


RNA-Seq of Dermal Fibroblasts from Patients with Hypermobile Ehlers–Danlos Syndrome and Hypermobility Spectrum Disorders Supports Their Categorization as a Single Entity with Involvement of Extracellular Matrix Degrading and Proinflammatory Pathomechanisms

 ,
,  ,
,  ,
,  ,
, 
Abstract
1. Introduction
2. Patients and Methods
2.1. Patients
2.2. Skin Biopsies, Cell Cultures, and Immunofluorescence Microscopy (IF)
2.3. RNA Extraction and Transcriptome Sequencing
2.4. Functional Gene Annotation and Enrichment Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CRITERION 1 Presence of generalized joint hypermobility (gJHM) |
| BEIGHTON SCORE |
|
| CRITERION 2 Two or more among features (A–C) MUST be present (A and B; A and C; B and C; A and B and C) |
|
|
|
| CRITERION 3 All the following prerequisites must be met: |
|
2.5. qPCR
3. Results
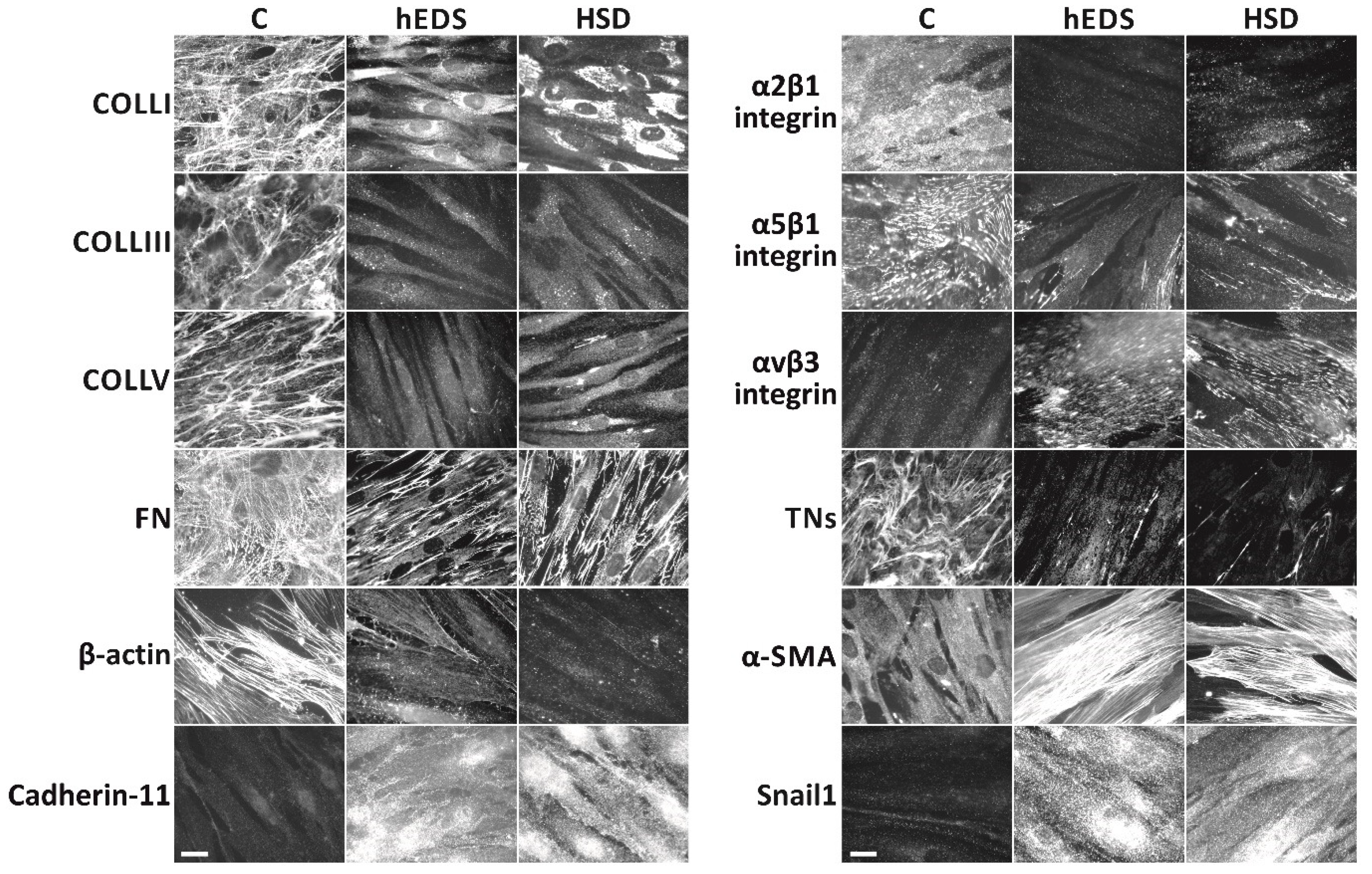
3.1. Comparison of the Cellular Phenotype of Dermal Fibroblasts Derived from hEDS and HSD Patients to That of Healthy Donors
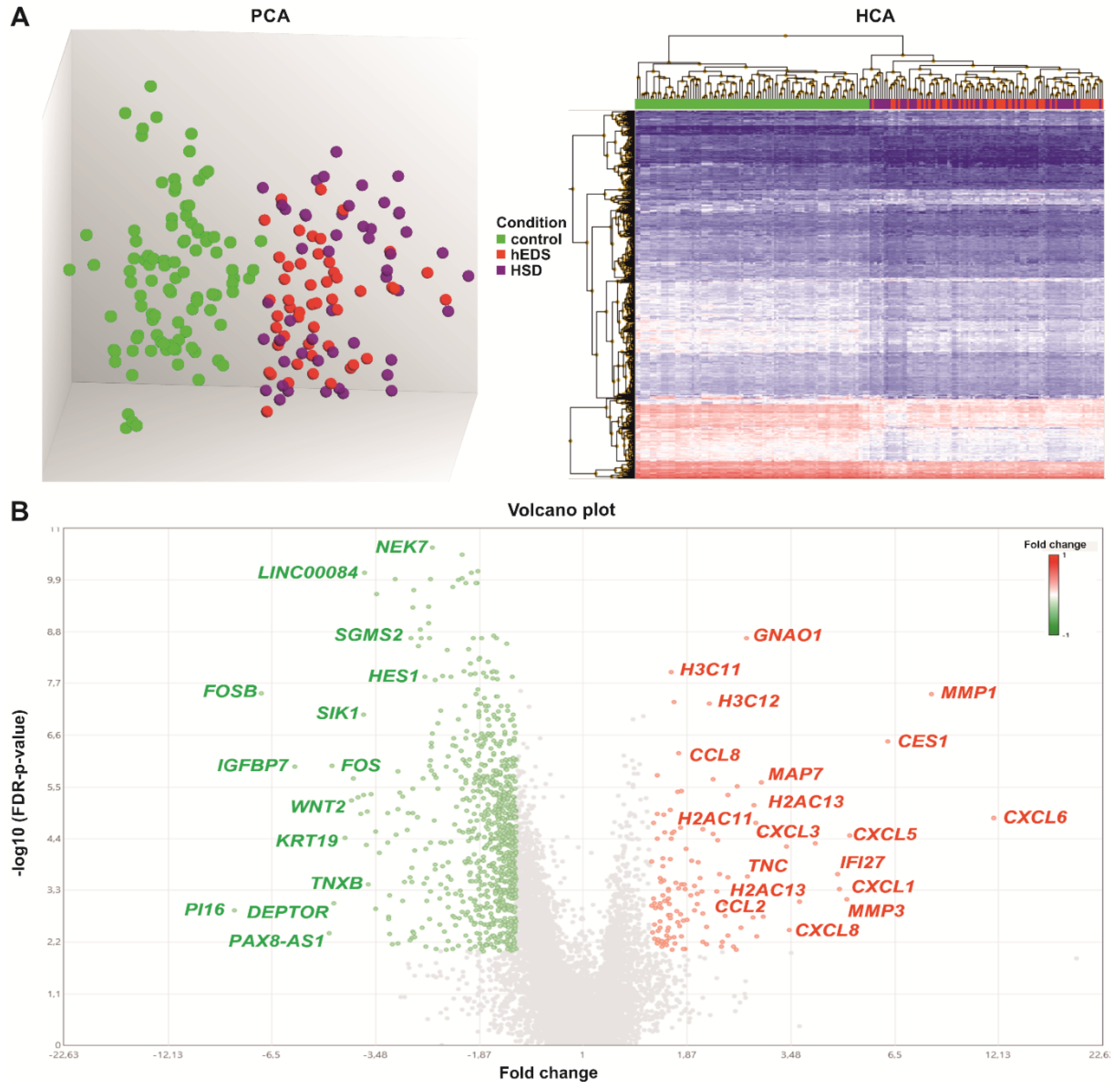
3.2. Global Analysis of mRNA/ncRNA Expression Profiles in hEDS and HSD Dermal Fibroblasts
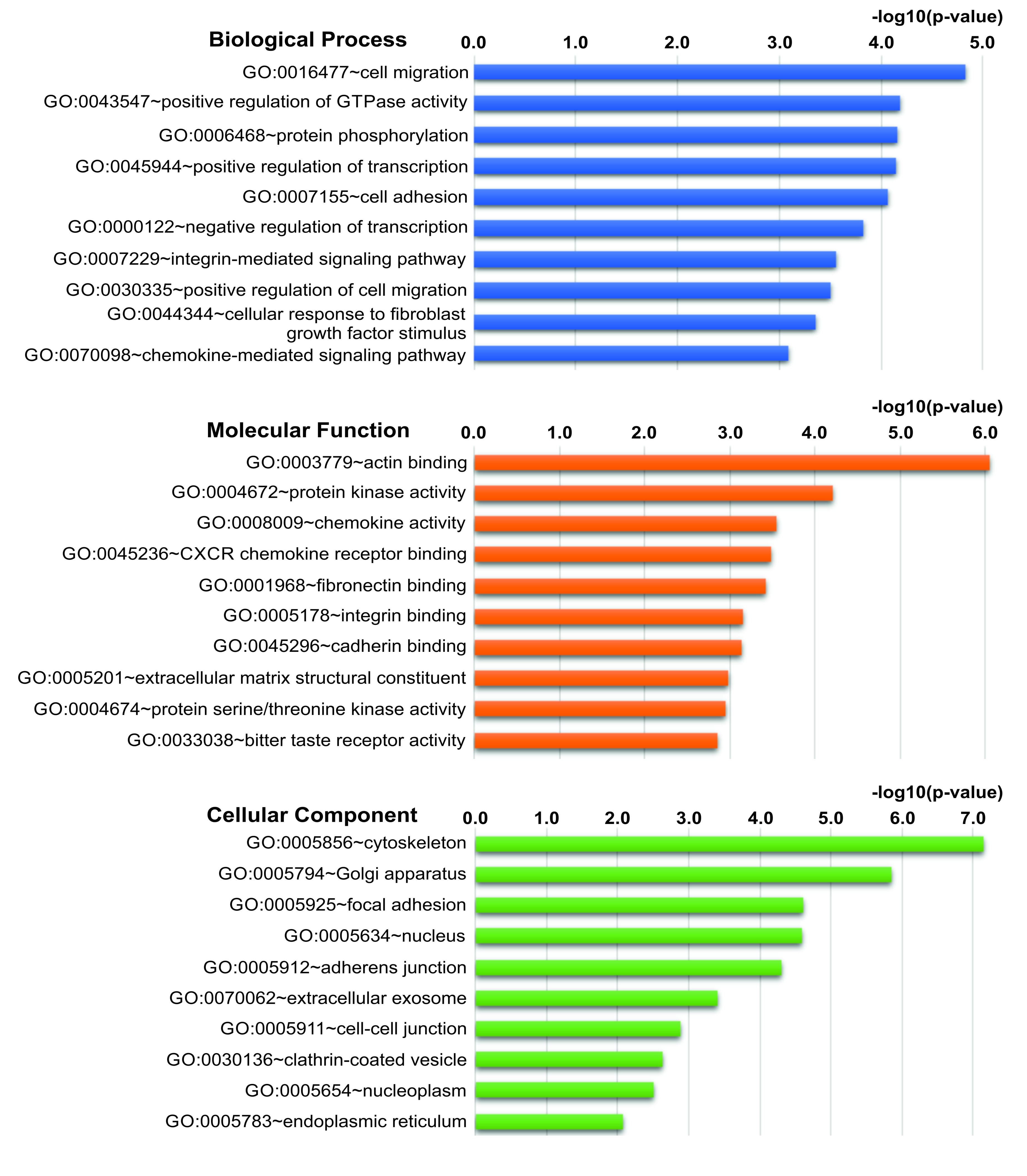
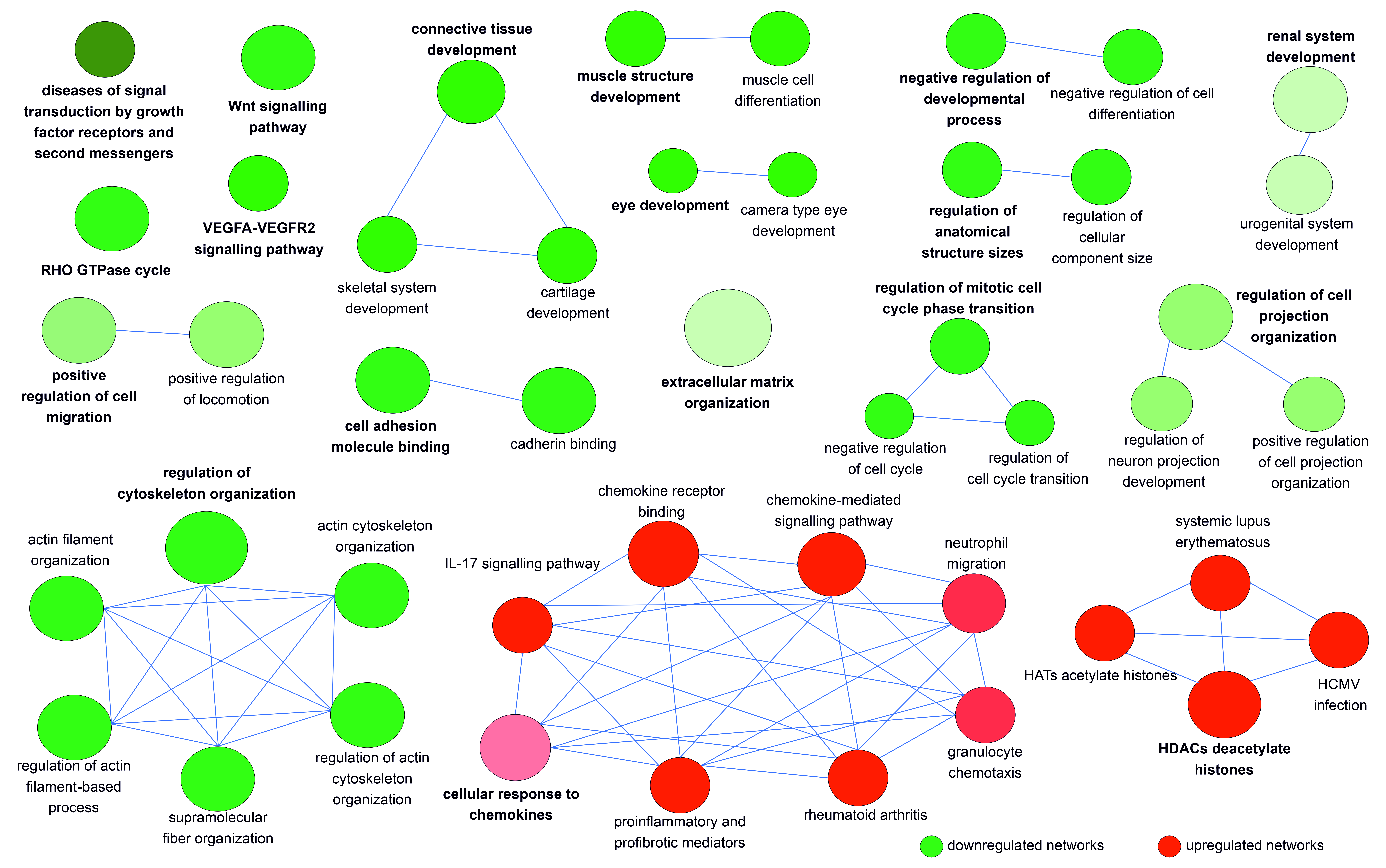
3.3. The DEGs in hEDS/HSD Are Involved in Biological Processes and Pathways That Are Potentially Relevant for the Disease Pathophysiology
