
Post-Translational Modifications of ATG4B in the Regulation of Autophagy


Abstract
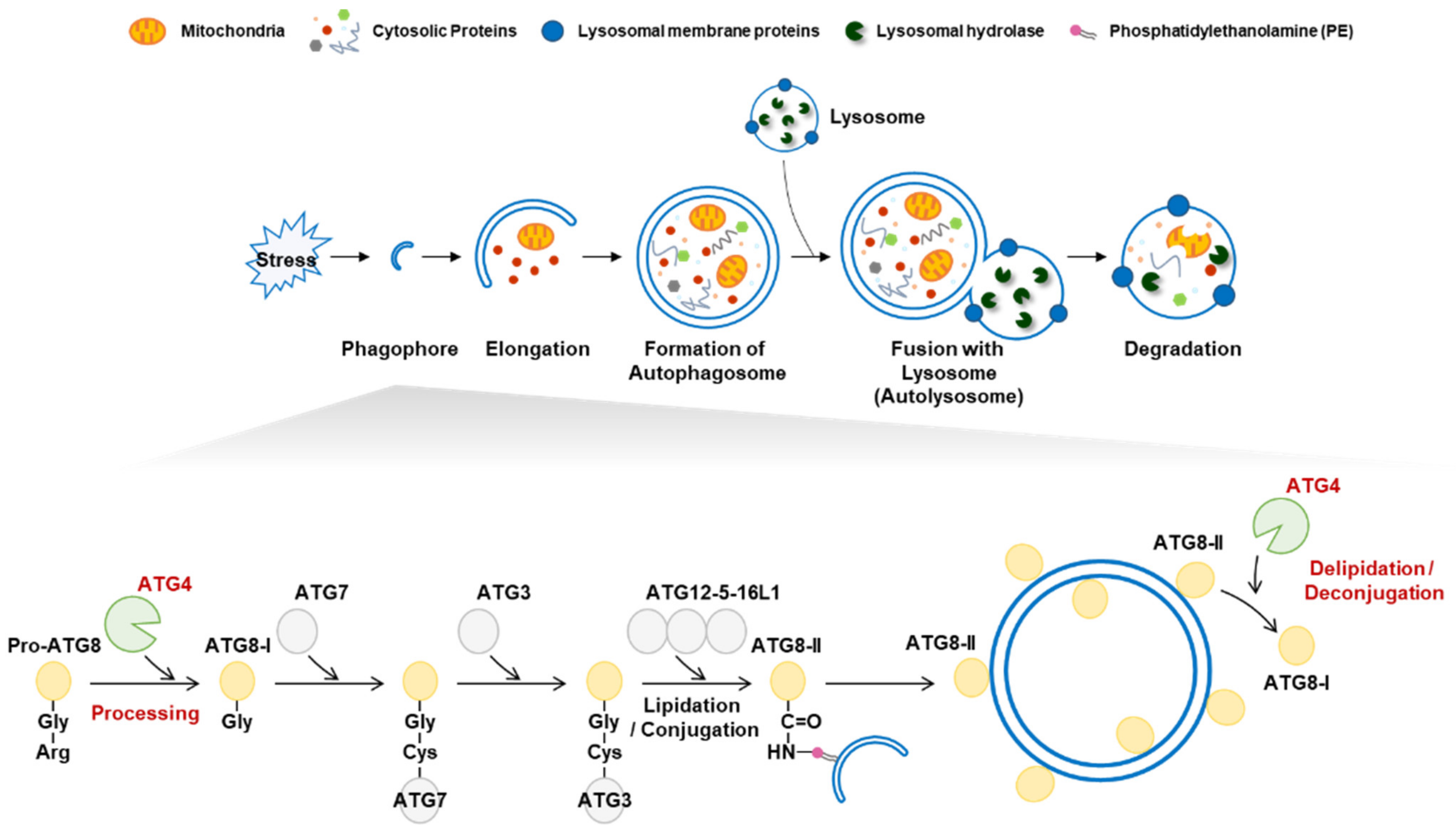
:1. Introduction
2. Structure and Function of ATG4
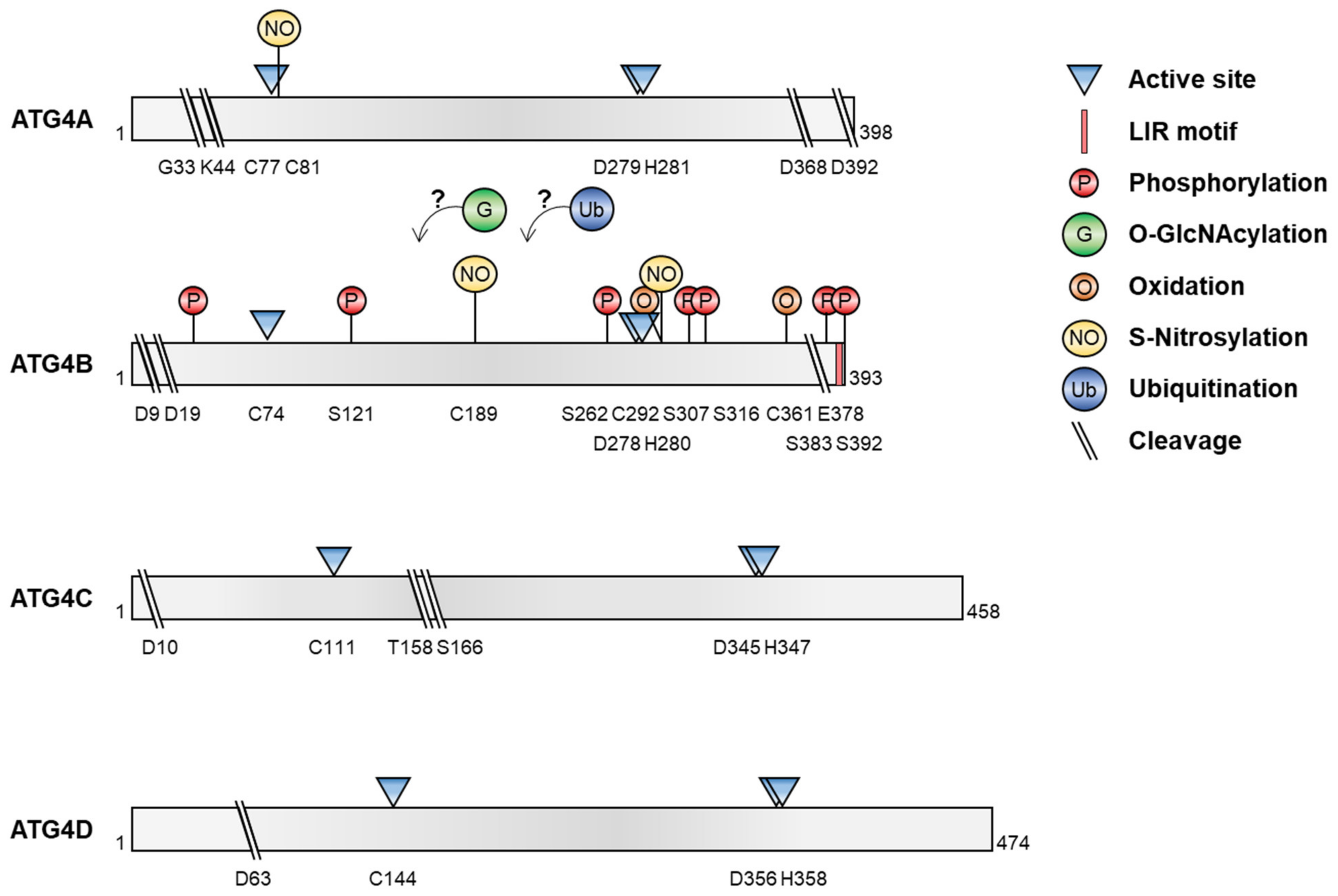
3. Post-Translational Modifications of ATG4
3.1. Phosphorylation
3.1.1. ATG4B Phosphorylation by MST4
3.1.2. Phosphorylation by AKT1 and AKT2
3.1.3. Phosphorylation by PFKP
3.1.4. Phosphorylation by Atg1/ULK1
3.2. ATG4 Dephosphorylation
3.3. O-GlcNAcylation of ATG4
3.4. ATG4 Oxidation
3.5. S-nitrosylation of ATG4
3.6. Ubiquitination of ATG4
3.7. Proteolytic Cleavage of ATG4
4. Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Y.; Du, J.; Wu, X.; Abdelrehem, A.; Ren, Y.; Liu, C.; Zhou, X.; Wang, S. Crosstalk between autophagy and microbiota in cancer progression. Mol. Cancer 2021, 20, 163. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Klionsky, D.J. The Emerging Roles of Autophagy in Human Diseases. Biomedicines 2021, 9, 1651. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J. Look people, “Atg” is an abbreviation for “autophagy-related”. That’s it. Autophagy 2012, 8, 1281–1282. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Martens, S.; Fracchiolla, D. Activation and targeting of ATG8 protein lipidation. Cell Discov. 2020, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Shpilka, T.; Weidberg, H.; Pietrokovski, S.; Elazar, Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011, 12, 226. [Google Scholar] [CrossRef]
- Fu, Y.; Huang, Z.; Hong, L.; Lu, J.H.; Feng, D.; Yin, X.M.; Li, M. Targeting ATG4 in Cancer Therapy. Cancers 2019, 11, 649. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Li, J.; Ouyang, L.; Liu, B.; Cheng, Y. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016, 373, 19–26. [Google Scholar] [CrossRef]
- Maruyama, T.; Noda, N.N. Autophagy-regulating protease Atg4: Structure, function, regulation and inhibition. J. Antibiot. 2017, 71, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Yu, S.; Jin, J.; Mugo, B.; Nguyen, N.; O’Brien, A.; Nag, S.; Lystad, A.H.; Melia, T.J. Delipidation of mammalian Atg8-family proteins by each of the four ATG4 proteases. Autophagy 2018, 14, 992–1010. [Google Scholar] [CrossRef]
- Hirata, E.; Ohya, Y.; Suzuki, K. Atg4 plays an important role in efficient expansion of autophagic isolation membranes by cleaving lipidated Atg8 in Saccharomyces cerevisiae. PLoS ONE 2017, 12, e0181047. [Google Scholar] [CrossRef] [Green Version]
