
Basal Autophagy Is Necessary for A Pharmacologic PPARα Transactivation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Animal Studies
2.3. Cell Culture and Drug Treatments
2.4. RNA Purification, cDNA Synthesis, and qPCR Analysis
2.5. Immunoblot Analysis
2.6. Statistical Analysis
3. Results
3.1. Autophagy Inhibition Impairs a Pharmacologic PPARα Transactivation
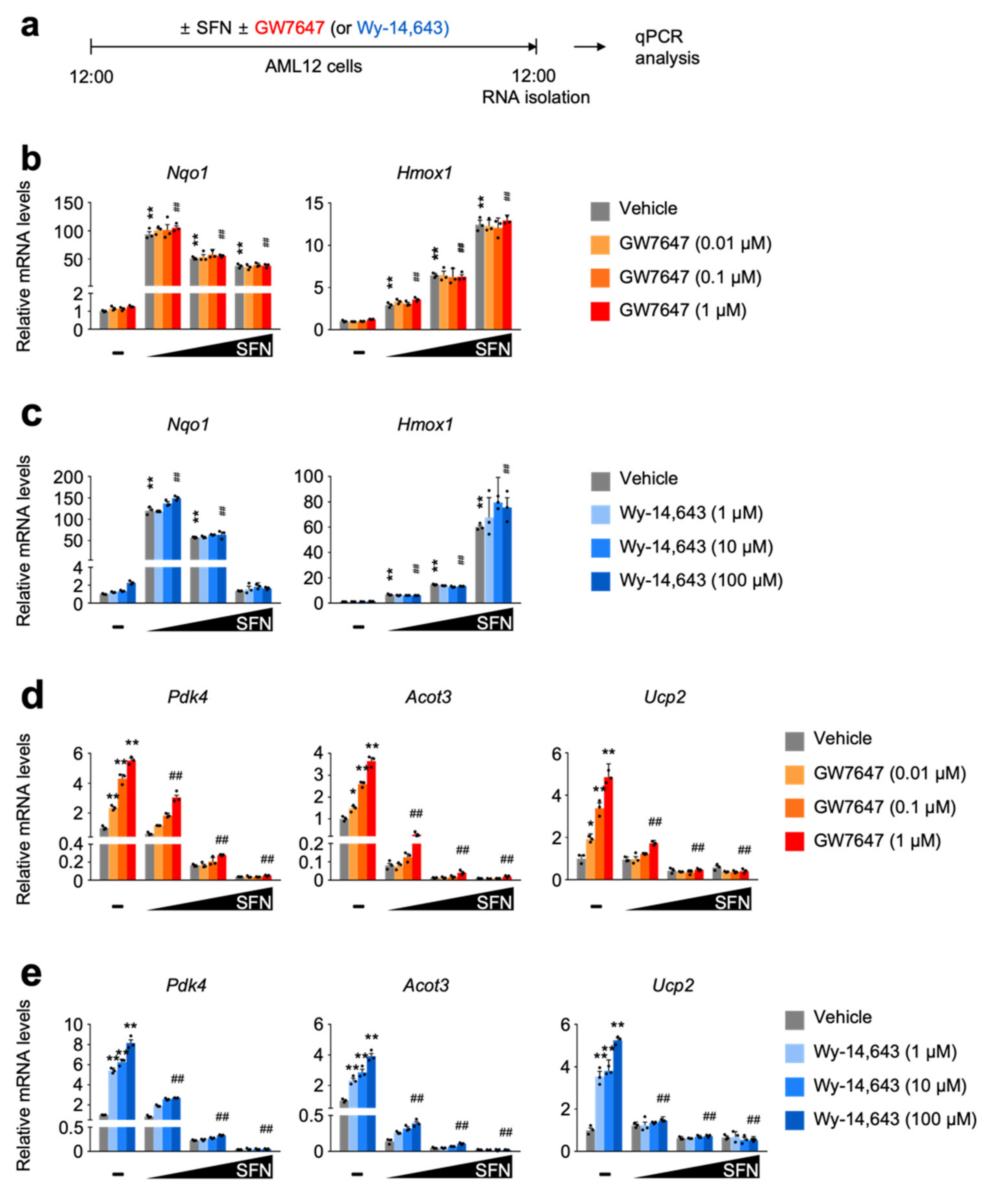
3.2. SFN-Mediated NRF2 Activation in a Dose-Dependent Manner Impairs a Pharmacologic PPARα Transactivation
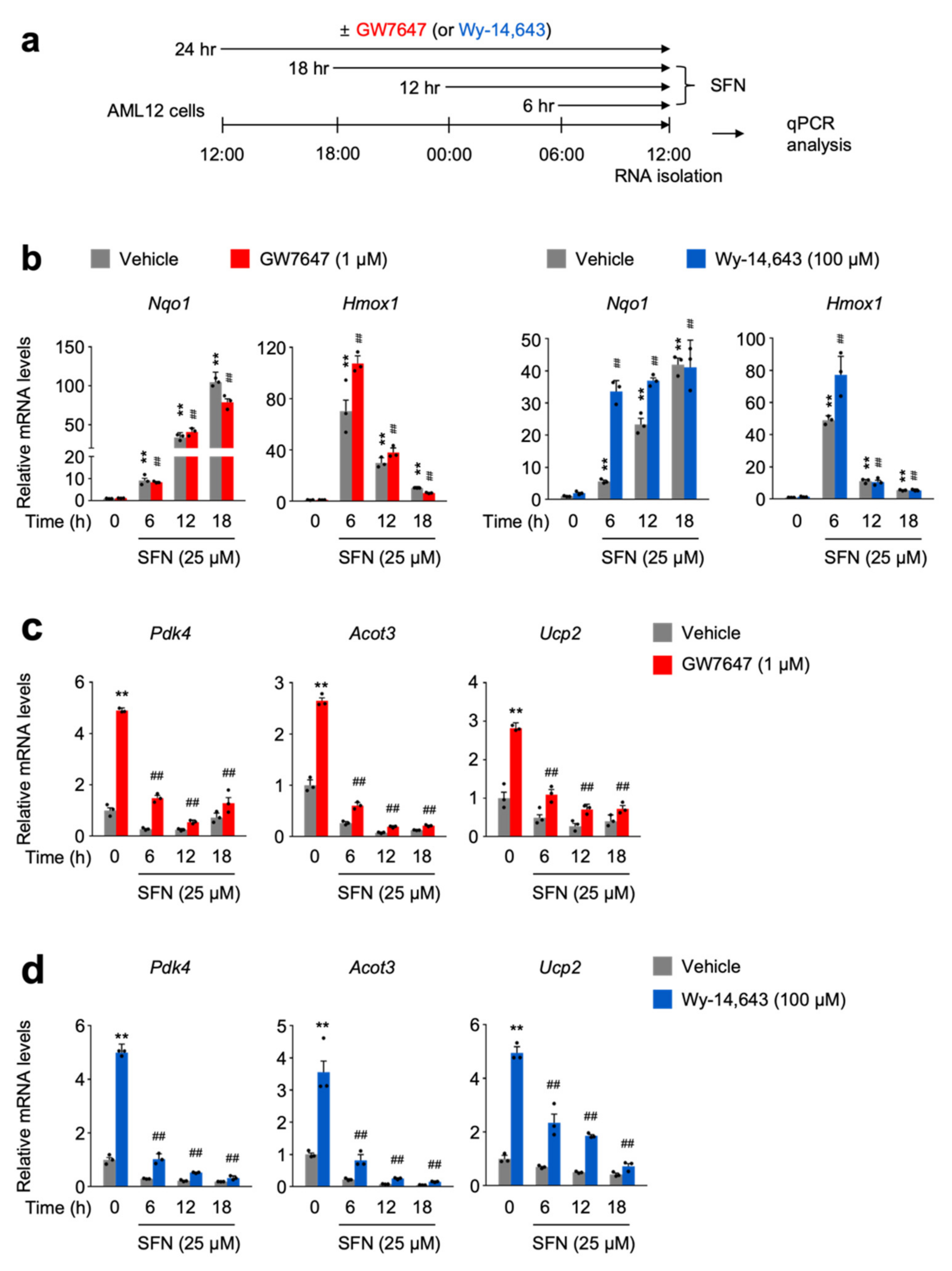
3.3. SFN-Mediated NRF2 Activation in a Time-Dependent Manner Impairs a Pharmacologic PPARα Transactivation
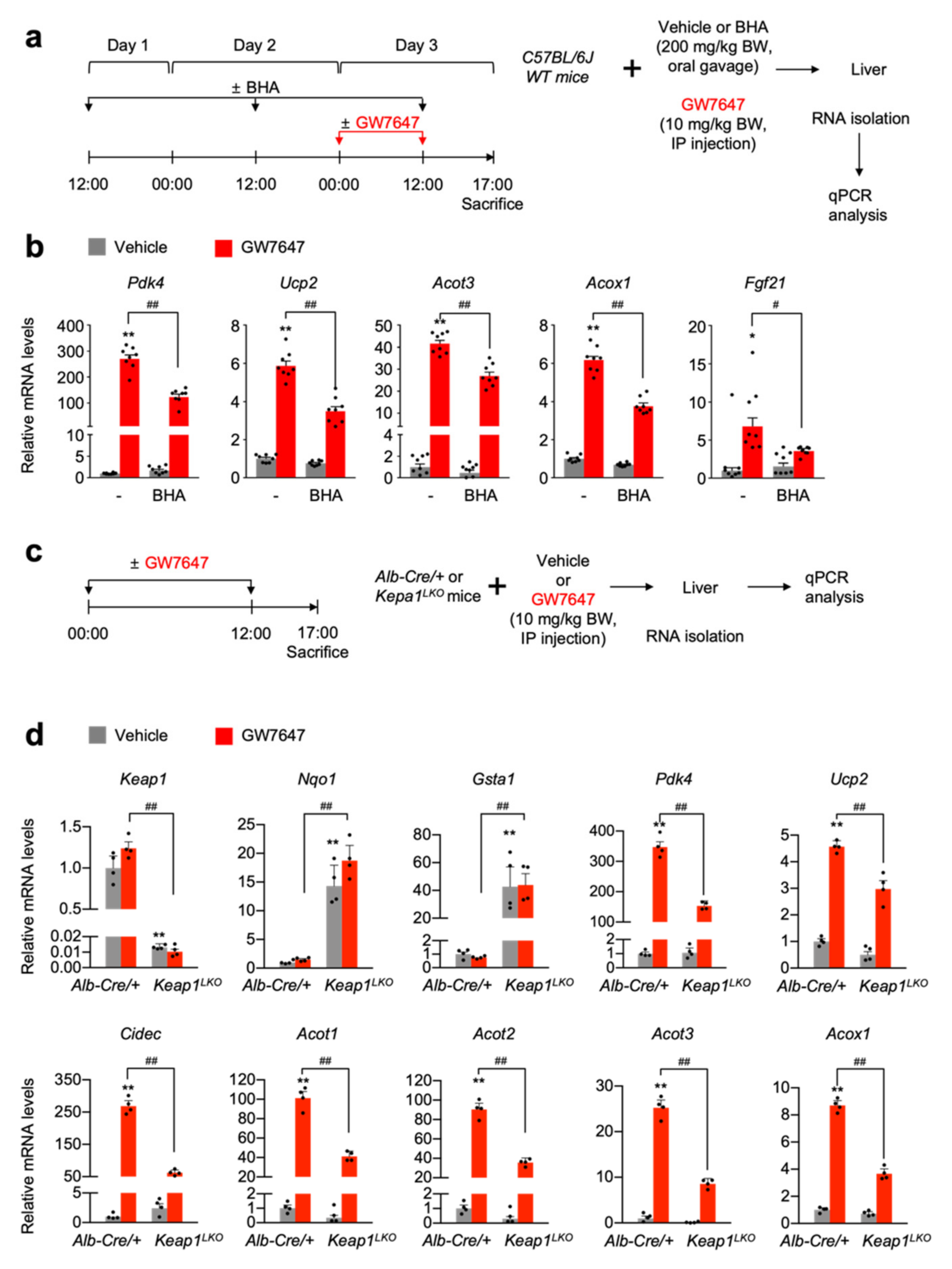
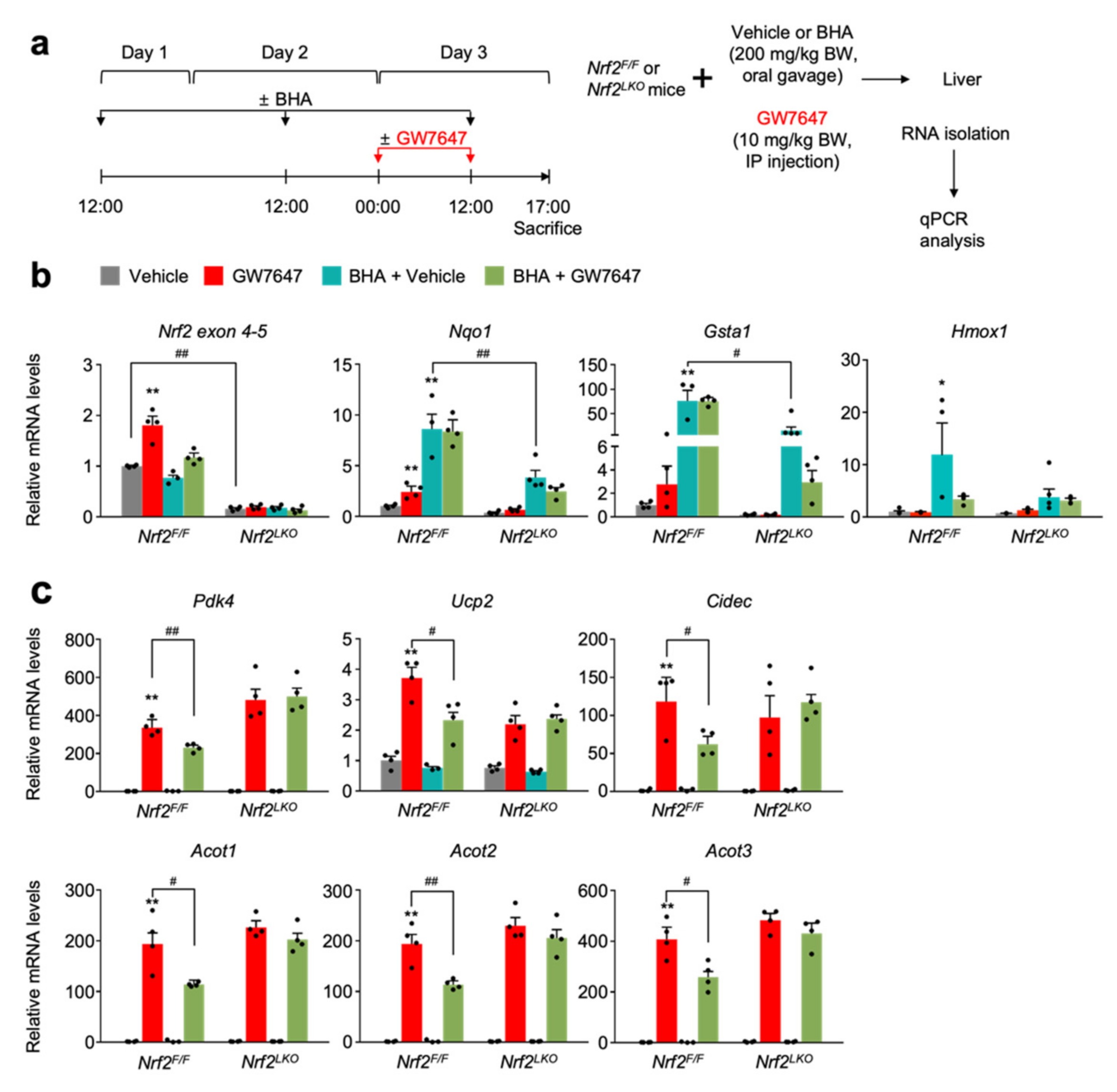
3.4. NRF2 Activation Is Necessary and Sufficient for Suppressing a Pharmacologic PPARα Transactivation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.; Eun, H.S.; Jo, E.K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; White, E. Autophagy and metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef]
- Silwal, P.; Paik, S.; Jeon, S.M.; Jo, E.K. Nuclear Receptors as Autophagy-Based Antimicrobial Therapeutics. Cells 2020, 9, 1979. [Google Scholar] [CrossRef]
- Yazdani, H.O.; Huang, H.; Tsung, A. Autophagy: Dual Response in the Development of Hepatocellular Carcinoma. Cells 2019, 8, 91. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Levine, B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014, 157, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D.C. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef]