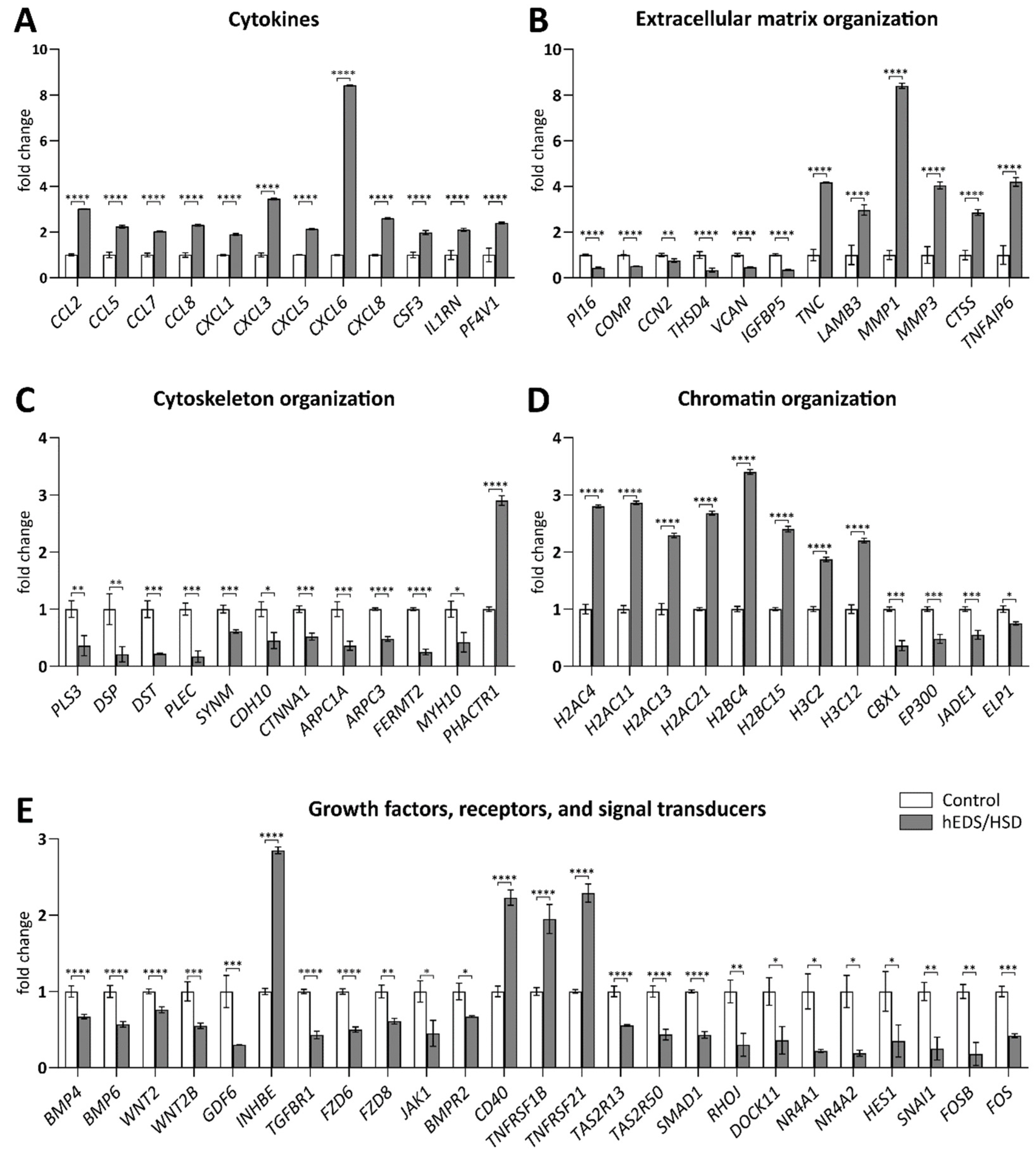
3.4. qPCR Validation of RNA-Seq Data
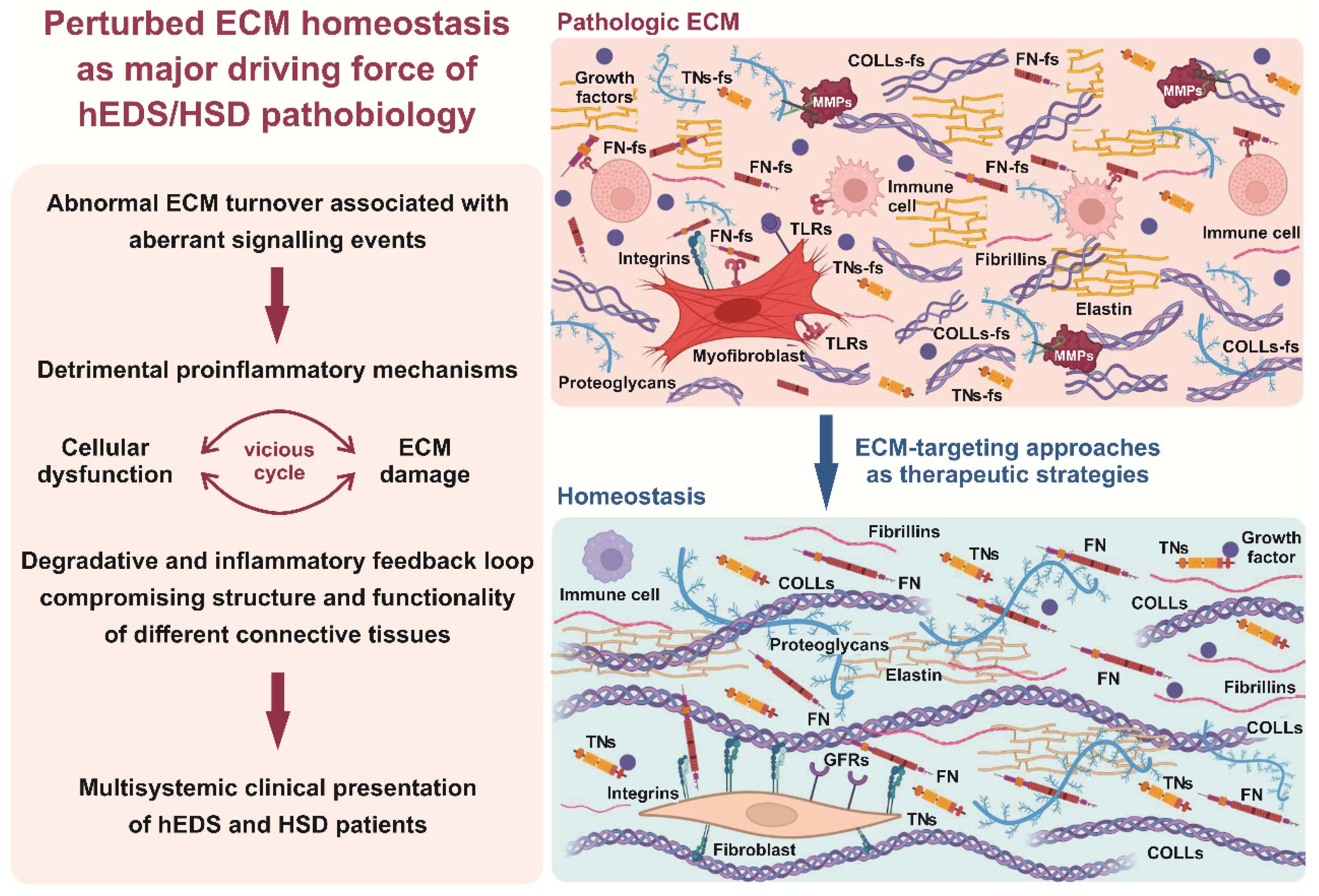
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malfait, F.; Castori, M.; Francomano, C.A.; Giunta, C.; Kosho, T.; Byers, P.H. The Ehlers–Danlos syndromes. Nat. Rev. Dis. Prim. 2020, 6, 64. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Castori, M.; Tinkle, B.; Levy, H.; Grahame, R.; Malfait, F.; Hakim, A. A framework for the classification of joint hypermobility and related conditions. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Tinkle, B.; Castori, M.; Berglund, B.; Cohen, H.; Grahame, R.; Kazkaz, H.; Levy, H. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome Type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 48–69. [Google Scholar] [CrossRef]
- Atwell, K.; Michael, W.; Dubey, J.; James, S.; Martonffy, A.; Anderson, S.; Rudin, N.; Schrager, S. Diagnosis and Management of Hypermobility Spectrum Disorders in Primary Care. J. Am. Board Fam. Med. 2021, 34, 838–848. [Google Scholar] [CrossRef]
- Hakim, A.J.; Tinkle, B.T.; Francomano, C.A. Ehlers–Danlos syndromes, hypermobility spectrum disorders, and associated co-morbidities: Reports from EDS ECHO. Am. J. Med. Genet. Part C Semin. Med. Genet. 2021, 187, 413–415. [Google Scholar] [CrossRef]
