
How to Slow down the Ticking Clock: Age-Associated Epigenetic Alterations and Related Interventions to Extend Life Span


Abstract
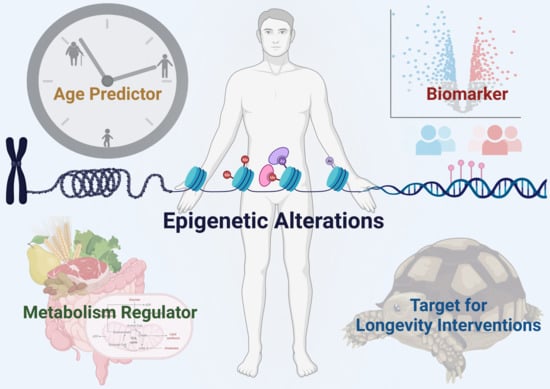
{kind=link}
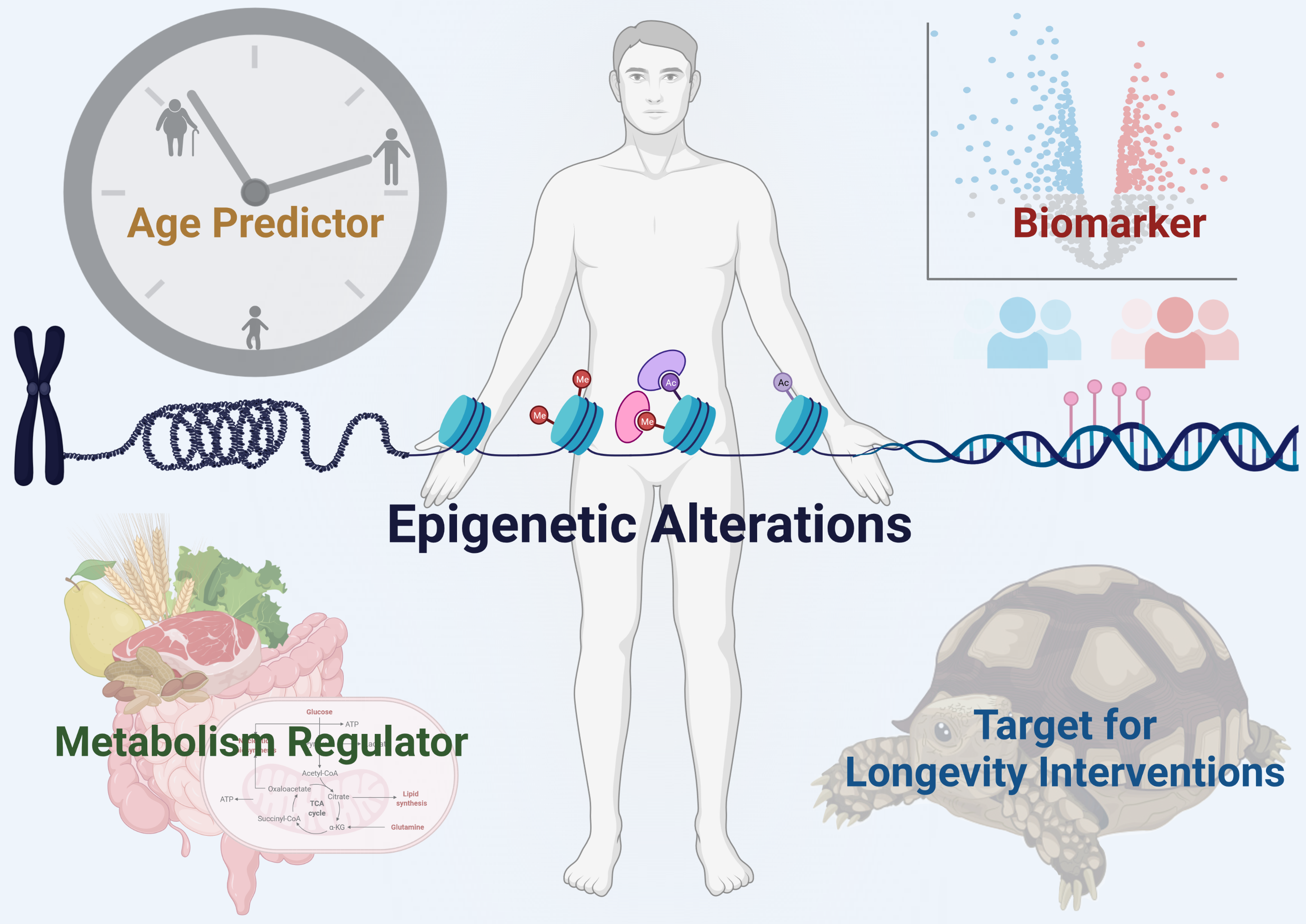
{kind=link}
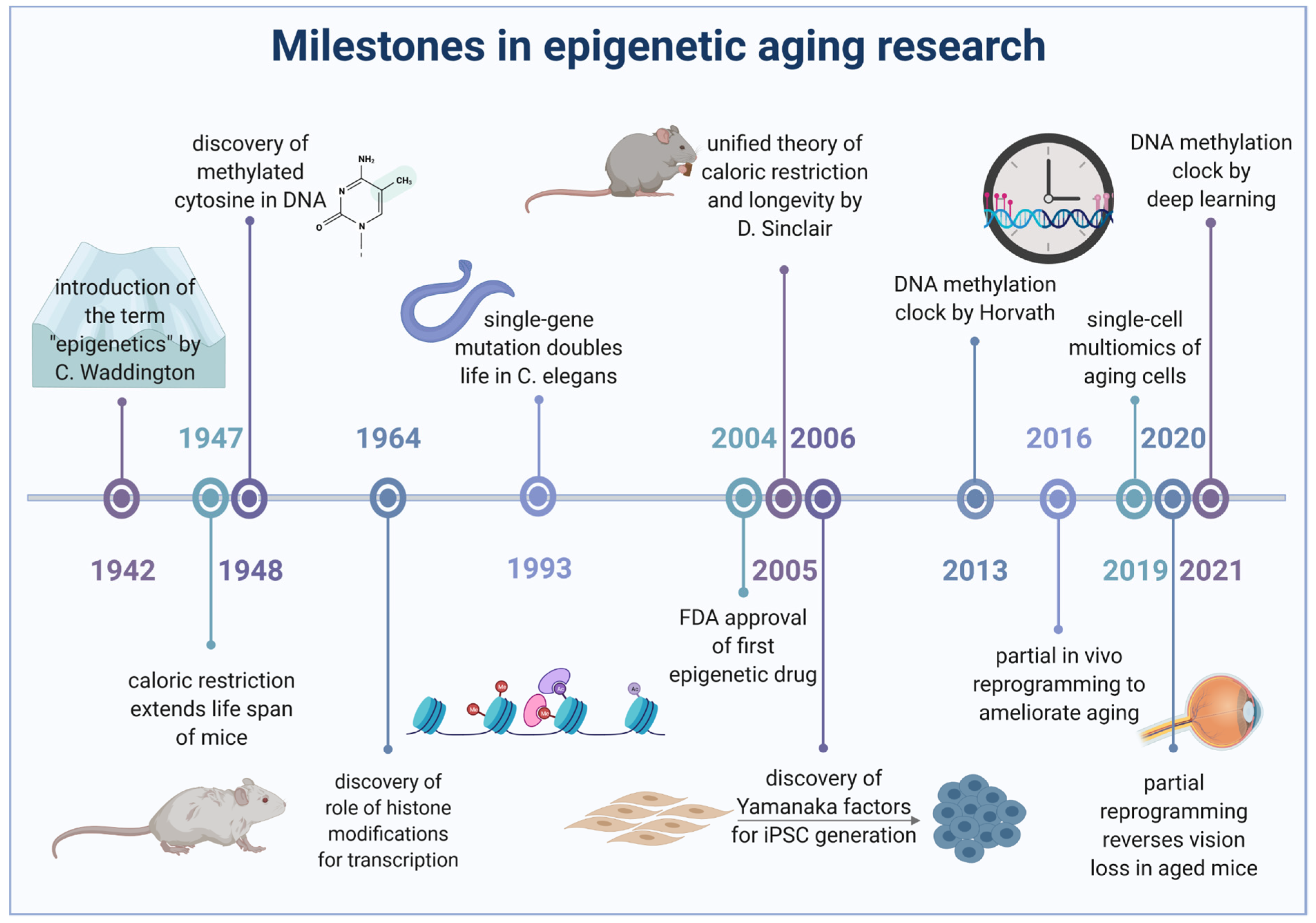
{kind=link}
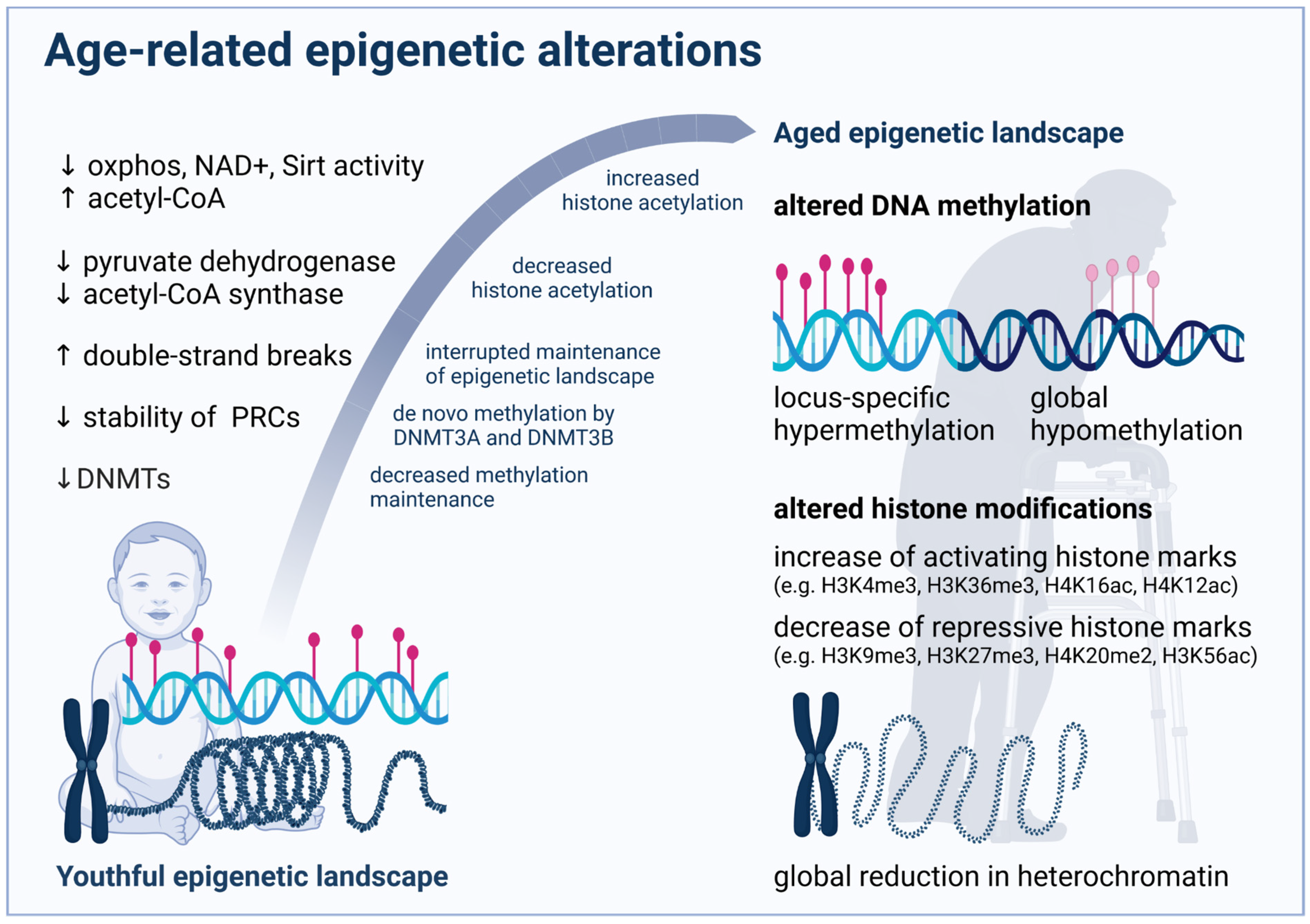
{kind=link}
{kind=link}
1. Introduction
2. Age-Related Changes in Histone Modifications
2.1. Alterations of Specific Histone Acetylation and Methylation Sites during Aging
2.2. Metabolic Regulation of Histone Modifications during Aging
3. Age-Related Changes in DNA Methylation
3.1. Epigenetic Clocks as Age Estimators
3.2. Physiological Relevance of DNA Methylation and Epigenetic Age
3.3. Interrelations of DNA Methylation and Other Hallmarks of Aging
4. Epigenetic Changes as Biomarker in Age-Related Diseases
4.1. Diabetes
4.2. Alzheimer’s Disease
4.3. Cardiovascular Diseases
4.4. Cancer
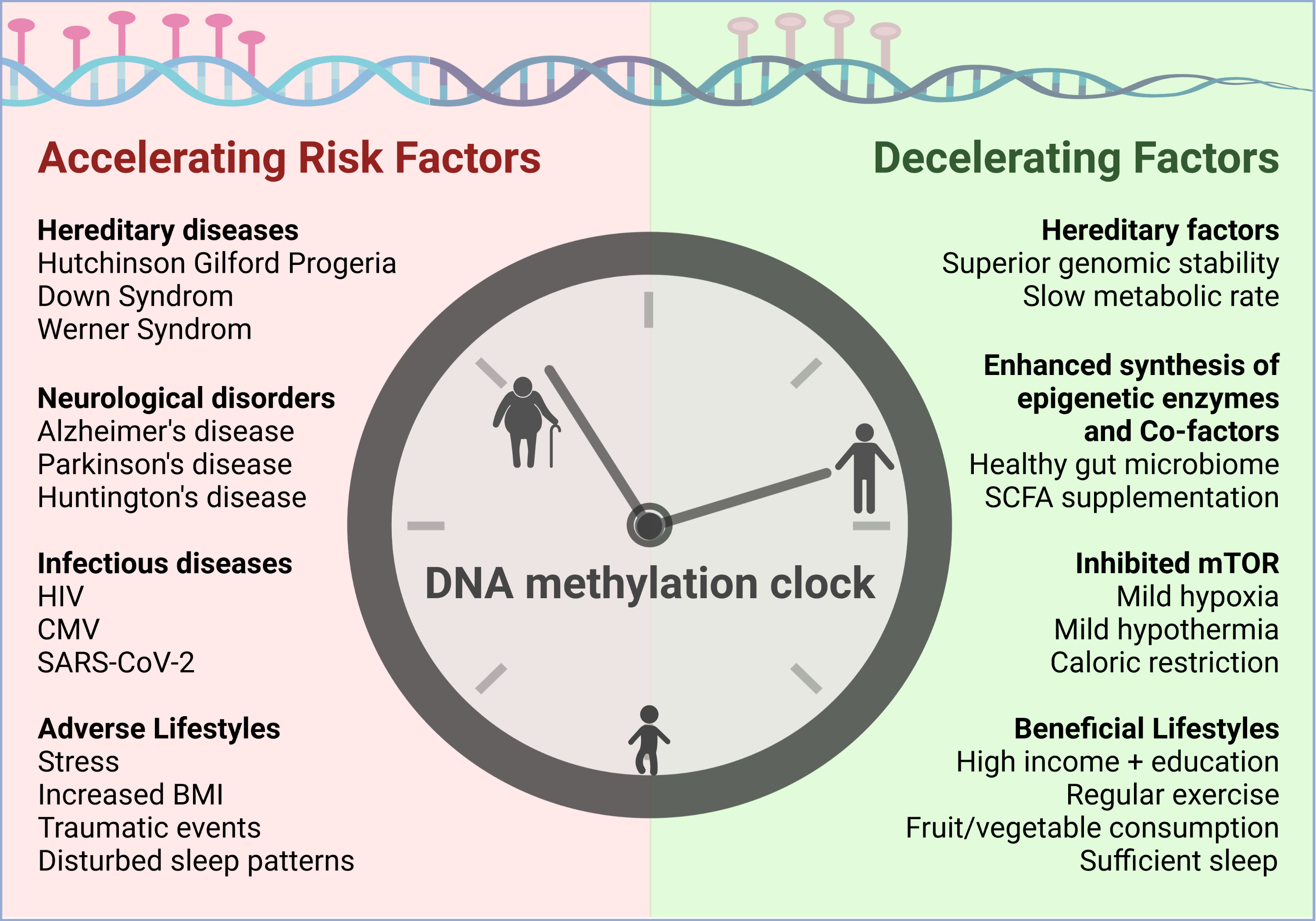
5. Risk Factors Accelerating Epigenetic Aging
5.1. Diseases Negatively Impacting Biological Age
5.2. Lifestyles Negatively Impacting Biological Age
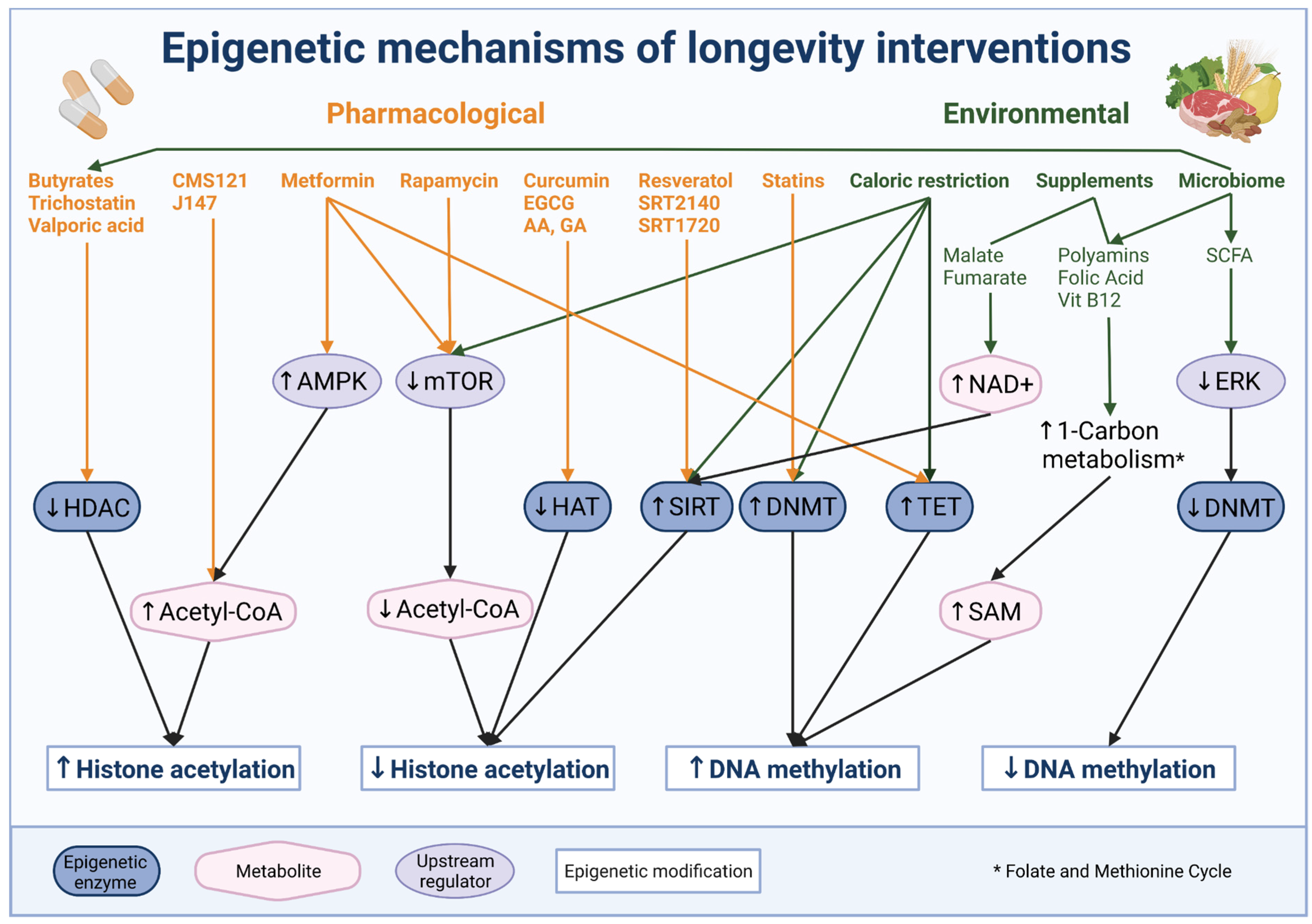
6. Epigenetic Therapy and Intervention to Extend Life Span
6.1. Environmental and Diet Interventions Targeting Epigenetic Mechanisms
6.2. Pharmacological Intervention Targeting or Impacting Epigenetic Mechanisms
6.3. Genetic Interventions Targeting Epigenetic Modifiers, Metabolic Linker and Epigenetic Reprogramming
7. Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- de Grey, A.; Rea, M. Ending Aging: The Rejuvenation Breakthroughs That Could Reverse Human Aging in Our Lifetime; St. Martin’s Press: New York, NY, USA, 2007. [Google Scholar]
- Kraushaar, L.E.; Bauer, P. Dismantling Anti-Ageing Medicine: Why Age-Relatedness of Cardiovascular Disease Is Proof of Robustness Rather Than of Ageing-Associated Vulnerability. Hear. Lung Circ. 2021, 30, 1702–1709. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The Aging Process. Proc. Natl. Acad. Sci. USA 1981, 78, 7124–7128. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Egervari, G.; Wang, Y.; Berger, S.L.; Lu, Z. Regulation of Chromatin and Gene Expression by Metabolic Enzymes and Metabolites. Nat. Rev. Mol. Cell Biol. 2018, 19, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Ma, R.; Wu, Y.; Zhai, Y.; Li, S. Reciprocal Regulation of Metabolic Reprogramming and Epigenetic Modifications in Cancer Front. Genet. 2018, 9, 394. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of development and the inheritance of acquired characters. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Faulkner, R.; Mirsky, A.E. Acetylation and methylation of histones and their possible role in the regulation of rna synthesis. Proc. Natl. Acad. Sci. USA 1964, 51, 786–794. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal Structure of the Nucleosome Core Particle at 2.8 Å Resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Gräff, J.; Tsai, L.-H. Histone Acetylation: Molecular Mnemonics on the Chromatin. Nat. Rev. Neurosci. 2013, 14, 97–111. [Google Scholar] [CrossRef]
- Dor, Y.; Cedar, H. Principles of DNA Methylation and Their Implications for Biology and Medicine. Lancet 2018, 392, 777–786. [Google Scholar] [CrossRef]
- Smith, Z.D.; Meissner, A. DNA Methylation: Roles in Mammalian Development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Skvortsova, K.; Zotenko, E.; Luu, P.-L.; Gould, C.M.; Nair, S.S.; Clark, S.J.; Stirzaker, C. Comprehensive Evaluation of Genome-Wide 5-Hydroxymethylcytosine Profiling Approaches in Human DNA. Epigenet. Chromatin 2017, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Fuks, F.; Hurd, P.J.; Wolf, D.; Nan, X.; Bird, A.P.; Kouzarides, T. The Methyl-CpG-Binding Protein MeCP2 Links DNA Methylation to Histone Methylation *. J. Biol. Chem. 2003, 278, 4035–4040. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D. Programmed Longevity, Youthspan, and Juventology. Aging Cell 2019, 18, e12843. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans Mutant That Lives Twice as Long as Wild Type. Nature 1993, 366, 461–464. [Google Scholar] [CrossRef]
- Grewal, S.I.S.; Moazed, D. Heterochromatin and Epigenetic Control of Gene Expression. Science 2003, 301, 798–802. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of Chromatin by Histone Modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The Evolving Metabolic Landscape of Chromatin Biology and Epigenetics. Nat. Rev. Genet. 2020, 21, 737–753. [Google Scholar] [CrossRef]
- Haithcock, E.; Dayani, Y.; Neufeld, E.; Zahand, A.J.; Feinstein, N.; Mattout, A.; Gruenbaum, Y.; Liu, J. Age-Related Changes of Nuclear Architecture in Caenorhabditis Elegans. Proc. Natl. Acad. Sci. USA 2005, 102, 16690–16695. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.G.; Hillenmeyer, S.; Lawrence, C.; Chang, C.; Hosier, S.; Lightfoot, W.; Mukherjee, E.; Jiang, N.; Schorl, C.; Brodsky, A.S.; et al. Chromatin Remodeling in the Aging Genome of Drosophila. Aging Cell 2010, 9, 971–978. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Chow, M.Z.Y.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 Lysine 16 Hypoacetylation Is Associated with Defective DNA Repair and Premature Senescence in Zmpste24-Deficient Mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef] [PubMed]
- Sedivy, J.M.; Banumathy, G.; Adams, P.D. Aging by Epigenetics—A Consequence of Chromatin Damage? Exp. Cell Res. 2008, 314, 1909–1917. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Vallania, F.; Warsinske, H.C.; Donato, M.; Schaffert, S.; Chang, S.E.; Dvorak, M.; Dekker, C.L.; Davis, M.M.; Utz, P.J.; et al. Single-Cell Chromatin Modification Profiling Reveals Increased Epigenetic Variations with Aging. Cell 2018, 173, 1385–1397.e14. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic Regulation of Ageing: Linking Environmental Inputs to Genomic Stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Pal, S.; Tyler, J.K. Epigenetics and Aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef]
- Zhang, W.; Qu, J.; Liu, G.-H.; Belmonte, J.C.I. The Ageing Epigenome and Its Rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.K.; Berger, S.L. Histone H4 Lysine 16 Acetylation Regulates Cellular Lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the Epigenetic Landscape of Normal Aging in Alzheimer’s Disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Feller, C.; Forne, I.; Schiller, E.; Sévin, D.C.; Schauer, T.; Regnard, C.; Straub, T.; Prestel, M.; Klima, C.; et al. Life Span Extension by Targeting a Link between Metabolism and Histone Acetylation in Drosophila. EMBO Rep. 2016, 17, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Suo, L.; Meng, Q.-G.; Pei, Y.; Yan, C.-L.; Fu, X.-W.; Bunch, T.D.; Zhu, S.-E. Changes in Acetylation on Lysine 12 of Histone H4 (AcH4K12) of Murine Oocytes during Maternal Aging May Affect Fertilization and Subsequent Embryo Development. Fertil. Steril. 2010, 93, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Bux, E.M.; Solis-Mezarino, V.; Kuhm, C.; Northoff, B.H.; Karin, I.; Klopstock, T.; Holdt, L.M.; Völker-Albert, M.; Imhof, A.; Peleg, S. Determining Histone H4 Acetylation Patterns in Human Peripheral Blood Mononuclear Cells Using Mass Spectrometry. Clin. Mass Spectrom. 2019, 15, 54–60. [Google Scholar] [CrossRef]
- Kim, C.; Jin, J.; Ye, Z.; Jadhav, R.R.; Gustafson, C.E.; Hu, B.; Cao, W.; Tian, L.; Weyand, C.M.; Goronzy, J.J. Histone Deficiency and Accelerated Replication Stress in T Cell Aging. J. Clin. Investig. 2021, 131, e143632. [Google Scholar] [CrossRef] [PubMed]
- Kronfol, M.M.; Jahr, F.M.; Dozmorov, M.G.; Phansalkar, P.S.; Xie, L.Y.; Aberg, K.A.; McRae, M.; Price, E.T.; Slattum, P.W.; Gerk, P.M.; et al. DNA Methylation and Histone Acetylation Changes to Cytochrome P450 2E1 Regulation in Normal Aging and Impact on Rates of Drug Metabolism in the Liver. Geroscience 2020, 42, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Xuan, H.; Green, C.D.; Han, Y.; Sun, N.; Shen, H.; McDermott, J.; Bennett, D.A.; Lan, F.; Han, J.-D.J. Repression of Human and Mouse Brain Inflammaging Transcriptome by Broad Gene-Body Histone Hyperacetylation. Proc. Natl. Acad. Sci. USA 2018, 115, 201800656. [Google Scholar] [CrossRef]
- Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac Separates Active from Poised Enhancers and Predicts Developmental State. Proc. Natl. Acad. Sci. USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Bravo, J.I.; Son, J.M.; Lee, C.; Benayoun, B.A. Remodeling of the H3 Nucleosomal Landscape during Mouse Aging. Transl. Med. Aging 2020, 4, 22–31. [Google Scholar] [CrossRef]
- Li, C.-L.; Pu, M.; Wang, W.; Lee, S.S. Locus-Specific H3K9me3 Gain in Aged Somatic Tissues in Caenorhabditis Elegans. bioRxiv 2021. [Google Scholar] [CrossRef]
- Greer, E.L.; Maures, T.J.; Ucar, D.; Hauswirth, A.G.; Mancini, E.; Lim, J.P.; Benayoun, B.A.; Shi, Y.; Brunet, A. Transgenerational Epigenetic Inheritance of Longevity in Caenorhabditis Elegans. Nature 2011, 479, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Sarg, B.; Koutzamani, E.; Helliger, W.; Rundquist, I.; Lindner, H.H. Postsynthetic Trimethylation of Histone H4 at Lysine 20 in Mammalian Tissues Is Associated with Aging *. J. Biol. Chem. 2002, 277, 39195–39201. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.M.; Jaber-Hijazi, F.; Cole, J.J.; Robertson, N.A.; Pawlikowski, J.S.; Norris, K.T.; Criscione, S.W.; Pchelintsev, N.A.; Piscitello, D.; Stong, N.; et al. Mapping H4K20me3 onto the Chromatin Landscape of Senescent Cells Indicates a Function in Control of Cell Senescence and Tumor Suppression through Preservation of Genetic and Epigenetic Stability. Genome Biol. 2016, 17, 158. [Google Scholar] [CrossRef] [PubMed]
- Carrozza, M.J.; Li, B.; Florens, L.; Suganuma, T.; Swanson, S.K.; Lee, K.K.; Shia, W.-J.; Anderson, S.; Yates, J.; Washburn, M.P.; et al. Histone H3 Methylation by Set2 Directs Deacetylation of Coding Regions by Rpd3S to Suppress Spurious Intragenic Transcription. Cell 2005, 123, 581–592. [Google Scholar] [CrossRef]
- Sen, P.; Dang, W.; Donahue, G.; Dai, J.; Dorsey, J.; Cao, X.; Liu, W.; Cao, K.; Perry, R.; Lee, J.Y.; et al. H3K36 Methylation Promotes Longevity by Enhancing Transcriptional Fidelity. Genes Dev. 2015, 29, 1362–1376. [Google Scholar] [CrossRef]
- McCauley, B.S.; Sun, L.; Yu, R.; Lee, M.; Liu, H.; Leeman, D.S.; Huang, Y.; Webb, A.E.; Dang, W. Altered Chromatin States Drive Cryptic Transcription in Aging Mammalian Stem Cells. Nat. Aging 2021, 1, 684–697. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Lin, S.; Britton, L.-M.; Krajewski, K.; Keogh, M.-C.; Garcia, B.A.; Strahl, B.D. Poly-Acetylated Chromatin Signatures Are Preferred Epitopes for Site-Specific Histone H4 Acetyl Antibodies. Sci. Rep. 2012, 2, 489. [Google Scholar] [CrossRef]
- Rothbart, S.B.; Dickson, B.M.; Raab, J.R.; Grzybowski, A.T.; Krajewski, K.; Guo, A.H.; Shanle, E.K.; Josefowicz, S.Z.; Fuchs, S.M.; Allis, C.D.; et al. An Interactive Database for the Assessment of Histone Antibody Specificity. Mol. Cell 2015, 59, 502–511. [Google Scholar] [CrossRef]
- Feller, C.; Forne, I.; Imhof, A.; Becker, P.B. Global and Specific Responses of the Histone Acetylome to Systematic Perturbation. Mol. Cell 2015, 57, 559–571. [Google Scholar] [CrossRef]
