
Efficacy of Clinically Used PARP Inhibitors in a Murine Model of Acute Lung Injury

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
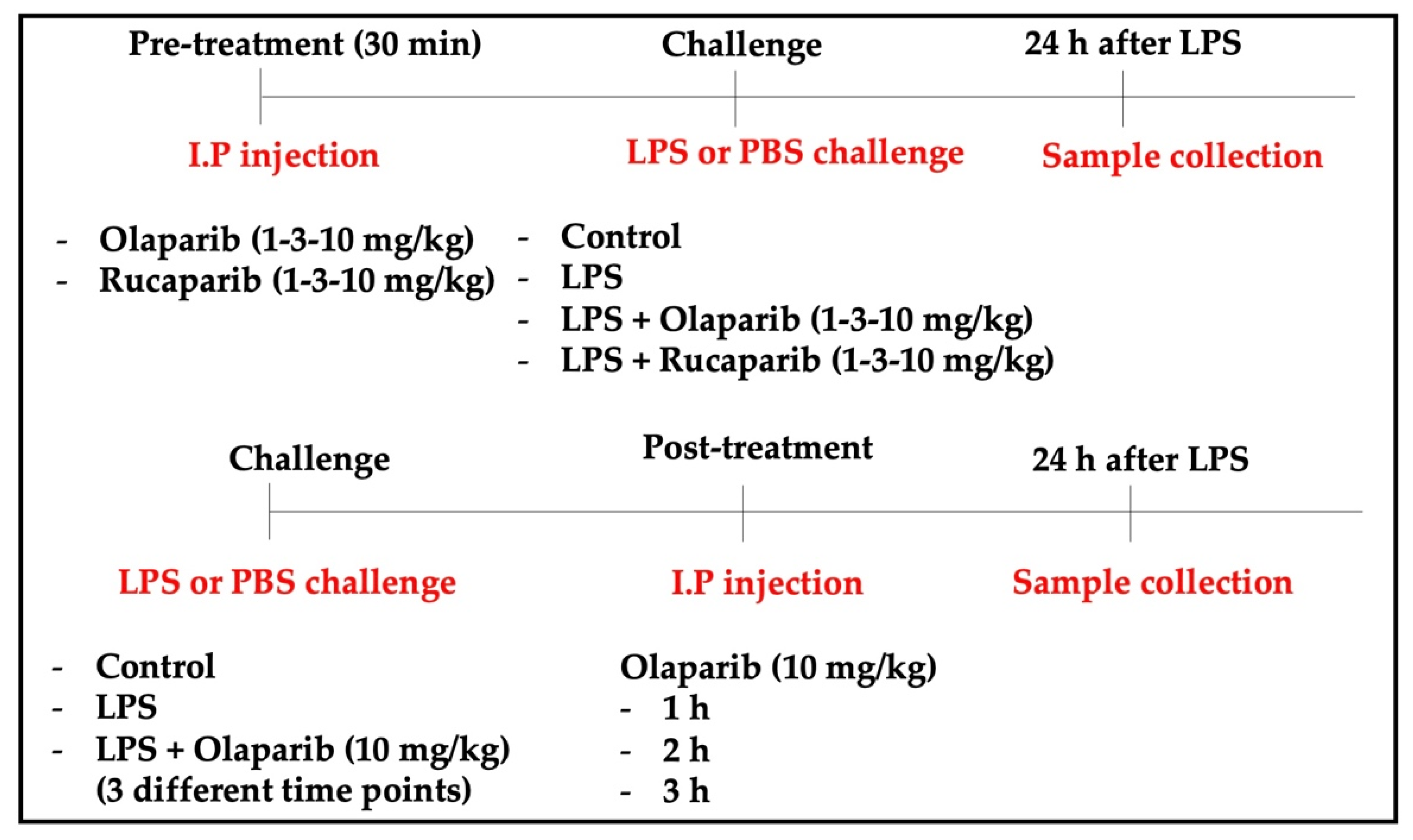
2. Materials and Methods
2.1. Cell Culture
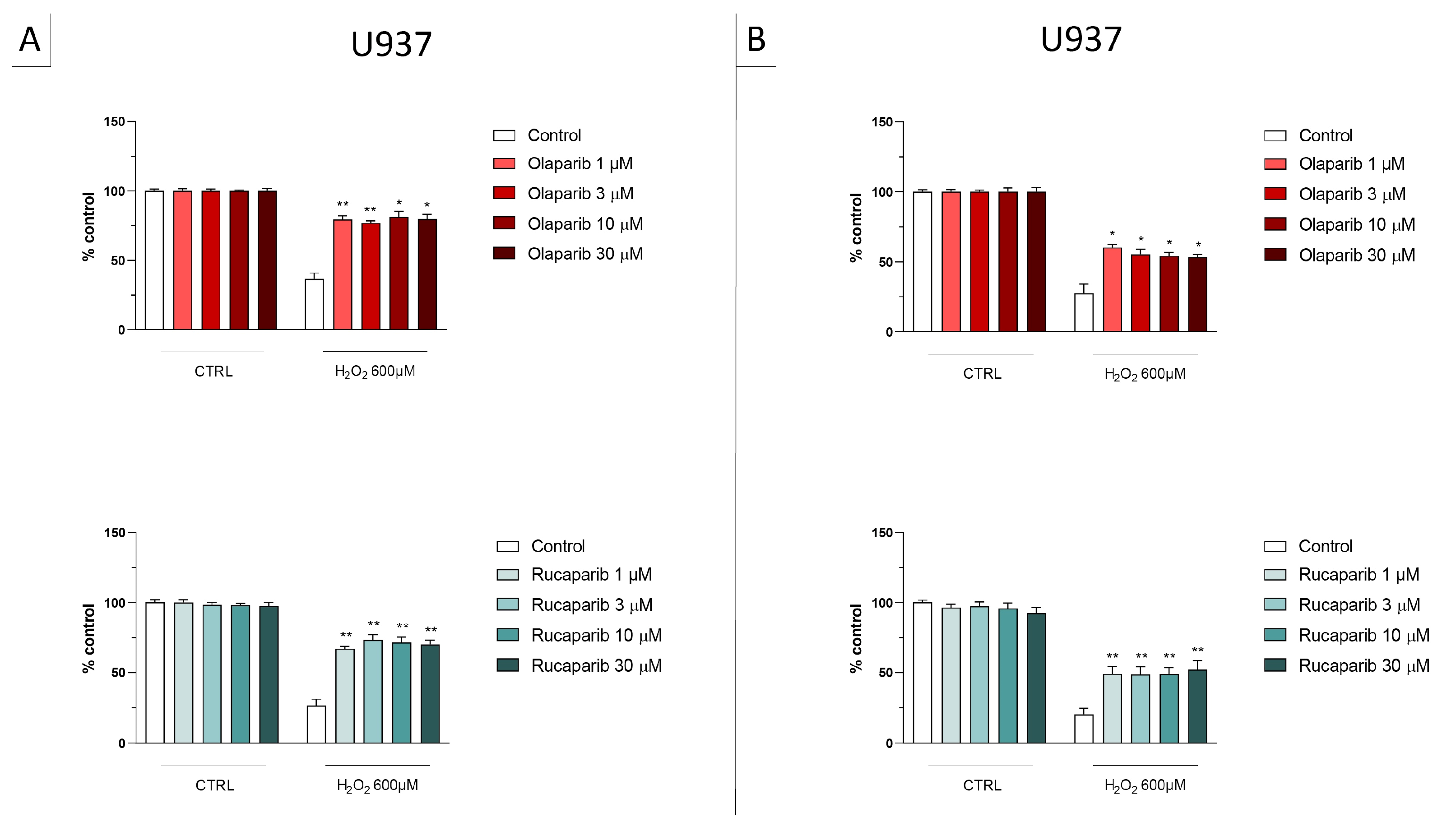
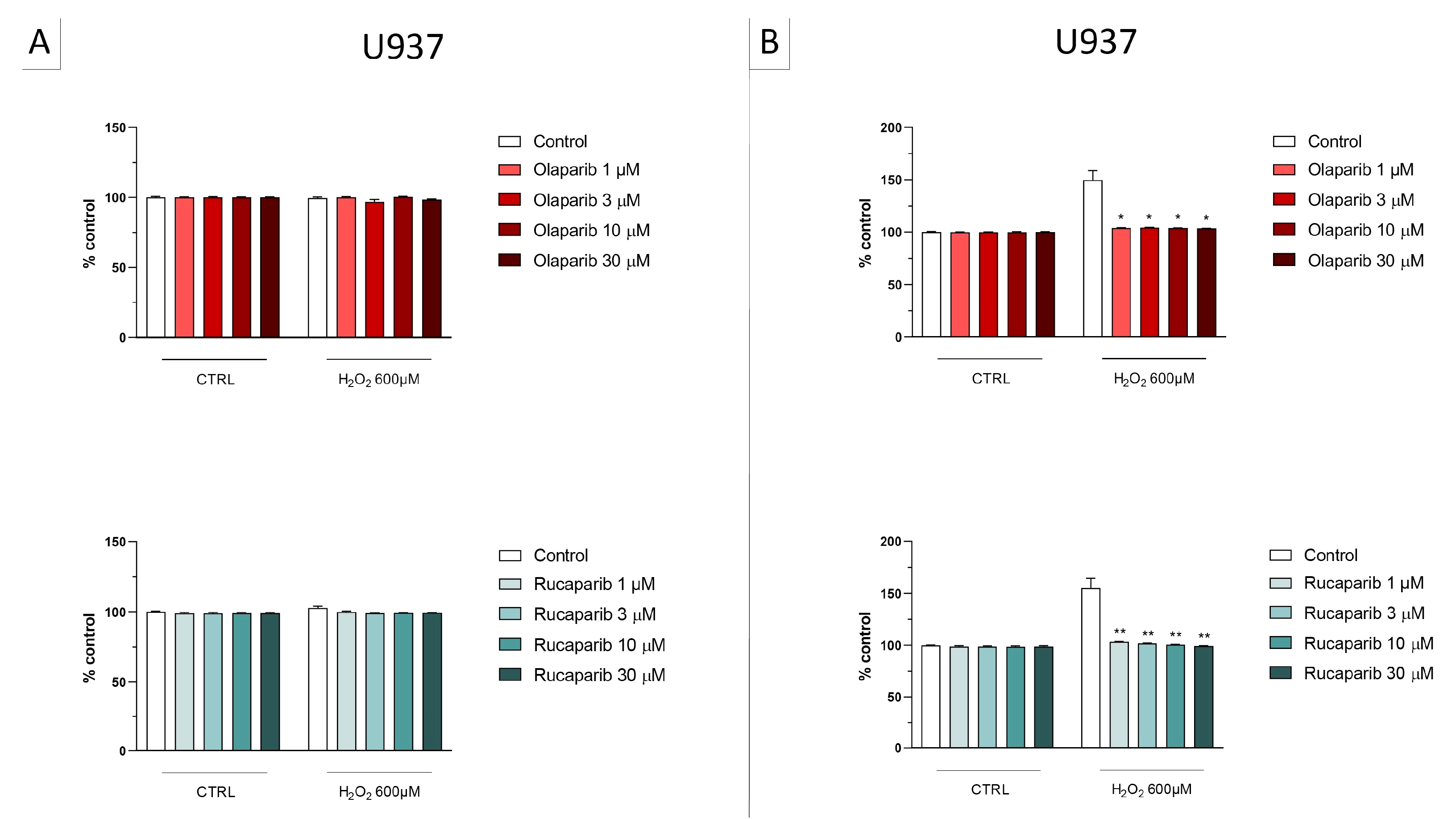
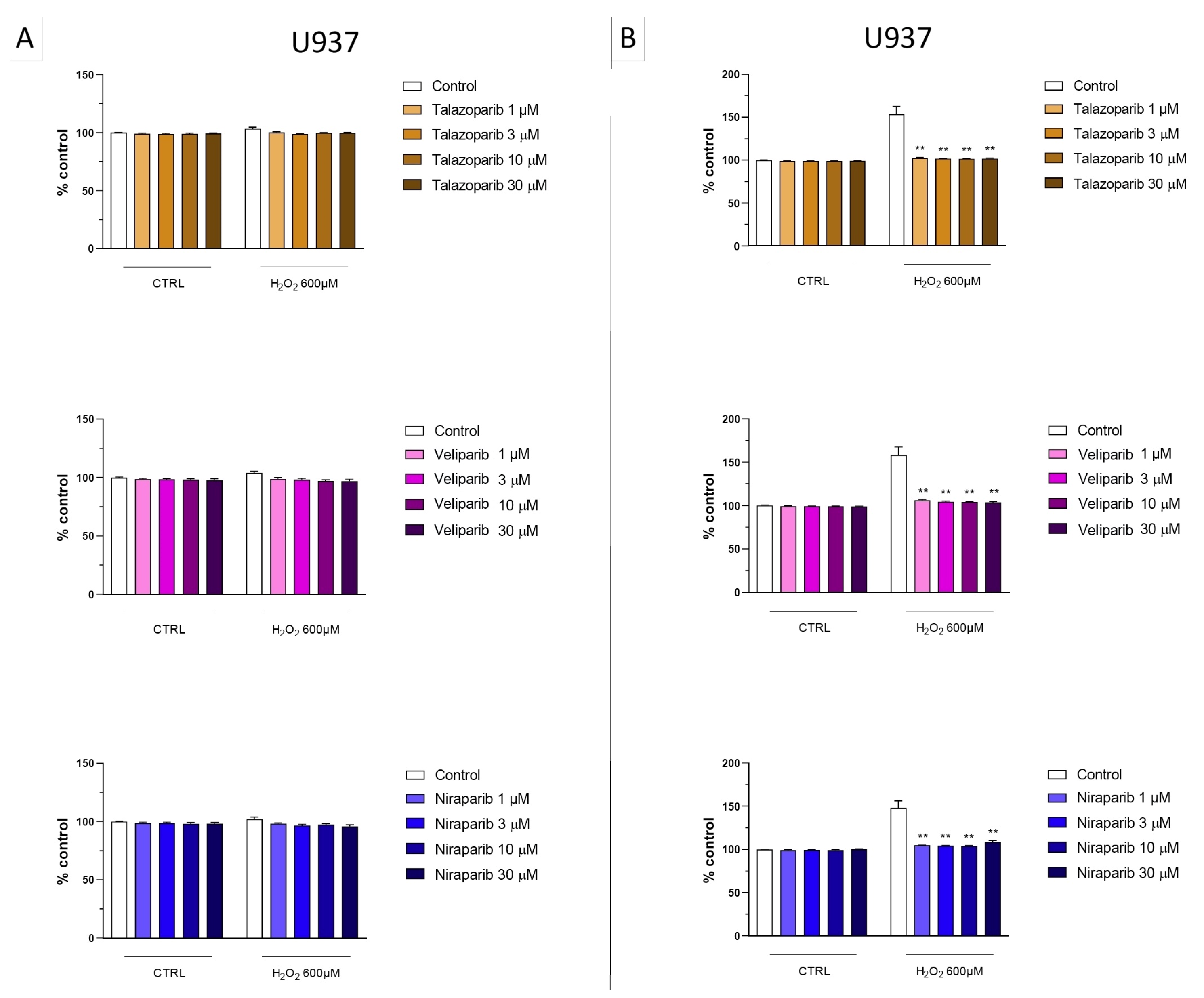
2.2. Cell Viability Assays
2.3. Quantification of DNA Damage In Vitro
2.4. Animal Model of ALI
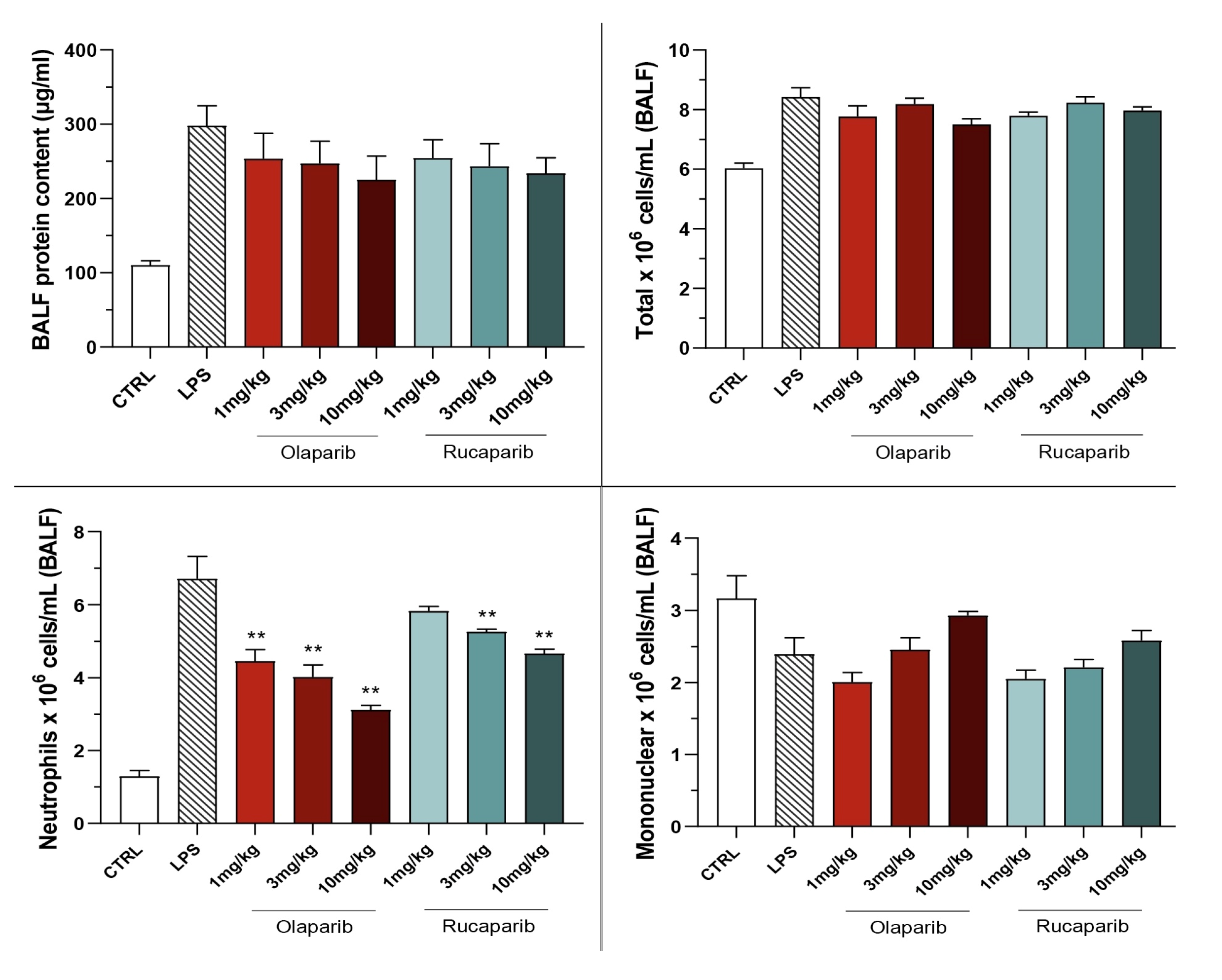
2.5. Bronchoalveolar Lavage Fluid (BALF) Collection and Analysis
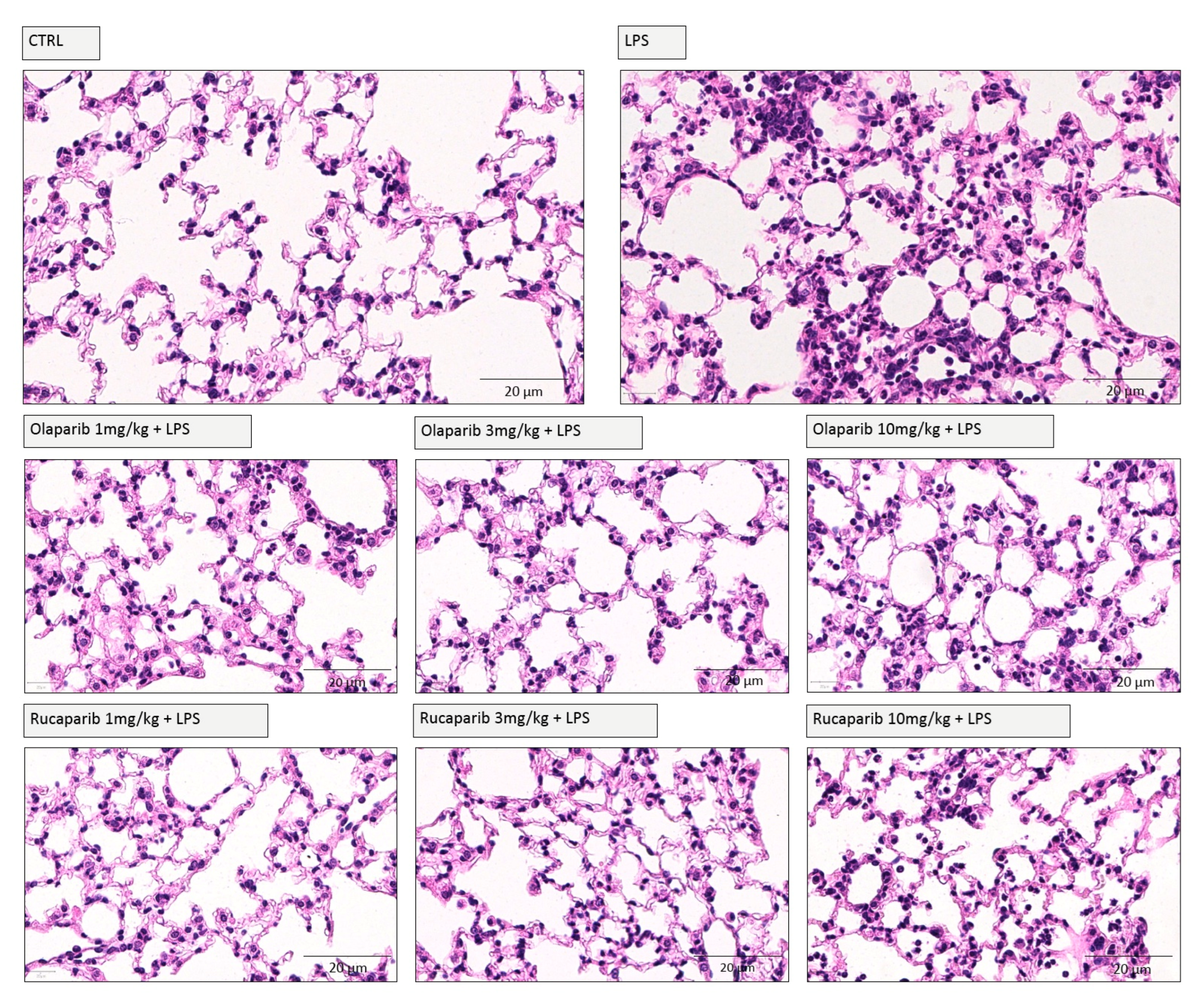
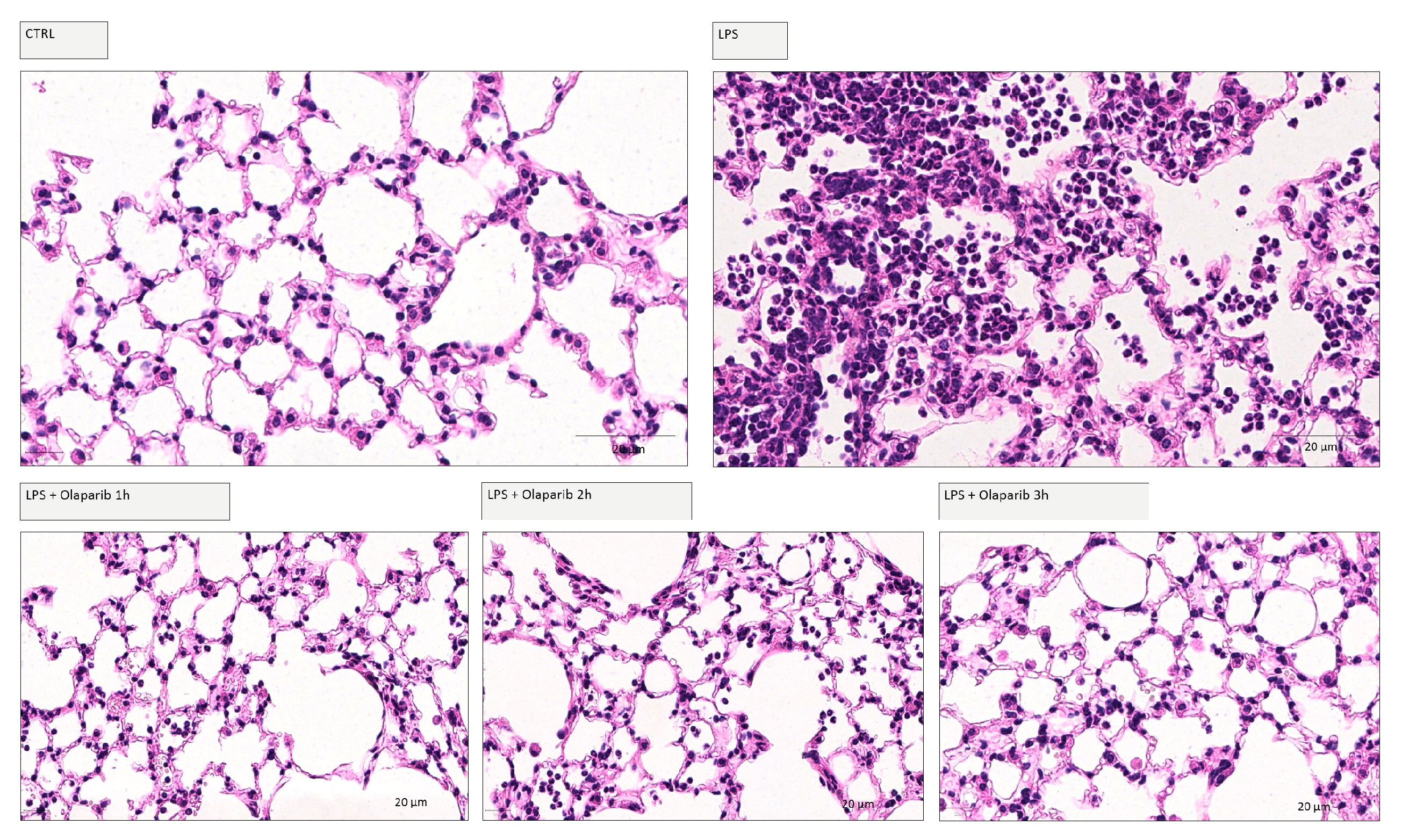
2.6. Histology
2.7. Histomorphometry
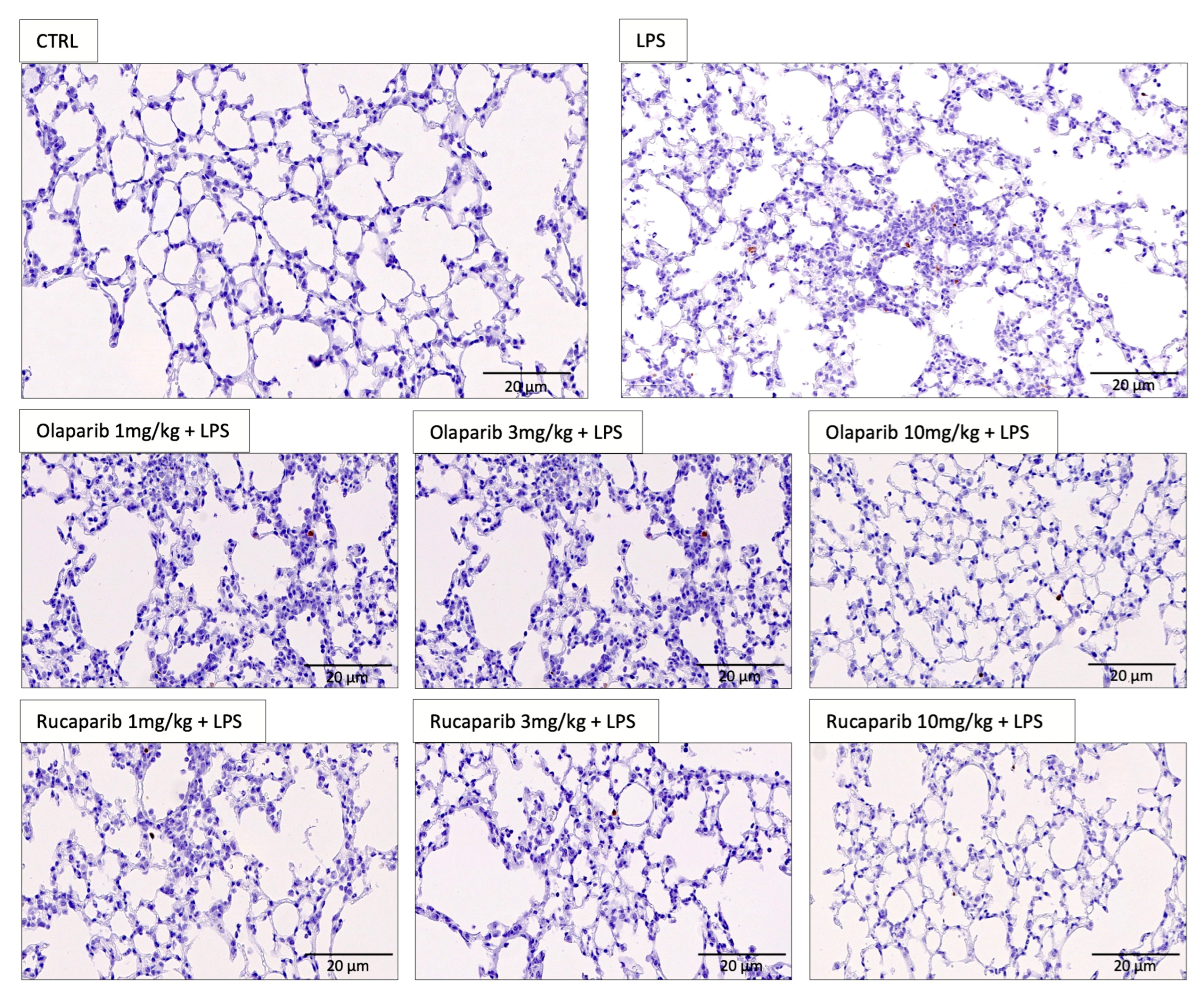
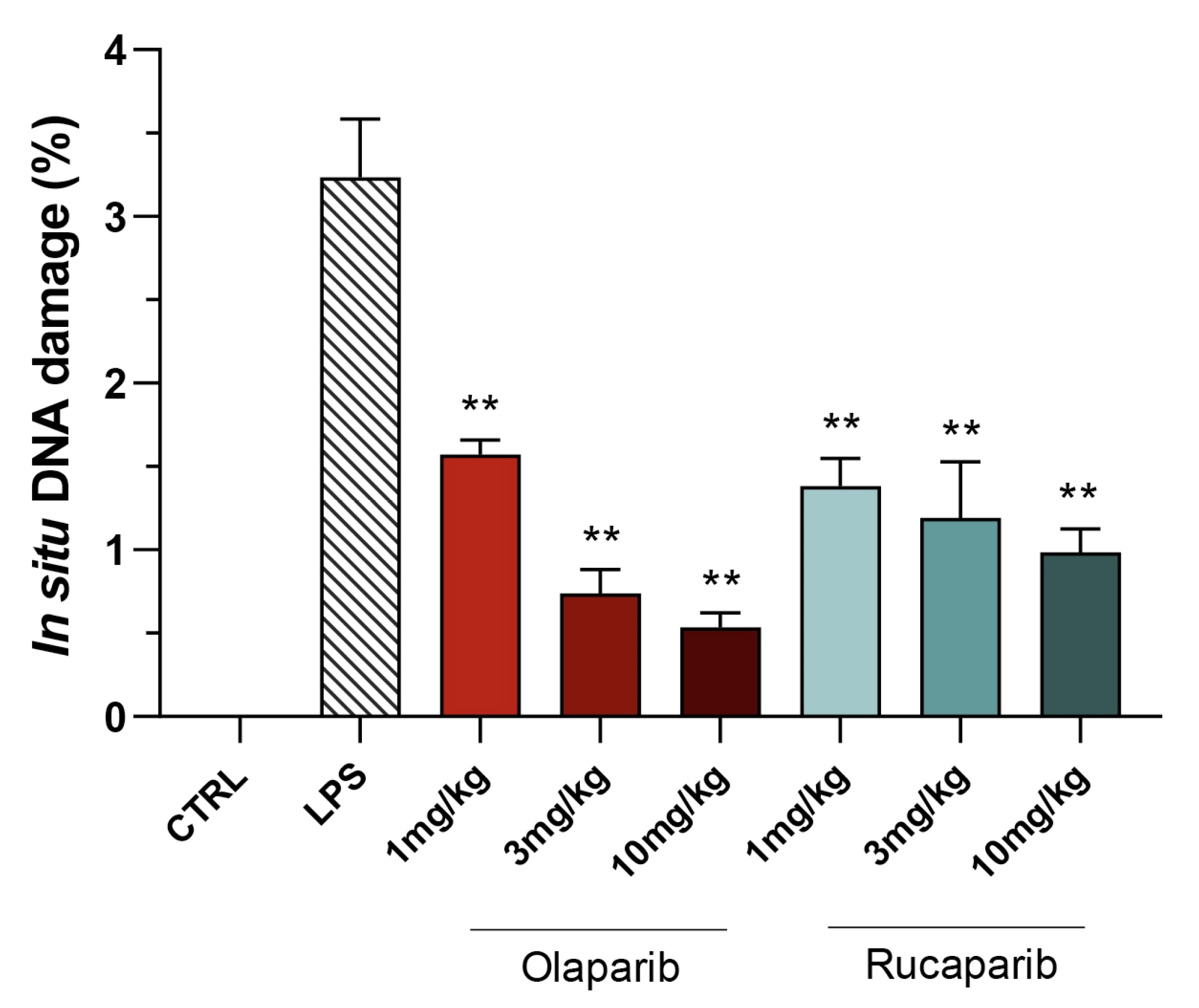
2.8. Quantification of DNA Damage In Vivo
2.9. Quantification of Pulmonary Extravasation
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. Western Blotting
2.12. Statistical Analysis
3. Results
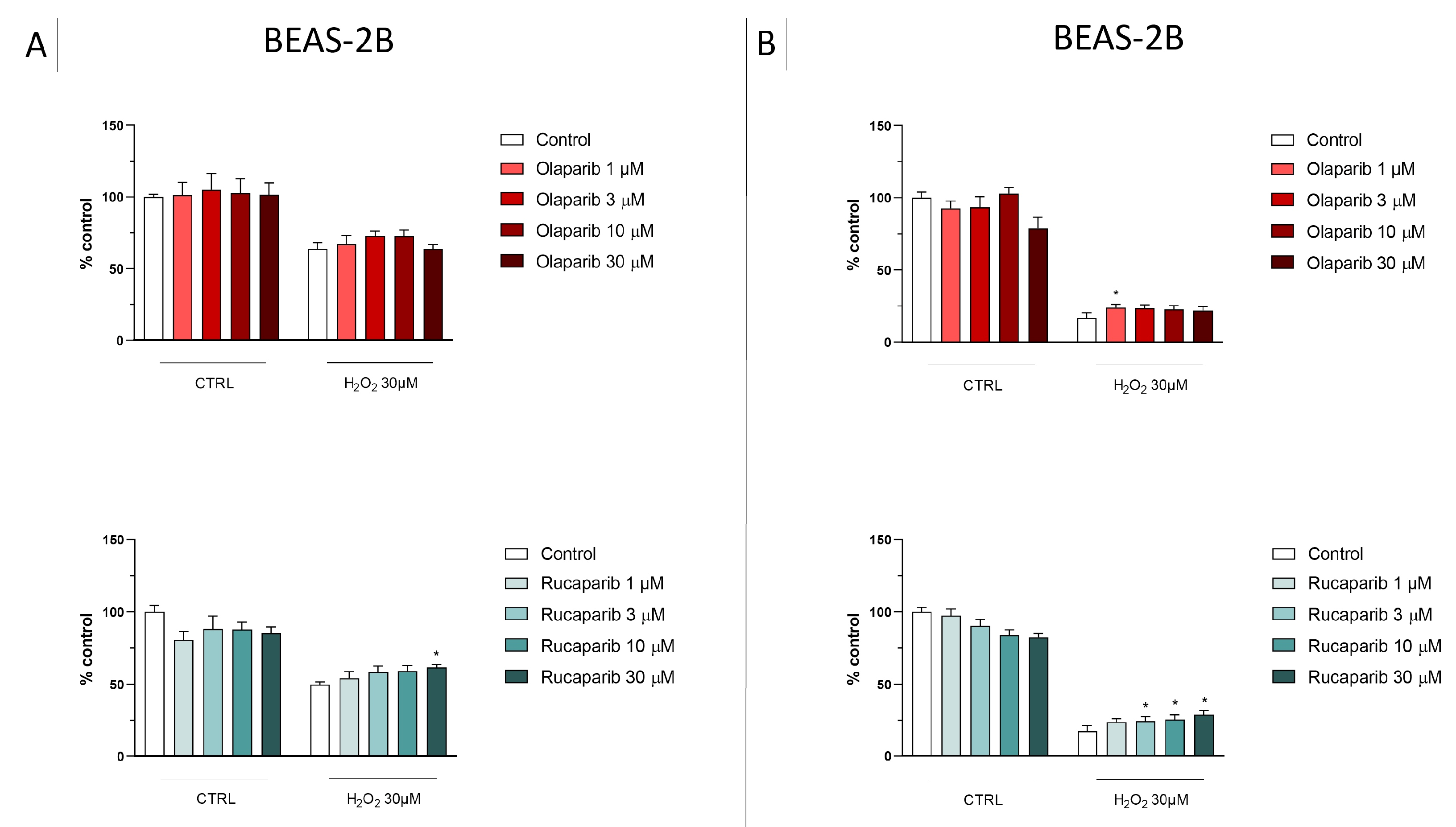
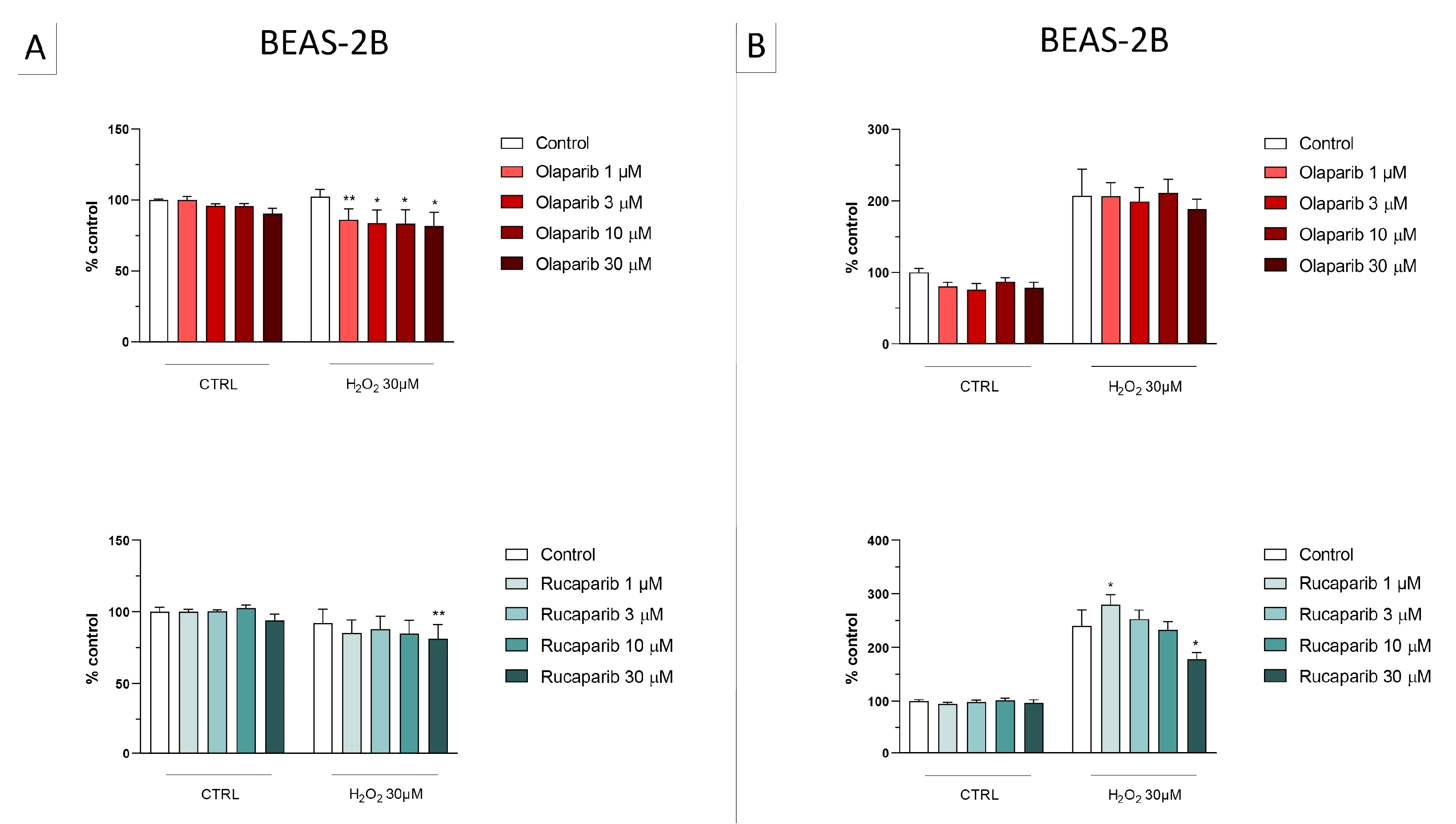
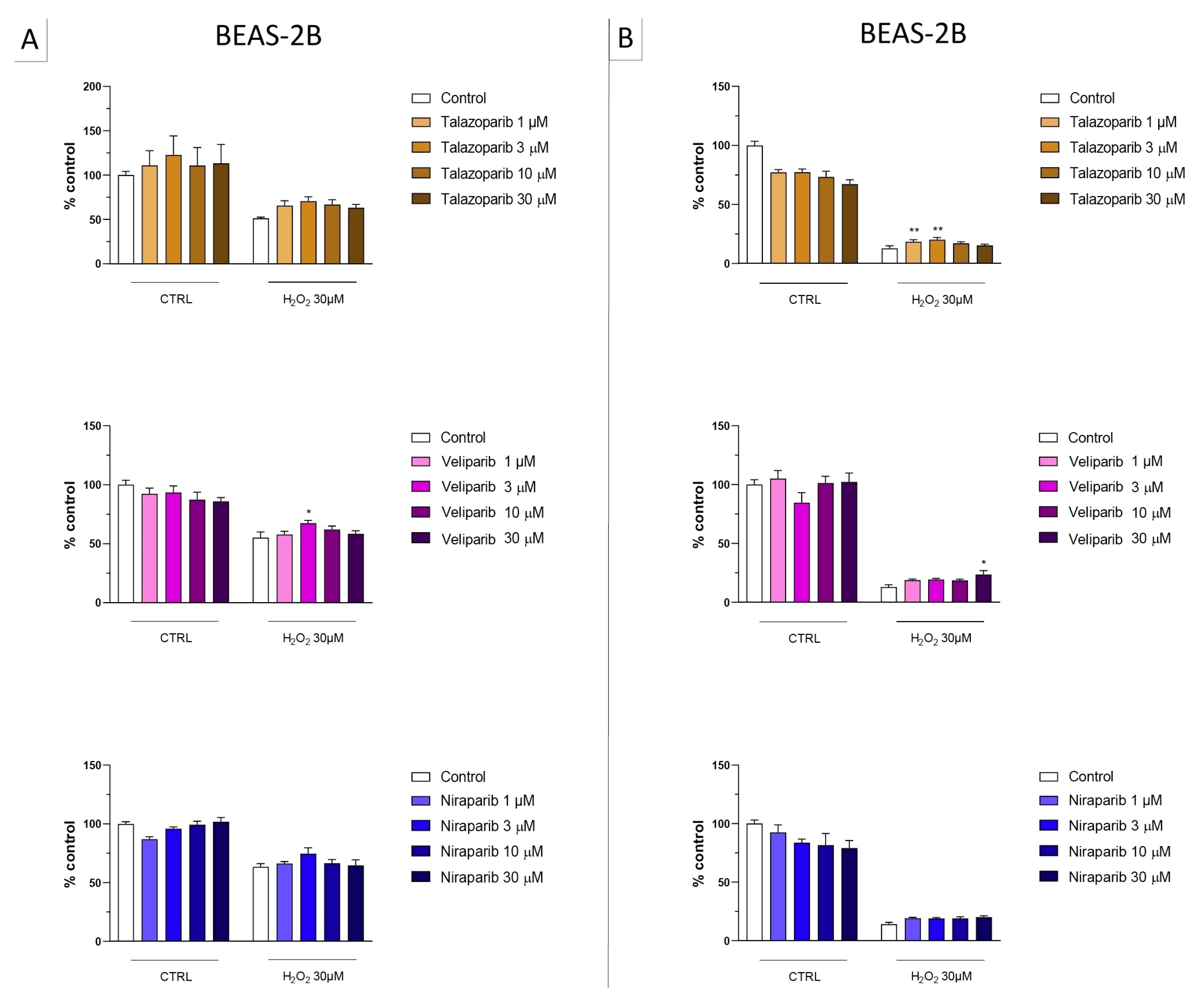
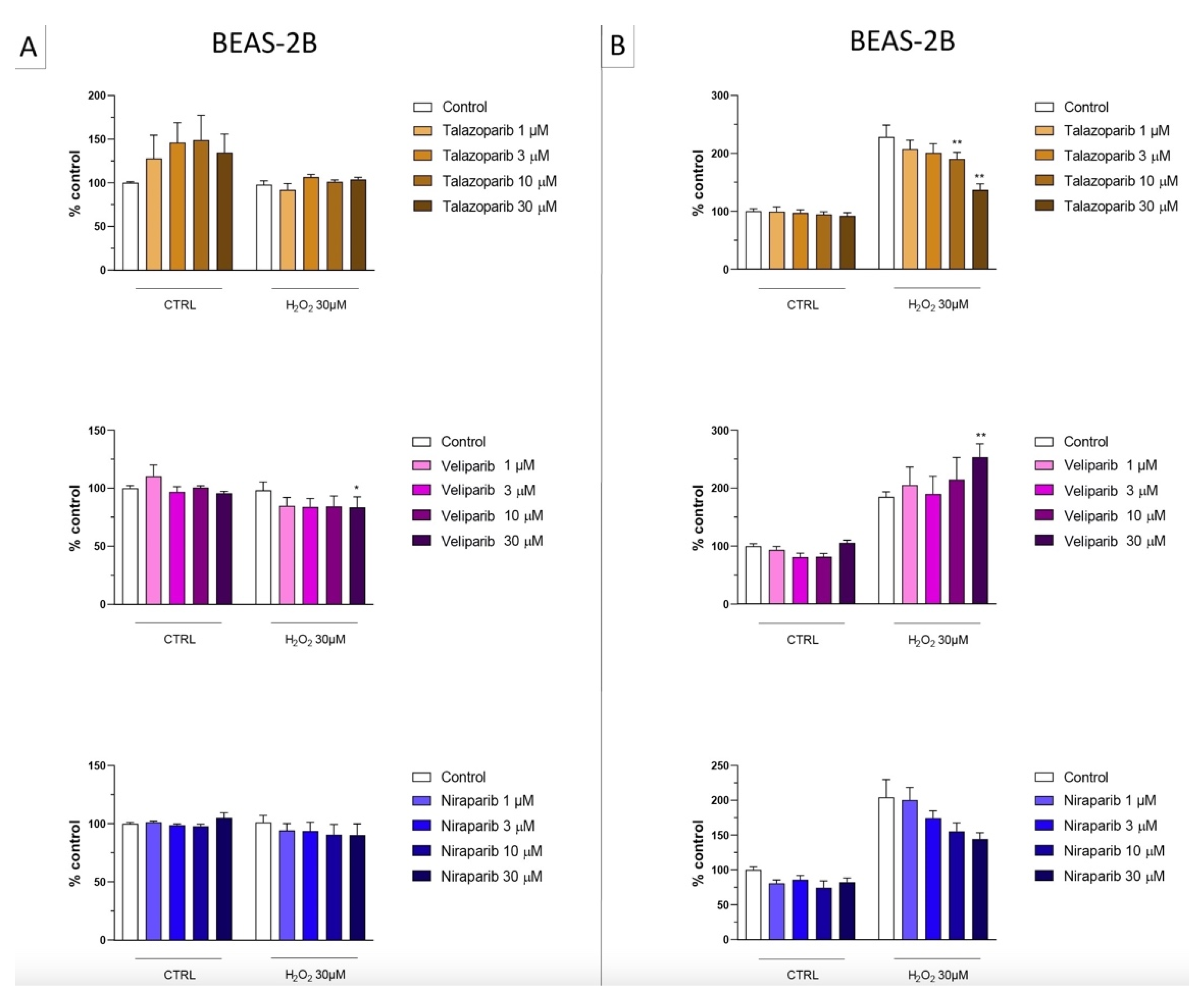
3.1. Cytoprotective Effects of PARP Inhibitors In Vitro
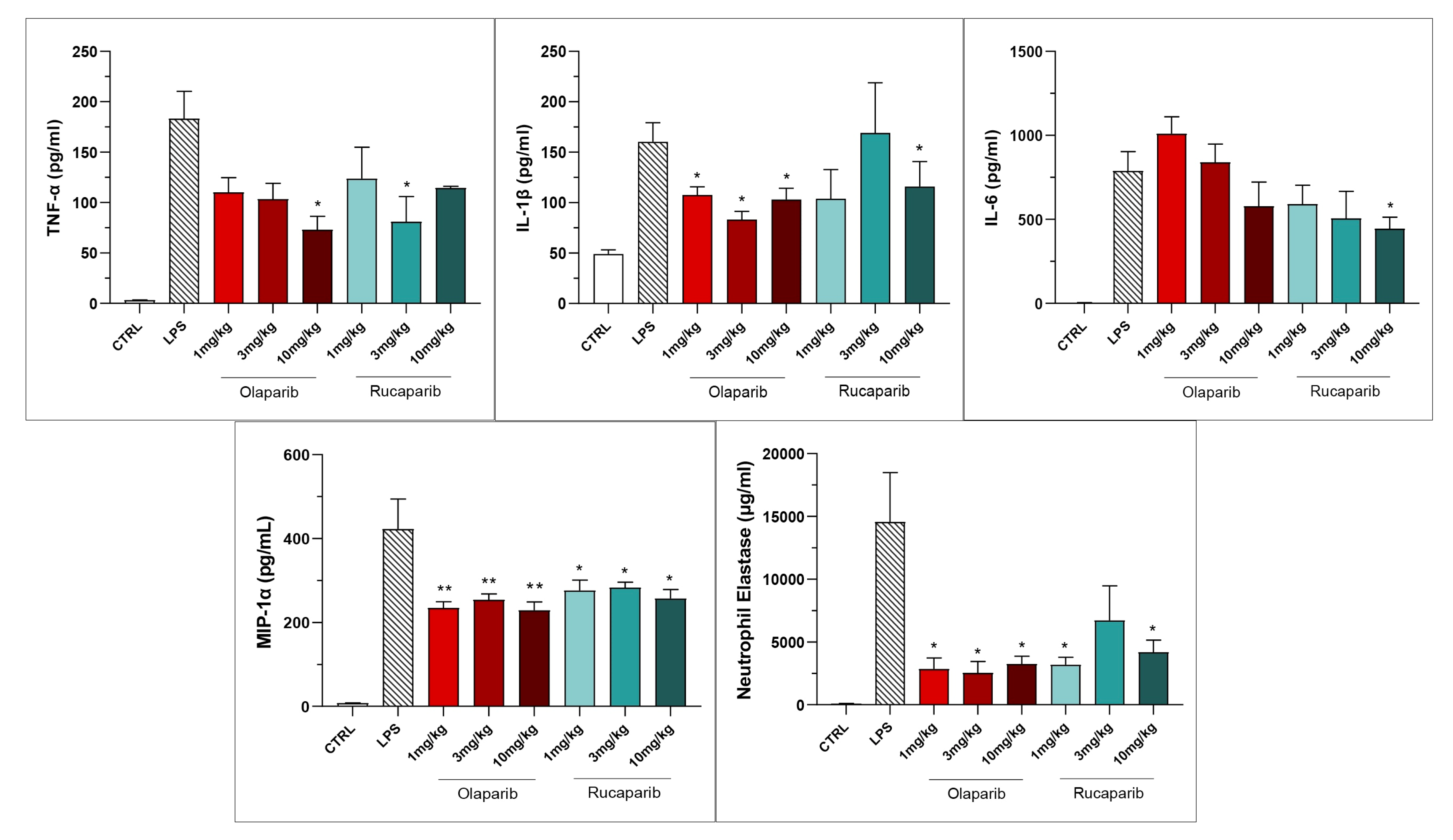
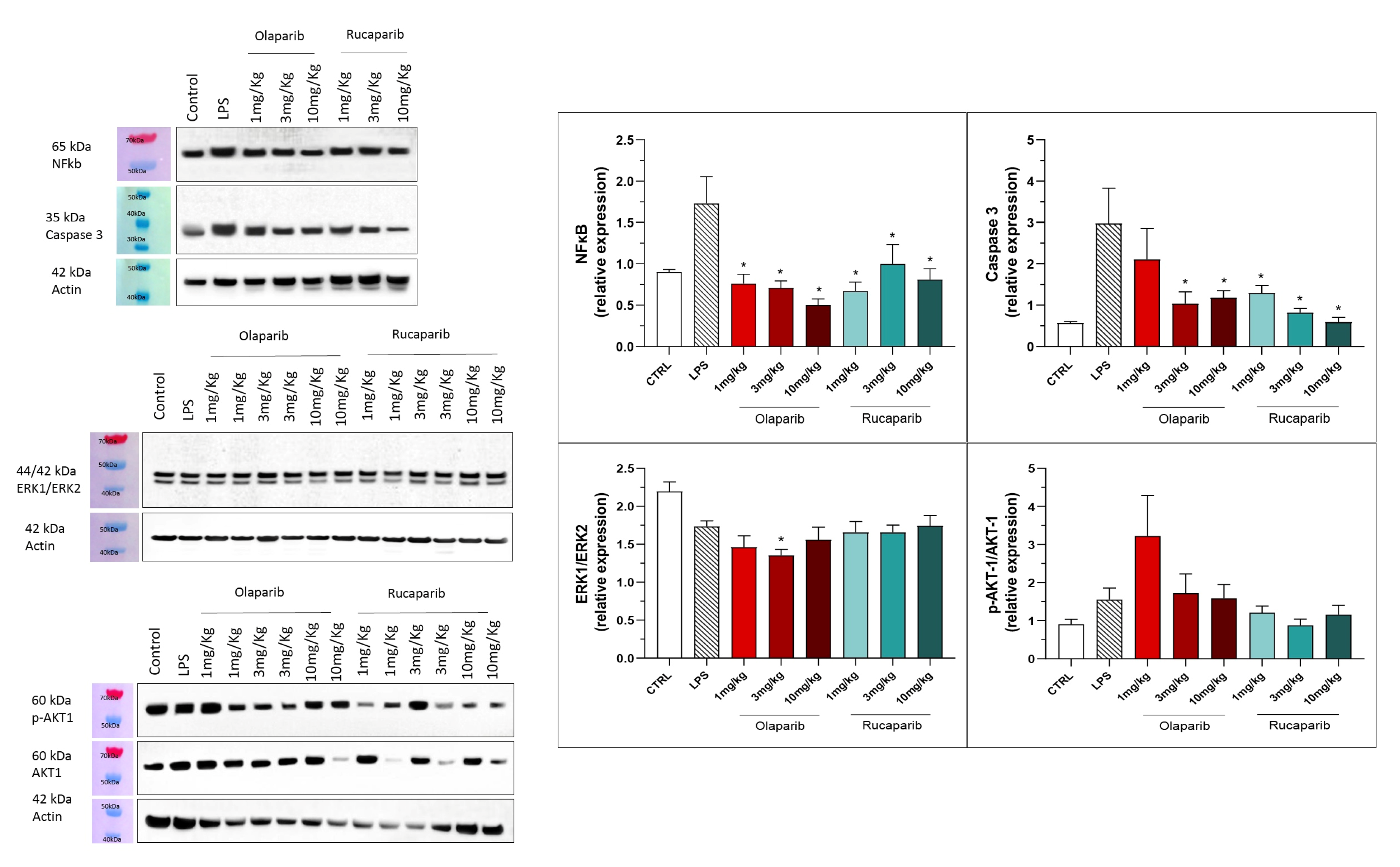
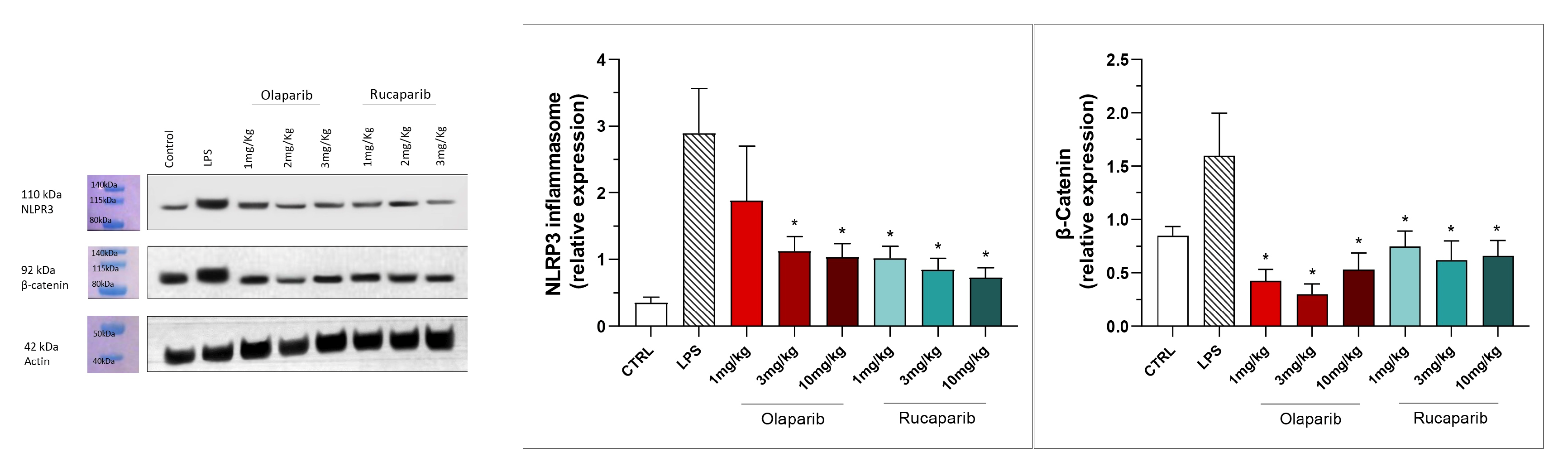
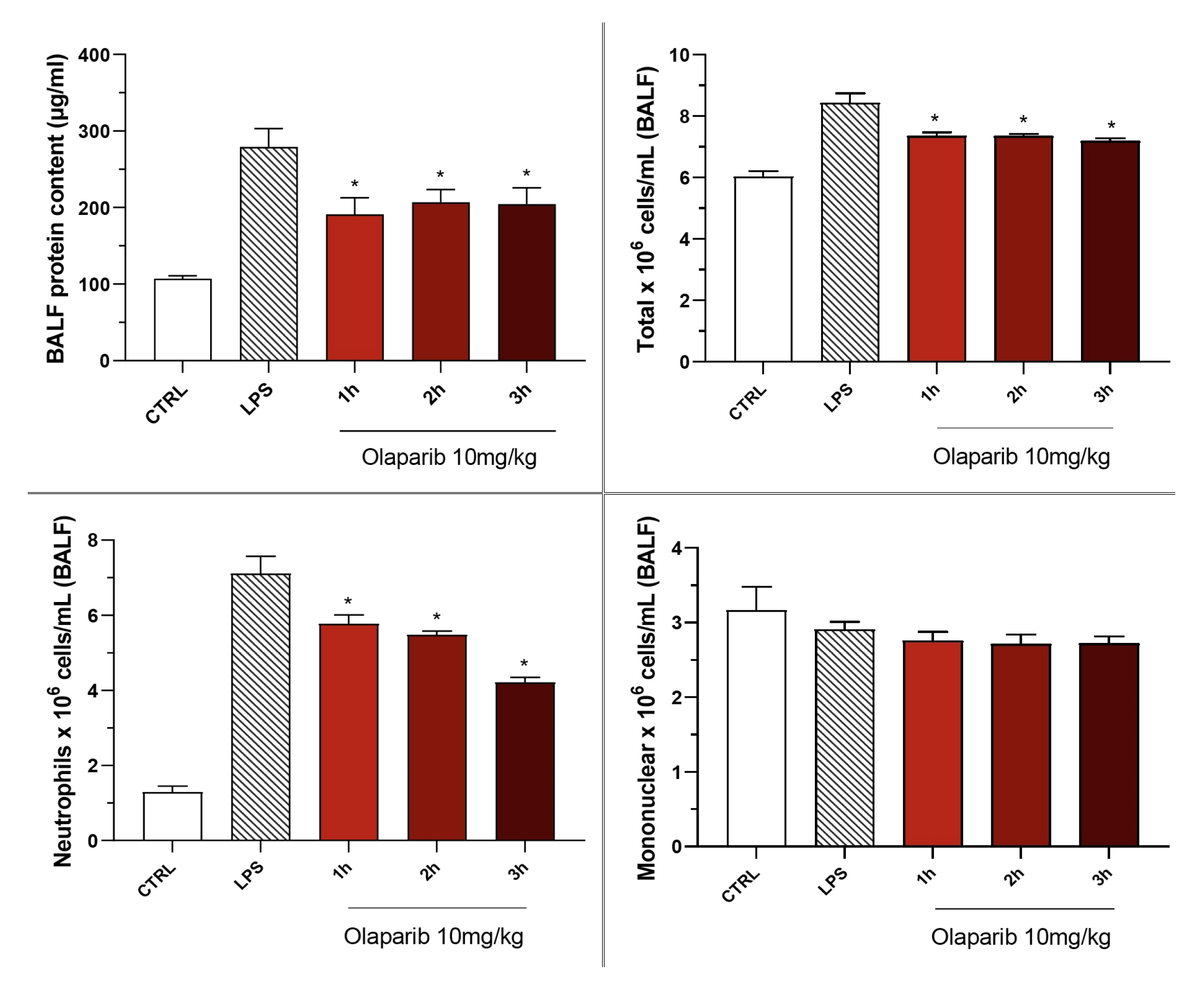
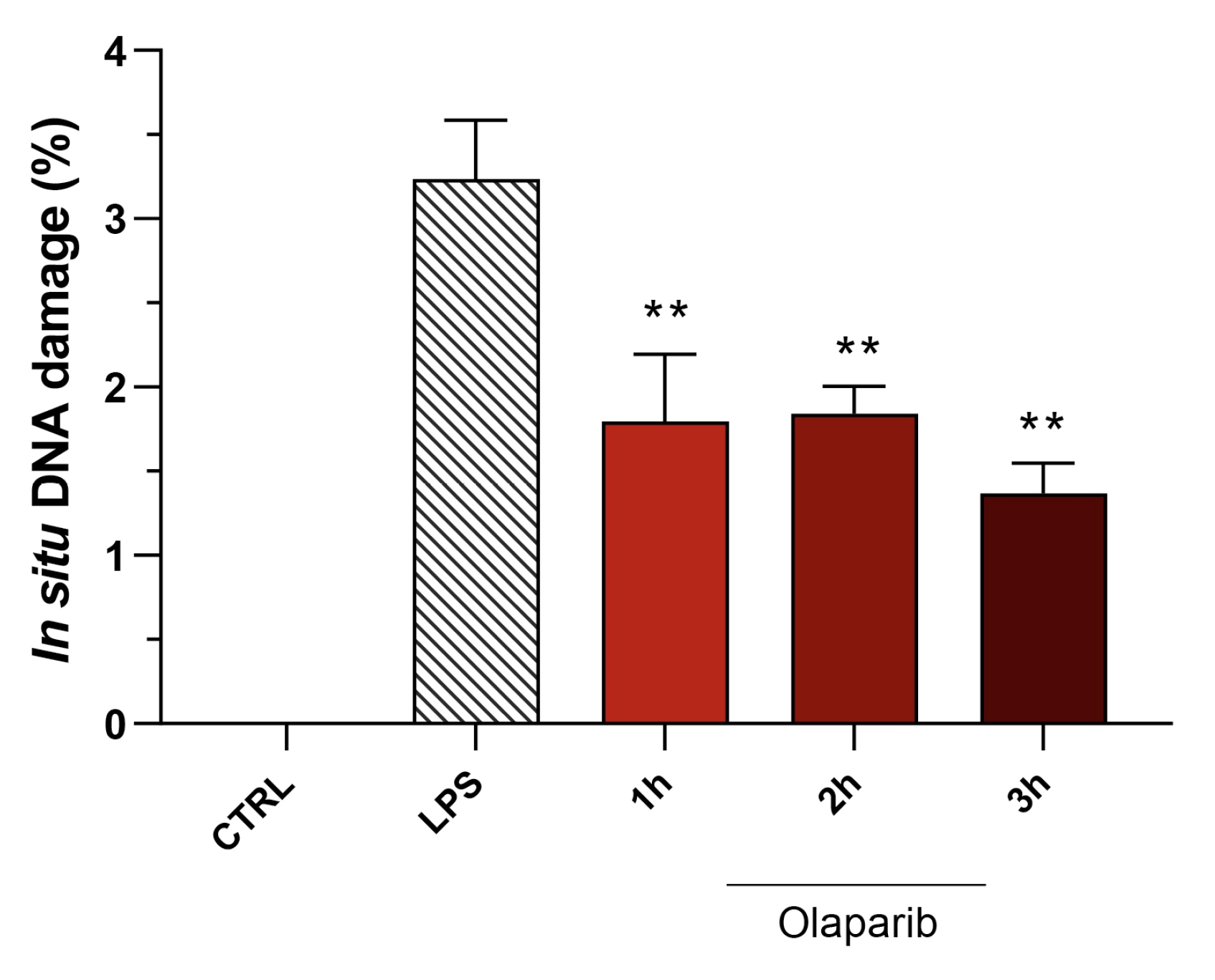
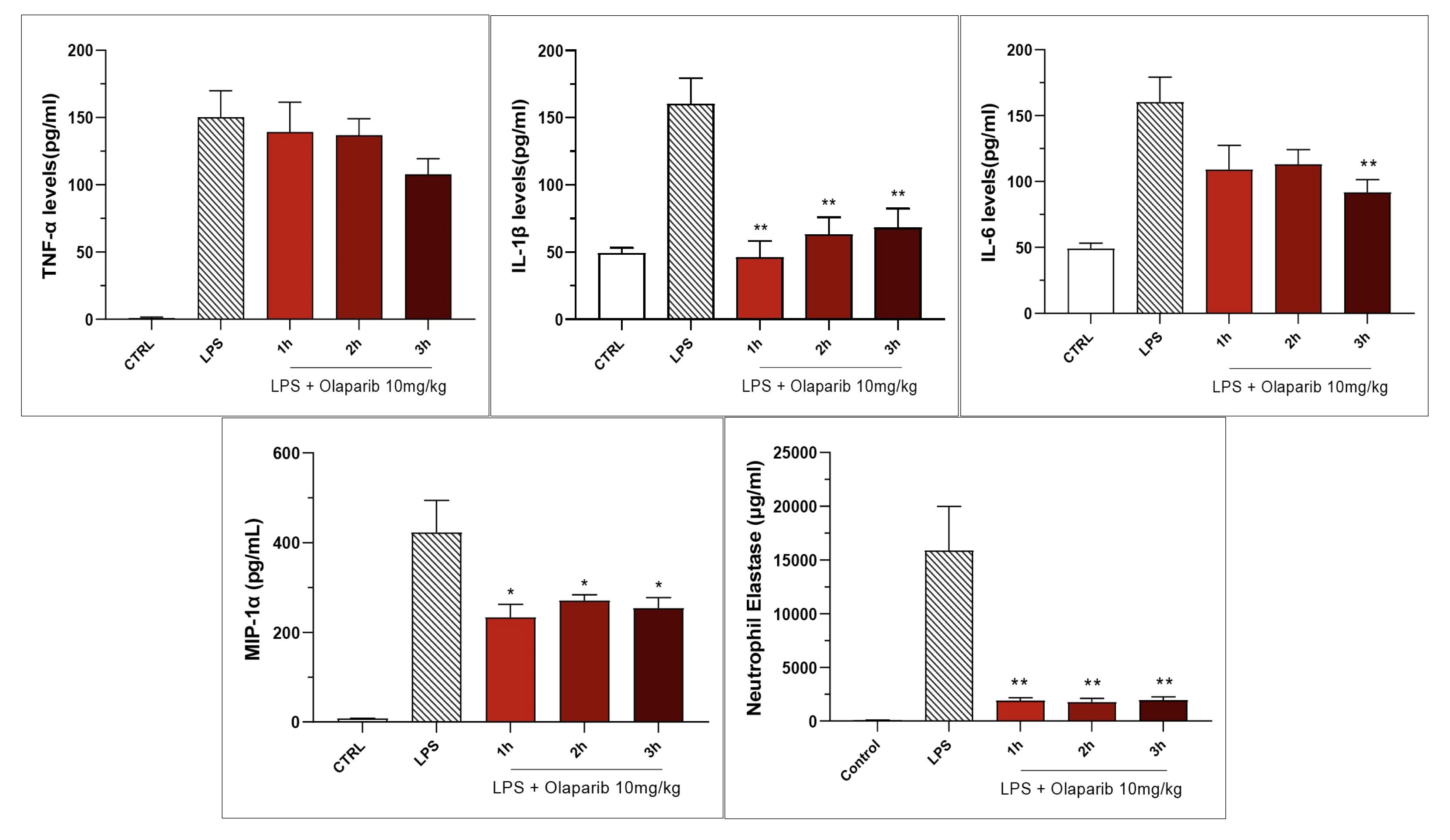
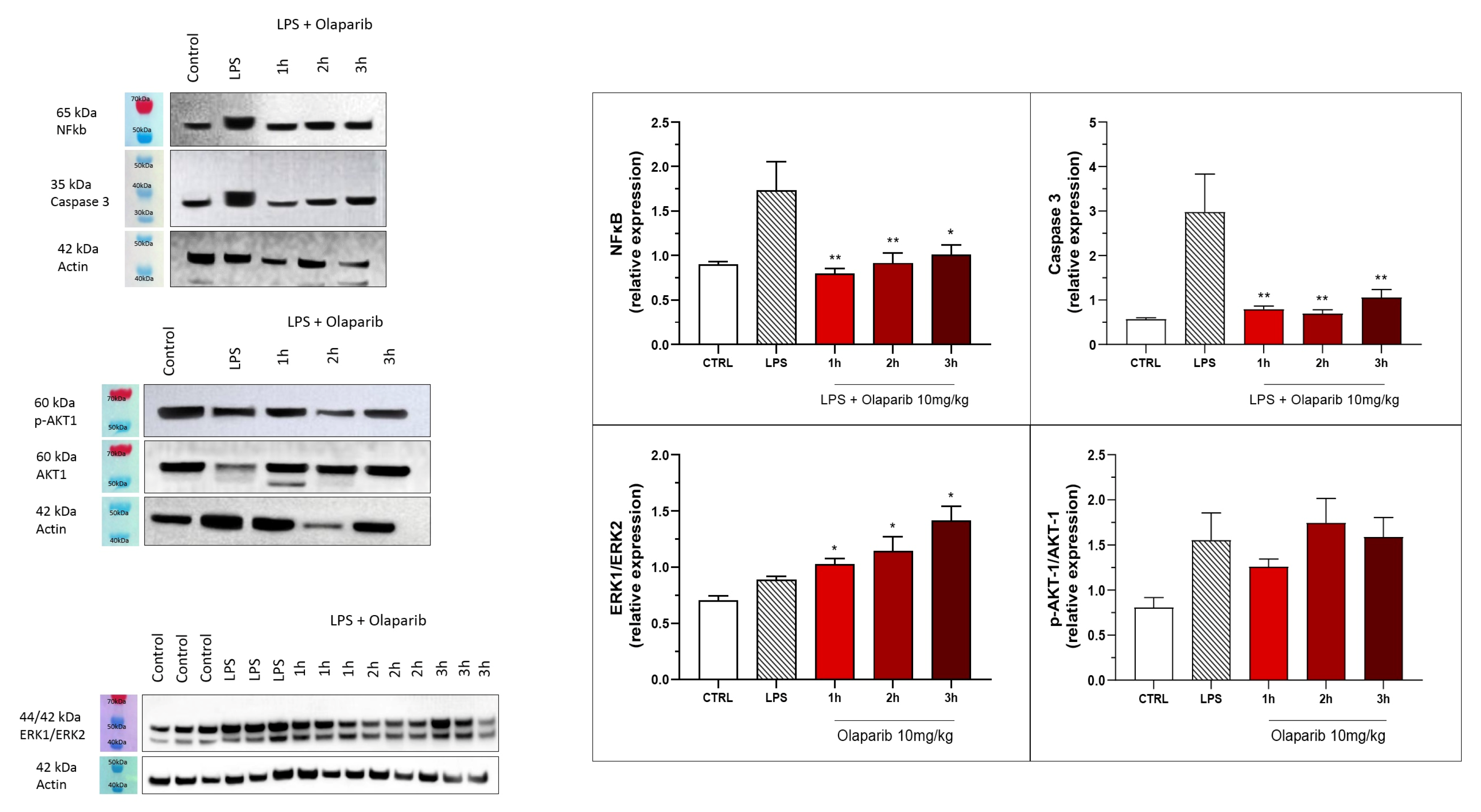
3.2. Cytoprotective Effects of PARP Inhibitors In Vivo
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jagtap, P.; Szabo, C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discov. 2005, 4, 421–440. [Google Scholar] [CrossRef]
- Pacher, P.; Szabo, C. Role of the peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease. Am. J. Pathol. 2008, 173, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Gupte, R.; Liu, Z.; Kraus, W.L. PARPs and ADP-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017, 31, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Pascal, J.M. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell Mol. Life Sci. 2020, 77, 19–33. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug. Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Kraus, W.L. The expanding universe of PARP1-mediated molecular and therapeutic mechanisms. Mol. Cell 2022, 82, 2315–2334. [Google Scholar] [CrossRef]
