1. Introduction
Synaptic plasticity forms the basis for learning and memory. This is achieved by regulating neuronal network functions that help an animal adjust to its ever-changing environment [
1,
2,
3,
4,
5]. For instance, the basal forebrain cholinergic network activity plays an important role in the hippocampal circuits underlying learning and memory, which is comprised of the tri-synaptic pathway of the dentate gyrus (DG), cornu ammonis 3 (CA3) and cornu ammonis 1 (CA1) [
6,
7]. Cholinergic projections from the medial septal (MS) and diagonal band of Broca (DBB) to the hippocampus also influence the formation of spatial memories [
8]. Consistent with the idea of extensive cholinergic involvement in learning and memory are the studies demonstrating that an alteration in hippocampal acetylcholine levels [
9] or nicotinic acetylcholine receptors (AChRs) during memory encoding and consolidation affects learning in various animal models [
10,
11,
12]. The role of cholinergic transmission in learning and memory is, therefore, of great significance for long-term memory consolidation and may also underlie many disorders, such as the Alzheimer’s disease (AD) and post-traumatic stress disorder (PTSD) [
13]. However, the precise mechanisms regulating cholinergic receptor function in the hippocampus remain poorly defined.
We have recently demonstrated that the multiple endocrine neoplasia type 1 (
MEN1) gene, which encodes the menin protein [
14,
15], may regulate the expression of nicotinic acetylcholine receptors (nAChRs) in the hippocampus by specifically targeting these receptors to synaptic sites. Menin, a 610 amino acid sequence, is a 67 kDa nuclear protein with 10 exons [
16,
17] that serves as a tumor suppressor of the parathyroid glands, pituitary gland, and pancreatic islet cells [
18]. While
MEN1 has long been known as a tumor suppressor gene, our novel findings vis-à-vis its involvement in cholinergic synapse formation and synaptic plasticity has since extended its role beyond tumorigenesis [
19,
20,
21,
22,
23,
24].
Menin is involved in the modification of histones, regulation of epigenetic cascades, and is associated with metabolic and autoimmune diseases [
25]. The
MEN1 gene has been exceptionally well conserved across evolution, and orthologues are found throughout the animal kingdom [
20,
26]. Furthermore,
MEN1 has also been shown to be required for cholinergic synaptogenesis and the regulation/targeting of nAChR at the glutamatergic synapses of the hippocampus [
20]. However, the precise involvement of menin in the regulation of hippocampal cholinergic receptors during learning and memory remains unknown. Because constitutive
MEN1 knockouts are lethal, an alternative approach was needed to generate a conditional knockout (CKO) model to deduce its precise function in the brain. In this study, we attempted to delete
MEN1 by either using a tamoxifen-inducible model by crossing Camk2-cre/ERT2 mice with
MEN1 floxed mice, or by using an AAV-9 viral vector transduction approach to specifically knockout MEN1 in the hippocampus of the animals. The data presented here underscore the importance of the
MEN1 gene in regulating the expression patterns of nicotinic acetylcholine receptors in the hippocampus, and its important role in fear conditioning learning and memory tests.
2. Methods
2.1. Animals
All study protocols involving mice were approved by the Animal Care and Use Committee of the University of Calgary and conducted in accordance with Tri-Council guidelines. All animals were allowed access to standard laboratory chow and water ad libitum and were housed in a 12 hr light/dark cycle under standard laboratory conditions. The MEN1 floxed mice were obtained from Jackson Labs stock no. 005109, and they had loxP sites flanking exons 3 to 8 of the MEN1 gene. Mice were homozygous for the allele without any abnormalities. The Camk2a-CreERT2 transgenic mice were obtained from Jackson Labs stock no. 012362. These expressed the tamoxifen-inducible Cre recombinase under the control of the mouse calcium/calmodulin-dependent protein kinase II alpha (Camk2a) promoter region. This was useful for studying gain or loss of function in Camk2a-expressing neurons in cortex, hippocampus, and striatum. The CaMK2a-CreERT2 transgenic mice were bred with MEN1 floxed mice and CreERT2 fusion gene activity was induced with tamoxifen administration.
Mouse genotyping was performed at the HBI Molecular Core Facility on ear notch samples using the Kapa Mouse Genotyping kit according to the manufacturer’s instructions. Ear tissue from pups between P21–27 was obtained. Here, P21 was chosen as the age for weaning and tamoxifen administration. This was based on the rationale that the absence of the mother from the cages made them independent juveniles. The experimental and the control groups received intraperitoneal injections of tamoxifen to turn on the activity of Cre recombinase. In the tamoxifen 3-day protocol, a concentration of 10mg/mL was prepared and a dose of 100 μL/mouse was injected for 3 consecutive days. The animals in the control group were also injected with the tamoxifen 3-day protocol. Mice were subjected to molecular experiments after a minimum of 1-week post-final injection so that all traces had been fully excreted (in urine/feces) from the mice [
27]. The CKO animals and controls were both subjected to the fear conditioning behavioral paradigm. After the reinjection of
MEN1 rescue virus, the animals and their control counterparts were maintained in a half-way house and monitored regularly for their physical health and subsequently tested again for fear conditioning. The sex distribution was kept constant in each experiment and n = 4 per condition (2 males and 2 females per condition, allowing for a sex-balanced n = 4 per condition).
2.2. Molecular Biology
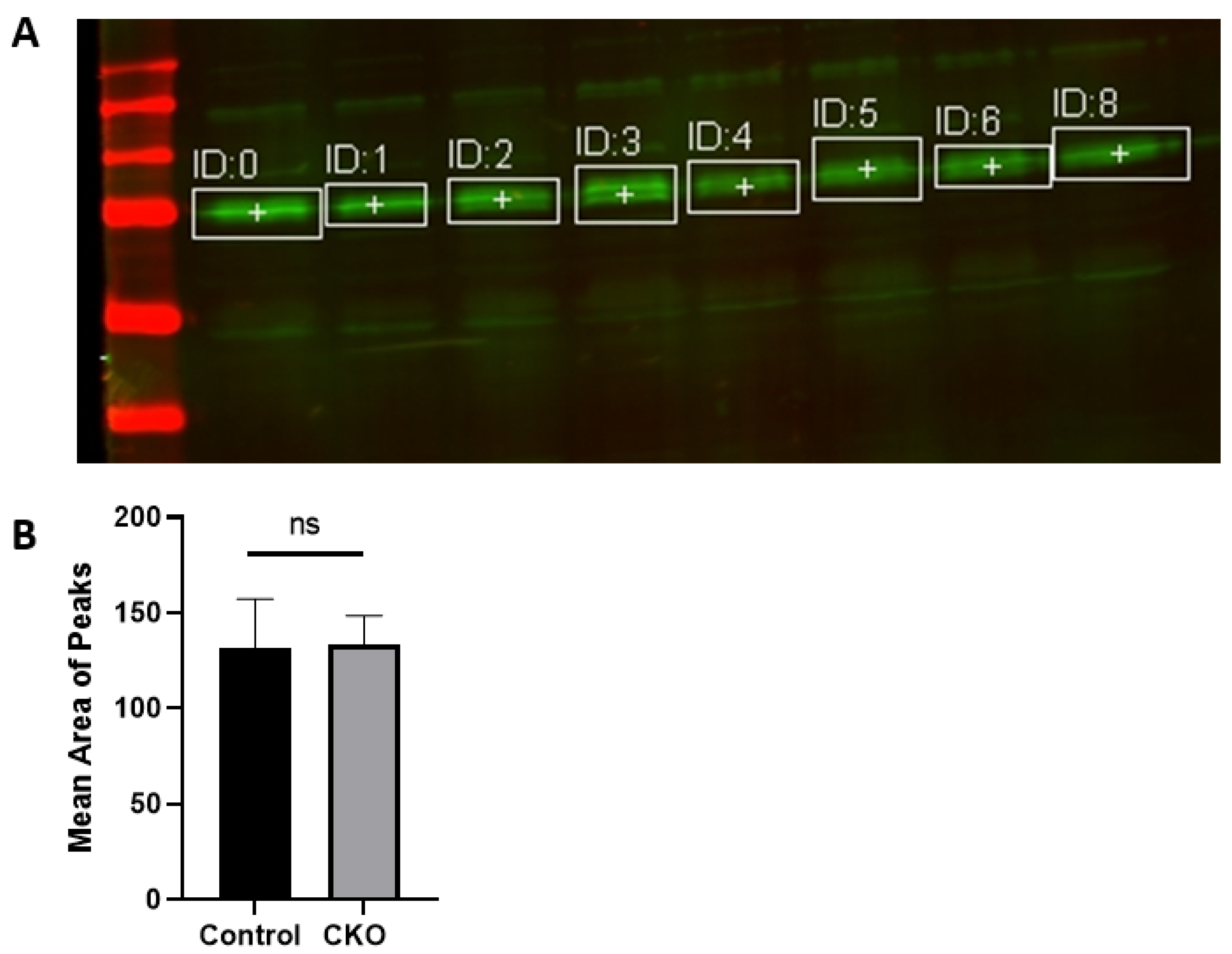
After 2 weeks of injections, the CKO and control mice were tested to determine if the Cre recombinase induced activity had led to menin deletion. Hippocampus tissue was obtained for western blots to confirm the absence of menin in the CKO mice compared to the controls. Membranes were visualized using a LI-COR Odyssey infra-red imager and analyzed using the gel analysis tool in ImageJ.
