [Google Scholar] [CrossRef]
- Bayne, S.; Li, H.; Jones, M.E.; Pinto, A.R.; van Sinderen, M.; Drummond, A.; Simpson, E.R.; Liu, J.P. Estrogen deficiency reversibly induces telomere shortening in mouse granulosa cells and ovarian aging In Vivo. Protein Cell 2011, 2, 333–346. [Google Scholar] [CrossRef]
- Bayne, S.; Jones, M.E.; Li, H.; Pinto, A.R.; Simpson, E.R.; Liu, J.P. Estrogen deficiency leads to telomerase inhibition, telomere shortening and reduced cell proliferation in the adrenal gland of mice. Cell Res. 2008, 18, 1141–1150. [Google Scholar] [CrossRef]
- Greider, C.W. Telomeres, telomerase and senescence. BioEssays News Rev. Mol. Cell. Dev. Biol. 1990, 12, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W. Telomeres and senescence: The history, the experiment, the future. Curr. Biol. CB 1998, 8, 178–181. [Google Scholar] [CrossRef]
- Xie, Z.; Jay, K.A.; Smith, D.L.; Zhang, Y.; Liu, Z.; Zheng, J.; Tian, R.; Li, H.; Blackburn, E.H. Early telomerase inactivation accelerates aging independently of telomere length. Cell 2015, 160, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Karlseder, J.; Smogorzewska, A.; de Lange, T. Senescence induced by altered telomere state, not telomere loss. Science 2002, 295, 2446–2449. [Google Scholar] [CrossRef]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; Von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA damage checkpoint response in telomere-initiated senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Guo, X.; Ferguson, D.O.; Chang, S. Multiple roles for MRE11 at uncapped telomeres. Nature 2009, 460, 914–918. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Serrano, R.; Tejera, A.; Gomez-Lopez, G.; Bobadilla, M.; Flores, J.M.; Bosch, F.; Blasco, M.A. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. eLife 2018, 7, e31299. [Google Scholar] [CrossRef]
- Xin, H.; Liu, D.; Wan, M.; Safari, A.; Kim, H.; Sun, W.; O’Connor, M.S.; Songyang, Z. TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 2007, 445, 559–562. [Google Scholar] [CrossRef]
- Wang, F.; Podell, E.R.; Zaug, A.J.; Yang, Y.; Baciu, P.; Cech, T.R.; Lei, M. The POT1-TPP1 telomere complex is a telomerase processivity factor. Nature 2007, 445, 506–510. [Google Scholar] [CrossRef]
- Miyoshi, T.; Kanoh, J.; Saito, M.; Ishikawa, F. Fission yeast Pot1-Tpp1 protects telomeres and regulates telomere length. Science 2008, 320, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.L.; Batista, L.F.; Freund, A.; Pech, M.F.; Venteicher, A.S.; Artandi, S.E. TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell 2012, 150, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, J.; Bell, C.F.; Weidenfeld, I.; Zaug, A.J.; Leinwand, L.A.; Cech, T.R. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 2012, 492, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.C.; Zaug, A.J.; Cech, T.R. Live Cell Imaging Reveals the Dynamics of Telomerase Recruitment to Telomeres. Cell 2016, 166, 1188–1197.e9. [Google Scholar] [CrossRef]
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334. [Google Scholar] [CrossRef]
- Guo, Y.; Kartawinata, M.; Li, J.; Pickett, H.A.; Teo, J.; Kilo, T.; Barbaro, P.M.; Keating, B.; Chen, Y.; Tian, L.; et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood 2014, 124, 2767–2774. [Google Scholar] [CrossRef]
- Kocak, H.; Ballew, B.J.; Bisht, K.; Eggebeen, R.; Hicks, B.D.; Suman, S.; O’Neil, A.; Giri, N.; Laboratory, N.D.C.G.R.; Group, N.D.C.S.W.; et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 2014, 28, 2090–2102. [Google Scholar] [CrossRef]
- Ahmad, T.; Sundar, I.K.; Tormos, A.M.; Lerner, C.A.; Gerloff, J.; Yao, H.; Rahman, I. Shelterin Telomere Protection Protein 1 Reduction Causes Telomere Attrition and Cellular Senescence via Sirtuin 1 Deacetylase in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2017, 56, 38–49. [Google Scholar] [CrossRef]