- McGillis, L.; Mittal, N.; Mina, D.S.; So, J.; Soowamber, M.; Weinrib, A.; Soever, L.; Rozenberg, D.; Liu, L.; Tse, Y.; et al. Utilization of the 2017 diagnostic criteria for hEDS by the Toronto GoodHope Ehlers–Danlos syndrome clinic: A retrospective review. Am. J. Med. Genet. Part A 2020, 182, 484–492. [Google Scholar] [CrossRef]
- Martinez, K.L.; Mauss, C.; Andrews, J.; Saboda, K.; Huynh, J.M.; Sanoja, A.J.; Jesudas, R.; Byers, P.H.; Laukaitis, C.M. Subtle differences in autonomic symptoms in people diagnosed with hypermobile Ehlers–Danlos syndrome and hypermobility spectrum disorders. Am. J. Med. Genet. Part A 2021, 185, 2012–2025. [Google Scholar] [CrossRef]
- Castori, M. Deconstructing and reconstructing joint hypermobility on an evo-devo perspective. Rheumatology 2021, 60, 2537–2544. [Google Scholar] [CrossRef]
- Aubry-Rozier, B.; Schwitzguebel, A.; Valerio, F.; Tanniger, J.; Paquier, C.; Berna, C.; Hügle, T.; Benaim, C. Are patients with hypermobile Ehlers–Danlos syndrome or hypermobility spectrum disorder so different? Rheumatol. Int. 2021, 41, 1785–1794. [Google Scholar] [CrossRef]
- Martin, A. An acquired or heritable connective tissue disorder? A review of hypermobile Ehlers Danlos Syndrome. Eur. J. Med. Genet. 2019, 62, 103672. [Google Scholar] [CrossRef] [PubMed]
- Yew, K.S.; Kamps-Schmitt, K.A.; Borge, R. Hypermobile Ehlers-Danlos Syndrome and Hypermobility Spectrum Disorders. Am. Fam. Physician 2021, 103, 481–492. [Google Scholar] [PubMed]