2.3. Camk2a-Cre/ERT2 Cross with tdTomato, a Mouse Reporter Line
There was a possibility that the mice might not have metabolized tamoxifen or that the Cre expression was leaky, thus, leading to the excision of the
MEN1 gene in both the controls and CKOs. To determine the efficacy of each tamoxifen protocol, a breeding pair between Ai14 mice, homozygous for the Rosa-CAG-LSL-tdTomato-WPRE conditional allele, and the CaMK2a-Cre/ERT2 line was set up to determine the optimal concentration of tamoxifen for inducing Cre activity. The Rosa-td-Tomato mice, homozygous for the Rosa-CAG-LSL-tdTomato-WPRE conditional allele, were obtained from Jackson Labs stock no. 007914 with loxP-flanked STOP cassette to prevent transcription of the red fluorescent protein variant tdTomato. Studies have shown that when bred to mice which expressed Cre recombinase, the STOP cassette is deleted in the Cre-expressing tissue with tdTomato fluorescence [
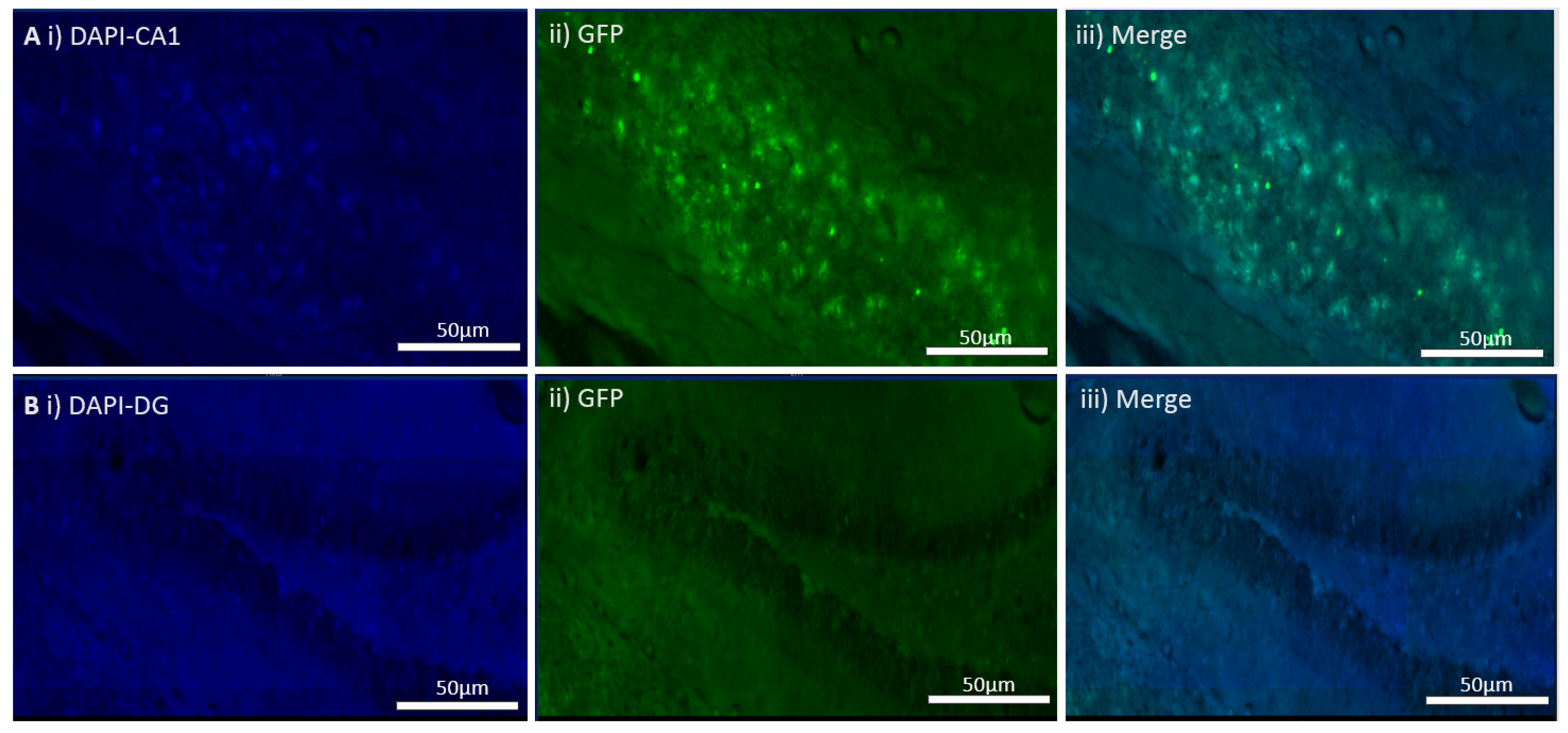
28]. At P21, the offspring from the cross were subjected to a tamoxifen 3-day protocol, where a concentration of 10mg/mL was prepared, and a dose of 100 μL/mouse was injected for 3 consecutive days along with a corn oil 3-day protocol. The animals in the control group were injected with corn oil 100 μL for 3 consecutive days. 2 weeks later, floating brain sections from the animals were live imaged for tdTomato’s expression. An n-value of 10 mice was used with equal sex distribution.
2.4. Virus Transduction of Neuronal Cultures
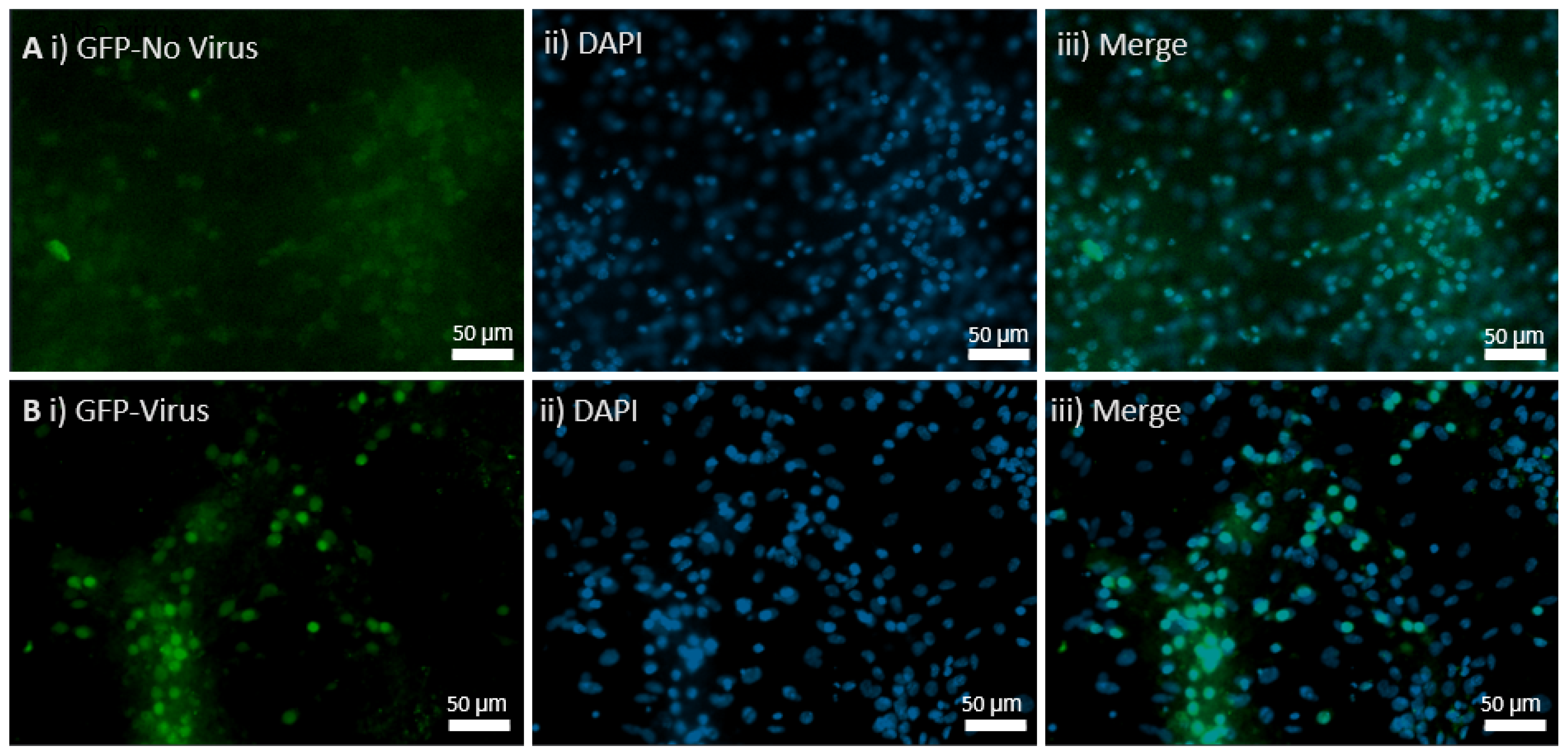
Ready-to-use AAV9 particles produced from pENN.AAV.hSyn.HI.eGFP-Cre.WPRE.SV40 (#105540) were ordered from Addgene. The AAV has synapsin-driven EGFP-Cre expression, and the packaging plasmid, pENN.AAV.hSyn.HI.eGFP-Cre.WPRE.SV40, encodes the adenoviral helper sequences, AAV rep gene, and AAV9 cap gene. The buffer used for this serotype was PBS + 0.001% Pluronic F-68. The purification method was iodixanol gradient ultracentrifugation. The vector titers were determined by quantitative PCR and the value was 3.1 × 1013 GC/mL. It was stored at −80 °C and thawed before use on ice. Controls received AAV9 pENN.AAV.hSyn.HI.eGFP.WPRE.SV40 for control GFP expression with a titer of 2.8 × 1013 GC/mL, and the in vivo CKO received the Cre AAV9 virus.