- Liaudet, L.; Pacher, P.; Mabley, J.G.; Virág, L.; Soriano, F.G.; Haskó, G.; Szabo, C. Activation of poly(ADP-ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. Am. J. Respir. Crit. Care Med. 2002, 165, 372–377. [Google Scholar] [CrossRef]
- Cuzzocrea, S.; McDonald, M.C.; Mazzon, E.; Dugo, L.; Serraino, I.; Threadgill, M.; Caputi, A.P.; Thiemermann, C. Effects of 5-aminoisoquinolinone, a water-soluble, potent inhibitor of the activity of poly (ADP-ribose) polymerase, in a rodent model of lung injury. Biochem. Pharmacol. 2002, 63, 293–304. [Google Scholar] [CrossRef]
- Berger, N.A.; Besson, V.C.; Boulares, A.H.; Bürkle, A.; Chiarugi, A.; Clark, R.S.; Curtin, N.J.; Cuzzocrea, S.; Dawson, T.M.; Dawson, V.L.; et al. Opportunities for the repurposing of PARP inhibitors for the therapy of non-oncological diseases. Br. J. Pharmacol. 2018, 175, 192–222. [Google Scholar] [CrossRef]
- Szabo, C.; Martins, V.; Liaudet, L. Poly(ADP-ribose) polymerase inhibition in acute lung injury. A reemerging concept. Am. J. Respir. Cell Mol. Biol. 2020, 63, 571–590. [Google Scholar] [CrossRef]
- Santos, S.S.; Brunialti, M.K.C.; Soriano, F.G.; Szabo, C.; Salomão, R. Repurposing of clinically approved poly-(ADP-ribose) polymerase inhibitors for the therapy of sepsis. Shock 2021, 56, 901–909. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.S.; Brunialti, M.K.C.; Rodrigues, L.O.C.P.; Liberatore, A.M.A.; Koh, I.H.J.; Martins, V.; Soriano, F.G.; Szabo, C.; Salomão, R. Effects of the PARP inhibitor olaparib on the response of human peripheral blood leukocytes to bacterial challenge or oxidative stress. Biomolecules 2022, 12, 788. [Google Scholar] [CrossRef]
- Mekhaeil, M.; Dev, K.K.; Conroy, M.J. Existing evidence for the repurposing of PARP-1 inhibitors in rare demyelinating diseases. Cancers 2022, 14, 687. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Lim, L.H.; Cuzzocrea, S.; Getting, S.J.; Zingarelli, B.; Flower, R.J.; Salzman, A.L.; Perretti, M. Inhibition of poly (ADP-ribose) synthetase attenuates neutrophil recruitment and exerts antiinflammatory effects. J. Exp. Med. 1997, 186, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Jagtap, P.; Soriano, F.G.; Virág, L.; Liaudet, L.; Mabley, J.; Szabó, E.; Hasko, G.; Marton, A.; Lorigados, C.B.; Gallyas, F., Jr.; et al. Novel phenanthridinone inhibitors of poly (adenosine 5’-diphosphate-ribose) synthetase: Potent cytoprotective and antishock agents. Crit. Care Med. 2002, 30, 1071–1082. [Google Scholar] [CrossRef]