- Bennett, S.E.; Walsh, N.; Moss, T.; Palmer, S. Developing a self-management intervention to manage hypermobility spectrum disorders (HSD) and hypermobile Ehlers-Danlos syndrome (hEDS): An analysis informed by behaviour change theory. Disabil. Rehabil. 2022, 44, 5231–5240. [Google Scholar] [CrossRef] [PubMed]
- Brock, I.; Prendergast, W.; Maitland, A. Mast cell activation disease and immunoglobulin deficiency in patients with hypermobile Ehlers-Danlos syndrome/hypermobility spectrum disorder. Am. J. Med. Genet. Part C Semin. Med. Genet. 2021, 187, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.Y.; Palsson, O.S.; Whitehead, W.E.; Sperber, A.D.; Tornblom, H.; Simren, M.; Aziz, I. Rome IV Functional Gastrointestinal Disorders and Health Impairment in Subjects With Hypermobility Spectrum Disorders or Hypermobile Ehlers-Danlos Syndrome. Clin. Gastroenterol. Hepatol. 2021, 19, 277–287.e3. [Google Scholar] [CrossRef]
- Vermeulen, S.; De Mits, S.; De Ridder, R.; Calders, P.; De Schepper, J.; Malfait, F.; Rombaut, L. Altered Multisegment Ankle and Foot Kinematics During Gait in Patients With Hypermobile Ehlers-Danlos Syndrome/Hypermobility Spectrum Disorder: A Case–Control Study. Arthritis Care Res. 2022, 74, 841–848. [Google Scholar] [CrossRef]