- Mews, P. Alcohol Metabolism Directly Fuels Histone Acetylation in the Brain. Nature 2019, 43, 144A. [Google Scholar]
- Sidoli, S.; Lopes, M.; Lund, P.J.; Goldman, N.; Fasolino, M.; Coradin, M.; Kulej, K.; Bhanu, N.V.; Vahedi, G.; Garcia, B.A. A Mass Spectrometry-Based Assay Using Metabolic Labeling to Rapidly Monitor Chromatin Accessibility of Modified Histone Proteins. Sci. Rep. 2019, 9, 13613. [Google Scholar] [CrossRef] [PubMed]
- Kerimoglu, C.; Pham, L.; Tonchev, A.B.; Sakib, M.S.; Xie, Y.; Sokpor, G.; Ulmke, P.A.; Kaurani, L.; Abbas, E.; Nguyen, H.; et al. H3 Acetylation Selectively Promotes Basal Progenitor Proliferation and Neocortex Expansion. Sci. Adv. 2021, 7, eabc6792. [Google Scholar] [CrossRef] [PubMed]
- Schwörer, S.; Becker, F.; Feller, C.; Baig, A.H.; Köber, U.; Henze, H.; Kraus, J.M.; Xin, B.; Lechel, A.; Lipka, D.B.; et al. Epigenetic Stress Responses Induce Muscle Stem-Cell Ageing by Hoxa9 Developmental Signals. Nature 2016, 540, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, T.; Workman, J.L. Chromatin and Metabolism. Annu. Rev. Biochem. 2018, 87, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, Epigenetics, and Metabolism Converge to Cell Senescence and Ageing: The Regulation and Intervention. Signal Transduct. Target Ther. 2021, 6, 245. [Google Scholar] [CrossRef]
- Peleg, S.; Feller, C.; Ladurner, A.G.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic Regulation of Gene Expression through Histone Acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional Silencing and Longevity Protein Sir2 Is an NAD-Dependent Histone Deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Izzo, L.T.; Affronti, H.C.; Wellen, K.E. The Bidirectional Relationship Between Cancer Epigenetics and Metabolism. Annu. Rev. Cancer Biol. 2020, 5, 235–257. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Pedro, J.M.B.-S.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef]
- Baker, D.J.; Peleg, S. Biphasic Modeling of Mitochondrial Metabolism Dysregulation during Aging. Trends Biochem. Sci. 2017, 42, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. The Metabolic Regulation of Aging. Nat. Med. 2015, 21, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial Energetics and Therapeutics. Annu. Rev. Pathol. 2010, 5, 297–348. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Wang, H.; Cai, Y.; Wang, H.; Niu, K.; Wu, X.; Ma, H.; Yang, Y.; Tong, W.; Liu, F.; et al. Epigenetic Drift of H3K27me3 in Aging Links Glycolysis to Healthy Longevity in Drosophila. elife 2018, 7, e35368. [Google Scholar] [CrossRef] [PubMed]
- Pontzer, H.; Yamada, Y.; Sagayama, H.; Ainslie, P.N.; Andersen, L.F.; Anderson, L.J.; Arab, L.; Baddou, I.; Bedu-Addo, K.; Blaak, E.E.; et al. Daily Energy Expenditure through the Human Life Course. Science 2021, 373, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Lesnefsky, E.J.; Hoppel, C.L. Oxidative Phosphorylation and Aging. Ageing Res. Rev. 2006, 5, 402–433. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, P.C. Acetyl-CoA Metabolism and Histone Acetylation in the Regulation of Aging and Lifespan. Antioxidants 2021, 10, 572. [Google Scholar] [CrossRef]
- McReynolds, M.R.; Chellappa, K.; Chiles, E.; Jankowski, C.; Shen, Y.; Chen, L.; Descamps, H.C.; Mukherjee, S.; Bhat, Y.R.; Lingala, S.R.; et al. NAD+ Flux Is Maintained in Aged Mice despite Lower Tissue Concentrations. Cell Syst. 2021, 12, 1160–1172.e4. [Google Scholar] [CrossRef]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age Related Changes in NAD+ Metabolism Oxidative Stress and Sirt1 Activity in Wistar Rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Massudi, H.; Grant, R.; Braidy, N.; Guest, J.; Farnsworth, B.; Guillemin, G.J. Age-Associated Changes In Oxidative Stress and NAD+ Metabolism In Human Tissue. PLoS ONE 2012, 7, e42357. [Google Scholar] [CrossRef]
- Gurd, B.J.; Peters, S.J.; Heigenhauser, G.J.F.; LeBlanc, P.J.; Doherty, T.J.; Paterson, D.H.; Kowalchuk, J.M. O2 Uptake Kinetics, Pyruvate Dehydrogenase Activity, and Muscle Deoxygenation in Young and Older Adults during the Transition to Moderate-Intensity Exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R577–R584. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. The Pyruvate Dehydrogenase Complex as a Therapeutic Target for Age-related Diseases. Aging Cell 2012, 11, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Mews, P.; Donahue, G.; Drake, A.M.; Luczak, V.; Abel, T.; Berger, S.L. Acetyl-CoA Synthetase Regulates Histone Acetylation and Hippocampal Memory. Nature 2017, 546, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Currais, A.; Huang, L.; Goldberg, J.; Petrascheck, M.; Ates, G.; Pinto-Duarte, A.; Shokhirev, M.N.; Schubert, D.; Maher, P. Elevating Acetyl-CoA Levels Reduces Aspects of Brain Aging. elife 2019, 8, e47866. [Google Scholar] [CrossRef]
- Kurian, J.; Bohl, V.; Behanan, M.; Mohsin, S.; Khan, M. Transcriptional Profiling of Cardiac Cells Links Age-Dependent Changes in Acetyl-CoA Signaling to Chromatin Modifications. Int. J. Mol. Sci. 2021, 22, 6987. [Google Scholar] [CrossRef]
- Eisenberg, T.; Schroeder, S.; Andryushkova, A.; Pendl, T.; Küttner, V.; Bhukel, A.; Mariño, G.; Pietrocola, F.; Harger, A.; Zimmermann, A.; et al. Nucleocytosolic Depletion of the Energy Metabolite Acetyl-Coenzyme a Stimulates Autophagy and Prolongs Lifespan. Cell Metab. 2014, 19, 431–444. [Google Scholar] [CrossRef]
- Pouikli, A.; Parekh, S.; Maleszewska, M.; Nikopoulou, C.; Baghdadi, M.; Tripodi, I.; Folz-Donahue, K.; Hinze, Y.; Mesaros, A.; Hoey, D.; et al. Chromatin Remodeling Due to Degradation of Citrate Carrier Impairs Osteogenesis of Aged Mesenchymal Stem Cells. Nat. Aging 2021, 1, 810–825. [Google Scholar] [CrossRef]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered Histone Acetylation Is Associated with Age-Dependent Memory Impairment in Mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef]
- Dong, Y.; Brewer, G.J. Global Metabolic Shifts in Age and Alzheimer’s Disease Mouse Brains Pivot at NAD+/NADH Redox Sites. J. Alzheimer’s Dis. 2019, 71, 119–140. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA Methylation-Based Biomarkers and the Epigenetic Clock Theory of Ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.P. “Epigenetic Clocks”: Theory and Applications in Human Biology. Am. J. Hum. Biol. 2021, 33, e23488. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.-B.; Gao, Y.; et al. Genome-Wide Methylation Profiles Reveal Quantitative Views of Human Aging Rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA Methylation GrimAge Strongly Predicts Lifespan and Healthspan. Aging (Albany NY) 2019, 11, 303–327. [Google Scholar] [CrossRef] [PubMed]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Blasio, A.M.D.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 Gene as a New Epigenetic Marker of Age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Weidner, C.I.; Costa, I.G.; Marioni, R.E.; Ferreira, M.R.P.; Deary, I.J.; Wagner, W. DNA Methylation Levels at Individual Age-Associated CpG Sites Can Be Indicative for Life Expectancy. Aging (Albany NY) 2016, 8, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An Epigenetic Biomarker of Aging for Lifespan and Healthspan. Aging (Albany NY) 2018, 10, 573–591. Aging (Albany NY) 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Salameh, Y.; Bejaoui, Y.; Hajj, N.E. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11, 171. [Google Scholar] [CrossRef]
- Pelegí-Sisó, D.; de Prado, P.; Ronkainen, J.; Bustamante, M.; González, J.R. Methylclock: A Bioconductor Package to Estimate DNA Methylation Age. Bioinformatics 2020, 37, 1759–1760. [Google Scholar] [CrossRef]
- Lowe, R.; Barton, C.; Jenkins, C.A.; Ernst, C.; Forman, O.; Fernandez-Twinn, D.S.; Bock, C.; Rossiter, S.J.; Faulkes, C.G.; Ozanne, S.E.; et al. Ageing-Associated DNA Methylation Dynamics Are a Molecular Readout of Lifespan Variation among Mammalian Species. Genome Biol. 2018, 19, 22. [Google Scholar] [CrossRef]
- Wang, M.; Lemos, B. Ribosomal DNA Harbors an Evolutionarily Conserved Clock of Biological Aging. Genome Res. 2019, 29, 325–333. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA Methylation Aging Clocks: Challenges and Recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef]
- Horvath, S.; Haghani, A.; Macoretta, N.; Ablaeva, J.; Zoller, J.A.; Li, C.Z.; Zhang, J.; Takasugi, M.; Zhao, Y.; Rydkina, E.; et al. DNA Methylation Clocks Tick in Naked Mole Rats but Queens Age More Slowly than Nonbreeders. Nat. Aging 2022, 2, 46–59. [Google Scholar] [CrossRef]
- Ruby, J.G.; Smith, M.; Buffenstein, R. Naked Mole-Rat Mortality Rates Defy Gompertzian Laws by Not Increasing with Age. elife 2018, 7, e31157. [Google Scholar] [CrossRef]
- Zuo, W.; Tang, X.; Hou, C. Why Naked Mole-Rats Have High Oxidative Damage but Live a Long Life: A Simple Explanation Based on the Oxidative Stress Theory of Aging. Adv. Geriatr. Med. Res. 2020, 2. [Google Scholar] [CrossRef]
- Binder, A.M.; Corvalan, C.; Mericq, V.; Pereira, A.; Santos, J.L.; Horvath, S.; Shepherd, J.; Michels, K.B. Faster Ticking Rate of the Epigenetic Clock Is Associated with Faster Pubertal Development in Girls. Epigenetics 2018, 13, 85–94. [Google Scholar] [CrossRef]
- Suarez, A.; Lahti, J.; Czamara, D.; Lahti-Pulkkinen, M.; Girchenko, P.; Andersson, S.; Strandberg, T.E.; Reynolds, R.M.; Kajantie, E.; Binder, E.B.; et al. The Epigenetic Clock and Pubertal, Neuroendocrine, Psychiatric, and Cognitive Outcomes in Adolescents. Clin. Epigenetics 2018, 10, 96. [Google Scholar] [CrossRef]
- Simpkin, A.J.; Howe, L.D.; Tilling, K.; Gaunt, T.R.; Lyttleton, O.; McArdle, W.L.; Ring, S.M.; Horvath, S.; Smith, G.D.; Relton, C.L. The Epigenetic Clock and Physical Development during Childhood and Adolescence: Longitudinal Analysis from a UK Birth Cohort. Int. J. Epidemiol. 2017, 46, 549–558. [Google Scholar] [CrossRef]
- Hayano, M.; Yang, J.-H.; Bonkowski, M.S.; Amorim, J.A.; Ross, J.M.; Coppotelli, G.; Griffin, P.T.; Chew, Y.C.; Guo, W.; Yang, X.; et al. DNA Break-Induced Epigenetic Drift as a Cause of Mammalian Aging. bioRxiv 2019. [Google Scholar] [CrossRef]
- Yang, J.-H.; Griffin, P.T.; Vera, D.L.; Apostolides, J.K.; Hayano, M.; Meer, M.V.; Salfati, E.L.; Su, Q.; Munding, E.M.; Blanchette, M.; et al. Erosion of the Epigenetic Landscape and Loss of Cellular Identity as a Cause of Aging in Mammals. bioRxiv 2019, 808642. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Down, T.A.; Maslau, S.; Andrew, T.; Yang, T.-P.; Beyan, H.; Whittaker, P.; McCann, O.T.; Finer, S.; Valdes, A.M.; et al. Human Aging-Associated DNA Hypermethylation Occurs Preferentially at Bivalent Chromatin Domains. Genome Res. 2010, 20, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-Dependent DNA Methylation of Genes That Are Suppressed in Stem Cells Is a Hallmark of Cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Mozhui, K.; Pandey, A.K. Conserved Effect of Aging on DNA Methylation and Association with EZH2 Polycomb Protein in Mice and Humans. Mech. Ageing Dev. 2017, 162, 27–37. [Google Scholar] [CrossRef][Green Version]