- Greider, C.W.; Blackburn, E.H. A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 1989, 337, 331–337. [Google Scholar] [CrossRef]
- Blackburn, E.H.; Greider, C.W.; Henderson, E.; Lee, M.S.; Shampay, J.; Shippen-Lentz, D. Recognition and elongation of telomeres by telomerase. Genome 1989, 31, 553–560. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef]
- Harle-Bachor, C.; Boukamp, P. Telomerase activity in the regenerative basal layer of the epidermis inhuman skin and in immortal and carcinoma-derived skin keratinocytes. Proc. Natl. Acad. Sci. USA 1996, 93, 6476–6481. [Google Scholar] [CrossRef]
- Counter, C.M.; Hahn, W.C.; Wei, W.; Caddle, S.D.; Beijersbergen, R.L.; Lansdorp, P.M.; Sedivy, J.M.; Weinberg, R.A. Dissociation among In Vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc. Natl. Acad. Sci. USA 1998, 95, 14723–14728. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Blasco, M.A.; Gottlieb, G.J.; Horner, J.W., 2nd; Greider, C.W.; DePinho, R.A. Essential role of mouse telomerase in highly proliferative organs. Nature 1998, 392, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Collins, K. Mammalian telomeres and telomerase. Curr. Opin. Cell Biol. 2000, 12, 378–383. [Google Scholar] [CrossRef]
- Mitchell, J.R.; Collins, K. Human telomerase activation requires two independent interactions between telomerase RNA and telomerase reverse transcriptase. Mol. Cell 2000, 6, 361–371. [Google Scholar] [CrossRef]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [Google Scholar] [CrossRef]
- Maida, Y.; Yasukawa, M.; Masutomi, K. De Novo RNA Synthesis by RNA-Dependent RNA Polymerase Activity of Telomerase Reverse Transcriptase. Mol. Cell. Biol. 2016, 36, 1248–1259. [Google Scholar] [CrossRef]
- Yasukawa, M.; Ando, Y.; Yamashita, T.; Matsuda, Y.; Shoji, S.; Morioka, M.S.; Kawaji, H.; Shiozawa, K.; Machitani, M.; Abe, T.; et al. CDK1 dependent phosphorylation of hTERT contributes to cancer progression. Nat. Commun. 2020, 11, 1557. [Google Scholar] [CrossRef]
- Rudolph, K.L.; Chang, S.; Millard, M.; Schreiber-Agus, N.; DePinho, R.A. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000, 287, 1253–1258. [Google Scholar] [CrossRef]
- Lin, S.; Nascimento, E.M.; Gajera, C.R.; Chen, L.; Neuhofer, P.; Garbuzov, A.; Wang, S.; Artandi, S.E. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature 2018, 556, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, C.; Li, H.; Wang, J.Q.; Liu, J.P. Molecular regulation of telomerase activity in aging. Protein Cell 2012, 2, 726–738. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: Old actors and new players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef]
- Xu, D.; Li, H.; Liu, J.P. Inhibition of telomerase by targeting MAP kinase signaling. Methods Mol. Biol. 2007, 405, 147–165. [Google Scholar] [CrossRef]
- Xu, D.; Dwyer, J.; Li, H.; Duan, W.; Liu, J.P. Ets2 maintains hTERT gene expression and breast cancer cell proliferation by interacting with c-Myc. J. Biol. Chem. 2008, 283, 23567–23580. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.; Li, H.; Xu, D.; Liu, J.P. Transcriptional regulation of telomerase activity: Roles of the the ets transcription factor family. Ann. N. Y. Acad. Sci. 2007, 1114, 36–47. [Google Scholar] [CrossRef]
- Dwyer, J.M.; Liu, J.P. Ets2 Transcription Factor, Telomerase Activity and Breast Cancer. Clin. Exp. Pharmacol. Physiol. 2009, 37, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Xu, D.; Li, J.; Berndt, M.C.; Liu, J.P. Transforming growth factor beta suppresses human telomerase reverse transcriptase (hTERT) by Smad3 interactions with c-Myc and the hTERT gene. J. Biol. Chem. 2006, 281, 25588–25600. [Google Scholar] [CrossRef]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, S.; Zhu, J. ETS variant transcription factor 5 and c-Myc cooperate in derepressing the human telomerase gene promoter via composite ETS/E-box motifs. J. Biol. Chem. 2020, 295, 10062–10075. [Google Scholar] [CrossRef]