- Boulares, A.H.; Zoltoski, A.J.; Sherif, Z.A.; Jolly, P.; Massaro, D.; Smulson, M.E. Gene knockout or pharmacological inhibition of poly(ADP-ribose) polymerase-1 prevents lung inflammation in a murine model of asthma. Am. J. Respir. Cell Mol. Biol. 2003, 28, 322–329. [Google Scholar] [CrossRef]
- Veres, B.; Gallyas, F., Jr.; Varbiro, G.; Berente, Z.; Osz, E.; Szekeres, G.; Szabo, C.; Sumegi, B. Decrease of the inflammatory response and induction of the Akt/protein kinase B pathway by poly-(ADP-ribose) polymerase 1 inhibitor in endotoxin-induced septic shock. Biochem. Pharmacol. 2003, 65, 1373–1382. [Google Scholar] [CrossRef]
- Murakami, K.; Enkhbaatar, P.; Shimoda, K.; Cox, R.A.; Burke, A.S.; Hawkins, H.K.; Traber, L.D.; Schmalstieg, F.C.; Salzman, A.L.; Mabley, J.G.; et al. Inhibition of poly (ADP-ribose) polymerase attenuates acute lung injury in an ovine model of sepsis. Shock 2004, 21, 126–133. [Google Scholar] [CrossRef]
- Veres, B.; Radnai, B.; Gallyas, F., Jr.; Varbiro, G.; Berente, Z.; Osz, E.; Sumegi, B. Regulation of kinase cascades and transcription factors by a poly(ADP-ribose) polymerase-1 inhibitor, 4-hydroxyquinazoline, in lipopolysaccharide-induced inflammation in mice. J. Pharmacol. Exp. Ther. 2004, 310, 247–255. [Google Scholar] [CrossRef]
- Virág, L.; Bai, P.; Bak, I.; Pacher, P.; Mabley, J.G.; Liaudet, L.; Bakondi, E.; Gergely, P.; Kollai, M.; Szabo, C. Effects of poly(ADP-ribose) polymerase inhibition on inflammatory cell migration in a murine model of asthma. Med. Sci. Monit. 2004, 10, BR77–BR83. [Google Scholar]
- Kao, S.J.; Liu, D.D.; Su, C.F.; Chen, H.I. Niacinamide abrogates the organ dysfunction and acute lung injury caused by endotoxin. J. Cardiovasc. Pharmacol. 2007, 50, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Mota, R.; Sánchez-Bueno, F.; Berenguer-Pina, J.J.; Hernández-Espinosa, D.; Parrilla, P.; Yélamos, J. Therapeutic treatment with poly(ADP-ribose) polymerase inhibitors attenuates the severity of acute pancreatitis and associated liver and lung injury. Br. J. Pharmacol. 2007, 151, 998–1005. [Google Scholar] [CrossRef]
- Hamahata, A.; Enkhbaatar, P.; Lange, M.; Yamaki, T.; Sakurai, H.; Shimoda, K.; Nakazawa, H.; Traber, L.D.; Traber, D.L. Administration of poly(ADP-ribose) polymerase inhibitor into bronchial artery attenuates pulmonary pathophysiology after smoke inhalation and burn in an ovine model. Burns 2012, 38, 1210–1215. [Google Scholar] [CrossRef]