Embryos were dissected from MEN1 floxed pregnant females on E18 for hippocampal neuronal cultures. The pregnant mice were anesthetized with isoflurane and then sacrificed by decapitation. The hippocampi from E18 embryos were dissected in solution (1 × HBSS containing 10 mM HEPES; 310 mOsm, pH 7.2), and then treated with papain (50 U/mL), 150 mM CaCl2, 100 µM L-cysteine, and 500 µM EDTA in neurobasal medium (NBM) for 20 m at 37 °C and 5% CO2 (in an incubator). Then, NBM supplemented with 4% FBS, 2% B27, 1% penicillin-streptomycin, and 1% L-glutamine (GIBCO) was used to wash the tissue 3 times. For trituration, fire-polished glass Pasteur pipettes were used and then plated on glass coverslips. These had been previously washed with nitric acid and coated with poly-D-lysine (30 µg/mL) and laminin (2 µg/mL). The culture media was changed to NBM supplemented with 2% B27, 1% penicillin-streptomycin, and 1% L-glutamine the very next day, and the samples were divided into 2 groups. The controls were supplemented with the media, and the experimental groups was transduced with the AAV9 Cre virus. The cultures were maintained at 37 °C with 5% CO2, and the media was changed every 3 to 4 days.
2.5. Immunocytochemistry and Microscopy
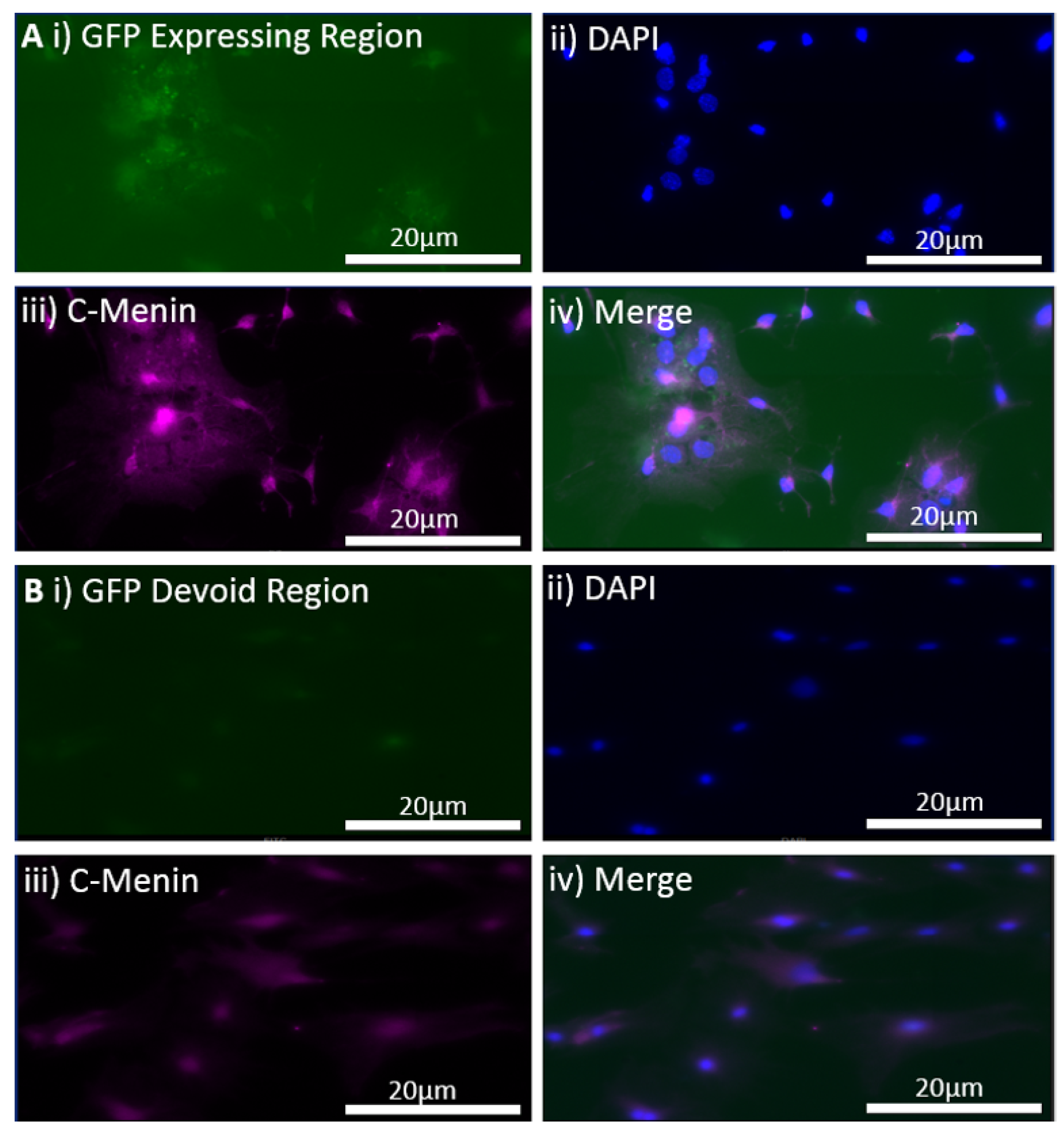
Hippocampal neuronal cultures from the control and the experimental groups were fixed on day in vitro (DIV) 21 with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MI, USA) in 1 × PBS for 30 min. The cells were permeabilized for 1 h with incubation medium (IM) containing 0.5% Triton and 10% goat serum in 1 × PBS. Primary antibody, menin C-terminal epitope [Bethyl Laboratories, A300-105, Montgomery, TX, USA], was used at 1:500 in IM for 1 h. Secondary antibodies, namely Alexa Fluor 488, 568, or 680 (Invitrogen, Vancouver, BC, Canada) were used at 1:100 in IM for 1 h followed by three 15 m washes in 1 × PBS. Cells were mounted using ProLong Gold antifade reagent with DAPI (Invitrogen, BC, Canada). A VS120 microscope at 40X magnification was used for imaging. Fluorophores were excited with 488, 568, and 680 lasers. Imaging parameters, including field of view size, laser intensity, exposure, and channel gain, were kept consistent throughout.