- Wu, K.J.; Grandori, C.; Amacker, M.; Simon-Vermot, N.; Polack, A.; Lingner, J.; Dalla-Favera, R. Direct activation of TERT transcription by c-MYC. Nat. Genet. 1999, 21, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Popov, N.; Hou, M.; Wang, Q.; Bjorkholm, M.; Gruber, A.; Menkel, A.R.; Henriksson, M. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc. Natl. Acad. Sci. USA 2001, 98, 3826–3831. [Google Scholar] [CrossRef] [PubMed]
- Bayne, S.; Jones, M.E.; Li, H.; Liu, J.P. Potential roles for estrogen regulation of telomerase activity in aging. Ann. N. Y. Acad. Sci. 2007, 1114, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Bayne, S.; Liu, J.P. Hormones and growth factors regulate telomerase activity in ageing and cancer. Mol. Cell. Endocrinol. 2005, 240, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wang, L.; Wang, Z.; Liu, J.P. Roles of Telomere Biology in Cell Senescence, Replicative and Chronological Ageing. Cells 2019, 8, 54. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Ding, D.; Hao, W.; Yang, F.; Wu, X.; Wang, M.; Xu, X.; Ju, Z.; Liu, J.P.; Song, Z.; et al. hTERT promotes tumor angiogenesis by activating VEGF via interactions with the Sp1 transcription factor. Nucleic Acids Res. 2016, 44, 8693–8703. [Google Scholar] [CrossRef]
- Liu, N.; Guo, X.H.; Liu, J.P.; Cong, Y.S. Role of telomerase in the tumour microenvironment. Clin. Exp. Pharmacol. Physiol. 2020, 47, 357–364. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.; Jiang, L.; Dua, K.; Hansbro, P.M.; Liu, G. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir. Res. 2020, 21, 182. [Google Scholar] [CrossRef]
- Wicik, Z.; Eyileten, C.; Jakubik, D.; Simoes, S.N.; Martins, D.C., Jr.; Pavao, R.; Siller-Matula, J.M.; Postula, M. ACE2 Interaction Networks in COVID-19: A Physiological Framework for Prediction of Outcome in Patients with Cardiovascular Risk Factors. J. Clin. Med. 2020, 9, 3743. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.D.; Hua, J.; Mui, A.; O’Connor, R.; Grotendorst, G.; Khalil, N. Release of biologically active TGF-beta1 by alveolar epithelial cells results in pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Liu, J.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Ding, L.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Degryse, A.L.; Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Jones, B.R.; Boomershine, C.S.; Ortiz, C.; Sherrill, T.P.; McMahon, F.B.; Gleaves, L.A.; et al. TGFbeta signaling in lung epithelium regulates bleomycin-induced alveolar injury and fibroblast recruitment. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, 887–897. [Google Scholar] [CrossRef]
- Zhang, T.; Zhou, J.; Yue, H.; Du, C.; Xiao, Z.; Zhao, W.; Li, N.; Wang, X.; Liu, X.; Li, Y.; et al. Glycogen synthase kinase-3beta promotes radiation-induced lung fibrosis by regulating beta-catenin/lin28 signaling network to determine type II alveolar stem cell transdifferentiation state. FASEB J. 2020, 34, 12466–12480. [Google Scholar] [CrossRef]
- Bonniaud, P.; Kolb, M.; Galt, T.; Robertson, J.; Robbins, C.; Stampfli, M.; Lavery, C.; Margetts, P.J.; Roberts, A.B.; Gauldie, J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J. Immunol. 2004, 173, 2099–2108. [Google Scholar] [CrossRef]
- Kang, J.H.; Jung, M.Y.; Yin, X.; Andrianifahanana, M.; Hernandez, D.M.; Leof, E.B. Cell-penetrating peptides selectively targeting SMAD3 inhibit profibrotic TGF-beta signaling. J. Clin. Investig. 2017, 127, 2541–2554. [Google Scholar] [CrossRef]