- Lee, T.I.; Jenner, R.G.; Boyer, L.A.; Guenther, M.G.; Levine, S.S.; Kumar, R.M.; Chevalier, B.; Johnstone, S.E.; Cole, M.F.; Isono, K.; et al. Control of Developmental Regulators by Polycomb in Human Embryonic Stem Cells. Cell 2006, 125, 301–313. [Google Scholar] [CrossRef]
- Bracken, A.P.; Dietrich, N.; Pasini, D.; Hansen, K.H.; Helin, K. Genome-Wide Mapping of Polycomb Target Genes Unravels Their Roles in Cell Fate Transitions. Genes Dev. 2006, 20, 1123–1136. [Google Scholar] [CrossRef]
- Viré, E.; Brenner, C.; Deplus, R.; Blanchon, L.; Fraga, M.; Didelot, C.; Morey, L.; Eynde, A.V.; Bernard, D.; Vanderwinden, J.-M.; et al. The Polycomb Group Protein EZH2 Directly Controls DNA Methylation. Nature 2006, 439, 871–874. [Google Scholar] [CrossRef]
- Lynch, M.D.; Smith, A.J.H.; Gobbi, M.D.; Flenley, M.; Hughes, J.R.; Vernimmen, D.; Ayyub, H.; Sharpe, J.A.; Sloane-Stanley, J.A.; Sutherland, L.; et al. An Interspecies Analysis Reveals a Key Role for Unmethylated CpG Dinucleotides in Vertebrate Polycomb Complex Recruitment. EMBO J. 2012, 31, 317–329. [Google Scholar] [CrossRef]
- Reddington, J.P.; Perricone, S.M.; Nestor, C.E.; Reichmann, J.; Youngson, N.A.; Suzuki, M.; Reinhardt, D.; Dunican, D.S.; Prendergast, J.G.; Mjoseng, H.; et al. Redistribution of H3K27me3 upon DNA Hypomethylation Results in De-Repression of Polycomb Target Genes. Genome Biol. 2013, 14, R25. [Google Scholar] [CrossRef]
- Blackledge, N.P.; Farcas, A.M.; Kondo, T.; King, H.W.; McGouran, J.F.; Hanssen, L.L.P.; Ito, S.; Cooper, S.; Kondo, K.; Koseki, Y.; et al. Variant PRC1 Complex-Dependent H2A Ubiquitylation Drives PRC2 Recruitment and Polycomb Domain Formation. Cell 2014, 157, 1445–1459. [Google Scholar] [CrossRef]
- Jung, M.; Pfeifer, G.P. Aging and DNA Methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Lunnon, K.; Smith, R.; Hannon, E.; Jager, P.L.D.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic Profiling Implicates Cortical Deregulation of ANK1 in Alzheimer’s Disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Jager, P.L.D.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s Disease: Early Alterations in Brain DNA Methylation at ANK1, BIN1, RHBDF2 and Other Loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.R.; Smith, R.G.; Macdonald, R.; Marzi, S.J.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Mill, J.; Lunnon, K. The Histone Modification H3K4me3 Is Altered at the ANK1 Locus in Alzheimer’s Disease Brain. Futur Sci. OA 2021, 7, FSO665. [Google Scholar] [CrossRef] [PubMed]
- Putiri, E.L.; Tiedemann, R.L.; Liu, C.; Choi, J.-H.; Robertson, K.D. Impact of Human MLL/COMPASS and Polycomb Complexes on the DNA Methylome. Oncotarget 2014, 5, 6338–6352. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. Proteostasis and Aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef] [PubMed]
- Treaster, S.B.; Ridgway, I.D.; Richardson, C.A.; Gaspar, M.B.; Chaudhuri, A.R.; Austad, S.N. Superior Proteome Stability in the Longest Lived Animal. Age 2013, 36, 9597. [Google Scholar] [CrossRef]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging Is Associated with Hypermethylation of Autophagy Genes in Macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Cheray, M.; Füllgrabe, J.; Salli, M.; Engskog-Vlachos, P.; Keane, L.; Cunha, V.; Lupa, A.; Li, W.; Ma, Q.; et al. The DNA Methyltransferase DNMT3A Contributes to Autophagy Long-Term Memory. Autophagy 2020, 17, 1259–1277. [Google Scholar] [CrossRef]
- Ng, K.-M.; Mok, P.Y.; Butler, A.W.; Ho, J.C.Y.; Choi, S.-W.; Lee, Y.-K.; Lai, W.-H.; Au, K.-W.; Lau, Y.-M.; Wong, L.-Y.; et al. Amelioration of X-Linked Related Autophagy Failure in Danon Disease With DNA Methylation Inhibitor. Circulation 2016, 134, 1373–1389. [Google Scholar] [CrossRef]
- Tserel, L.; Kolde, R.; Limbach, M.; Tretyakov, K.; Kasela, S.; Kisand, K.; Saare, M.; Vilo, J.; Metspalu, A.; Milani, L.; et al. Age-Related Profiling of DNA Methylation in CD8+ T Cells Reveals Changes in Immune Response and Transcriptional Regulator Genes. Sci. Rep. 2015, 5, 13107. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.D.; Wiener, H.W.; Smith, A.K.; Nishitani, S.; Absher, D.M.; Arnett, D.K.; Aslibekyan, S.; Conneely, K.N. Non-Linear Patterns in Age-Related DNA Methylation May Reflect CD4+ T Cell Differentiation. Epigenetics 2017, 12, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.K.; Delaney, C.; Toubai, T.; Ghosh, A.; Reddy, P.; Banerjee, R.; Yung, R. Aging Is Associated with Increased Regulatory T-cell Function. Aging Cell 2014, 13, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.; Krasnienkov, D. Telomere Length as a Marker of Biological Age: State-of-the-Art, Open Issues, and Future Perspectives. Front. Genet. 2021, 11, 630186. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomere States and Cell Fates. Nature 2000, 408, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, K.; Vera, E.; Martínez-Nevado, E.; Sanpera, C.; Blasco, M.A. Telomere Shortening Rate Predicts Species Life Span. Proc. Natl. Acad. Sci. USA 2019, 116, 15122–15127. [Google Scholar] [CrossRef]
- Lu, A.T.; Xue, L.; Salfati, E.L.; Chen, B.H.; Ferrucci, L.; Levy, D.; Joehanes, R.; Murabito, J.M.; Kiel, D.P.; Tsai, P.-C.; et al. GWAS of Epigenetic Aging Rates in Blood Reveals a Critical Role for TERT. Nat. Commun. 2018, 9, 387. [Google Scholar] [CrossRef]
- Mendelsohn, A.R.; Larrick, J.W. Telomerase May Paradoxically Accelerate Aging of the DNA Methylome. Rejuv. Res. 2018, 21, 168–172. [Google Scholar] [CrossRef]
- Lowe, D.; Horvath, S.; Raj, K. Epigenetic Clock Analyses of Cellular Senescence and Ageing. Oncotarget 2016, 7, 8524–8531. [Google Scholar] [CrossRef]
- Franzen, J.; Zirkel, A.; Blake, J.; Rath, B.; Benes, V.; Papantonis, A.; Wagner, W. Senescence-associated DNA Methylation Is Stochastically Acquired in Subpopulations of Mesenchymal Stem Cells. Aging Cell 2017, 16, 183–191. [Google Scholar] [CrossRef]
- Kabacik, S.; Horvath, S.; Cohen, H.; Raj, K. Epigenetic Ageing Is Distinct from Senescence-Mediated Ageing and Is Not Prevented by Telomerase Expression. Aging (Albany NY) 2018, 10, 2800–2815. [Google Scholar] [CrossRef] [PubMed]
- Wagner, W. The Link Between Epigenetic Clocks for Aging and Senescence. Front. Genet. 2019, 10, 303. [Google Scholar] [CrossRef] [PubMed]
- Hatazawa, Y.; Ono, Y.; Hirose, Y.; Kanai, S.; Fujii, N.L.; Machida, S.; Nishino, I.; Shimizu, T.; Okano, M.; Kamei, Y.; et al. Reduced Dnmt3a Increases Gdf5 Expression with Suppressed Satellite Cell Differentiation and Impaired Skeletal Muscle Regeneration. FASEB J. 2018, 32, 1452–1467. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Ehnert, S.; Culmes, M.; Bachmann, A.; Seeliger, C.; Schyschka, L.; Wang, Z.; Rahmanian-Schwarz, A.; Stöckle, U.; Sousa, P.A.D.; et al. 5-Azacytidine Improves the Osteogenic Differentiation Potential of Aged Human Adipose-Derived Mesenchymal Stem Cells by DNA Demethylation. PLoS ONE 2014, 9, e90846. [Google Scholar] [CrossRef] [PubMed]
- Bigot, A.; Duddy, W.J.; Ouandaogo, Z.G.; Negroni, E.; Mariot, V.; Ghimbovschi, S.; Harmon, B.; Wielgosik, A.; Loiseau, C.; Devaney, J.; et al. Age-Associated Methylation Suppresses SPRY1, Leading to a Failure of Re-Quiescence and Loss of the Reserve Stem Cell Pool in Elderly Muscle. Cell Rep. 2015, 13, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic Profiling of Young and Aged HSCs Reveals Concerted Changes during Aging That Reinforce Self-Renewal. Cell Stem. Cell 2014, 14, 673–688. [Google Scholar] [CrossRef]
- Jeong, M.; Park, H.J.; Celik, H.; Ostrander, E.L.; Reyes, J.M.; Guzman, A.; Rodriguez, B.; Lei, Y.; Lee, Y.; Ding, L.; et al. Loss of Dnmt3a Immortalizes Hematopoietic Stem Cells In Vivo. Cell Rep. 2018, 23, 1–10. [Google Scholar] [CrossRef]
- Hashizume, O.; Ohnishi, S.; Mito, T.; Shimizu, A.; Ishikawa, K.; Iashikawa, K.; Nakada, K.; Soda, M.; Mano, H.; Togayachi, S.; et al. Epigenetic Regulation of the Nuclear-Coded GCAT and SHMT2 Genes Confers Human Age-Associated Mitochondrial Respiration Defects. Sci. Rep. 2015, 5, 10434. [Google Scholar] [CrossRef] [PubMed]
- Maddocks, O.D.K.; Labuschagne, C.F.; Adams, P.D.; Vousden, K.H. Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol. Cell 2016, 61, 210–221. [Google Scholar] [CrossRef]
- Kornicka, K.; Marycz, K.; Marędziak, M.; Tomaszewski, K.A.; Nicpoń, J. The Effects of the DNA Methyltranfserases Inhibitor 5-Azacitidine on Ageing, Oxidative Stress and DNA Methylation of Adipose Derived Stem Cells. J. Cell Mol. Med. 2017, 21, 387–401. [Google Scholar] [CrossRef]
- Shock, L.S.; Thakkar, P.V.; Peterson, E.J.; Moran, R.G.; Taylor, S.M. DNA Methyltransferase 1, Cytosine Methylation, and Cytosine Hydroxymethylation in Mammalian Mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Bianchessi, V.; Vinci, M.C.; Nigro, P.; Rizzi, V.; Farina, F.; Capogrossi, M.C.; Pompilio, G.; Gualdi, V.; Lauri, A. Methylation Profiling by Bisulfite Sequencing Analysis of the MtDNA Non-Coding Region in Replicative and Senescent Endothelial Cells. Mitochondrion 2016, 27, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Chang, C.; Huang, G.; Zhou, Z. Epigenetics in Allergy and Autoimmunity. Adv. Exp. Med. Biol. 2020, 1253, 223–257. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.A.H.; Ansari, S.A.; Mensah-Brown, E.P.K.; Emerald, B.S. The Role of DNA Methylation in the Pathogenesis of Type 2 Diabetes Mellitus. Clin. Epigenetics 2020, 12, 104. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Xia, L.; Tu, K.; Duan, M.; Kukurba, K.; Li-Pook-Than, J.; Xie, D.; Snyder, M. Longitudinal Personal DNA Methylome Dynamics in a Human with a Chronic Condition. Nat. Med. 2018, 24, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Toperoff, G.; Aran, D.; Kark, J.D.; Rosenberg, M.; Dubnikov, T.; Nissan, B.; Wainstein, J.; Friedlander, Y.; Levy-Lahad, E.; Glaser, B.; et al. Genome-Wide Survey Reveals Predisposing Diabetes Type 2-Related DNA Methylation Variations in Human Peripheral Blood. Hum. Mol. Genet. 2012, 21, 371–383. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.-C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-Wide Association Study of Body Mass Index, and the Adverse Outcomes of Adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef]
- Chambers, J.C.; Loh, M.; Lehne, B.; Drong, A.; Kriebel, J.; Motta, V.; Wahl, S.; Elliott, H.R.; Rota, F.; Scott, W.R.; et al. Epigenome-Wide Association of DNA Methylation Markers in Peripheral Blood from Indian Asians and Europeans with Incident Type 2 Diabetes: A Nested Case-Control Study. Lancet Diabetes Endocrinol. 2015, 3, 526–534. [Google Scholar] [CrossRef]
- Walaszczyk, E.; Luijten, M.; Spijkerman, A.M.W.; Bonder, M.J.; Lutgers, H.L.; Snieder, H.; Wolffenbuttel, B.H.R.; van Vliet-Ostaptchouk, J.V. DNA Methylation Markers Associated with Type 2 Diabetes, Fasting Glucose and HbA1c Levels: A Systematic Review and Replication in a Case–Control Sample of the Lifelines Study. Diabetologia 2018, 61, 354–368. [Google Scholar] [CrossRef]