2.6. In Vivo Virus Transduction in CA1 Hippocampus of MEN1 Floxed Mice
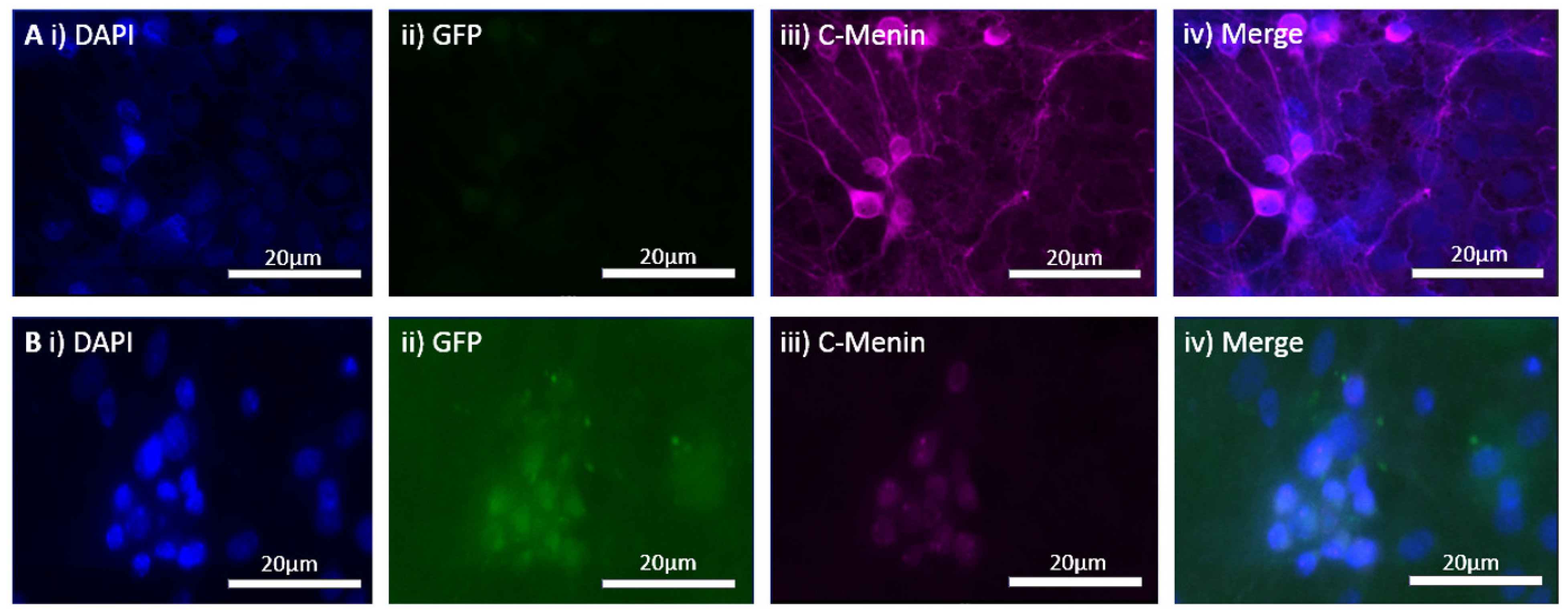
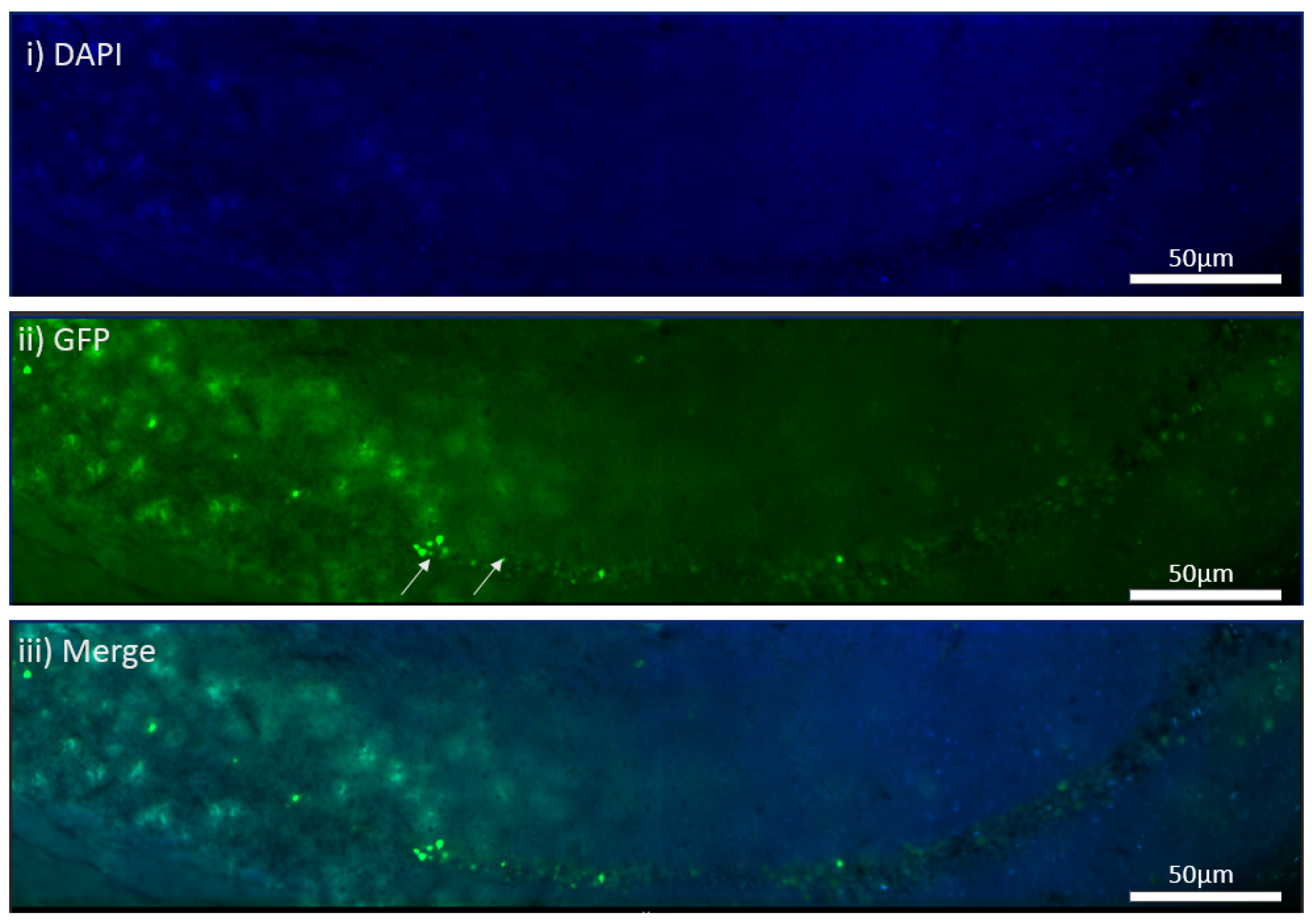
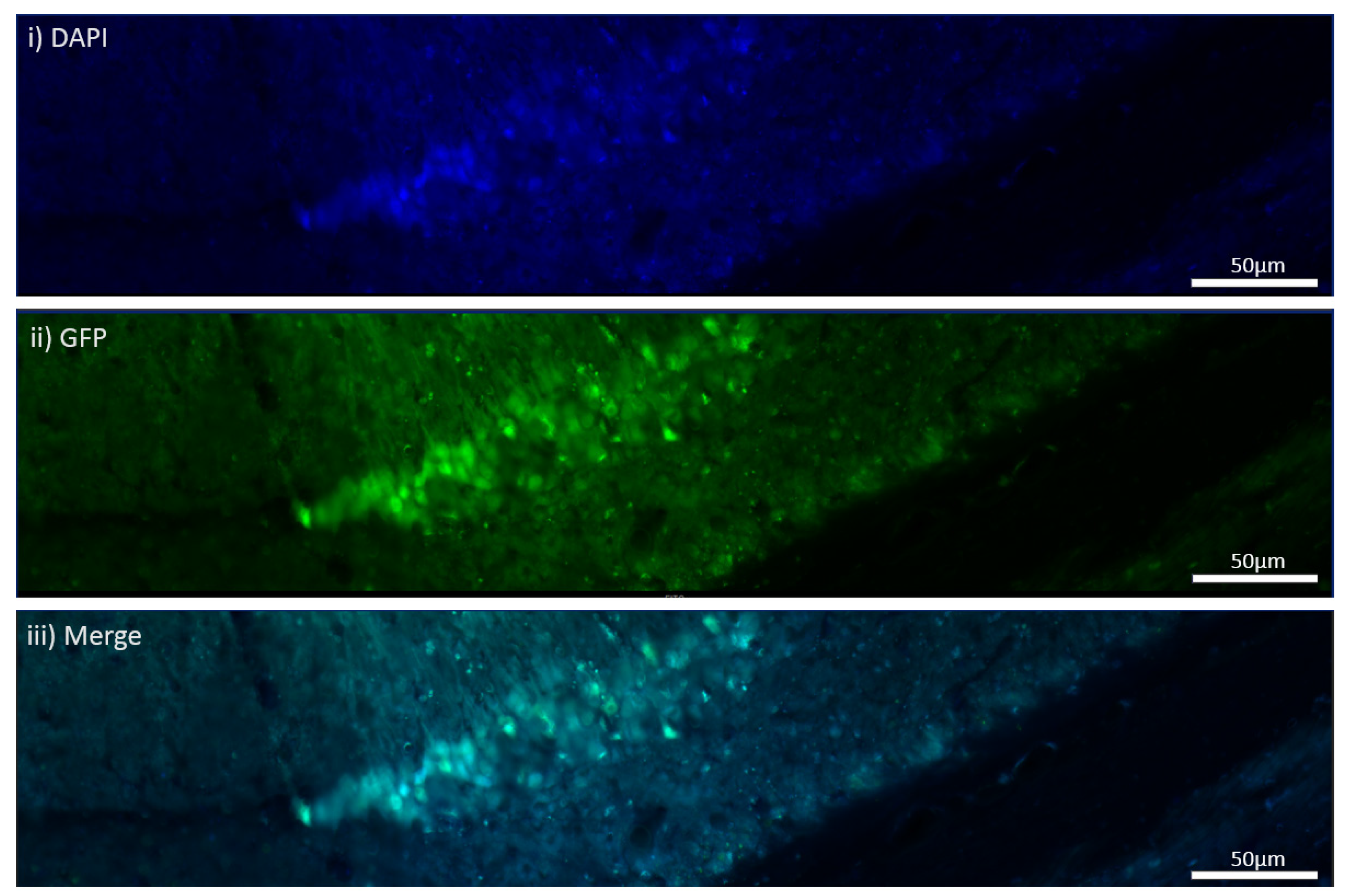
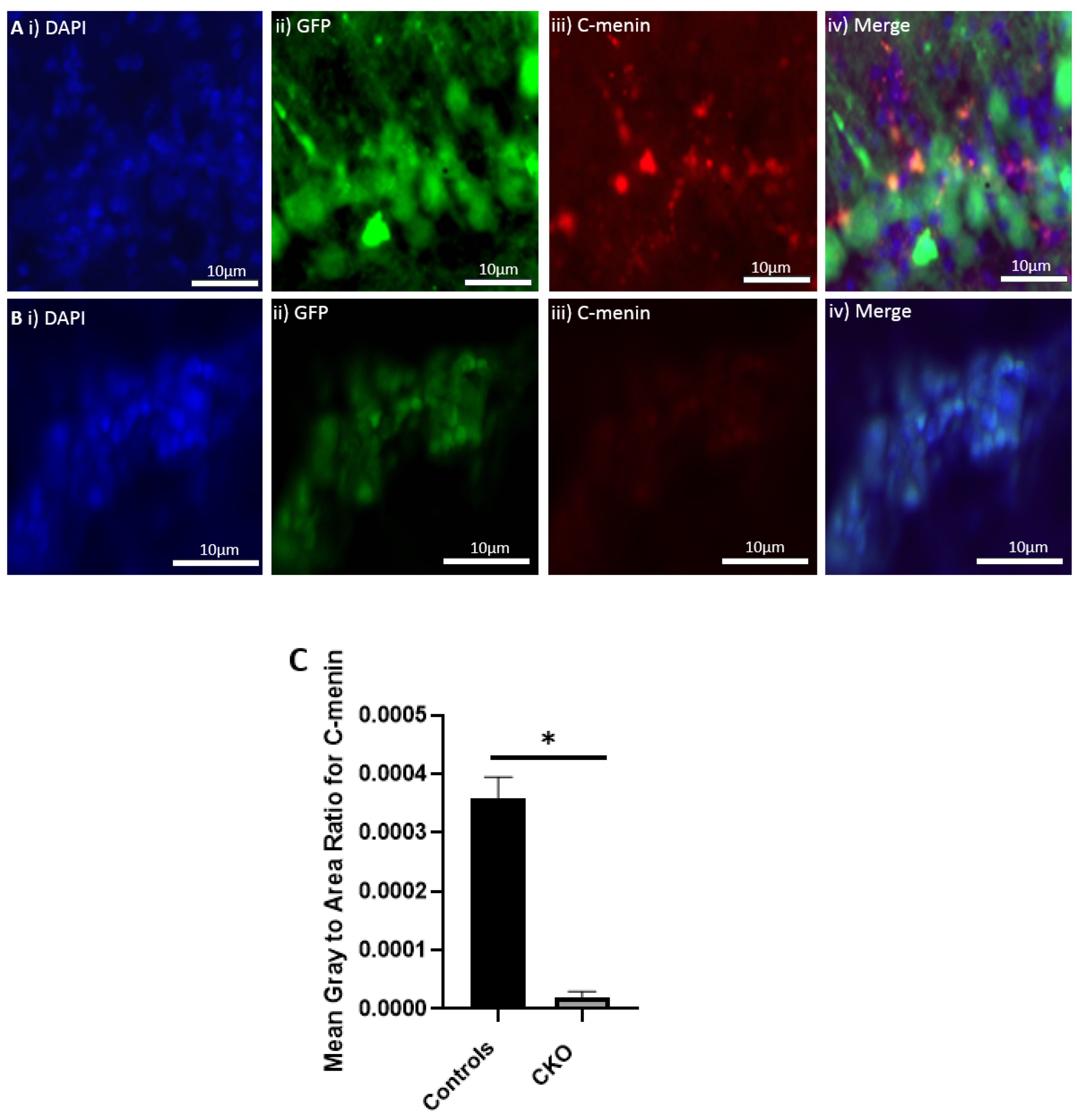
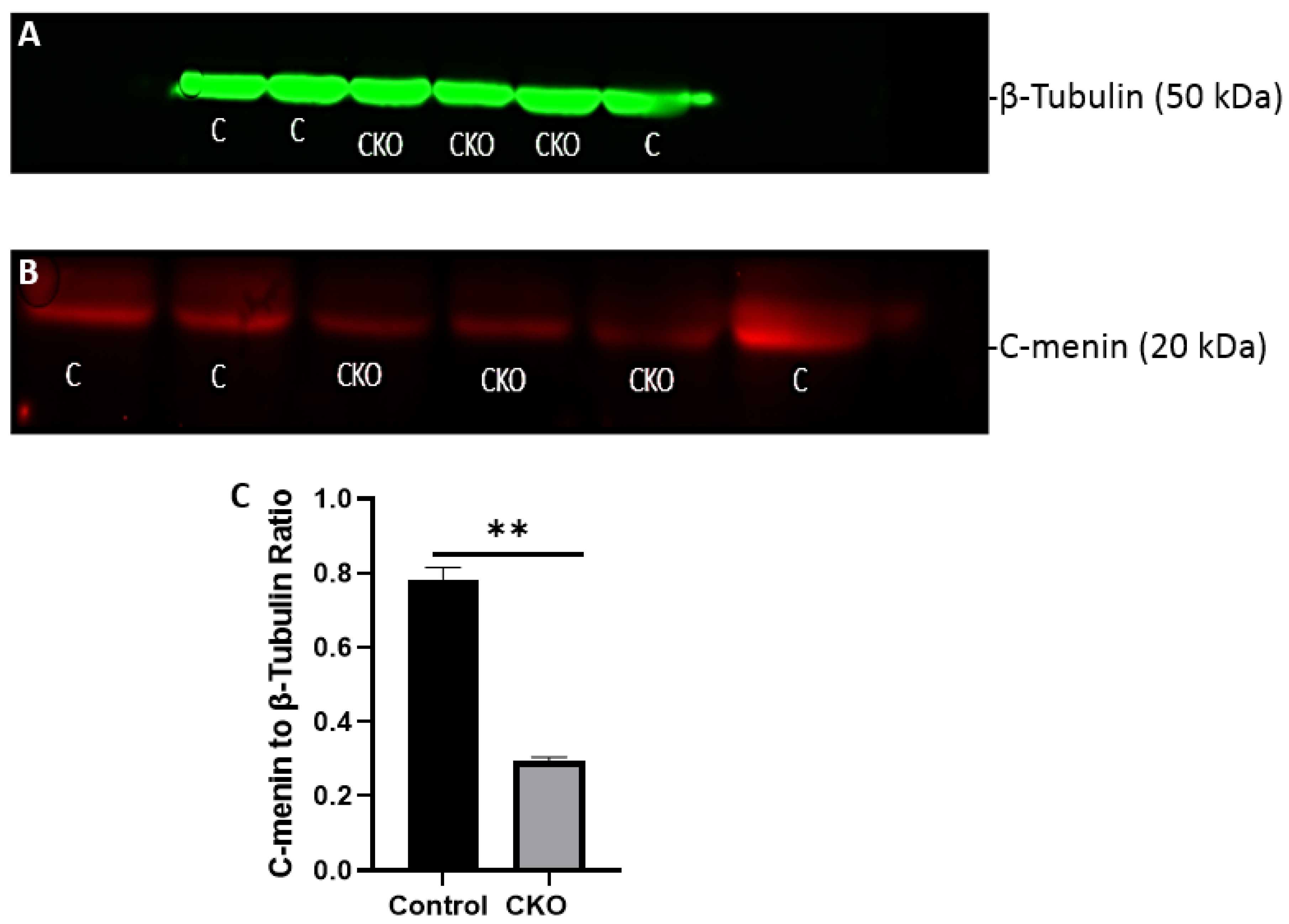
The MEN1 floxed mice were injected with the adeno-associated virus (AAV) under the synapsin promoter for neuron-specific MEN1 deletion. The hippocampal CA1 neurons and a control AAV were used to express GFP under the synapsin promoter bilaterally into the dorsal hippocampi (2 mm behind bregma, 2 mm lateral, and 1.6 mm below the dural surface for CA1). The injections were performed with glass micropipettes with the tip diameter ranging between 10 to 20 μm and injected slowly with a pressure-injection system. The recovery time post-injection was 4 weeks. To validate this model, immunostaining was performed, where the brains of the controls and CKOs were fixed with 4% paraformaldehyde (PFA) overnight, transferred to 2% sucrose, and then snap-frozen using dry ice and OCT. Brain slices were prepared using cryostat with thickness of 16 μm. The sections were tagged with C-menin, as previously described, to confirm menin deletion.