- Cassar, L.; Li, H.; Pinto, A.R.; Nicholls, C.; Bayne, S.; Liu, J.P. Bone morphogenetic protein-7 inhibits telomerase activity, telomere maintenance, and cervical tumor growth. Cancer Res. 2008, 68, 9157–9166. [Google Scholar] [CrossRef]
- Cassar, L.; Li, H.; Jiang, F.X.; Liu, J.P. TGF-beta induces telomerase-dependent pancreatic tumor cell cycle arrest. Mol. Cell. Endocrinol. 2010, 320, 97–105. [Google Scholar] [CrossRef]
- Cassar, L.; Nicholls, C.; Pinto, A.R.; Chen, R.; Wang, L.; Li, H.; Liu, J.P. TGF-beta receptor mediated telomerase inhibition, telomere shortening and breast cancer cell senescence. Protein Cell 2017, 8, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Cassar, L.; Nicholls, C.; Pinto, A.R.; Li, H.; Liu, J.P. Bone morphogenetic protein-7 induces telomerase inhibition, telomere shortening, breast cancer cell senescence, and death via Smad3. FASEB J. 2009, 23, 1880–1892. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Z.; Liu, J.P. Identification of peptidomimetic telomere dysfunction inhibitor (TELODIN) through telomere dysfunction-induced foci (TIF) assay. STAR Protoc. 2021, 2, 100620. [Google Scholar] [CrossRef]
- Prowse, K.R.; Greider, C.W. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc. Natl. Acad. Sci. USA 1995, 92, 4818–4822. [Google Scholar] [CrossRef]
- Davis, R.J.; Welcker, M.; Clurman, B.E. Tumor suppression by the Fbw7 ubiquitin ligase: Mechanisms and opportunities. Cancer Cell 2014, 26, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.H.; Ko, J.; Donoghue, D.J. The importance of regulatory ubiquitination in cancer and metastasis. Cell Cycle 2017, 16, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun 2016, 7, 11190. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Zhu, Y.; Langhi, L.G.P.; Tchkonia, T.; Kruger, P.; Fielder, E.; Victorelli, S.; Ruswhandi, R.A.; Giorgadze, N.; Pirtskhalava, T.; et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab. 2019, 29, 1061–1077.e8. [Google Scholar] [CrossRef]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Jeon, O.H.; Kim, C.; Laberge, R.M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New agents that target senescent cells: The flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef]
- He, Y.; Li, W.; Lv, D.; Zhang, X.; Zhang, X.; Ortiz, Y.T.; Budamagunta, V.; Campisi, J.; Zheng, G.; Zhou, D. Inhibition of USP7 activity selectively eliminates senescent cells in part via restoration of p53 activity. Aging Cell 2020, 19, e13117. [Google Scholar] [CrossRef]
- Weber, L. Patented inhibitors of p53-Mdm2 interaction (2006–2008). Expert Opin. Ther. Pat. 2010, 20, 179–191. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef]
- Miyata, Y. Hsp90 inhibitor geldanamycin and its derivatives as novel cancer chemotherapeutic agents. Curr. Pharm. Des. 2005, 11, 1131–1138. [Google Scholar] [CrossRef]
- Cherif, H.; Bisson, D.G.; Jarzem, P.; Weber, M.; Ouellet, J.A.; Haglund, L. Curcumin and o-Vanillin Exhibit Evidence of Senolytic Activity in Human IVD Cells In Vitro. J. Clin. Med. 2019, 8, 433. [Google Scholar] [CrossRef]
- Li, W.; He, Y.; Zhang, R.; Zheng, G.; Zhou, D. The curcumin analog EF24 is a novel senolytic agent. Aging 2019, 11, 771–782. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef]