- Wani, W.Y.; Boyer-Guittaut, M.; Dodson, M.; Chatham, J.; Darley-Usmar, V.; Zhang, J. Regulation of autophagy by protein post-translational modification. Lab. Investig. 2015, 95, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Kumanomidou, T.; Mizushima, T.; Komatsu, M.; Suzuki, A.; Tanida, I.; Sou, Y.S.; Ueno, T.; Kominami, E.; Tanaka, K.; Yamane, T. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J. Mol. Biol. 2006, 355, 612–618. [Google Scholar] [CrossRef]
- Sugawara, K.; Suzuki, N.N.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J. Biol. Chem. 2005, 280, 40058–40065. [Google Scholar] [CrossRef] [Green Version]
- Satoo, K.; Noda, N.N.; Kumeta, H.; Fujioka, Y.; Mizushima, N.; Ohsumi, Y.; Inagaki, F. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009, 28, 1341–1350. [Google Scholar] [CrossRef]
- Skytte Rasmussen, M.; Mouilleron, S.; Kumar Shrestha, B.; Wirth, M.; Lee, R.; Bowitz Larsen, K.; Abudu Princely, Y.; O’Reilly, N.; Sjøttem, E.; Tooze, S.A.; et al. ATG4B contains a C-terminal LIR motif important for binding and efficient cleavage of mammalian orthologs of yeast Atg8. Autophagy 2017, 13, 834–853. [Google Scholar] [CrossRef] [Green Version]
- Betin, V.M.; MacVicar, T.D.; Parsons, S.F.; Anstee, D.J.; Lane, J.D. A cryptic mitochondrial targeting motif in Atg4D links caspase cleavage with mitochondrial import and oxidative stress. Autophagy 2012, 8, 664–676. [Google Scholar] [CrossRef] [Green Version]
- Betin, V.M.; Lane, J.D. Atg4D at the interface between autophagy and apoptosis. Autophagy 2009, 5, 1057–1059. [Google Scholar] [CrossRef] [Green Version]
- Betin, V.M.; Lane, J.D. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J. Cell Sci. 2009, 122, 2554–2566. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hou, Y.; Wang, J.; Chen, X.; Shao, Z.M.; Yin, X.M. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J. Biol. Chem. 2011, 286, 7327–7338. [Google Scholar] [CrossRef] [Green Version]
- Agrotis, A.; Pengo, N.; Burden, J.J.; Ketteler, R. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy 2019, 15, 976–997. [Google Scholar] [CrossRef] [Green Version]