- Copetti, M.; Morlino, S.; Colombi, M.; Grammatico, P.; Fontana, A.; Castori, M. Severity classes in adults with hypermobile Ehlers–Danlos syndrome/hypermobility spectrum disorders: A pilot study of 105 Italian patients. Rheumatology 2019, 58, 1722–1730. [Google Scholar] [CrossRef]
- Demes, J.S.; McNair, B.; Taylor, M.R. Use of complementary therapies for chronic pain management in patients with reported Ehlers-Danlos syndrome or hypermobility spectrum disorders. Am. J. Med. Genet. Part A 2020, 182, 2611–2623. [Google Scholar] [CrossRef]
- Zoppi, N.; Chiarelli, N.; Binetti, S.; Ritelli, M.; Colombi, M. Dermal fibroblast-to-myofibroblast transition sustained by αvß3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Biochim. et Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 1010–1023. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Dordoni, C.; Ritelli, M.; Venturini, M.; Castori, M.; Colombi, M. Transcriptome-Wide Expression Profiling in Skin Fibroblasts of Patients with Joint Hypermobility Syndrome/Ehlers-Danlos Syndrome Hypermobility Type. PLoS ONE 2016, 11, e0161347. [Google Scholar] [CrossRef]
- Zoppi, N.; Chiarelli, N.; Ritelli, M.; Colombi, M. Multifaced Roles of the αvβ3 Integrin in Ehlers–Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts. Int. J. Mol. Sci. 2018, 19, 982. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, N.; Zoppi, N.; Venturini, M.; Capitanio, D.; Gelfi, C.; Ritelli, M.; Colombi, M. Matrix Metalloproteinases Inhibition by Doxycycline Rescues Extracellular Matrix Organization and Partly Reverts Myofibroblast Differentiation in Hypermobile Ehlers-Danlos Syndrome Dermal Fibroblasts: A Potential Therapeutic Target? Cells 2021, 10, 3236. [Google Scholar] [CrossRef] [PubMed]
- Chiarelli, N.; Zoppi, N.; Ritelli, M.; Venturini, M.; Capitanio, D.; Gelfi, C.; Colombi, M. Biological insights in the pathogenesis of hypermobile Ehlers-Danlos syndrome from proteome profiling of patients’ dermal myofibroblasts. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1867, 166051. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Venturini, M.; Cinquina, V.; Chiarelli, N.; Colombi, M. Multisystemic manifestations in a cohort of 75 classical Ehlers-Danlos syndrome patients: Natural history and nosological perspectives. Orphanet J. Rare Dis. 2020, 15, 197. [Google Scholar] [CrossRef]
- Rymen, D.; Ritelli, M.; Zoppi, N.; Cinquina, V.; Giunta, C.; Rohrbach, M.; Colombi, M. Clinical and Molecular Characterization of Classical-Like Ehlers-Danlos Syndrome Due to a Novel TNXB Variant. Genes 2019, 10, 843. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Cinquina, V.; Venturini, M.; Pezzaioli, L.; Formenti, A.M.; Chiarelli, N.; Colombi, M. Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome. Genes 2019, 10, 135. [Google Scholar] [CrossRef]