- Ebato, C.; Uchida, T.; Arakawa, M.; Komatsu, M.; Ueno, T.; Komiya, K.; Azuma, K.; Hirose, T.; Tanaka, K.; Kominami, E.; et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008, 8, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.S.; Chung, K.W.; Won Kim, J.; Kim, J.; Komatsu, M.; Tanaka, K.; Nguyen, Y.H.; Kang, T.M.; Yoon, K.H.; Kim, J.W.; et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008, 8, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Masiero, E.; Agatea, L.; Mammucari, C.; Blaauw, B.; Loro, E.; Komatsu, M.; Metzger, D.; Reggiani, C.; Schiaffino, S.; Sandri, M. Autophagy is required to maintain muscle mass. Cell Metab. 2009, 10, 507–515. [Google Scholar] [CrossRef]
- Zhang, Y.; Goldman, S.; Baerga, R.; Zhao, Y.; Komatsu, M.; Jin, S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 19860–19865. [Google Scholar] [CrossRef]
- Singh, R.; Xiang, Y.; Wang, Y.; Baikati, K.; Cuervo, A.M.; Luu, Y.K.; Tang, Y.; Pessin, J.E.; Schwartz, G.J.; Czaja, M.J. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Investig. 2009, 119, 3329–3339. [Google Scholar] [CrossRef]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Tontonoz, P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 2008, 454, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-Sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef]