- Mariño, G.; Fernández, A.F.; Cabrera, S.; Lundberg, Y.W.; Cabanillas, R.; Rodríguez, F.; Salvador-Montoliu, N.; Vega, J.A.; Germanà, A.; Fueyo, A.; et al. Autophagy is essential for mouse sense of balance. J. Clin. Investig. 2010, 120, 2331–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, R.; Savelieva, K.; Baker, K.; Hansen, G.; Vogel, P. Histopathological and neurological features of Atg4b knockout mice. Vet. Pathol. 2011, 48, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Salvador-Montoliu, N.; Fueyo, A.; Knecht, E.; Mizushima, N.; López-Otín, C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. J. Biol. Chem. 2007, 282, 18573–18583. [Google Scholar] [CrossRef] [Green Version]
- Ali, M.A.; Matboli, M.; El-Khazragy, N.; Saber, O.; El-Nakeep, S.; Abdelzaher, H.M.; Shafei, A.E.; Mostafa, R. Investigating miRNA-661 and ATG4-B mRNA expression as potential biomarkers for hepatocellular carcinoma. Biomark. Med. 2018, 12, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Ping, Y.F.; Jiang, Y.X.; Luo, X.; Zhang, X.; Bian, X.W.; Yu, P.W. ATG4A promotes tumor metastasis by inducing the epithelial-mesenchymal transition and stem-like properties in gastric cells. Oncotarget 2016, 7, 39279–39292. [Google Scholar] [CrossRef] [Green Version]
- Rothe, K.; Lin, H.; Lin, K.B.; Leung, A.; Wang, H.M.; Malekesmaeili, M.; Brinkman, R.R.; Forrest, D.L.; Gorski, S.M.; Jiang, X. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood 2014, 123, 3622–3634. [Google Scholar] [CrossRef]
- Liu, P.F.; Leung, C.M.; Chang, Y.H.; Cheng, J.S.; Chen, J.J.; Weng, C.J.; Tsai, K.W.; Hsu, C.J.; Liu, Y.C.; Hsu, P.C.; et al. ATG4B promotes colorectal cancer growth independent of autophagic flux. Autophagy 2014, 10, 1454–1465. [Google Scholar] [CrossRef] [Green Version]
- Andaloussi, A.E.; Habib, S.; Soylemes, G.; Laknaur, A.; Elhusseini, H.; Al-Hendy, A.; Ismail, N. Defective expression of ATG4D abrogates autophagy and promotes growth in human uterine fibroids. Cell Death Discov. 2017, 3, 17041. [Google Scholar] [CrossRef]
- Liu, P.F.; Tsai, K.L.; Hsu, C.J.; Tsai, W.L.; Cheng, J.S.; Chang, H.W.; Shiau, C.W.; Goan, Y.G.; Tseng, H.H.; Wu, C.H.; et al. Drug Repurposing Screening Identifies Tioconazole as an ATG4 Inhibitor that Suppresses Autophagy and Sensitizes Cancer Cells to Chemotherapy. Theranostics 2018, 8, 830–845. [Google Scholar] [CrossRef]
- Kurdi, A.; Cleenewerck, M.; Vangestel, C.; Lyssens, S.; Declercq, W.; Timmermans, J.P.; Stroobants, S.; Augustyns, K.; De Meyer, G.R.Y.; Van Der Veken, P.; et al. ATG4B inhibitors with a benzotropolone core structure block autophagy and augment efficiency of chemotherapy in mice. Biochem. Pharmacol. 2017, 138, 150–162. [Google Scholar] [CrossRef]