- Wang, G.; Huang, X.; Li, Y.; Guo, K.; Ning, P.; Zhang, Y. PARP-1 inhibitor, DPQ, attenuates LPS-induced acute lung injury through inhibiting NF-κB-mediated inflammatory response. PLoS ONE 2013, 8, e79757. [Google Scholar] [CrossRef] [PubMed]
- Hatachi, G.; Tsuchiya, T.; Miyazaki, T.; Matsumoto, K.; Yamasaki, N.; Okita, N.; Nanashima, A.; Higami, Y.; Nagayasu, T. The poly(adenosine diphosphate-ribose) polymerase inhibitor PJ34 reduces pulmonary ischemia-reperfusion injury in rats. Transplantation 2014, 98, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.F.; Zoheir, K.M.; Ansari, M.A.; Korashy, H.M.; Bakheet, S.A.; Ashour, A.E.; Al-Shabanah, O.A.; Al-harbi, M.M.; Attia, S.M. The role of poly(ADP-ribose) polymerase-1 inhibitor in carrageenan-induced lung inflammation in mice. Mol. Immunol. 2015, 63, 394–405. [Google Scholar] [CrossRef]
- Tuncer, S.K.; Altinel, S.; Toygar, M.; Istanbulluoglu, H.; Ates, K.; Ogur, R.; Altinel, O.; Karslioglu, Y.; Topal, T.; Korkmaz, A.; et al. Poly-ADP-ribose polymerase inhibition provides protection against lung injury in a rat paraquat toxicity model. Inflammopharmacology 2016, 24, 155–161. [Google Scholar] [CrossRef]
- Kapoor, K.; Singla, E.; Sahu, B.; Naura, A.S. PARP inhibitor, olaparib ameliorates acute lung and kidney injury upon intratracheal administration of LPS in mice. Mol. Cell Biochem. 2015, 400, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Sethi, G.S.; Sharma, S.; Naura, A.S. PARP inhibition by olaparib alleviates chronic asthma-associated remodeling features via modulating inflammasome signaling in mice. IUBMB Life 2019, 71, 1003–1013. [Google Scholar] [CrossRef]
- Ahmad, A.; Vieira, J.C.; de Mello, A.H.; de Lima, T.M.; Ariga, S.K.; Barbeiro, D.F.; Barbeiro, H.V.; Szczesny, B.; Törö, G.; Druzhyna, N.; et al. The PARP inhibitor olaparib exerts beneficial effects in mice subjected to cecal ligature and puncture and in cells subjected to oxidative stress without impairing DNA integrity: A potential opportunity for repurposing a clinically used oncological drug for the experimental therapy of sepsis. Pharmacol. Res. 2019, 145, 104263. [Google Scholar]
- Sahu, B.; Narota, A.; Naura, A.S. Pharmacological inhibition of poly (ADP-ribose) polymerase by olaparib, prevents acute lung injury associated cognitive deficits potentially through suppression of inflammatory response. Eur. J. Pharmacol. 2020, 877, 173091. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Ren, X.; Wang, Q.; Zhang, Y.; Du, J. Pharmacological inhibition of poly (ADP-ribose) polymerase by olaparib ameliorates influenza-virus-induced pneumonia in mice. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic dissection of PARP1 trapping and the impact on in vivo tolerability and efficacy of PARP inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping poly(ADP-ribose) polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef]
- Oei, S.L.; Ziegler, M. ATP for the DNA ligation step in base excision repair is generated from poly(ADP-ribose). J. Biol. Chem. 2000, 275, 23234–23239. [Google Scholar] [CrossRef]
- Wright, R.H.; Lioutas, A.; Le Dily, F.; Soronellas, D.; Pohl, A.; Bonet, J.; Nacht, A.S.; Samino, S.; Font-Mateu, J.; Vicent, G.P.; et al. ADP-ribose-derived nuclear ATP synthesis by NUDIX5 is required for chromatin remodeling. Science 2016, 352, 1221–1225. [Google Scholar] [CrossRef]
- Sims, J.L.; Berger, S.J.; Berger, N.A. Poly(ADP-ribose) polymerase inhibitors preserve nicotinamide adenine dinucleotide and adenosine 5’-triphosphate pools in DNA damaged cells: Mechanism of stimulation of unscheduled DNA synthesis. Biochemistry 1983, 22, 5188–5194. [Google Scholar] [CrossRef]
- Schraufstatter, I.U.; Hinshaw, D.B.; Hyslop, P.A.; Spragg, R.G.; Cochrane, C.G. Oxidant injury of cells. DNA strand-breaks activate polyadenosine diphosphate-ribose polymerase and lead to depletion of nicotinamide adenine dinucleotide. J. Clin. Investig. 1986, 77, 1312–1320. [Google Scholar] [CrossRef]
- Szabo, C.; Zingarelli, B.; O’Connor, M.; Salzman, A.L. DNA strand breakage, activation of poly (ADP-ribose) synthetase, and cellular energy depletion are involved in the cytotoxicity of macrophages and smooth muscle cells exposed to peroxynitrite. Proc. Natl. Acad. Sci. USA 1996, 93, 1753–1758. [Google Scholar] [CrossRef]
- Virág, L.; Salzman, A.L.; Szabo, C. Poly(ADP-ribose) synthetase activation mediates mitochondrial injury during oxidant-induced cell death. J. Immunol. 1998, 161, 3753–3759. [Google Scholar] [PubMed]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. USA 1999, 96, 13978–13982. [Google Scholar] [CrossRef]
- Kovács, D.; Vántus, V.B.; Vámos, E.; Kálmán, N.; Schicho, R.; Gallyas, F.; Radnai, B. Olaparib: A clinically applied PARP inhibitor protects from experimental Crohn’s disease and maintains barrier integrity by improving bioenergetics through rescuing glycolysis in colonic epithelial cells. Oxid. Med. Cell Longev. 2021, 2021, 7308897. [Google Scholar] [CrossRef] [PubMed]