- Vlassara, H.; Cai, W.; Chen, X.; Serrano, E.J.; Shobha, M.S.; Uribarri, J.; Woodward, M.; Striker, G.E. Managing Chronic Inflammation in the Aging Diabetic Patient With CKD by Diet or Sevelamer Carbonate: A Modern Paradigm Shift. J. Gerontol. Ser. 2012, 67, 1410–1416. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; Reddy, M.A.; Miao, F.; Shanmugam, N.; Yee, J.-K.; Hawkins, D.; Ren, B.; Natarajan, R. Role of the Histone H3 Lysine 4 Methyltransferase, SET7/9, in the Regulation of NF-ΚB-Dependent Inflammatory Genes Relevance to Diabetes and Inflammation *. J. Biol. Chem. 2008, 283, 26771–26781. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, L.M.; Reddy, M.A.; Lanting, L.L.; Wang, M.; Meng, L.; Natarajan, R. Epigenetic Histone H3 Lysine 9 Methylation in Metabolic Memory and Inflammatory Phenotype of Vascular Smooth Muscle Cells in Diabetes. Proc. Natl. Acad. Sci. USA 2008, 105, 9047–9052. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.; Groop, L. Epigenetics: A Molecular Link Between Environmental Factors and Type 2 Diabetes. Diabetes 2009, 58, 2718–2725. [Google Scholar] [CrossRef]
- Sibbett, R.A.; Altschul, D.M.; Marioni, R.E.; Deary, I.J.; Starr, J.M.; Russ, T.C. DNA Methylation-Based Measures of Accelerated Biological Ageing and the Risk of Dementia in the Oldest-Old: A Study of the Lothian Birth Cohort. BMC Psychiatry 2020, 20, 91. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Bennett, D.A.; Horvath, S. Epigenetic Age of the Pre-Frontal Cortex Is Associated with Neuritic Plaques, Amyloid Load, and Alzheimer’s Disease Related Cognitive Functioning. Aging 2015, 7, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Shinagawa, S.; Nagata, T.; Shimada, K.; Shibata, N.; Ohnuma, T.; Kasanuki, K.; Arai, H.; Yamada, H.; Nakayama, K.; et al. Development of Biomarkers Based on DNA Methylation in the NCAPH2/LMF2 Promoter Region for Diagnosis of Alzheimer’s Disease and Amnesic Mild Cognitive Impairment. PLoS ONE 2016, 11, e0146449. [Google Scholar] [CrossRef] [PubMed]
- Lardenoije, R.; Roubroeks, J.A.Y.; Pishva, E.; Leber, M.; Wagner, H.; Iatrou, A.; Smith, A.R.; Smith, R.G.; Eijssen, L.M.T.; Kleineidam, L.; et al. Alzheimer’s Disease-Associated (Hydroxy)Methylomic Changes in the Brain and Blood. Clin. Epigenetics 2019, 11, 164. [Google Scholar] [CrossRef]
- Gräff, J.; Rei, D.; Guan, J.-S.; Wang, W.-Y.; Seo, J.; Hennig, K.M.; Nieland, T.J.F.; Fass, D.M.; Kao, P.F.; Kahn, M.; et al. An Epigenetic Blockade of Cognitive Functions in the Neurodegenerating Brain. Nature 2012, 483, 222–226. [Google Scholar] [CrossRef]
- Janczura, K.J.; Volmar, C.-H.; Sartor, G.C.; Rao, S.J.; Ricciardi, N.R.; Lambert, G.; Brothers, S.P.; Wahlestedt, C. Inhibition of HDAC3 Reverses Alzheimer’s Disease-Related Pathologies in Vitro and in the 3xTg-AD Mouse Model. Proc. Natl. Acad. Sci. USA 2018, 115, 201805436. [Google Scholar] [CrossRef]
- Fischer, A.; Sananbenesi, F.; Wang, X.; Dobbin, M.; Tsai, L.-H. Recovery of Learning and Memory Is Associated with Chromatin Remodelling. Nature 2007, 447, 178–182. [Google Scholar] [CrossRef]
- Govindarajan, N.; Agis-Balboa, R.C.; Walter, J.; Sananbenesi, F.; Fischer, A. Sodium Butyrate Improves Memory Function in an Alzheimer’s Disease Mouse Model When Administered at an Advanced Stage of Disease Progression. J. Alzheimer’s Dis. JAD 2011, 26, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Penney, J.; Tsai, L.-H. Histone Deacetylases in Memory and Cognition. Sci. Signal. 2014, 7, re12. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, M.; Miller, C.A.; Fass, D.M.; Hennig, K.M.; Haggarty, S.J.; Sweatt, J.D.; Rumbaugh, G. Inhibitors of Class 1 Histone Deacetylases Reverse Contextual Memory Deficits in a Mouse Model of Alzheimer’s Disease. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2010, 35, 870–880. [Google Scholar] [CrossRef] [PubMed]
- Benito, E.; Urbanke, H.; Ramachandran, B.; Barth, J.; Halder, R.; Awasthi, A.; Jain, G.; Capece, V.; Burkhardt, S.; Navarro-Sala, M.; et al. HDAC Inhibitor–Dependent Transcriptome and Memory Reinstatement in Cognitive Decline Models. J. Clin. Investig. 2015, 125, 3572–3584. [Google Scholar] [CrossRef]
- Kitahara, M.; Inoue, T.; Mani, H.; Takamatsu, Y.; Ikegami, R.; Tohyama, H.; Maejima, H. Exercise and Pharmacological Inhibition of Histone Deacetylase Improves Cognitive Function Accompanied by an Increase of Gene Expressions Crucial for Neuronal Plasticity in the Hippocampus. Neurosci. Lett. 2021, 749, 135749. [Google Scholar] [CrossRef] [PubMed]
- Perna, L.; Zhang, Y.; Mons, U.; Holleczek, B.; Saum, K.-U.; Brenner, H. Epigenetic Age Acceleration Predicts Cancer, Cardiovascular, and All-Cause Mortality in a German Case Cohort. Clin. Epigenetics 2016, 8, 64. [Google Scholar] [CrossRef]
- Lind, L.; Ingelsson, E.; Sundström, J.; Siegbahn, A.; Lampa, E. Methylation-based Estimated Biological Age and Cardiovascular Disease. Eur. J. Clin. Investig. 2018, 48, e12872. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Sanlés, A.; Sayols-Baixeras, S.; Subirana, I.; Degano, I.R.; Elosua, R. Association between DNA Methylation and Coronary Heart Disease or Other Atherosclerotic Events: A Systematic Review. Atherosclerosis 2017, 263, 325–333. [Google Scholar] [CrossRef]
- Infante, T.; Forte, E.; Schiano, C.; Punzo, B.; Cademartiri, F.; Cavaliere, C.; Salvatore, M.; Napoli, C. Evidence of Association of Circulating Epigenetic-Sensitive Biomarkers with Suspected Coronary Heart Disease Evaluated by Cardiac Computed Tomography. PLoS ONE 2019, 14, e0210909. [Google Scholar] [CrossRef]
- Zaina, S.; Heyn, H.; Carmona, F.J.; Varol, N.; Sayols, S.; Condom, E.; Ramírez-Ruz, J.; Gomez, A.; Gonçalves, I.; Moran, S.; et al. DNA Methylation Map of Human Atherosclerosis. Circ. Cardiovasc. Genet. 2018, 7, 692–700. [Google Scholar] [CrossRef]
- del Pilar Valencia-Morales, M.; Zaina, S.; Heyn, H.; Carmona, F.J.; Varol, N.; Sayols, S.; Condom, E.; Ramírez-Ruz, J.; Gomez, A.; Moran, S.; et al. The DNA Methylation Drift of the Atherosclerotic Aorta Increases with Lesion Progression. BMC Med. Genom. 2015, 8, 7. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, J.; Garg, G.; Kumar, A.; Patowary, A.; Karthikeyan, G.; Ramakrishnan, L.; Brahmachari, V.; Sengupta, S. Detection of Altered Global DNA Methylation in Coronary Artery Disease Patients. DNA Cell Biol. 2008, 27, 357–365. [Google Scholar] [CrossRef]
- de la Rocha, C.; Zaina, S.; Lund, G. Is Any Cardiovascular Disease-Specific DNA Methylation Biomarker Within Reach? Curr. Atheroscler. Rep. 2020, 22, 62. [Google Scholar] [CrossRef] [PubMed]
- Navas-Acien, A.; Domingo-Relloso, A.; Subedi, P.; Riffo-Campos, A.L.; Xia, R.; Gomez, L.; Haack, K.; Goldsmith, J.; Howard, B.V.; Best, L.G.; et al. Blood DNA Methylation and Incident Coronary Heart Disease: Evidence From the Strong Heart Study. JAMA Cardiol. 2021. [Google Scholar] [CrossRef]
- Jiang, D.; Wang, Y.; Chang, G.; Duan, Q.; You, L.; Sun, M.; Hu, C.; Gao, L.; Wu, S.; Tao, H.; et al. DNA Hydroxymethylation Combined with Carotid Plaques as a Novel Biomarker for Coronary Atherosclerosis. Aging (Albany NY) 2019, 11, 3170–3181. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Peng, W.; Qu, K.; Lin, X.; Zeng, Z.; Chen, J.; Wei, D.; Wang, Z. TET2: A Novel Epigenetic Regulator and Potential Intervention Target for Atherosclerosis. DNA Cell Biol. 2018, 37, 517–523. [Google Scholar] [CrossRef]
- Lee, C.-J.; Ahn, H.; Jeong, D.; Pak, M.; Moon, J.H.; Kim, S. Impact of Mutations in DNA Methylation Modification Genes on Genome-Wide Methylation Landscapes and Downstream Gene Activations in Pan-Cancer. BMC Med. Genom. 2020, 13, 27. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An Immeasurable Source of Knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Li, M.; Alsager, J.S.; Wang, Z.; Cheng, L.; Shan, B. Epigenetic Upregulation of HOXC10 in Non-Small Lung Cancer Cells. Aging 2020, 12, 16921–16935. [Google Scholar] [CrossRef]
- Pakneshan, P.; Têtu, B.; Rabbani, S.A. Demethylation of Urokinase Promoter as a Prognostic Marker in Patients with Breast Carcinoma. Clin. Cancer Res. 2004, 10, 3035–3041. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Barger, C.J.; Link, P.A.; Mhawech-Fauceglia, P.; Miller, A.; Akers, S.N.; Odunsi, K.; Karpf, A.R. DNA Hypomethylation-Mediated Activation of Cancer/Testis Antigen 45 (CT45) Genes Is Associated with Disease Progression and Reduced Survival in Epithelial Ovarian Cancer. Epigenetics 2015, 10, 736–748. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter Hypermethylation and BRCA1 Inactivation in Sporadic Breast and Ovarian Tumors. JNCI J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chen, Y.; Petersen, I. Expression and Promoter DNA Methylation of MLH1 in Colorectal Cancer and Lung Cancer. Pathol. Res. Pr. 2017, 213, 333–338. [Google Scholar] [CrossRef]
- Herfarth, K.K.-F.; Brent, T.P.; Danam, R.P.; Remack, J.S.; Kodner, I.J.; Wells, S.A.; Goodfellow, P.J. A Specific CpG Methylation Pattern of the MGMT Promoter Region Associated with Reduced MGMT Expression in Primary Colorectal Cancers. Mol. Carcinog. 1999, 24, 90–98. [Google Scholar] [CrossRef]
- Tang, X.; Wu, W.; Sun, S.-Y.; Wistuba, I.I.; Hong, W.K.; Mao, L. Hypermethylation of the Death-Associated Protein Kinase Promoter Attenuates the Sensitivity to TRAIL-Induced Apoptosis in Human Non-Small Cell Lung Cancer Cells. Mol. Cancer Res. Mcr. 2004, 2, 685–691. [Google Scholar]
- Martinez, R.; Setien, F.; Voelter, C.; Casado, S.; Quesada, M.P.; Schackert, G.; Esteller, M. CpG Island Promoter Hypermethylation of the Pro-Apoptotic Gene Caspase-8 Is a Common Hallmark of Relapsed Glioblastoma Multiforme. Carcinogenesis 2007, 28, 1264–1268. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic Silencing of TH1-Type Chemokines Shapes Tumour Immunity and Immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef]
- Raneros, A.B.; Martín-Palanco, V.; Fernandez, A.F.; Rodriguez, R.M.; Fraga, M.F.; Lopez-Larrea, C.; Suarez-Alvarez, B. Methylation of NKG2D Ligands Contributes to Immune System Evasion in Acute Myeloid Leukemia. Genes Immun. 2015, 16, 71–82. [Google Scholar] [CrossRef]
- Serrano, A.; Castro-Vega, I.; Redondo, M. Role of Gene Methylation in Antitumor Immune Response: Implication for Tumor Progression. Cancers 2011, 3, 1672–1690. [Google Scholar] [CrossRef]
- Moran, S.; Martínez-Cardús, A.; Sayols, S.; Musulén, E.; Balañá, C.; Estival-Gonzalez, A.; Moutinho, C.; Heyn, H.; Diaz-Lagares, A.; de Moura, M.C.; et al. Epigenetic Profiling to Classify Cancer of Unknown Primary: A Multicentre, Retrospective Analysis. Lancet Oncol. 2016, 17, 1386–1395. [Google Scholar] [CrossRef]
- Gai, W.; Sun, K. Epigenetic Biomarkers in Cell-Free DNA and Applications in Liquid Biopsy. Genes 2019, 10, 32. [Google Scholar] [CrossRef] [PubMed]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive Human Cell-Type Methylation Atlas Reveals Origins of Circulating Cell-Free DNA in Health and Disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Li, Q.; Chen, Q.; Zhou, Y.; Park, S.; Lee, G.; Grimes, B.; Krysan, K.; Yu, M.; Wang, W.; et al. CancerLocator: Non-Invasive Cancer Diagnosis and Tissue-of-Origin Prediction Using Methylation Profiles of Cell-Free DNA. Genome Biol. 2017, 18, 53. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, Q.; Kang, S.; Same, M.; Zhou, Y.; Sun, C.; Liu, C.-C.; Matsuoka, L.; Sher, L.; Wong, W.H.; et al. CancerDetector: Ultrasensitive and Non-Invasive Cancer Detection at the Resolution of Individual Reads Using Cell-Free DNA Methylation Sequencing Data. Nucleic Acids Res. 2018, 46, gky423. [Google Scholar] [CrossRef] [PubMed]