- Akin, D.; Wang, S.K.; Habibzadegah-Tari, P.; Law, B.; Ostrov, D.; Li, M.; Yin, X.M.; Kim, J.S.; Horenstein, N.; Dunn, W.A., Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014, 10, 2021–2035. [Google Scholar] [CrossRef]
- Huang, T.; Kim, C.K.; Alvarez, A.A.; Pangeni, R.P.; Wan, X.; Song, X.; Shi, T.; Yang, Y.; Sastry, N.; Horbinski, C.M.; et al. MST4 Phosphorylation of ATG4B Regulates Autophagic Activity, Tumorigenicity, and Radioresistance in Glioblastoma. Cancer Cell 2017, 32, 840–855.e848. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Hong, L.; Xu, J.; Zhong, G.; Gu, Q.; Gu, Q.; Guan, Y.; Zheng, X.; Dai, Q.; Luo, X.; et al. Discovery of a small molecule targeting autophagy via ATG4B inhibition and cell death of colorectal cancer cells in vitro and in vivo. Autophagy 2019, 15, 295–311. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Guo, M.; Li, J.; Zheng, Y.; Zhang, S.; Xie, T.; Liu, B. Systems biology-based discovery of a potential Atg4B agonist (Flubendazole) that induces autophagy in breast cancer. Mol. Biosyst. 2015, 11, 2860–2866. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Wilkie-Grantham, R.P.; Yanagi, T.; Shu, C.W.; Matsuzawa, S.; Reed, J.C. ATG4B (Autophagin-1) phosphorylation modulates autophagy. J. Biol. Chem. 2015, 290, 26549–26561. [Google Scholar] [CrossRef] [Green Version]
- Ni, Z.; He, J.; Wu, Y.; Hu, C.; Dai, X.; Yan, X.; Li, B.; Li, X.; Xiong, H.; Li, Y.; et al. AKT-mediated phosphorylation of ATG4B impairs mitochondrial activity and enhances the Warburg effect in hepatocellular carcinoma cells. Autophagy 2018, 14, 685–701. [Google Scholar] [CrossRef] [Green Version]
- Pengo, N.; Prak, K.; Costa, J.R.; Luft, C.; Agrotis, A.; Freeman, J.; Gewinner, C.A.; Chan, A.W.E.; Selwood, D.L.; Kriston-Vizi, J.; et al. Identification of Kinases and Phosphatases That Regulate ATG4B Activity by siRNA and Small Molecule Screening in Cells. Front. Cell Dev. Biol. 2018, 6, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Sun, L.; Yan, G.; Yan, X. PFKP facilitates ATG4B phosphorylation during amino acid deprivation-induced autophagy. Cell. Signal. 2021, 82, 109956. [Google Scholar] [CrossRef] [PubMed]