- Oliver, F.J.; Ménissier-de Murcia, J.; Nacci, C.; Decker, P.; Andriantsitohaina, R.; Muller, S.; de la Rubia, G.; Stoclet, J.C.; de Murcia, G. Resistance to endotoxic shock as a consequence of defective NF-kappaB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999, 18, 4446–4454. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Hottiger, M.O. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell Mol. Life Sci. 2002, 59, 1534–1553. [Google Scholar] [CrossRef] [PubMed]
- Haskó, G.; Mabley, J.G.; Németh, Z.H.; Pacher, P.; Deitch, E.A.; Szabó, C. Poly(ADP-ribose) polymerase is a regulator of chemokine production: Relevance for the pathogenesis of shock and inflammation. Mol. Med. 2002, 8, 283–289. [Google Scholar] [CrossRef]
- Mabley, J.G.; Horváth, E.M.; Murthy, K.G.; Zsengellér, Z.; Vaslin, A.; Benko, R.; Kollai, M.; Szabó, C. Gender differences in the endotoxin-induced inflammatory and vascular responses: Potential role of poly(ADP-ribose) polymerase activation. J. Pharmacol. Exp. Ther. 2005, 315, 812–820. [Google Scholar] [CrossRef]
- Soriano, F.G.; Liaudet, L.; Szabó, E.; Virág, L.; Mabley, J.G.; Pacher, P.; Szabó, C. Resistance to acute septic peritonitis in poly(ADP-ribose) polymerase-1-deficient mice. Shock 2002, 17, 286–292. [Google Scholar] [CrossRef]
- Faraoni, I.; Consalvo, M.I.; Aloisio, F.; Fabiani, E.; Giansanti, M.; Di Cristino, F.; Falconi, G.; Tentori, L.; Di Veroli, A.; Curzi, P.; et al. Cytotoxicity and differentiating effect of the poly(ADP-ribose) polymerase inhibitor olaparib in myelodysplastic syndromes. Cancers 2019, 11, 1373. [Google Scholar] [CrossRef]
- Gill, S.E.; Yamashita, C.M.; Veldhuizen, R.A. Lung remodeling associated with recovery from acute lung injury. Cell Tissue Res. 2017, 367, 495–509. [Google Scholar] [CrossRef]
- Noone, P.M.; Reddy, S.P. Recent advances in dead cell clearance during acute lung injury and repair. Fac. Rev. 2021, 10, 33. [Google Scholar] [CrossRef]
- Kondratska, O.; Grushka, N.; Pavlovych, S.; Krasutska, N.; Tsyhankov, S.; Yanchii, R. Effects of poly (ADP-ribose) polymerase inhibition on DNA integrity and gene expression in ovarian follicular cells in mice with endotoxemia. Iran. Biomed. J. 2022, 26, 44–52. [Google Scholar] [PubMed]
- Huang, X.; Halicka, H.D.; Traganos, F.; Tanaka, T.; Kurose, A.; Darzynkiewicz, Z. Cytometric assessment of DNA damage in relation to cell cycle phase and apoptosis. Cell Prolif. 2005, 38, 223–243. [Google Scholar] [CrossRef]