- Lee, J.M. Transcriptional coordination of hepatic autophagy by nutrient-sensing nuclear receptor PPARα and FXR. Ann. Pediatr. Endocrinol. Metab. 2016, 21, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Lee, Y.S.; Sin, D.M.; Lee, S.; Lee, M.K.; Lee, Y.M.; Hong, J.T.; Yun, Y.P.; Yoo, H.S. Sulforaphane inhibits mitotic clonal expansion during adipogenesis through cell cycle arrest. Obesity 2012, 20, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Tabbi-Anneni, I.; Gunda, V.; Wang, L. Transcription factor Nrf2 regulates SHP and lipogenic gene expression in hepatic lipid metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 299, G1211–G1221. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Xu, C.; Shen, G.; Jain, M.R.; Khor, T.O.; Gopalkrishnan, A.; Lin, W.; Reddy, B.; Chan, J.Y.; Kong, A.N. Gene expression profiles induced by cancer chemopreventive isothiocyanate sulforaphane in the liver of C57BL/6J mice and C57BL/6J/Nrf2 (−/−) mice. Cancer Lett. 2006, 243, 170–192. [Google Scholar] [CrossRef] [PubMed]
- Lee, C. Collaborative Power of Nrf2 and PPARgamma Activators against Metabolic and Drug-Induced Oxidative Injury. Oxid. Med. Cell Longev. 2017, 2017, 1378175. [Google Scholar] [CrossRef]