- Pengo, N.; Agrotis, A.; Prak, K.; Jones, J.; Ketteler, R. A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nat. Commun. 2017, 8, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, Y.K.; Park, N.Y.; Park, S.J.; Kim, B.G.; Shin, J.H.; Jo, D.S.; Bae, D.J.; Suh, Y.A.; Chang, J.H.; Lee, E.K.; et al. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget 2016, 7, 57186–57196. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yang, Z.; Gu, Q.; Xia, F.; Fu, Y.; Liu, P.; Yin, X.M.; Li, M. The protease activity of human ATG4B is regulated by reversible oxidative modification. Autophagy 2020, 16, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y.; Wang, L.; Wang, P.; Xue, Y.; Li, X.; Qiao, X.; Zhang, X.; Xu, T.; Liu, G.; et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy 2017, 13, 1145–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, E.; Okumura, C.Y.; Sheffy-Levin, S.; Varsano, T.; Shu, V.C.; Qi, J.; Niesman, I.R.; Yang, H.J.; López-Otín, C.; Yang, W.Y.; et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 2012, 8, e1003007. [Google Scholar] [CrossRef] [Green Version]
- Ramazi, S.; Zahiri, J. Posttranslational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Licheva, M.; Raman, B.; Kraft, C.; Reggiori, F. Phosphoregulation of the autophagy machinery by kinases and phosphatases. Autophagy 2022, 18, 104–123. [Google Scholar] [CrossRef]
- Dai, J.; Jin, W.H.; Sheng, Q.H.; Shieh, C.H.; Wu, J.R.; Zeng, R. Protein phosphorylation and expression profiling by Yin-yang multidimensional liquid chromatography (Yin-yang MDLC) mass spectrometry. J. Proteome Res. 2007, 6, 250–262. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Villén, J.; Beausoleil, S.A.; Gerber, S.A.; Gygi, S.P. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. USA 2007, 104, 1488–1493. [Google Scholar] [CrossRef] [Green Version]
- Gnad, F.; Ren, S.; Cox, J.; Olsen, J.V.; Macek, B.; Oroshi, M.; Mann, M. PHOSIDA (phosphorylation site database): Management, structural and evolutionary investigation, and prediction of phosphosites. Genome Biol. 2007, 8, R250. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Rho, H.S.; Newman, R.H.; Zhang, J.; Zhu, H.; Qian, J. PhosphoNetworks: A database for human phosphorylation networks. Bioinformatics 2014, 30, 141–142. [Google Scholar] [CrossRef]
- Pasquin, S.; Chehboun, S.; Dejda, A.; Meliani, Y.; Savin, V.; Warner, G.J.; Bosse, R.; Tormo, A.; Mayer, G.; Sharma, M.; et al. Effect of human very low-density lipoproteins on cardiotrophin-like cytokine factor 1 (CLCF1) activity. Sci. Rep. 2018, 8, 3990. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Wandelmer, J.; Kriegenburg, F.; Rohringer, S.; Schuschnig, M.; Gómez-Sánchez, R.; Zens, B.; Abreu, S.; Hardenberg, R.; Hollenstein, D.; Gao, J.; et al. Atg4 proteolytic activity can be inhibited by Atg1 phosphorylation. Nat. Commun. 2017, 8, 295. [Google Scholar] [CrossRef]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [Green Version]