- Mitchell, L.A.; De Iuliis, G.N.; Aitken, R.J. The TUNEL assay consistently underestimates DNA damage in human spermatozoa and is influenced by DNA compaction and cell vitality: Development of an improved methodology. Int. J. Androl. 2011, 34, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.L.; Savenka, A.V.; Basnakian, A.G. TUNEL assay: A powerful tool for kidney injury evaluation. Int. J. Mol. Sci. 2021, 22, 412. [Google Scholar] [CrossRef] [PubMed]
- Garcia Soriano, F.; Virág, L.; Jagtap, P.; Szabó, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(ADP-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113. [Google Scholar] [CrossRef]
- Nie, Y.; Nirujogi, T.S.; Ranjan, R.; Reader, B.F.; Chung, S.; Ballinger, M.N.; Englert, J.A.; Christman, J.W.; Karpurapu, M. PolyADP-ribosylation of NFATc3 and NF-κB transcription factors modulate macrophage inflammatory gene expression in LPS-induced acute lung injury. J. Innate Immun. 2021, 13, 83–93. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, E.; Wang, C.; Zhang, P.; Huang, L. PARP-1 inhibition repressed imbalance of Th17 and Treg cells in preterm rats with intrauterine infection-induced acute respiratory distress syndrome by reducing the expression level of IL-6. J. Healthc. Eng. 2022, 2022, 1255674. [Google Scholar] [CrossRef]
- Chiu, L.Y.; Huang, D.Y.; Lin, W.W. PARP-1 regulates inflammasome activity by poly-ADP-ribosylation of NLRP3 and interaction with TXNIP in primary macrophages. Cell Mol. Life Sci. 2022, 79, 108. [Google Scholar] [CrossRef]
- Sun, Y.; Wu, J.; Dong, X.; Zhang, J.; Meng, C.; Liu, G. MicroRNA-506-3p increases the response to PARP inhibitors and cisplatin by targeting EZH2/β-catenin in serous ovarian cancers. Transl. Oncol. 2021, 14, 100987. [Google Scholar] [CrossRef]
- Nakamura, N.; Fujihara, H.; Kawaguchi, K.; Yamada, H.; Nakayama, R.; Yasukawa, M.; Kishi, Y.; Hamada, Y.; Masutani, M. Possible action of olaparib for preventing invasion of oral squamous cell carcinoma in vitro and in vivo. Int. J. Mol. Sci. 2022, 23, 2527. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.P.; Bachli, E.B.; Schoedon, G. The Wnt pathway: A macrophage effector molecule that triggers inflammation. Curr. Atheroscler. Rep. 2009, 11, 236–242. [Google Scholar] [CrossRef]