- Galardi, F.; Luca, F.D.; Romagnoli, D.; Biagioni, C.; Moretti, E.; Biganzoli, L.; Leo, A.D.; Migliaccio, I.; Malorni, L.; Benelli, M. Cell-Free DNA-Methylation-Based Methods and Applications in Oncology. Biomolecules 2020, 10, 1677. [Google Scholar] [CrossRef]
- Klein, E.A.; Hubbell, E.; Maddala, T.; Aravanis, A.; Beausang, J.F.; Filippova, D.; Gross, S.; Jamshidi, A.; Kurtzman, K.; Shen, L.; et al. Development of a Comprehensive Cell-Free DNA (CfDNA) Assay for Early Detection of Multiple Tumor Types: The Circulating Cell-Free Genome Atlas (CCGA) Study. J. Clin. Oncol. 2018, 36, 12021. [Google Scholar] [CrossRef]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Consortium, C.; Investigators, C.; Liu, M.C.; Oxnard, G.R.; Klein, E.A.; et al. Sensitive and Specific Multi-Cancer Detection and Localization Using Methylation Signatures in Cell-Free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef]
- Locke, W.J.; Guanzon, D.; Ma, C.; Liew, Y.J.; Duesing, K.R.; Fung, K.Y.C.; Ross, J.P. DNA Methylation Cancer Biomarkers: Translation to the Clinic. Front. Genet. 2019, 10, 1150. [Google Scholar] [CrossRef]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-Repair Gene MGMT and the Clinical Response of Gliomas to Alkylating Agents. N. Engl. J. Med. 2000, 343, 1350–1354. [Google Scholar] [CrossRef]
- Shen, L.; Kondo, Y.; Ahmed, S.; Boumber, Y.; Konishi, K.; Guo, Y.; Chen, X.; Vilaythong, J.N.; Issa, J.-P.J. Drug Sensitivity Prediction by CpG Island Methylation Profile in the NCI-60 Cancer Cell Line Panel. Cancer Res. 2007, 67, 11335–11343. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yang, Y.; Lu, Y.; Herman, J.G.; Brock, M.V.; Zhao, P.; Guo, M. Predictive Value of CHFR and MLH1 Methylation in Human Gastric Cancer. Gastric Cancer 2015, 18, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, T.; Tischkowitz, M.; Ameziane, N.; Hodgson, S.V.; Mathew, C.G.; Joenje, H.; Mok, S.C.; D’Andrea, A.D. Disruption of the Fanconi Anemia–BRCA Pathway in Cisplatin-Sensitive Ovarian Tumors. Nat. Med. 2003, 9, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.L.; Rosell, R.; Taron, M.; Sanchez-Ronco, M.; Alberola, V.; de las Peñas, R.; Sanchez, J.M.; Moran, T.; Camps, C.; Massuti, B.; et al. 14-3-3σ Methylation in Pretreatment Serum Circulating DNA of Cisplatin-Plus-Gemcitabine-Treated Advanced Non–Small-Cell Lung Cancer Patients Predicts Survival: The Spanish Lung Cancer Group. J. Clin. Oncol. 2005, 23, 9105–9112. [Google Scholar] [CrossRef]
- de Caceres, I.I.; Cortes-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguán-García, C.; Cejas, P.; López-Ríos, F.; Paz-Ares, L.; de CastroCarpeño, J.; et al. IGFBP-3 Hypermethylation-Derived Deficiency Mediates Cisplatin Resistance in Non-Small-Cell Lung Cancer. Oncogene 2010, 29, 1681–1690. [Google Scholar] [CrossRef]
- Faller, W.J.; Rafferty, M.; Hegarty, S.; Gremel, G.; Ryan, D.; Fraga, M.F.; Esteller, M.; Dervan, P.A.; Gallagher, W.M. Metallothionein 1E Is Methylated in Malignant Melanoma and Increases Sensitivity to Cisplatin-Induced Apoptosis. Melanoma Res. 2010, 20, 392–400. [Google Scholar] [CrossRef]
- Sigalotti, L.; Fratta, E.; Coral, S.; Tanzarella, S.; Danielli, R.; Colizzi, F.; Fonsatti, E.; Traversari, C.; Altomonte, M.; Maio, M. Intratumor Heterogeneity of Cancer/Testis Antigens Expression in Human Cutaneous Melanoma Is Methylation-Regulated and Functionally Reverted by 5-Aza-2′-Deoxycytidine. Cancer Res. 2004, 64, 9167–9171. [Google Scholar] [CrossRef]
- Kiziltepe, T.; Hideshima, T.; Catley, L.; Raje, N.; Yasui, H.; Shiraishi, N.; Okawa, Y.; Ikeda, H.; Vallet, S.; Pozzi, S.; et al. 5-Azacytidine, a DNA Methyltransferase Inhibitor, Induces ATR-Mediated DNA Double-Strand Break Responses, Apoptosis, and Synergistic Cytotoxicity with Doxorubicin and Bortezomib against Multiple Myeloma Cells. Mol. Cancer Ther. 2007, 6, 1718–1727. [Google Scholar] [CrossRef]
- Adair, S.J.; Hogan, K.T. Treatment of Ovarian Cancer Cell Lines with 5-Aza-2′-Deoxycytidine Upregulates the Expression of Cancer-Testis Antigens and Class I Major Histocompatibility Complex-Encoded Molecules. Cancer Immunol. Immunother. 2009, 58, 589–601. [Google Scholar] [CrossRef]
- Cruz, C.R.; Gerdemann, U.; Leen, A.M.; Shafer, J.A.; Ku, S.; Tzou, B.; Horton, T.M.; Sheehan, A.; Copeland, A.; Younes, A.; et al. Improving T-Cell Therapy for Relapsed EBV-Negative Hodgkin Lymphoma by Targeting Upregulated MAGE-A. Clin. Cancer Res. 2011, 17, 7058–7066. [Google Scholar] [CrossRef]
- Silverman, L.R.; Demakos, E.P.; Peterson, B.L.; Kornblith, A.B.; Holland, J.C.; Odchimar-Reissig, R.; Stone, R.M.; Nelson, D.; Powell, B.L.; DeCastro, C.M.; et al. Randomized Controlled Trial of Azacitidine in Patients With the Myelodysplastic Syndrome: A Study of the Cancer and Leukemia Group B. J. Clin. Oncol. 2002, 20, 2429–2440. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Issa, J.J.; Rosenfeld, C.S.; Bennett, J.M.; Albitar, M.; DiPersio, J.; Klimek, V.; Slack, J.; de Castro, C.; Ravandi, F.; et al. Decitabine Improves Patient Outcomes in Myelodysplastic Syndromes. Cancer 2006, 106, 1794–1803. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of Acetylation at Lys16 and Trimethylation at Lys20 of Histone H4 Is a Common Hallmark of Human Cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Csh. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer Epigenetics: Moving Forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef]
- Nepali, K.; Liou, J.-P. Recent Developments in Epigenetic Cancer Therapeutics: Clinical Advancement and Emerging Trends. J. Biomed. Sci. 2021, 28, 27. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting Epigenetic Regulators for Cancer Therapy: Mechanisms and Advances in Clinical Trials. Signal. Transduct. Target Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Zhao, S.; Allis, C.D.; Wang, G.G. The Language of Chromatin Modification in Human Cancers. Nat. Rev. Cancer 2021, 413–430. [Google Scholar] [CrossRef]
- Falkenberg, K.J.; Johnstone, R.W. Histone Deacetylases and Their Inhibitors in Cancer, Neurological Diseases and Immune Disorders. Nat. Rev. Drug Discov. 2014, 13, 673–691. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and Emerging HDAC Inhibitors for Cancer Treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Collins, L.B.; Wali, A.; Bigler, R.; Sun, W.; Bultman, S.J. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol. Cell 2012, 48, 612–626. [Google Scholar] [CrossRef] [PubMed]
- Becker, L.; Nogueira, M.S.; Klima, C.; de Angelis, M.H.; Peleg, S. Rapid and Transient Oxygen Consumption Increase Following Acute HDAC/KDAC Inhibition in Drosophila Tissue. Sci. Rep. 2018, 8, 4199. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine Acetylation Targets Protein Complexes and Co-Regulates Major Cellular Functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, L.; Lenart, A.; Tan, Q.; Vaupel, J.W.; Aviv, A.; McGue, M.; Christensen, K. DNA Methylation Age Is Associated with Mortality in a Longitudinal Danish Twin Study. Aging Cell 2016, 15, 149–154. [Google Scholar] [CrossRef]
- Marioni, R.E.; Shah, S.; McRae, A.F.; Chen, B.H.; Colicino, E.; Harris, S.E.; Gibson, J.; Henders, A.K.; Redmond, P.; Cox, S.R.; et al. DNA Methylation Age of Blood Predicts All-Cause Mortality in Later Life. Genome Biol. 2015, 16, 25. [Google Scholar] [CrossRef]
- Fransquet, P.D.; Wrigglesworth, J.; Woods, R.L.; Ernst, M.E.; Ryan, J. The Epigenetic Clock as a Predictor of Disease and Mortality Risk: A Systematic Review and Meta-Analysis. Clin. Epigenetics 2019, 11, 62. [Google Scholar] [CrossRef]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Blasio, A.M.D.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated Epigenetic Aging in Down Syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef]
- Maierhofer, A.; Flunkert, J.; Oshima, J.; Martin, G.M.; Haaf, T.; Horvath, S. Accelerated Epigenetic Aging in Werner Syndrome. Aging 2017, 9, 1143–1152. [Google Scholar] [CrossRef]
- Horvath, S.; Oshima, J.; Martin, G.M.; Lu, A.T.; Quach, A.; Cohen, H.; Felton, S.; Matsuyama, M.; Lowe, D.; Kabacik, S.; et al. Epigenetic Clock for Skin and Blood Cells Applied to Hutchinson Gilford Progeria Syndrome and Ex Vivo Studies. Aging (Albany NY) 2018, 10, 1758–1775. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Ritz, B.R. Increased Epigenetic Age and Granulocyte Counts in the Blood of Parkinson’s Disease Patients. Aging 2015, 7, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Langfelder, P.; Kwak, S.; Aaronson, J.; Rosinski, J.; Vogt, T.F.; Eszes, M.; Faull, R.L.M.; Curtis, M.A.; Waldvogel, H.J.; et al. Huntington’s Disease Accelerates Epigenetic Aging of Human Brain and Disrupts DNA Methylation Levels. Aging (Albany NY) 2016, 8, 1485–1504. [Google Scholar] [CrossRef]
- Gross, A.M.; Jaeger, P.A.; Kreisberg, J.F.; Licon, K.; Jepsen, K.L.; Khosroheidari, M.; Morsey, B.M.; Swindells, S.; Shen, H.; Ng, C.T.; et al. Methylome-Wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol. Cell 2016, 62, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Kananen, L.; Nevalainen, T.; Jylhävä, J.; Marttila, S.; Hervonen, A.; Jylhä, M.; Hurme, M. Cytomegalovirus Infection Accelerates Epigenetic Aging. Exp. Gerontol. 2015, 72, 227–229. [Google Scholar] [CrossRef]
- Mongelli, A.; Barbi, V.; Zamperla, M.G.; Atlante, S.; Forleo, L.; Nesta, M.; Massetti, M.; Pontecorvi, A.; Nanni, S.; Farsetti, A.; et al. Evidence for Biological Age Acceleration and Telomere Shortening in COVID-19 Survivors. Int. J. Mol. Sci. 2021, 22, 6151. [Google Scholar] [CrossRef]
- Wolf, E.J.; Maniates, H.; Nugent, N.; Maihofer, A.X.; Armstrong, D.; Ratanatharathorn, A.; Ashley-Koch, A.E.; Garrett, M.; Kimbrel, N.A.; Lori, A.; et al. Traumatic Stress and Accelerated DNA Methylation Age: A Meta-Analysis. Psychoneuroendocrino 2018, 92, 123–134. [Google Scholar] [CrossRef]
- Zannas, A.S.; Arloth, J.; Carrillo-Roa, T.; Iurato, S.; Röh, S.; Ressler, K.J.; Nemeroff, C.B.; Smith, A.K.; Bradley, B.; Heim, C.; et al. Lifetime Stress Accelerates Epigenetic Aging in an Urban, African American Cohort: Relevance of Glucocorticoid Signaling. Genome Biol. 2015, 16, 266. [Google Scholar] [CrossRef]
- Davis, E.G.; Humphreys, K.L.; McEwen, L.M.; Sacchet, M.D.; Camacho, M.C.; MacIsaac, J.L.; Lin, D.T.S.; Kobor, M.S.; Gotlib, I.H. Accelerated DNA Methylation Age in Adolescent Girls: Associations with Elevated Diurnal Cortisol and Reduced Hippocampal Volume. Transl. Psychiatry 2017, 7, e1223. [Google Scholar] [CrossRef]
- Lawn, R.B.; Anderson, E.L.; Suderman, M.; Simpkin, A.J.; Gaunt, T.R.; Teschendorff, A.E.; Widschwendter, M.; Hardy, R.; Kuh, D.; Relton, C.L.; et al. Psychosocial Adversity and Socioeconomic Position during Childhood and Epigenetic Age: Analysis of Two Prospective Cohort Studies. Hum. Mol. Genet. 2018, 27, 1301–1308. [Google Scholar] [CrossRef]
- Han, L.K.M.; Aghajani, M.; Clark, S.L.; Chan, R.F.; Hattab, M.W.; Shabalin, A.A.; Zhao, M.; Kumar, G.; Xie, L.Y.; Jansen, R.; et al. Epigenetic Aging in Major Depressive Disorder. Am. J. Psychiatry 2018, 175, 774–782. [Google Scholar] [CrossRef]