2.7. Behavioral Paradigm for Contextual Fear Conditioning
The behavioral tests were adapted from the Cumming School of Medicine Optogenetics facility at the University of Calgary. Mice reach sexual maturity between six to eight weeks and, based on the timing of stereotactic injections and recovery time, the time frame for behavior testing was deemed to be two months [
29]. The protocol for contextual fear conditioning was two days long, and this test is used to assess hippocampal-dependent learning and memory [
30]. On day one, mice were placed in the conditioning chamber, habituated to their surroundings for 2 min, and then were given three consecutive shocks (1 s, 0.5 mA). This was recorded with a camera mounted inside the chamber. On day two, the procedure and context remained the same to test conditioned fear of the shock, but no shocks were administered. Using the ANY-maze software (Stoelting Co., Wood Dale, IL, USA), freezing (defined by the complete absence of motion), as a measure of fear in the rodent model was scored as percentage of time spent frozen and analyzed with GraphPad Prism 8 (Dotmatics).
2.8. AAV Production for MEN1 Rescue Virus
The mouse
MEN1 cDNA rom Horizon Discovery (NCBI accession NM_001168490.1) was PCR-amplified and inserted in place of Cre recombinase at the BspEI and HindIII restriction sites in pENN-AAV-hSyn-HI-eGFP-cre (Addgene plasmid, Teddington, UK #105540) using the NEBuilder HiFi DNA assembly kit, and the pAAV-hSyn-HI-eGFP-MEN1 was generated. The AAV viral vectors, which had the AAV9 capsid, were generated based on methods previously described [
31]. The 293FT cells (Thermofisher, Waltham, MA, USA) were co-transfected with 129 µg pHELPER (Agilent, CA, USA ), 238 µg pAAV 2/9n rep-cap plasmid (Addgene plasmid #112865) and pAAV.hSyn-HI-eGFP-MEN1 with the PEIpro transfection reagent. The AAVs were precipitated using 40%PEG/2.5 M NaCl, and the pooled cells were harvested after 5 days in 500 mM NaCl, 40 mM Tris Base, and 10 mM MgCl2 buffer. The lysate was incubated with 100 U/mL salt-active nucleases (Arcticzymes, Tromsø. Norway) at 37 °C for 1 h and then centrifuged at 2000×
g for 15 min. Using the iodixanol step gradient containing 15, 25, 40, and 60% iodixanol in OptiSeal tubes followed by centrifugation at 350,000×
g, the AAV was purified from the lysate. Then, using the 10 cc syringe and 16-gauge needle, the AAVs were harvested in 1XPBS containing 0.001% Pluronic F68 (Gibco, Grand Island, NE, USA) and filtered. A qPCR adeno-associated virus titration kit was used for calculating the titer (Expedeon, Heidelberg, Germany).
2.9. In Vitro MEN1 Virus Transduction for Testing MEN1 Rescue Virus Efficacy
Embryos were dissected from MEN1 floxed pregnant females on E18 for hippocampal neuronal cultures. The pregnant mice were sacrificed by decapitation after being anesthetized with isoflurane. The hippocampi from E18 embryos were dissected in solution (1 × HBSS containing 10 mM HEPES; 310 mOsm, pH 7.2), and then treated with papain (50 U/mL), 150 mM CaCl2, 100 µM L-cysteine, and 500 µM EDTA in neurobasal medium (NBM) for 20 m at 37 °C and 5% CO2 (incubator). Then, NBM supplemented with 4% FBS, 2% B27, 1% penicillin-streptomycin, and 1% L-glutamine (GIBCO) was used to wash the tissue 3 times. For trituration, fire-polished glass Pasteur pipettes were used and then plated on the glass coverslips. These had been previously washed with nitric acid and coated with poly-D-lysine (30 µg/mL) and laminin (2 µg/mL). The culture media was changed to NBM supplemented with 2% B27, 1% penicillin-streptomycin, and 1% L-glutamine the very next day, and the samples were divided into 2 groups. The controls were supplemented with the media, and the experimental groups were transduced with the MEN1 rescue virus. The cultures were maintained at 37 °C with 5% CO2, and the media was changed every 3 to 4 days.
Hippocampal neuronal cultures from the control and the experimental groups were fixed on DIV21 with 4% paraformaldehyde (Sigma-Aldrich, St. Louis, MI, USA) in 1 × PBS for 30 min. The cells were permeabilized for 1 h with incubation medium (IM) containing 0.5% Triton and 10% goat serum in 1 × PBS. Primary antibody, menin C-terminal epitope (Bethyl Laboratories, A300-105, Montgomery, TX, USA), was used at 1:500 in IM for 1 h. Secondary antibodies, namely Alexa Fluor 488, 568, or 680 (Invitrogen, Vancouver, BC, Canada) were used at 1:100 in IM for 1 h followed by three 15 m washes in 1 × PBS. Cells were mounted using ProLong Gold antifade reagent with DAPI (Invitrogen, BC, Canada). A VS120 microscope at the 40X magnification was used for imaging. Fluorophores were excited with 488, 568, and 680 lasers. Imaging parameters, including field of view size, laser intensity, and channel gain, were kept consistent throughout.
2.10. Reinjection of MEN1 Rescue Virus into the CKO Animals
The CKO mice underwent stereotaxic injections with MEN1 AAV rescue virus (pAAV-hSyn-HI-eGFP-MEN1), and the controls were injected with PBS bilaterally into the dorsal hippocampi (2 mm behind bregma, 2 mm lateral, and 1.6 mm below the dural surface for CA1). The injections were performed with glass micropipettes with the tip diameter ranging between 10 to 20 μm, and injected slowly with a pressure-injection system. The recovery time post-injection was 4 weeks before the animals were tested again for contextual fear and conditioning task, as mentioned above.
5. Discussion
Cholinergic transmission and plasticity play an important role in learning and memory. Perturbation of this pathway not only affects these processes, but also cognition, both of which are hallmarks of neurodegenerative diseases, such as AD. How cholinergic synaptic plasticity plays a role in the targeting of nAChRs to specific hippocampal synaptic sites remains, however, largely unknown. We previously demonstrated that a knockout of
MEN1 both in vitro and in vivo affects the localization of nAChRs [
20,
21]. Using the shRNA approach to knockdown menin expression, we have shown that upon menin deletion, the nAChR alpha 7 and alpha 5 were downregulated as determined by qPCR [
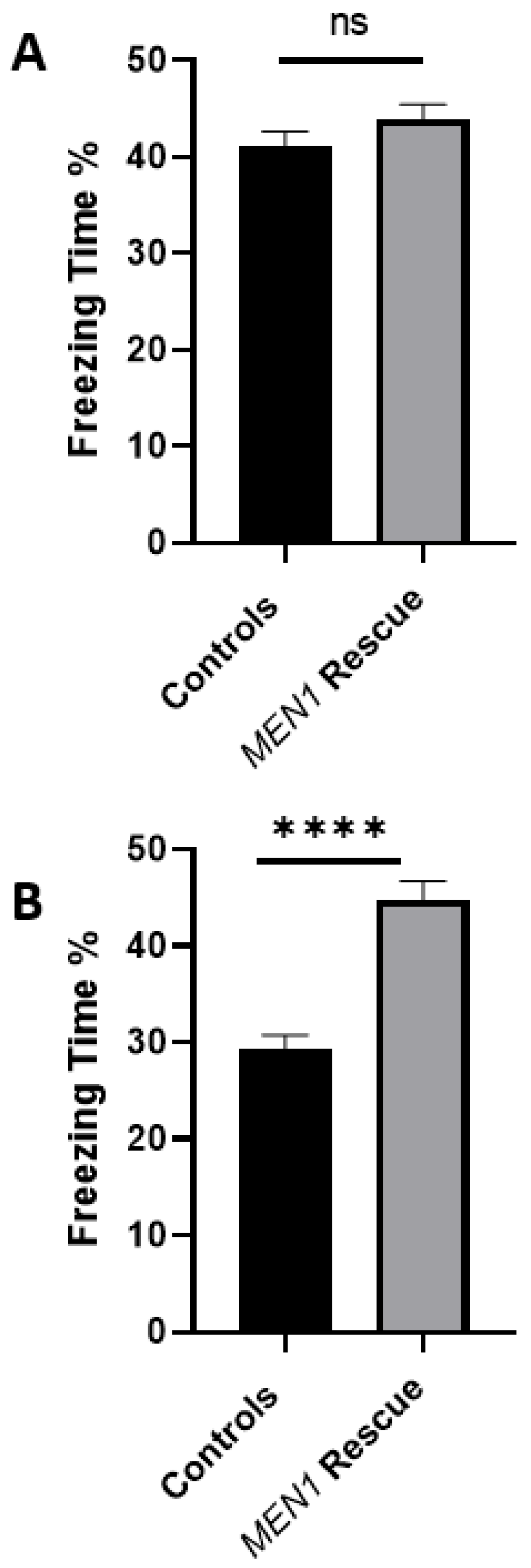
21]. However, an ultimate test was needed to seek unequivocal evidence for the involvement of menin in learning and memory of freely behaving animals; this study has achieved just that. Moreover, we have further demonstrated that the reintroduction of MEN1 using a viral vector transfection in the live animals rescues the mice from learning and memory deficits induced by the knockout of the same gene.