- Dovinova, I.; Kvandova, M.; Balis, P.; Gresova, L.; Majzunova, M.; Horakova, L.; Chan, J.Y.; Barancik, M. The role of Nrf2 and PPARgamma in the improvement of oxidative stress in hypertension and cardiovascular diseases. Physiol. Res. 2020, 69, S541–S553. [Google Scholar] [CrossRef]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef]
- Kong, X.; Thimmulappa, R.; Craciun, F.; Harvey, C.; Singh, A.; Kombairaju, P.; Reddy, S.P.; Remick, D.; Biswal, S. Enhancing Nrf2 pathway by disruption of Keap1 in myeloid leukocytes protects against sepsis. Am. J. Respir. Crit. Care Med. 2011, 184, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Okawa, H.; Motohashi, H.; Kobayashi, A.; Aburatani, H.; Kensler, T.W.; Yamamoto, M. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem. Biophys. Res. Commun. 2006, 339, 79–88. [Google Scholar] [CrossRef]
- Brown, P.J.; Stuart, L.W.; Hurley, K.P.; Lewis, M.C.; Winegar, D.A.; Wilson, J.G.; Wilkison, W.O.; Ittoop, O.R.; Willson, T.M. Identification of a subtype selective human PPARα agonist through parallel-array synthesis. Bioorg. Med. Chem. Lett. 2001, 11, 1225–1227. [Google Scholar] [CrossRef]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The role of autophagy during the early neonatal starvation period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Santilli, A.A.; Scotese, A.C.; Tomarelli, R.M. A potent antihypercholesterolemic agent: (4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio) acetic acid (Wy-14643). Experientia 1974, 30, 1110–1111. [Google Scholar] [CrossRef]