- Papinski, D.; Schuschnig, M.; Reiter, W.; Wilhelm, L.; Barnes, C.A.; Maiolica, A.; Hansmann, I.; Pfaffenwimmer, T.; Kijanska, M.; Stoffel, I.; et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol. Cell 2014, 53, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Egan, D.F.; Chun, M.G.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Hart, G.W.; Housley, M.P.; Slawson, C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature 2007, 446, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Krause, M.W.; Love, D.C. Bittersweet memories: Linking metabolism to epigenetics through O-GlcNAcylation. Nat. Rev. Mol. Cell Biol. 2012, 13, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.H.; Weng, C.L.; Lin, K.I. O-GlcNAcylation and its role in the immune system. J. Biomed. Sci. 2020, 27, 57. [Google Scholar] [CrossRef] [PubMed]
- Hardivillé, S.; Hart, G.W. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Lewis, B.A. O-GlcNAcylation at promoters, nutrient sensors, and transcriptional regulation. Biochim. Biophys. Acta 2013, 1829, 1202–1206. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.L.; Lindner, A.B. Protein Posttranslational Modifications: Roles in Aging and Age-Related Disease. Oxid. Med. Cell Longev. 2017, 2017, 5716409. [Google Scholar] [CrossRef] [Green Version]
- Ryan, B.J.; Nissim, A.; Winyard, P.G. Oxidative post-translational modifications and their involvement in the pathogenesis of autoimmune diseases. Redox Biol. 2014, 2, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, R.H.; Chen, Y.H.; Huang, T.Y. Ubiquitin-mediated regulation of autophagy. J. Biomed. Sci. 2019, 26, 80. [Google Scholar] [CrossRef]
- Su, T.; Yang, M.; Wang, P.; Zhao, Y.; Ma, C. Interplay between the Ubiquitin Proteasome System and Ubiquitin-Mediated Autophagy in Plants. Cells 2020, 9, 2219. [Google Scholar] [CrossRef]
- Klein, T.; Eckhard, U.; Dufour, A.; Solis, N.; Overall, C.M. Proteolytic Cleavage-Mechanisms, Function, and ”Omic” Approaches for a Near-Ubiquitous Posttranslational Modification. Chem. Rev. 2018, 118, 1137–1168. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Target | Modification | Regulator | Site (* Predicted Site) | Consequence | Cell Type/Model | Reference (PMID) |