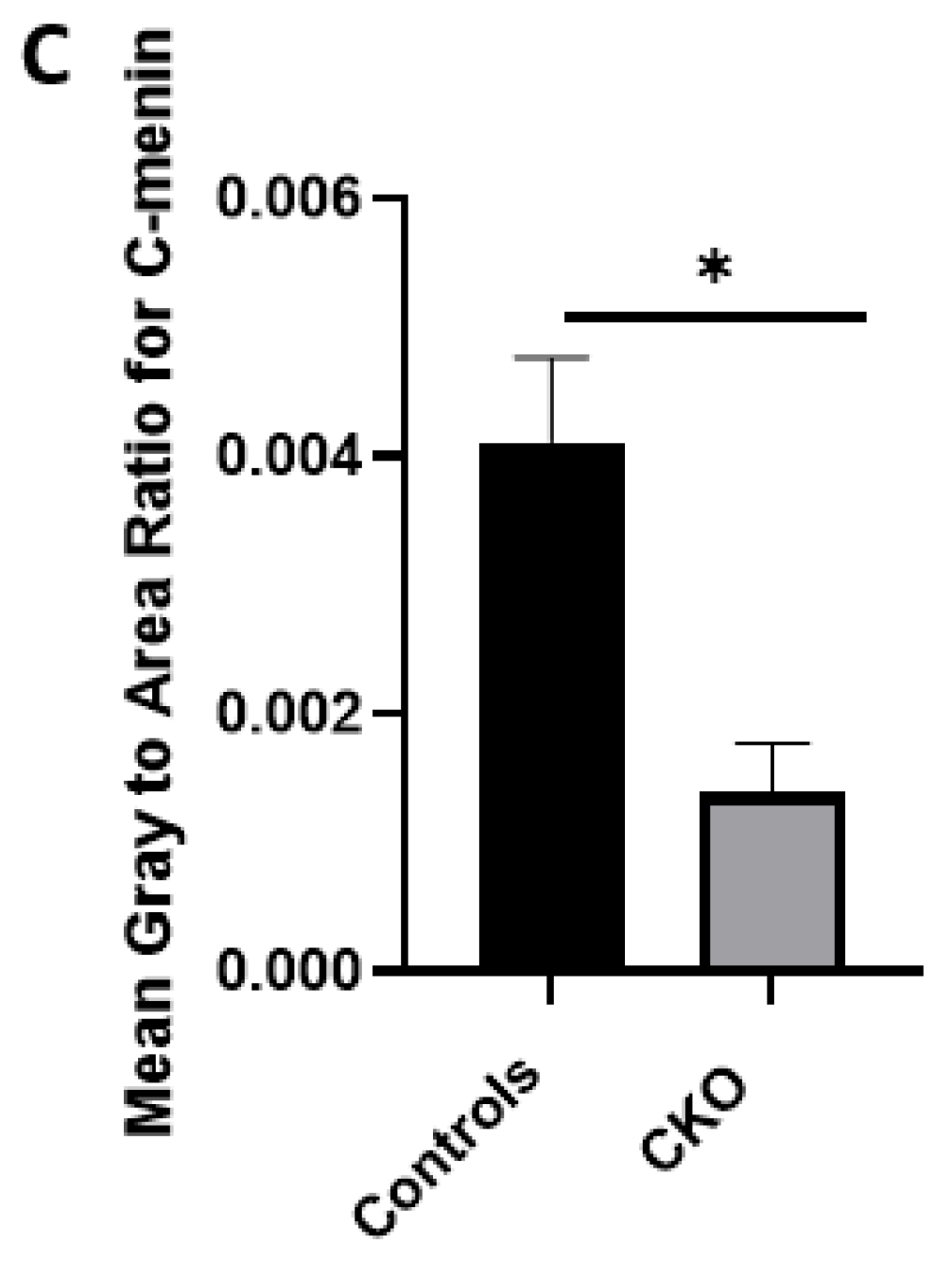
Using different conditional knockout approaches, the current study has, on the one hand, identified the potential challenges which can arise when creating a gene knock down model in the CNS, while also underscoring the importance of alternative approaches. The tamoxifen-inducible approach, where the CamK2 Cre-Ert2 line was crossed with the MEN1 floxed mice, did not yield a reasonable CKO model to work with, since no differences were observed at the protein level. Specifically, we discovered that when the CamK2 Cre-Ert2 line was crossed with the tdTomato reporter line, no difference in the expression of Cre recombinase was discernable, neither in the controls nor in the CKOs. Another shortcoming of this approach was that even though Camk2a-expressing neurons were present in the hippocampus, this also led to the deletion of menin from other regions, including the cortex and cerebellum. Since the aim of creating a CKO model was to test the role of menin specifically in the hippocampus, the loss of the gene from other regions was undesirable. Thus, a more direct approach was needed for specifically deleting MEN1 from the hippocampus with complete spatiotemporal control. We then opted for a viral transduction approach and successfully employed it to delete menin exclusively in the hippocampal neurons with spatiotemporal control. Menin deletion was confirmed in both in vitro hippocampal cultures and in vivo—exclusively in the CA1 region of the hippocampus. Specifically, we demonstrated that there was a reduction in menin protein in the CKO model compared to the controls—both in the hippocampal culture and in the live animals.
Previous studies have used the Cre recombinase-loxP conditional knockout approach to study the tumor suppressor role of
MEN1 gene in the pituitary glands and pancreas [
36]. A recent study showed the role of neuron-specific menin deletion on p35 and CdK5 pathways in the dysregulation and impairment of learning and memory [
37]. This team created a conditional knockout model by crossing the
MEN1 floxed mice with CamKIIα-Cre line T29-1 transgenic mice that have the mouse Camk2a promoter for driving Cre recombinase expression in the forebrain, including the CA1 pyramidal cell layer of the hippocampus [
37]. The downside of using this approach was that it did not allow for temporal control over menin deletion and the Cre recombinase led to menin deletion after birth. To overcome the lack of temporal control in inducing menin deletion, and the possibility of chromosomal effects, we used the approach of developing an inducible
MEN1 CKO model with the Camk2CreErt2 line. This was so that the leaky expression of Cre would not present itself as a problem. The tamoxifen-inducible model, CamK2CreErt2 crossed by
MEN1 floxed mice, also allowed for temporal control on the expression of Cre recombinase, which leads to
MEN1 deletion. Both the Camk2 Cre Ert 2 mice and the
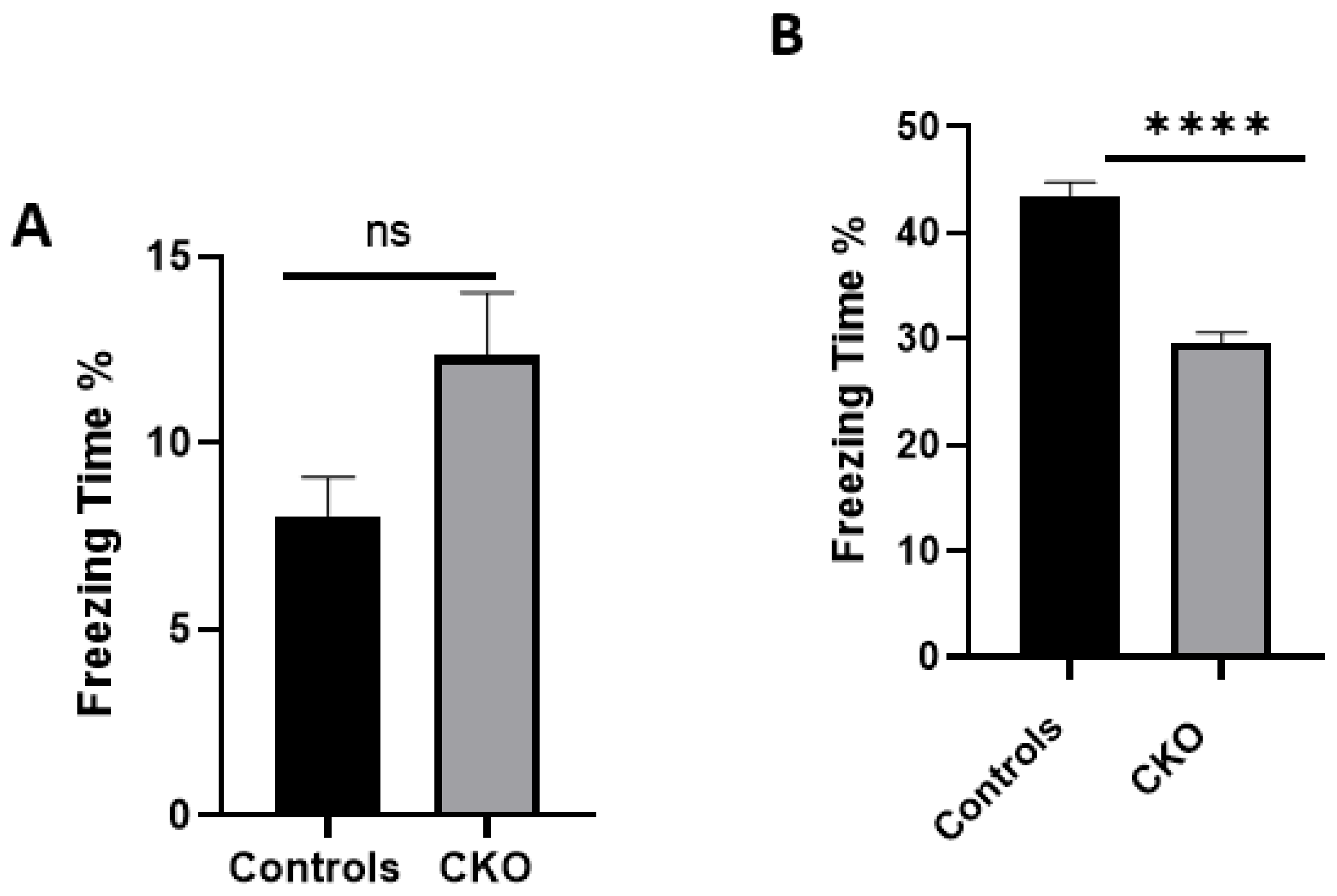
MEN1 floxed mice were first genotyped to confirm the presence of Cre and the mutant menin. Since the tamoxifen protocol was already established, the next step was to quantify menin in the CKOs and controls after the mice were injected with tamoxifen. However, we were surprised to find equal fluorescence intensity in both the controls and CKOs in the WB; this approach was, therefore, deemed unsuitable.