- Carroll, J.E.; Irwin, M.R.; Levine, M.; Seeman, T.E.; Absher, D.; Assimes, T.; Horvath, S. Epigenetic Aging and Immune Senescence in Women With Insomnia Symptoms: Findings From the Women’s Health Initiative Study. Biol. Psychiatry 2017, 81, 136–144. [Google Scholar] [CrossRef] [PubMed]
- White, A.J.; Kresovich, J.K.; Xu, Z.; Sandler, D.P.; Taylor, J.A. Shift Work, DNA Methylation and Epigenetic Age. Int. J. Epidemiol. 2019, 48, 1536–1544. [Google Scholar] [CrossRef]
- Cedernaes, J.; Osler, M.E.; Voisin, S.; Broman, J.-E.; Vogel, H.; Dickson, S.L.; Zierath, J.R.; Schiöth, H.B.; Benedict, C. Acute Sleep Loss Induces Tissue-Specific Epigenetic and Transcriptional Alterations to Circadian Clock Genes in Men. J. Clin. Endocrinol. Metab. 2015, 100, E1255–E1261. [Google Scholar] [CrossRef] [PubMed]
- Boks, M.P.; van Mierlo, H.C.; Rutten, B.P.F.; Radstake, T.R.D.J.; Witte, L.D.; Geuze, E.; Horvath, S.; Schalkwyk, L.C.; Vinkers, C.H.; Broen, J.C.A.; et al. Longitudinal Changes of Telomere Length and Epigenetic Age Related to Traumatic Stress and Post-Traumatic Stress Disorder. Psychoneuroendocrino 2015, 51, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Verhoeven, J.E.; Yang, R.; Wolkowitz, O.M.; Bersani, F.S.; Lindqvist, D.; Mellon, S.H.; Yehuda, R.; Flory, J.D.; Lin, J.; Abu-Amara, D.; et al. Epigenetic Age in Male Combat-Exposed War Veterans: Associations with Posttraumatic Stress Disorder Status. Mol. Neuropsychiatry 2018, 4, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Mehta, D.; Bruenig, D.; Lawford, B.; Harvey, W.; Carrillo-Roa, T.; Morris, C.P.; Jovanovic, T.; Young, R.M.; Binder, E.B.; Voisey, J. Accelerated DNA Methylation Aging and Increased Resilience in Veterans: The Biological Cost for Soldiering On. Neurobiol. Stress 2018, 8, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Simons, R.L.; Lei, M.K.; Beach, S.R.H.; Philibert, R.A.; Cutrona, C.E.; Gibbons, F.X.; Barr, A. Economic Hardship and Biological Weathering: The Epigenetics of Aging in a U.S. Sample of Black Women. Soc. Sci. Med. 2016, 150, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Erhart, W.; Brosch, M.; Ammerpohl, O.; von Schönfels, W.; Ahrens, M.; Heits, N.; Bell, J.T.; Tsai, P.-C.; Spector, T.D.; et al. Obesity Accelerates Epigenetic Aging of Human Liver. Proc. Natl. Acad. Sci. USA 2014, 111, 15538–15543. [Google Scholar] [CrossRef]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic Clock Analysis of Diet, Exercise, Education, and Lifestyle Factors. Aging 2017, 9, 419–446. [Google Scholar] [CrossRef]
- Nevalainen, T.; Kananen, L.; Marttila, S.; Jylhävä, J.; Mononen, N.; Kähönen, M.; Raitakari, O.T.; Hervonen, A.; Jylhä, M.; Lehtimäki, T.; et al. Obesity Accelerates Epigenetic Aging in Middle-Aged but Not in Elderly Individuals. Clin. Epigenetics 2017, 9, 20. [Google Scholar] [CrossRef]
- de Toro-Martín, J.; Guénard, F.; Tchernof, A.; Hould, F.-S.; Lebel, S.; Julien, F.; Marceau, S.; Vohl, M.-C. Body Mass Index Is Associated with Epigenetic Age Acceleration in the Visceral Adipose Tissue of Subjects with Severe Obesity. Clin. Epigenetics 2019, 11, 172. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Sierra, J.V.; Helbing, A.H.B.; Williams, E.G.; Ashbrook, D.G.; Roy, S.; Williams, R.W.; Mozhui, K. Body Weight and High-fat Diet Are Associated with Epigenetic Aging in Female Members of the BXD Murine Family. Aging Cell 2020, 19, e13207. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Antebi, A.; Bartke, A.; Barzilai, N.; Brown-Borg, H.M.; Caruso, C.; Curiel, T.J.; de Cabo, R.; Franceschi, C.; Gems, D.; et al. Interventions to Slow Aging in Humans: Are We Ready? Aging Cell 2015, 14, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.J.; Robertson, N.A.; Rather, M.I.; Thomson, J.P.; McBryan, T.; Sproul, D.; Wang, T.; Brock, C.; Clark, W.; Ideker, T.; et al. Diverse Interventions That Extend Mouse Lifespan Suppress Shared Age-Associated Epigenetic Changes at Critical Gene Regulatory Regions. Genome Biol. 2017, 18, 58. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.B.; Kane, A.E.; Mitchell, S.J.; MacArthur, M.R.; Warner, E.; Vogel, D.S.; Mitchell, J.R.; Howlett, S.E.; Bonkowski, M.S.; Sinclair, D.A. Age and Life Expectancy Clocks Based on Machine Learning Analysis of Mouse Frailty. Nat. Commun. 2020, 11, 4618. [Google Scholar] [CrossRef] [PubMed]
- Keller, K.; Kane, A.; Heinze-Milne, S.; Grandy, S.A.; Howlett, S.E. Chronic Treatment With the ACE Inhibitor Enalapril Attenuates the Development of Frailty and Differentially Modifies Pro- and Anti-Inflammatory Cytokines in Aging Male and Female C57BL/6 Mice. J. Gerontol. Ser. 2018, 74, 1149–1157. [Google Scholar] [CrossRef]
- Chen, Z.; Raj, A.; Prateek, G.V.; Francesco, A.D.; Liu, J.; Keyes, B.E.; Kolumam, G.; Jojic, V.; Freund, A. Automated, High-Dimensional Evaluation of Physiological Aging and Resilience in Outbred Mice. bioRxiv 2021. [Google Scholar] [CrossRef]
- Søraas, A.; Matsuyama, M.; de Lima, M.; Wald, D.; Buechner, J.; Gedde-Dahl, T.; Søraas, C.L.; Chen, B.; Ferrucci, L.; Dahl, J.A.; et al. Epigenetic Age Is a Cell-intrinsic Property in Transplanted Human Hematopoietic Cells. Aging Cell 2019, 18, e12897. [Google Scholar] [CrossRef]
- Horvath, S.; Singh, K.; Raj, K.; Khairnar, S.; Sanghavi, A.; Shrivastava, A.; Zoller, J.A.; Li, C.Z.; Herenu, C.B.; Canatelli-Mallat, M.; et al. Reversing Age: Dual Species Measurement of Epigenetic Age with a Single Clock. bioRxiv 2020. [Google Scholar] [CrossRef]
- Childebayeva, A.; Harman, T.; Weinstein, J.; Goodrich, J.M.; Dolinoy, D.C.; Day, T.A.; Bigham, A.W.; Brutsaert, T.D. DNA Methylation Changes Are Associated With an Incremental Ascent to High Altitude. Front. Genet. 2019, 10, 1062. [Google Scholar] [CrossRef] [PubMed]
- Thienpont, B.; Steinbacher, J.; Zhao, H.; D’Anna, F.; Kuchnio, A.; Ploumakis, A.; Ghesquière, B.; Dyck, L.V.; Boeckx, B.; Schoonjans, L.; et al. Tumour Hypoxia Causes DNA Hypermethylation by Reducing TET Activity. Nature 2016, 537, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.P.; Miles, S.L. Silencing HIF-1α Induces TET2 Expression and Augments Ascorbic Acid Induced 5-Hydroxymethylation of DNA in Human Metastatic Melanoma Cells. Biochem. Bioph. Res. Commun. 2017, 490, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Camuzi, D.; de Amorim, Í.S.S.; Pinto, L.F.R.; Trivilin, L.O.; Mencalha, A.L.; Lima, S.C.S. Regulation Is in the Air: The Relationship between Hypoxia and Epigenetics in Cancer. Cells 2019, 8, 300. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, M.; WuWong, D.J.; Horvath, S.; Matsuyama, S. Epigenetic Clock Analysis of Human Fibroblasts in Vitro: Effects of Hypoxia, Donor Age, and Expression of HTERT and SV40 LargeT. Aging (Albany NY) 2019, 11, 3012–3022. [Google Scholar] [CrossRef] [PubMed]
- Arsham, A.M.; Howell, J.J.; Simon, M.C. A Novel Hypoxia-Inducible Factor-Independent Hypoxic Response Regulating Mammalian Target of Rapamycin and Its Targets *. J. Biol. Chem. 2003, 278, 29655–29660. [Google Scholar] [CrossRef]
- Green, C.L.; Lamming, D.W.; Fontana, L. Molecular Mechanisms of Dietary Restriction Promoting Health and Longevity. Nat. Rev. Mol. Cell Biol. 2021, 23, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Daniel, M.; Tollefsbol, T.O. Epigenetic Regulation of Caloric Restriction in Aging. BMC Med. 2011, 9, 98. [Google Scholar] [CrossRef]
- Maegawa, S.; Lu, Y.; Tahara, T.; Lee, J.T.; Madzo, J.; Liang, S.; Jelinek, J.; Colman, R.J.; Issa, J.-P.J. Caloric Restriction Delays Age-Related Methylation Drift. Nat. Commun. 2017, 8, 539. [Google Scholar] [CrossRef]
- Kim, C.H.; Lee, E.K.; Choi, Y.J.; An, H.J.; Jeong, H.O.; Park, D.; Kim, B.C.; Yu, B.P.; Bhak, J.; Chung, H.Y. Short-term Calorie Restriction Ameliorates Genomewide, Age-related Alterations in DNA Methylation. Aging Cell 2016, 15, 1074–1081. [Google Scholar] [CrossRef]
- Unnikrishnan, A.; Hadad, N.; Masser, D.R.; Jackson, J.; Freeman, W.M.; Richardson, A. Revisiting the Genomic Hypomethylation Hypothesis of Aging. Ann. N. Y. Acad. Sci. 2018, 1418, 69–79. [Google Scholar] [CrossRef]
- Liao, C.-Y.; Rikke, B.A.; Johnson, T.E.; Diaz, V.; Nelson, J.F. Genetic Variation in the Murine Lifespan Response to Dietary Restriction: From Life Extension to Life Shortening. Aging Cell 2010, 9, 92–95. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Galluzzi, L.; Freije, J.M.P.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Williams, R.W.; Auwerx, J. Metabolic Networks of Longevity. Cell 2010, 142, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Wu, W.K.K.; Wang, H.; Li, X. Serine and One-Carbon Metabolism, a Bridge That Links MTOR Signaling and DNA Methylation in Cancer. Pharmacol. Res. 2019, 149, 104352. [Google Scholar] [CrossRef] [PubMed]
- Cohen, H.Y.; Miller, C.; Bitterman, K.J.; Wall, N.R.; Hekking, B.; Kessler, B.; Howitz, K.T.; Gorospe, M.; de Cabo, R.; Sinclair, D.A. Calorie Restriction Promotes Mammalian Cell Survival by Inducing the SIRT1 Deacetylase. Science 2004, 305, 390–392. [Google Scholar] [CrossRef]
- Kawakami, K.; Nakamura, A.; Goto, S. Dietary Restriction Increases Site-Specific Histone H3 Acetylation in Rat Liver: Possible Modulation by Sirtuins. Biochem. Bioph. Res. Commun. 2012, 418, 836–840. [Google Scholar] [CrossRef]
- Sato, S.; Solanas, G.; Peixoto, F.O.; Bee, L.; Symeonidi, A.; Schmidt, M.S.; Brenner, C.; Masri, S.; Benitah, S.A.; Sassone-Corsi, P. Circadian Reprogramming in the Liver Identifies Metabolic Pathways of Aging. Cell 2017, 170, 664–677.e11. [Google Scholar] [CrossRef]
- Edwards, C.B.; Copes, N.; Brito, A.G.; Canfield, J.; Bradshaw, P.C. Malate and Fumarate Extend Lifespan in Caenorhabditis Elegans. PLoS ONE 2013, 8, e58345. [Google Scholar] [CrossRef]
- McDonnell, E.; Crown, S.B.; Fox, D.B.; Kitir, B.; Ilkayeva, O.R.; Olsen, C.A.; Grimsrud, P.A.; Hirschey, M.D. Lipids Reprogram Metabolism to Become a Major Carbon Source for Histone Acetylation. Cell Rep. 2016, 17, 1463–1472. [Google Scholar] [CrossRef]
- Hahn, O.; Grönke, S.; Stubbs, T.M.; Ficz, G.; Hendrich, O.; Krueger, F.; Andrews, S.; Zhang, Q.; Wakelam, M.J.; Beyer, A.; et al. Dietary Restriction Protects from Age-Associated DNA Methylation and Induces Epigenetic Reprogramming of Lipid Metabolism. Genome Biol. 2017, 18, 56. [Google Scholar] [CrossRef]
- Han, S.; Schroeder, E.A.; Silva-García, C.G.; Hebestreit, K.; Mair, W.B.; Brunet, A. Mono-Unsaturated Fatty Acids Link H3K4me3 Modifiers to C. Elegans Lifespan. Nature 2017, 544, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Tollefsbol, T.O. Glucose Restriction Can Extend Normal Cell Lifespan and Impair Precancerous Cell Growth through Epigenetic Control of HTERT and P16 Expression. Faseb J. 2010, 24, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Grandison, R.C.; Piper, M.D.W.; Partridge, L. Amino-Acid Imbalance Explains Extension of Lifespan by Dietary Restriction in Drosophila. Nature 2009, 462, 1061–1064. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Buehner, G.; Chang, Y.; Harper, J.M.; Sigler, R.; Smith-Wheelock, M. Methionine-deficient Diet Extends Mouse Lifespan, Slows Immune and Lens Aging, Alters Glucose, T4, IGF-I and Insulin Levels, and Increases Hepatocyte MIF Levels and Stress Resistance. Aging Cell 2005, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N. Role of Methionine on Epigenetic Modification of DNA Methylation and Gene Expression in Animals. Anim. Nutr. 2018, 4, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Soda, K.; Dobashi, Y.; Kano, Y.; Tsujinaka, S.; Konishi, F. Polyamine-Rich Food Decreases Age-Associated Pathology and Mortality in Aged Mice. Exp. Gerontol. 2009, 44, 727–732. [Google Scholar] [CrossRef]