- Reddy, J.K.; Moody, D.E.; Azarnoff, D.L.; Tomarelli, R.M. Hepatic effects of some [4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio] acetic acid (WY-14,643) analogs in the mouse. Arch. Int. Pharmacodyn. Ther. 1977, 225, 51–57. [Google Scholar]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef]
- McMahon, M.; Itoh, K.; Yamamoto, M.; Chanas, S.A.; Henderson, C.J.; McLellan, L.I.; Wolf, C.R.; Cavin, C.; Hayes, J.D. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001, 61, 3299–3307. [Google Scholar] [PubMed]
- Wilms, H.; Sievers, J.; Rickert, U.; Rostami-Yazdi, M.; Mrowietz, U.; Lucius, R. Dimethylfumarate inhibits microglial and astrocytic inflammation by suppressing the synthesis of nitric oxide, IL-1beta, TNF-α and IL-6 in an in-vitro model of brain inflammation. J. Neuroinflamm. 2010, 7, 30. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Maher, J.M.; Suzuki, T.; Kawatani, Y.; Motohashi, H.; Yamamoto, M. Genetic analysis of cytoprotective functions supported by graded expression of Keap1. Mol. Cell. Biol. 2010, 30, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Iershov, A.; Nemazanyy, I.; Alkhoury, C.; Girard, M.; Barth, E.; Cagnard, N.; Montagner, A.; Chretien, D.; Rugarli, E.I.; Guillou, H.; et al. The class 3 PI3K coordinates autophagy and mitochondrial lipid catabolism by controlling nuclear receptor PPARα. Nat. Commun. 