- Yang, D.; Tian, X.; Ye, Y.; Liang, Y.; Zhao, J.; Wu, T.; Lu, N. Identification of GL-V9 as a novel senolytic agent against senescent breast cancer cells. Life Sci. 2021, 272, 119196. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Triana-Martinez, F.; Picallos-Rabina, P.; Da Silva-Alvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreiros, A.; Barradas, M.; et al. Identification and characterization of Cardiac Glycosides as senolytic compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Gualda, E.; Paez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R., 3rd; Gonzalez-Lopez, C.; Ou, H.L.; Miron-Barroso, S.; Zhang, Z.; Lerida-Viso, A.; et al. Galacto-conjugation of Navitoclax as an efficient strategy to increase senolytic specificity and reduce platelet toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of senescent cells by beta-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Guiho, R.; Herranz, N.; Uren, A.; Withers, D.J.; Martinez-Barbera, J.P.; Tietze, L.F.; Gil, J. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 2020, 19, e13133. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.N.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.; et al. Using proteolysis-targeting chimera technology to reduce navitoclax platelet toxicity and improve its senolytic activity. Nat. Commun. 2020, 11, 1996. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Recalde, U.; Lorenzo-Gomez, I.; Blanco, F.J.; Loza, M.I.; Grassi, D.; Shirinsky, V.; Shirinsky, I.; Lotz, M.; Robbins, P.D.; Dominguez, E.; et al. Fibrates as drugs with senolytic and autophagic activity for osteoarthritis therapy. EBioMedicine 2019, 45, 588–605. [Google Scholar] [CrossRef]
- Ozsvari, B.; Nuttall, J.R.; Sotgia, F.; Lisanti, M.P. Azithromycin and Roxithromycin define a new family of “senolytic” drugs that target senescent human fibroblasts. Aging 2018, 10, 3294–3307. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Yang, E.J.; Park, J.T.; Kim, J.R.; Kim, E.C.; Jung, K.J.; Park, S.C.; Lee, Y.S. Identification of SYK inhibitor, R406 as a novel senolytic agent. Aging 2020, 12, 8221–8240. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, Z.N.; Zubova, S.G.; Bukreeva, E.I.; Pospelov, V.A.; Pospelova, T.V.; Blagosklonny, M.V. Rapamycin decelerates cellular senescence. Cell Cycle 2009, 8, 1888–1895. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Davidova, E.; Rohlenova, K.; Stursa, J.; Werner, L.; Andera, L.; Dong, L.; Terp, M.G.; Hodny, Z.; Ditzel, H.J.; et al. Selective elimination of senescent cells by mitochondrial targeting is regulated by ANT2. Cell Death Differ. 2019, 26, 276–290. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschenes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Peilin, W.; Songsong, T.; Chengyu, Z.; Zhi, C.; Chunhui, M.; Yinxian, Y.; Lei, Z.; Min, M.; Zongyi, W.; Mengkai, Y.; et al. Directed elimination of senescent cells attenuates development of osteoarthritis by inhibition of c-IAP and XIAP. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2618–2632. [Google Scholar] [CrossRef]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, X.; Wang, L.; Zhang, K.; Liu, J.; Liu, J.-P. Molecular Mechanisms of Alveolar Epithelial Stem Cell Senescence and Senescence-Associated Differentiation Disorders in Pulmonary Fibrosis. Cells 2022, 11, 877. https://doi.org/10.3390/cells11050877
Hong X, Wang L, Zhang K, Liu J, Liu J-P. Molecular Mechanisms of Alveolar Epithelial Stem Cell Senescence and Senescence-Associated Differentiation Disorders in Pulmonary Fibrosis. Cells. 2022; 11(5):877. https://doi.org/10.3390/cells11050877
Chicago/Turabian StyleHong, Xiaojing, Lihui Wang, Kexiong Zhang, Jun Liu, and Jun-Ping Liu. 2022. "Molecular Mechanisms of Alveolar Epithelial Stem Cell Senescence and Senescence-Associated Differentiation Disorders in Pulmonary Fibrosis" Cells 11, no. 5: 877. https://doi.org/10.3390/cells11050877
APA StyleHong, X., Wang, L., Zhang, K., Liu, J., & Liu, J.-P. (2022). Molecular Mechanisms of Alveolar Epithelial Stem Cell Senescence and Senescence-Associated Differentiation Disorders in Pulmonary Fibrosis. Cells, 11(5), 877. https://doi.org/10.3390/cells11050877