- Soda, K. Spermine and Gene Methylation: A Mechanism of Lifespan Extension Induced by Polyamine-Rich Diet. Amino Acids 2020, 52, 213–224. [Google Scholar] [CrossRef]
- Sae-Lee, C.; Corsi, S.; Barrow, T.M.; Kuhnle, G.G.C.; Bollati, V.; Mathers, J.C.; Byun, H. Dietary Intervention Modifies DNA Methylation Age Assessed by the Epigenetic Clock. Mol. Nutr. Food Res. 2018, 62, 1800092. [Google Scholar] [CrossRef]
- Fitzgerald, K.N.; Hodges, R.; Hanes, D.; Stack, E.; Cheishvili, D.; Szyf, M.; Henkel, J.; Twedt, M.W.; Giannopoulou, D.; Herdell, J.; et al. Potential Reversal of Epigenetic Age Using a Diet and Lifestyle Intervention: A Pilot Randomized Clinical Trial. Aging 2021, 13, 9419–9432. [Google Scholar] [CrossRef]
- Longo, V.D.; Kennedy, B.K. Sirtuins in Aging and Age-Related Disease. Cell 2006, 126, 257–268. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 Complex and SIR2 Alone Promote Longevity in Saccharomyces Cerevisiae by Two Different Mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Mercken, E.M.; Mitchell, S.J.; Martin-Montalvo, A.; Minor, R.K.; Almeida, M.; Gomes, A.P.; Scheibye-Knudsen, M.; Palacios, H.H.; Licata, J.J.; Zhang, Y.; et al. SRT2104 Extends Survival of Male Mice on a Standard Diet and Preserves Bone and Muscle Mass. Aging Cell 2014, 13, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Martin-Montalvo, A.; Mercken, E.M.; Palacios, H.H.; Ward, T.M.; Abulwerdi, G.; Minor, R.K.; Vlasuk, G.P.; Ellis, J.L.; Sinclair, D.A.; et al. The SIRT1 Activator SRT1720 Extends Lifespan and Improves Health of Mice Fed a Standard Diet. Cell Rep. 2014, 6, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ Repletion Improves Mitochondrial and Stem Cell Function and Enhances Life Span in Mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef]
- Xu, W.S.; Parmigiani, R.B.; Marks, P.A. Histone Deacetylase Inhibitors: Molecular Mechanisms of Action. Oncogene 2007, 26, 5541–5552. [Google Scholar] [CrossRef]
- Gräff, J.; Tsai, L.-H. The Potential of HDAC Inhibitors as Cognitive Enhancers. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 311–330. [Google Scholar] [CrossRef]
- Yang, F.; Zhao, N.; Ge, D.; Chen, Y. Next-Generation of Selective Histone Deacetylase Inhibitors. Rsc. Adv. 2019, 9, 19571–19583. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy With Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef]
- Vaca, H.R.; Celentano, A.M.; Toscanini, M.A.; Heimburg, T.; Ghazy, E.; Zeyen, P.; Hauser, A.-T.; Oliveira, G.; Elissondo, M.C.; Jung, M.; et al. The Potential for Histone Deacetylase (HDAC) Inhibitors as Cestocidal Drugs. PLoS Negl. Trop. D 2021, 15, e0009226. [Google Scholar] [CrossRef]
- Tao, D.; Lu, J.; Sun, H.; Zhao, Y.-M.; Yuan, Z.-G.; Li, X.-X.; Huang, B.-Q. Trichostatin A Extends the Lifespan of Drosophila Melanogaster by Elevating Hsp22 Expression. Acta Biochim. Et Biophys. Sin. 2004, 36, 618–622. [Google Scholar] [CrossRef]
- Evason, K.; Collins, J.J.; Huang, C.; Hughes, S.; Kornfeld, K. Valproic Acid Extends Caenorhabditis Elegans Lifespan. Aging Cell 2008, 7, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-Beta-Hydroxybutyrate Extends Lifespan in C. Elegans. Aging 2014, 6, 621–644. [Google Scholar] [CrossRef] [PubMed]
- Vaiserman, A.M.; Koshel’, N.M.; Zabuga, O.G.; Kolyada, A.K.; Roshina, N.V.; Pasyukova, E.G. Geroprotective Potential of Sodium Butyrate in Drosophila Melanogaster: Long-Term Effects. Adv. Gerontol. 2013, 3, 297–301. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, H.; Lu, J.; Li, X.; Chen, X.; Tao, D.; Huang, W.; Huang, B. Lifespan Extension and Elevated Hsp Gene Expression in Drosophila Caused by Histone Deacetylase Inhibitors. J. Exp. Biol. 2005, 208, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Schölz, C.; Weinert, B.T.; Wagner, S.A.; Beli, P.; Miyake, Y.; Qi, J.; Jensen, L.J.; Streicher, W.; McCarthy, A.R.; Westwood, N.J.; et al. Acetylation Site Specificities of Lysine Deacetylase Inhibitors in Human Cells. Nat. Biotechnol. 2015, 33, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Stilling, R.M.; van de Wouw, M.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. The Neuropharmacology of Butyrate: The Bread and Butter of the Microbiota-Gut-Brain Axis? Neurochem. Int. 2016, 99, 110–132. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhong, D.; Zhu, J.; An, Y.; Gao, M.; Zhu, S.; Dang, W.; Wang, X.; Yang, B.; Xie, Z. Inhibition of Histone Acetyltransferase GCN5 Extends Lifespan in Both Yeast and Human Cell Lines. Aging Cell 2020, 19, e13129. [Google Scholar] [CrossRef]
- Tezil, T.; Chamoli, M.; Ng, C.-P.; Simon, R.P.; Butler, V.J.; Jung, M.; Andersen, J.; Kao, A.W.; Verdin, E. Lifespan-Increasing Drug Nordihydroguaiaretic Acid Inhibits P300 and Activates Autophagy. Npj. Aging Mech. Dis. 2019, 5, 7. [Google Scholar] [CrossRef]
- Richie, J.P.; Mills, B.J.; Lang, C.A. Dietary Nordihydroguaiaretic Acid Increases the Life Span of the Mosquito. Proc. Soc. Exp. Biol. Med. 1986, 183, 81–85. [Google Scholar] [CrossRef]
- Strong, R.; Miller, R.A.; Astle, C.M.; Floyd, R.A.; Flurkey, K.; Hensley, K.L.; Javors, M.A.; Leeuwenburgh, C.; Nelson, J.F.; Ongini, E.; et al. Nordihydroguaiaretic Acid and Aspirin Increase Lifespan of Genetically Heterogeneous Male Mice. Aging Cell 2008, 7, 641–650. [Google Scholar] [CrossRef]
- Eisenberg, T.; Knauer, H.; Schauer, A.; Büttner, S.; Ruckenstuhl, C.; Carmona-Gutierrez, D.; Ring, J.; Schroeder, S.; Magnes, C.; Antonacci, L.; et al. Induction of Autophagy by Spermidine Promotes Longevity. Nat. Cell Biol. 2009, 11, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Fernandez, E.; Flurkey, K.; Han, M.; Javors, M.A.; Li, X.; Nadon, N.L.; Nelson, J.F.; et al. Rapamycin-Mediated Lifespan Increase in Mice Is Dose and Sex Dependent and Metabolically Distinct from Dietary Restriction. Aging Cell 2014, 13, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Tsui, B.; Kreisberg, J.F.; Robertson, N.A.; Gross, A.M.; Yu, M.K.; Carter, H.; Brown-Borg, H.M.; Adams, P.D.; Ideker, T. Epigenetic Aging Signatures in Mice Livers Are Slowed by Dwarfism, Calorie Restriction and Rapamycin Treatment. Genome Biol. 2017, 18, 57. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Qian, H.; Ertl, R.; Astle, C.M.; Wang, G.G.; Harrison, D.E.; Xu, X. Histone Modifications Change with Age, Dietary Restriction and Rapamycin Treatment in Mouse Brain. Oncotarget 2015, 6, 15882–15890. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; You, Z.; Xu, Y.; Zhou, L.; Guan, Z.; Peng, C.; Wong, C.C.L.; Su, H.; Zhou, T.; Xia, H.; et al. MTORC1 Phosphorylates Acetyltransferase P300 to Regulate Autophagy and Lipogenesis. Mol. Cell 2017, 68, 323–335.e6. [Google Scholar] [CrossRef] [PubMed]
- Vadla, R.; Haldar, D. Mammalian Target of Rapamycin Complex 2 (MTORC2) Controls Glycolytic Gene Expression by Regulating Histone H3 Lysine 56 Acetylation. Cell Cycle 2018, 17, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.-J.; et al. Metformin Improves Healthspan and Lifespan in Mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- Cabreiro, F.; Au, C.; Leung, K.-Y.; Vergara-Irigaray, N.; Cochemé, H.M.; Noori, T.; Weinkove, D.; Schuster, E.; Greene, N.D.E.; Gems, D. Metformin Retards Aging in C. Elegans by Altering Microbial Folate and Methionine Metabolism. Cell 2013, 153, 228–239. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic Effects of Metformin: From Molecular Mechanisms to Clinical Implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of AMP-Activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells *. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, Y.; Lee, B.B.; Cho, E.Y.; Han, J.; Shim, Y.M.; Kim, D.-H. Metformin Reduces Histone H3K4me3 at the Promoter Regions of Positive Cell Cycle Regulatory Genes in Lung Cancer Cells. Cancers 2021, 13, 739. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Hu, D.; Chen, H.; Shi, G.; Fetahu, I.S.; Wu, F.; Rabidou, K.; Fang, R.; Tan, L.; Xu, S.; et al. Glucose-Regulated Phosphorylation of TET2 by AMPK Reveals a Pathway Linking Diabetes to Cancer. Nature 2018, 559, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.C.; Mamotte, C.D. Pleiotropic and Adverse Effects of Statins—Do Epigenetics Play a Role? J. Pharmacol. Exp. Ther. 2017, 362, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-J.; Tang, L.-Y.; Reddy, M.N.; Shen, C.-K.J. DNA Methyltransferase Gene DDnmt2 and Longevity of Drosophila *. J. Biol. Chem. 2005, 280, 861–864. [Google Scholar] [CrossRef]
- Liu, L.; van Groen, T.; Kadish, I.; Li, Y.; Wang, D.; James, S.R.; Karpf, A.R.; Tollefsbol, T.O. Insufficient DNA Methylation Affects Healthy Aging and Promotes Age-Related Health Problems. Clin. Epigenetics 2011, 2, 349–360. [Google Scholar] [CrossRef]
- Yu, R.; Cao, X.; Sun, L.; Zhu, J.; Wasko, B.M.; Liu, W.; Crutcher, E.; Liu, H.; Jo, M.C.; Qin, L.; et al. Inactivating Histone Deacetylase HDA Promotes Longevity by Mobilizing Trehalose Metabolism. Nat. Commun. 2021, 12, 1981. [Google Scholar] [CrossRef]
- Rogina, B.; Helfand, S.L.; Frankel, S. Longevity Regulation by Drosophila Rpd3 Deacetylase and Caloric Restriction. Science 2002, 298, 1745. [Google Scholar] [CrossRef]
- Frankel, S.; Woods, J.; Ziafazeli, T.; Rogina, B. RPD3 Histone Deacetylase and Nutrition Have Distinct but Interacting Effects on Drosophila Longevity. Aging 2015, 7, 1112–1128. [Google Scholar] [CrossRef]
- Kanfi, Y.; Naiman, S.; Amir, G.; Peshti, V.; Zinman, G.; Nahum, L.; Bar-Joseph, Z.; Cohen, H.Y. The Sirtuin SIRT6 Regulates Lifespan in Male Mice. Nature 2012, 483, 218–221. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.-I. Sirt1 Extends Life Span and Delays Aging in Mice through the Regulation of Nk2 Homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Burnett, C.; Valentini, S.; Cabreiro, F.; Goss, M.; Somogyvári, M.; Piper, M.D.; Hoddinott, M.; Sutphin, G.L.; Leko, V.; McElwee, J.J.; et al. Absence of Effects of Sir2 Overexpression on Lifespan in C. Elegans and Drosophila. Nature 2011, 477, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Brunet, A. Histone Methylation Makes Its Mark on Longevity. Trends Cell Biol. 2012, 22, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Maures, T.J.; Hauswirth, A.G.; Green, E.M.; Leeman, D.S.; Maro, G.S.; Han, S.; Banko, M.R.; Gozani, O.; Brunet, A. Members of the H3K4 Trimethylation Complex Regulate Lifespan in a Germline-Dependent Manner in C. Elegans. Nature 2010, 466, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Siebold, A.P.; Banerjee, R.; Tie, F.; Kiss, D.L.; Moskowitz, J.; Harte, P.J. Polycomb Repressive Complex 2 and Trithorax Modulate Drosophila Longevity and Stress Resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Merkwirth, C.; Jovaisaite, V.; Durieux, J.; Matilainen, O.; Jordan, S.D.; Quiros, P.M.; Steffen, K.K.; Williams, E.G.; Mouchiroud, L.; Tronnes, S.U.; et al. Two Conserved Histone Demethylases Regulate Mitochondrial Stress-Induced Longevity. Cell 2016, 165, 1209–1223. [Google Scholar] [CrossRef] [PubMed]