- Hakim, A.; Grahame, R. Joint hypermobility. Best Pract. Res. Clin. Rheumatol. 2003, 17, 989–1004. [Google Scholar] [CrossRef]
- Gensemer, C.; Burks, R.; Kautz, S.; Judge, D.P.; Lavallee, M.; Norris, R.A. Hypermobile Ehlers-Danlos syndromes: Complex phenotypes, challenging diagnoses, and poorly understood causes. Dev. Dyn. 2021, 250, 318–344. [Google Scholar] [CrossRef]
- Loeys, B.L.; Dietz, H.C.; Braverman, A.C.; Callewaert, B.L.; De Backer, J.; Devereux, R.B.; Hilhorst-Hofstee, Y.; Jondeau, G.; Faivre, L.; Milewicz, D.M.; et al. The revised Ghent nosology for the Marfan syndrome. J. Med. Genet. 2010, 47, 476–485. [Google Scholar] [CrossRef]
- Juul-Kristensen, B.; Schmedling, K.; Rombaut, L.; Lund, H.; Engelbert, R.H.H. Measurement properties of clinical assessment methods for classifying generalized joint hypermobility-A systematic review. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 116–147. [Google Scholar] [CrossRef]
- Demmler, J.C.; Atkinson, M.D.; Reinhold, E.J.; Choy, E.; Lyons, R.A.; Brophy, S. Diagnosed prevalence of Ehlers-Danlos syndrome and hypermobility spectrum disorder in Wales, UK: A national electronic cohort study and case–control comparison. BMJ Open 2019, 9, e031365. [Google Scholar] [CrossRef] [PubMed]
- The Ehlers Danlos Society. HEDGE Study-Hypermobile Ehlers-Danlos Genetic Evaluation Study. Available online: https://www.ehlers-danlos.com/hedge/ (accessed on 26 October 2022).