Although the reason remains unclear, it has been reported elsewhere that lines, such as Nestin-Cre, GFAP-Cre, CaMKIIα-Cre, and Synapsin1-Cre, sometimes undergo unexpected germline recombination [
38,
39]. Other possible reasons could be genetic background effects, spontaneous mutations in the ERT2 or tamoxifen binding site, or unexpected genetic recombination [
39]. Thus, we chose a more directed approach of deleting
MEN1 via virus transduction, which is discussed below.
An unexpected anomaly in the present study was the leaky expression of Cre in the hippocampus of the mice. The rationale for the ectopic expression could be that the titer of the virus was perhaps too high, resulting in its leakage into the vicinity of the hippocampus. Another possibility could be that the viral particles in the transduced neurons were jumping across the synapses. This explanation is, however, highly unlikely since it is not possible for AAVs to produce new viral particles [
40]. The AAVs are specifically designed to infect only one node of the synaptic circuitry, and the concern of the virus infecting the neighboring neurons was, therefore, unwarranted [
40].
There was a reduction in C-menin signal in the CKO compared to the controls both in vitro and in vivo. The reduction in menin in the CKO is indicative of reduced menin from neurons. The delivery of Cre to the hippocampus via the AAV approach is, thus, a viable approach both in vitro and in vivo. Generating the
MEN1 CKO model via injecting a neuron-specific Cre activated virus into the floxed
MEN1 to knock out
MEN1 function in the CA1 region allowed for precise spatial and temporal control. The
MEN1 was deleted in the CA1 region to draw a parallel between the 5XFAD model, where CA1 is one of the first regions where basal synaptic transmission and LTP start to deteriorate at around 6 months of age [
41,
42]. Furthermore, various subsets of the ventral CA1 neuron population play a role in the encoding and retrieval of contextual fear memories [
43], which we deemed to be the evidence for a functional hippocampal neuron-specific knockout. The main goal of developing a CKO mouse model by specifically knocking down menin in the CA1 region was to determine its role in hippocampal-dependent learning and memory. A CKO model where menin expression is knocked down and validated with different approaches was needed before moving on to determine the role of the
MEN1 gene and menin protein in learning and memory.
The data presented here demonstrate that
MEN1 CKO in a specific region of the CA1 had significant deficits in the hippocampal-dependent learning and memory task of contextual fear and conditioning. We had hypothesized that the CKO mice will exhibit deficits in hippocampal-dependent learning and memory, since
MEN1 gene deletion was shown previously to perturb the α7 nicotinic receptor expression [
21]. Moreover, we had sought to also determine if
MEN1 reintroduction in the specific CA1 region, where this gene had initially been conditionally knocked out, could recover the observed behavioral deficit. Indeed, the data presented here are consistent with the hypothesis, thus, further establishing the role of
MEN1 and its encoded protein menin, in hippocampal-dependent, and neuronally contingent learning and memory. For the very first time, to our knowledge, a mouse model of menin CKO in the CA1 specific region was developed, and the effects of menin deletion from the CA1 on hippocampal-dependent learning and memory was demonstrated. The limitation of the experiment is that it remains unclear whether MEN1 overexpression alone causes an increase in freezing (in wildtype animals) and/or broad perturbations of motor behavior, which could influence performance on the fear conditioning test. Thus, there is a possibility that this may not be a true rescue experiment, since that control was not included and this possibility cannot be excluded based on the data reported.
Previously, menin has been shown to play a role in the regulation of nicotinic receptors at the postsynaptic site. Thus, if one were to delete menin as an ascribed regulator of synaptic clustering and recruitment of α7 nAChRs at the hippocampal synapses, then one could deduce its precise role in the targeting of these receptors. Furthermore, the CA1 sub-region undergoes disruption in the AD model; however, the underlying mechanisms as to why this perturbation occurs remain unknown [
44]. Previously, CA1 region-specific alterations in the neuronal excitability have been reported in an AD mouse model [
44]. Thus, to specifically highlight the importance of nicotinic machinery in the mouse model, the role of menin, which leads to perturbation of nicotinic receptors in the CA1 region, was essential.
A difference between the control and the experimental group was apparent for the contextual fear conditioning task. It has previously been shown that the aversive stimuli from hippocampal contextual representations during fear learning invoke the CA1 region [
45]. Consistent with these observations, we found that after the deletion of menin from the CA1 region, the performance of the CKO mice in contextual and fear conditioning was impaired compared to the controls. This demonstrated that menin deletion led to the perturbation of circuitry in the CA1 region. Because the previous data from our lab had shown a direct correlation between menin and the α7 nAChRs, it, therefore, seems reasonable to conclude that the
MEN1 knockout-mediated loss of memory function had invoked these receptors. Future experiments will, however, be required to demonstrate this unequivocally, and may have to invoke either direct electrophysiology on brain slices, or optogenetic approaches, to determine the effects of menin deletion on the targeting of α7 nAChRs in freely behaving animals.
The scores from the contextual fear conditioning task suggested that the CKO mice had their learning and memory impaired after
MEN1 deletion. Furthermore, our results corroborate with a recent study which demonstrated that
MEN1 CKO mice exhibited spatial learning and memory deficits [
37]. This study reported that
MEN1 conditional knockout had deficits in the contextual fear and conditioning test, whereas they also did not find any difference in the cued conditional learning and memory. Our data expands on those findings, since the earlier CKO model employed in the Zhuang et al. study correlated the behavioral deficits in the CKO animals with a reduction in dendritic branching [
37]. However, the approach to delete menin specifically from the CA1 region, and its reintroduction into the same region, shows the necessity and sufficiency of
MEN1 in learning and memory, which in turn is likely contingent upon synaptic clustering and recruitment of α7 nAChRs in the hippocampus. As anticipated, we have demonstrated that the
MEN1 CKO renders the animals dysfunctional vis-à-vis learning, memory, and cognitive function, which involves the disruption of cholinergic structures and function in the hippocampus. The reintroduction of
MEN1 in these CKO animals also restored the behavioral deficit concomitant with the recovery of menin function, which likely involved restoration of cholinergic transmission. Neurological disorders and diseases with underlying perturbation of the hippocampal nAChRs can be better understood by delineating the neural and cellular basis of how neural pathways and signaling mechanisms are altered. Thus, the role of menin in hippocampus-dependent learning and memory involving cholinergic circuits needs to be further studied. Another important takeaway from this study is that the genes that are ascribed some specific functions may in fact serve myriad other roles in organs different than those of their original target sites, thus, underscoring the importance of multifunctionality vis-à-vis genetic diversity.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