- Maures, T.J.; Greer, E.L.; Hauswirth, A.G.; Brunet, A. he H3K27 Demethylase UTX-1 Regulates C. Elegans Lifespan in a Germline-independent, Insulin-dependent Manner. Aging Cell 2011, 10, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Guillermo, A.R.R.; Chocian, K.; Gavriilidis, G.; Vandamme, J.; Salcini, A.E.; Mellor, J.; Woollard, A. H3K27 Modifiers Regulate Lifespan in C. Elegans in a Context-Dependent Manner. BMC Biol. 2021, 19, 59. [Google Scholar] [CrossRef]
- Murn, J.; Shi, Y. The Winding Path of Protein Methylation Research: Milestones and New Frontiers. Nat. Rev. Mol. Cell Biol. 2017, 18, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Wu, X.; Zhou, J.; Li, X.; Huang, X.; Li, J.; Wu, J.; Bian, Q.; Wang, Y.; Tian, Y. NuRD Mediates Mitochondrial Stress–Induced Longevity via Chromatin Remodeling in Response to Acetyl-CoA Level. Sci. Adv. 2020, 6, eabb2529. [Google Scholar] [CrossRef]
- Pegoraro, G.; Kubben, N.; Wickert, U.; Göhler, H.; Hoffmann, K.; Misteli, T. Aging-Related Chromatin Defects via Loss of the NURD Complex. Nat. Cell Biol. 2009, 11, 1261–1267. [Google Scholar] [CrossRef] [PubMed]
- Wahlestedt, M.; Norddahl, G.L.; Sten, G.; Ugale, A.; Frisk, M.-A.M.; Mattsson, R.; Deierborg, T.; Sigvardsson, M.; Bryder, D. An Epigenetic Component of Hematopoietic Stem Cell Aging Amenable to Reprogramming into a Young State. Blood 2013, 121, 4257–4264. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]
- Buganim, Y.; Faddah, D.A.; Cheng, A.W.; Itskovich, E.; Markoulaki, S.; Ganz, K.; Klemm, S.L.; van Oudenaarden, A.; Jaenisch, R. Single-Cell Expression Analyses during Cellular Reprogramming Reveal an Early Stochastic and a Late Hierarchic Phase. Cell 2012, 150, 1209–1222. [Google Scholar] [CrossRef]
- Nashun, B.; Hill, P.W.; Hajkova, P. Reprogramming of Cell Fate: Epigenetic Memory and the Erasure of Memories Past. EMBO J. 2015, 34, 1296–1308. [Google Scholar] [CrossRef]
- Polo, J.M.; Anderssen, E.; Walsh, R.M.; Schwarz, B.A.; Nefzger, C.M.; Lim, S.M.; Borkent, M.; Apostolou, E.; Alaei, S.; Cloutier, J.; et al. A Molecular Roadmap of Reprogramming Somatic Cells into IPS Cells. Cell 2012, 151, 1617–1632. [Google Scholar] [CrossRef]
- Liu, G.-H.; Barkho, B.Z.; Ruiz, S.; Diep, D.; Qu, J.; Yang, S.-L.; Panopoulos, A.D.; Suzuki, K.; Kurian, L.; Walsh, C.; et al. Recapitulation of Premature Aging with IPSCs from Hutchinson-Gilford Progeria Syndrome. Nature 2011, 472, 221–225. [Google Scholar] [CrossRef]
- Lapasset, L.; Milhavet, O.; Prieur, A.; Besnard, E.; Babled, A.; Aït-Hamou, N.; Leschik, J.; Pellestor, F.; Ramirez, J.-M.; Vos, J.D.; et al. Rejuvenating Senescent and Centenarian Human Cells by Reprogramming through the Pluripotent State. Genes Dev. 2011, 25, 2248–2253. [Google Scholar] [CrossRef]
- Shahini, A.; Rajabian, N.; Choudhury, D.; Shahini, S.; Vydiam, K.; Nguyen, T.; Kulczyk, J.; Santarelli, T.; Ikhapoh, I.; Zhang, Y.; et al. Ameliorating the Hallmarks of Cellular Senescence in Skeletal Muscle Myogenic Progenitors in Vitro and in Vivo. Sci. Adv. 2021, 7, eabe5671. [Google Scholar] [CrossRef]
- Abad, M.; Mosteiro, L.; Pantoja, C.; Cañamero, M.; Rayon, T.; Ors, I.; Graña, O.; Megías, D.; Domínguez, O.; Martínez, D.; et al. Reprogramming in Vivo Produces Teratomas and IPS Cells with Totipotency Features. Nature 2013, 502, 340–345. [Google Scholar] [CrossRef]
- Ohnishi, K.; Semi, K.; Yamamoto, T.; Shimizu, M.; Tanaka, A.; Mitsunaga, K.; Okita, K.; Osafune, K.; Arioka, Y.; Maeda, T.; et al. Premature Termination of Reprogramming In Vivo Leads to Cancer Development through Altered Epigenetic Regulation. Cell 2014, 156, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733.e12. [Google Scholar] [CrossRef] [PubMed]
- Olova, N.; Simpson, D.J.; Marioni, R.E.; Chandra, T. Partial Reprogramming Induces a Steady Decline in Epigenetic Age before Loss of Somatic Identity. Aging Cell 2019, 18, e12877. [Google Scholar] [CrossRef] [PubMed]
- Larocca, D.; Lee, J.; West, M.D.; Labat, I.; Sternberg, H. No Time to Age: Uncoupling Aging from Chronological Time. Genes 2021, 12, 611. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, T.J.; Quarta, M.; Mukherjee, S.; Colville, A.; Paine, P.; Doan, L.; Tran, C.M.; Chu, C.R.; Horvath, S.; Qi, L.S.; et al. Transient Non-Integrative Expression of Nuclear Reprogramming Factors Promotes Multifaceted Amelioration of Aging in Human Cells. Nat. Commun. 2020, 11, 1545. [Google Scholar] [CrossRef]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to Recover Youthful Epigenetic Information and Restore Vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-Cell Genome-Wide Bisulfite Sequencing for Assessing Epigenetic Heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef]
- Cao, J.; Cusanovich, D.A.; Ramani, V.; Aghamirzaie, D.; Pliner, H.A.; Hill, A.J.; Daza, R.M.; McFaline-Figueroa, J.L.; Packer, J.S.; Christiansen, L.; et al. Joint Profiling of Chromatin Accessibility and Gene Expression in Thousands of Single Cells. Science 2018, 361, 1380–1385. [Google Scholar] [CrossRef]
- Angermueller, C.; Clark, S.J.; Lee, H.J.; Macaulay, I.C.; Teng, M.J.; Hu, T.X.; Krueger, F.; Smallwood, S.; Ponting, C.P.; Voet, T.; et al. Parallel Single-Cell Sequencing Links Transcriptional and Epigenetic Heterogeneity. Nat. Methods 2016, 13, 229–232. [Google Scholar] [CrossRef]
- Hernando-Herraez, I.; Evano, B.; Stubbs, T.; Commere, P.-H.; Bonder, M.J.; Clark, S.; Andrews, S.; Tajbakhsh, S.; Reik, W. Ageing Affects DNA Methylation Drift and Transcriptional Cell-to-Cell Variability in Mouse Muscle Stem Cells. Nat. Commun. 2019, 10, 4361. [Google Scholar] [CrossRef]
- Strzelecka, P.M.; Damm, F. Haematopoietic Ageing through the Lens of Single-Cell Technologies. Dis. Model Mech. 2021, 14, dmm047340. [Google Scholar] [CrossRef] [PubMed]
- Trapp, A.; Kerepesi, C.; Gladyshev, V.N. Profiling Epigenetic Age in Single Cells. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pitt, J.N.; Strait, N.L.; Vayndorf, E.M.; Blue, B.W.; Tran, C.H.; Davis, B.E.M.; Huang, K.; Johnson, B.J.; Lim, K.M.; Liu, S.; et al. WormBot, an Open-Source Robotics Platform for Survival and Behavior Analysis in C. Elegans. Geroscience 2019, 41, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Deng, L.; Xue, Y.; Suzuki, K.; Zhang, W.; Yu, Y.; Wu, J.; Sun, L.; Gong, X.; Luan, H.; et al. Visualization of Aging-Associated Chromatin Alterations with an Engineered TALE System. Cell Res. 2017, 27, 483–504. [Google Scholar] [CrossRef] [PubMed]
- Putin, E.; Mamoshina, P.; Aliper, A.; Korzinkin, M.; Moskalev, A.; Kolosov, A.; Ostrovskiy, A.; Cantor, C.; Vijg, J.; Zhavoronkov, A. Deep Biomarkers of Human Aging: Application of Deep Neural Networks to Biomarker Development. Aging Albany Ny 2016, 8, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Mamoshina, P.; Kochetov, K.; Putin, E.; Cortese, F.; Aliper, A.; Lee, W.-S.; Ahn, S.-M.; Uhn, L.; Skjodt, N.; Kovalchuk, O.; et al. Population Specific Biomarkers of Human Aging: A Big Data Study Using South Korean, Canadian, and Eastern European Patient Populations. J. Gerontol. Ser. Biol. Sci. Med. Sci. 2018, 73, 1482–1490. [Google Scholar] [CrossRef]
- Galkin, F.; Mamoshina, P.; Kochetov, K.; Sidorenko, D.; Zhavoronkov, A. DeepMAge: A Methylation Aging Clock Developed with Deep Learning. Aging Dis. 2021, 12, 1252–1262. [Google Scholar] [CrossRef]
- Zhavoronkov, A.; Mamoshina, P. Deep Aging Clocks: The Emergence of AI-Based Biomarkers of Aging and Longevity. Trends Pharm. Sci. 2019, 40, 546–549. [Google Scholar] [CrossRef]
- Galkin, F.; Mamoshina, P.; Aliper, A.; Putin, E.; Moskalev, V.; Gladyshev, V.N.; Zhavoronkov, A. Human Gut Microbiome Aging Clock Based on Taxonomic Profiling and Deep Learning. Iscience 2020, 23, 101199. [Google Scholar] [CrossRef]
- Sharma, M.; Li, Y.; Stoll, M.L.; Tollefsbol, T.O. The Epigenetic Connection Between the Gut Microbiome in Obesity and Diabetes. Front. Genet. 2020, 10, 1329. [Google Scholar] [CrossRef]
- Kaur, H.; Singh, Y.; Singh, S.; Singh, R.B. Gut Microbiome-Mediated Epigenetic Regulation of Brain Disorder and Application of Machine Learning for Multi-Omics Data Analysis. Genome 2021, 64, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Mischke, M.; Plösch, T. More than Just a Gut Instinct–the Potential Interplay between a Baby’s Nutrition, Its Gut Microbiome, and the Epigenome. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, R1065–R1069. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.M.; Ali, M.M. Methyl Donor Micronutrients That Modify DNA Methylation and Cancer Outcome. Nutrients 2019, 11, 608. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Amaretti, A.; Raimondi, S. Folate Production by Probiotic Bacteria. Nutrients 2011, 3, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Abujamra, A.L.; Loew, J.E.; Forman, L.W.; Perrine, S.P.; Faller, D.V. Histone Deacetylase Inhibitors Reverse CpG Methylation by Regulating DNMT1 through ERK Signaling. Anticancer. Res. 2011, 31, 2723–2732. [Google Scholar] [PubMed]
- Krautkramer, K.A.; Kreznar, J.H.; Romano, K.A.; Vivas, E.I.; Barrett-Wilt, G.A.; Rabaglia, M.E.; Keller, M.P.; Attie, A.D.; Rey, F.E.; Denu, J.M. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol. Cell 2016, 64, 982–992. [Google Scholar] [CrossRef] [PubMed]
- Demehri, F.R.; Frykman, P.K.; Cheng, Z.; Ruan, C.; Wester, T.; Nordenskjöld, A.; Kawaguchi, A.; Hui, T.T.; Granström, A.L.; Funari, V.; et al. Altered Fecal Short Chain Fatty Acid Composition in Children with a History of Hirschsprung-Associated Enterocolitis. J. Pediatr. Surg. 2016, 51, 81–86. [Google Scholar] [CrossRef]
- Yuille, S.; Reichardt, N.; Panda, S.; Dunbar, H.; Mulder, I.E. Human Gut Bacteria as Potent Class I Histone Deacetylase Inhibitors in Vitro through Production of Butyric Acid and Valeric Acid. PLoS ONE 2018, 13, e0201073. [Google Scholar] [CrossRef]
- Smith, P.; Willemsen, D.; Popkes, M.; Metge, F.; Gandiwa, E.; Reichard, M.; Valenzano, D.R. Regulation of Life Span by the Gut Microbiota in the Short-Lived African Turquoise Killifish. elife 2017, 6, e27014. [Google Scholar] [CrossRef]
- Kong, F.; Deng, F.; Li, Y.; Zhao, J. Identification of Gut Microbiome Signatures Associated with Longevity Provides a Promising Modulation Target for Healthy Aging. Gut Microbes 2018, 10, 210–215. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galow, A.-M.; Peleg, S. How to Slow down the Ticking Clock: Age-Associated Epigenetic Alterations and Related Interventions to Extend Life Span. Cells 2022, 11, 468. https://doi.org/10.3390/cells11030468
Galow A-M, Peleg S. How to Slow down the Ticking Clock: Age-Associated Epigenetic Alterations and Related Interventions to Extend Life Span. Cells. 2022; 11(3):468. https://doi.org/10.3390/cells11030468
Chicago/Turabian StyleGalow, Anne-Marie, and Shahaf Peleg. 2022. "How to Slow down the Ticking Clock: Age-Associated Epigenetic Alterations and Related Interventions to Extend Life Span" Cells 11, no. 3: 468. https://doi.org/10.3390/cells11030468
APA StyleGalow, A.-M., & Peleg, S. (2022). How to Slow down the Ticking Clock: Age-Associated Epigenetic Alterations and Related Interventions to Extend Life Span. Cells, 11(3), 468. https://doi.org/10.3390/cells11030468