|---|---|---|---|---|---|---|
| ATG4A | Oxidation | ROS (H2O2) | C81 | Activation | CHO cell | [38] (17347651) |
| Cleavage | Caspase-3 | EEFD368 | - | HeLa cells/ In vitro assay | [39] (21121091) | |
| LEED392 | - | |||||
| Calpain 1 | EKSK44LL | - | ||||
| WILG33KQ | - | |||||
| ATG4B | Phosphorylation | - | S383, S392 | Activation | HEK293T, MEF cells | [40] (26378241) |
| MST4 | S383 | Activation | Glioma stem-like cells | [35] (29232556) | ||
| AKT1 | S34 | non-canonical function | HepG2 cells | [41] (29165041) | ||
| AKT2 | S121, S262 | Activation | HEK293T cells | [42] (30443548) | ||
| PRKP | S34 | Activation | HEK293T cells | [43] (33607258) | ||
| ULK1 | S316 | Inhibition | MEF, HeLa, HEK293T cells | [44] (28821708) | ||
| Dephosphorylation | PP2A | S316 | Activation | HEK293T cells | [44] (28821708) | |
| O-GlcNAcylation | O-GlcNAc transferase | - | Activation | SH-SY5Y cells | [45] (27527864) | |
| Oxidation | ROS (H2O2) | C292, C361 | Inhibition | HEK293, HeLa cells | [46] (31880198) | |
| S-Nitrosylation | NO | C189, C292 | Inhibition | SH-SY5Y/ hippocampus | [47] (28633005) | |
| Ubiquitination | RNF5 | - | Inhibition | MEF cells | [48] (23093945) | |
| Cleavage | Caspase-3 | EFED19 | - | HeLa cells/ In vitro assay | [39] (21121091) | |
| LTYD9 | - | |||||
| Calpain 1 | ERLE378RF | - | ||||
| ATG4C | Cleavage | Caspase-3 | DEVD10 | - | HeLa cells/ In vitro assay | [39] (21121091) |
| Calpain 1 | KKFT158AS | - | ||||
| ASLS166GE | - | |||||
| ATG4D | Cleavage | Caspase-3 | DEVD63K | Activation | HeLa, A431, HEK293 cells | [21] (19549685) |
| HeLa cells | [19] (22441018) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, N.Y.; Jo, D.S.; Cho, D.-H. Post-Translational Modifications of ATG4B in the Regulation of Autophagy. Cells 2022, 11, 1330. https://doi.org/10.3390/cells11081330
Park NY, Jo DS, Cho D-H. Post-Translational Modifications of ATG4B in the Regulation of Autophagy. Cells. 2022; 11(8):1330. https://doi.org/10.3390/cells11081330
Chicago/Turabian StylePark, Na Yeon, Doo Sin Jo, and Dong-Hyung Cho. 2022. "Post-Translational Modifications of ATG4B in the Regulation of Autophagy" Cells 11, no. 8: 1330. https://doi.org/10.3390/cells11081330
APA StylePark, N. Y., Jo, D. S., & Cho, D.-H. (2022). Post-Translational Modifications of ATG4B in the Regulation of Autophagy. Cells, 11(8), 1330. https://doi.org/10.3390/cells11081330