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019, 286, 2830–2869. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated Molecular Patterns Derived from the Extracellular Matrix Provide Temporal Control of Innate Immunity. J. Histochem. Cytochem. 2018, 66, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Hazell, G.; Peachey, A.M.G.; Teasdale, J.E.; Sala-Newby, G.B.; Angelini, G.; Newby, A.C.; White, S.J. PI16 is a shear stress and inflammation-regulated inhibitor of MMP2. Sci. Rep. 2016, 6, 39553. [Google Scholar] [CrossRef]
- Stone, A.; Loeser, R.; Vanderman, K.; Long, D.; Clark, S.; Ferguson, C. Pro-inflammatory stimulation of meniscus cells increases production of matrix metalloproteinases and additional catabolic factors involved in osteoarthritis pathogenesis. Osteoarthr. Cartil. 2014, 22, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Pérez-García, S.; Calamia, V.; Hermida-Gómez, T.; Gutiérrez-Cañas, I.; Carrión, M.; Villanueva-Romero, R.; Castro, D.; Martínez, C.; Juarranz, Y.; Blanco, F.; et al. Proteomic Analysis of Synovial Fibroblasts and Articular Chondrocytes Co-Cultures Reveals Valuable VIP-Modulated Inflammatory and Degradative Proteins in Osteoarthritis. Int. J. Mol. Sci. 2021, 22, 6441. [Google Scholar] [CrossRef]
- Reed, K.S.; Ulici, V.; Kim, C.; Chubinskaya, S.; Loeser, R.F.; Phanstiel, D.H. Transcriptional response of human articular chondrocytes treated with fibronectin fragments: An in vitro model of the osteoarthritis phenotype. Osteoarthr. Cartil. 2020, 29, 235–247. [Google Scholar] [CrossRef]
- Lee, A.Y.S. Clinical use of anti-histone antibodies in idiopathic and drug-induced lupus. Immunol. Med. 2022, 45, 180–185. [Google Scholar] [CrossRef]
- Petrera, A.; Von Toerne, C.; Behler, J.; Huth, C.; Thorand, B.; Hilgendorff, A.; Hauck, S.M. Multiplatform Approach for Plasma Proteomics: Complementarity of Olink Proximity Extension Assay Technology to Mass Spectrometry-Based Protein Profiling. J. Proteome Res. 2021, 20, 751–762. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritelli, M.; Chiarelli, N.; Cinquina, V.; Zoppi, N.; Bertini, V.; Venturini, M.; Colombi, M. RNA-Seq of Dermal Fibroblasts from Patients with Hypermobile Ehlers–Danlos Syndrome and Hypermobility Spectrum Disorders Supports Their Categorization as a Single Entity with Involvement of Extracellular Matrix Degrading and Proinflammatory Pathomechanisms. Cells 2022, 11, 4040. https://doi.org/10.3390/cells11244040
Ritelli M, Chiarelli N, Cinquina V, Zoppi N, Bertini V, Venturini M, Colombi M. RNA-Seq of Dermal Fibroblasts from Patients with Hypermobile Ehlers–Danlos Syndrome and Hypermobility Spectrum Disorders Supports Their Categorization as a Single Entity with Involvement of Extracellular Matrix Degrading and Proinflammatory Pathomechanisms. Cells. 2022; 11(24):4040. https://doi.org/10.3390/cells11244040
Chicago/Turabian StyleRitelli, Marco, Nicola Chiarelli, Valeria Cinquina, Nicoletta Zoppi, Valeria Bertini, Marina Venturini, and Marina Colombi. 2022. "RNA-Seq of Dermal Fibroblasts from Patients with Hypermobile Ehlers–Danlos Syndrome and Hypermobility Spectrum Disorders Supports Their Categorization as a Single Entity with Involvement of Extracellular Matrix Degrading and Proinflammatory Pathomechanisms" Cells 11, no. 24: 4040. https://doi.org/10.3390/cells11244040
APA StyleRitelli, M., Chiarelli, N., Cinquina, V., Zoppi, N., Bertini, V., Venturini, M., & Colombi, M. (2022). RNA-Seq of Dermal Fibroblasts from Patients with Hypermobile Ehlers–Danlos Syndrome and Hypermobility Spectrum Disorders Supports Their Categorization as a Single Entity with Involvement of Extracellular Matrix Degrading and Proinflammatory Pathomechanisms. Cells, 11(24), 4040. https://doi.org/10.3390/cells11244040