2019, 10, 1566. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Kuma, A.; Sugiura, Y.; Ichimura, Y.; Obata, M.; Kitamura, H.; Okuda, S.; Lee, H.C.; Ikeda, K.; Kanegae, Y.; et al. Autophagy regulates lipid metabolism through selective turnover of NCoR1. Nat. Commun. 2019, 10, 1567. [Google Scholar] [CrossRef]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to PPARα Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef]
- Gordon, D.M.; Hong, S.H.; Kipp, Z.A.; Hinds, T.D., Jr. Identification of Binding Regions of Bilirubin in the Ligand-Binding Pocket of the Peroxisome Proliferator-Activated Receptor-A (PPARα). Molecules 2021, 26, 2975. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, E.Y.; Lee, J.M. Basal Autophagy Is Necessary for A Pharmacologic PPARα Transactivation. Cells 2022, 11, 754. https://doi.org/10.3390/cells11040754
Kim EY, Lee JM. Basal Autophagy Is Necessary for A Pharmacologic PPARα Transactivation. Cells. 2022; 11(4):754. https://doi.org/10.3390/cells11040754
Chicago/Turabian StyleKim, Eun Young, and Jae Man Lee. 2022. "Basal Autophagy Is Necessary for A Pharmacologic PPARα Transactivation" Cells 11, no. 4: 754. https://doi.org/10.3390/cells11040754
APA StyleKim, E. Y., & Lee, J. M. (2022). Basal Autophagy Is Necessary for A Pharmacologic PPARα Transactivation. Cells, 11(4), 754. https://doi.org/10.3390/cells11040754