- Houschyar, K.S.; Chelliah, M.P.; Rein, S.; Maan, Z.N.; Weissenberg, K.; Duscher, D.; Branski, L.K.; Siemers, F. Role of Wnt signaling during inflammation and sepsis: A review of the literature. Int. J. Artif. Organs 2018, 41, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hua, S. Transcription factor-mediated signaling pathways’ contribution to the pathology of acute lung injury and acute respiratory distress syndrome. Am. J. Transl. Res. 2020, 12, 5608–5618. [Google Scholar] [PubMed]
- Raslan, A.A.; Yoon, J.K. WNT signaling in lung repair and regeneration. Mol. Cells 2020, 43, 774–783. [Google Scholar]
- Hussain, M.; Xu, C.; Lu, M.; Wu, X.; Tang, L.; Wu, X. Wnt/β-catenin signaling links embryonic lung development and asthmatic airway remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3226–3242. [Google Scholar] [CrossRef] [PubMed]
- Radnai, B.; Antus, C.; Racz, B.; Engelmann, P.; Priber, J.K.; Tucsek, Z.; Veres, B.; Turi, Z.; Lorand, T.; Sumegi, B.; et al. Protective effect of the poly(ADP-ribose) polymerase inhibitor PJ34 on mitochondrial depolarization-mediated cell death in hepatocellular carcinoma cells involves attenuation of c-Jun N-terminal kinase-2 and protein kinase B/Akt activation. Mol. Cancer 2012, 11, 34. [Google Scholar] [CrossRef]
- Tapodi, A.; Bognar, Z.; Szabo, C.; Gallyas, F.; Sumegi, B.; Hocsak, E. PARP inhibition induces Akt-mediated cytoprotective effects through the formation of a mitochondria-targeted phospho-ATM-NEMO-Akt-mTOR signalosome. Biochem. Pharmacol. 2019, 162, 98–108. [Google Scholar] [CrossRef]
- Gallyas, F., Jr.; Sumegi, B.; Szabo, C. Role of Akt activation in PARP inhibitor resistance in cancer. Cancers 2020, 12, 532. [Google Scholar] [CrossRef]
- McCullough, L.D.; Zeng, Z.; Blizzard, K.K.; Debchoudhury, I.; Hurn, P.D. Ischemic nitric oxide and poly (ADP-ribose) polymerase-1 in cerebral ischemia: Male toxicity, female protection. J. Cereb. Blood Flow Metab. 2005, 25, 502–512. [Google Scholar] [CrossRef]
- Liu, F.; Lang, J.; Li, J.; Benashski, S.E.; Siegel, M.; Xu, Y.; McCullough, L.D. Sex differences in the response to poly(ADP-ribose) polymerase-1 deletion and caspase inhibition after stroke. Stroke 2011, 42, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Charriaut-Marlangue, C.; Leconte, C.; Csaba, Z.; Chafa, L.; Pansiot, J.; Talatizi, M.; Simon, K.; Moretti, R.; Marchand-Leroux, C.; Baud, O.; et al. Sex differences in the effects of PARP inhibition on microglial phenotypes following neonatal stroke. Brain Behav. Immun. 2018, 73, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, X.; Xu, S.; Zhang, M.; Wu, Z.; Zhang, X.; Xu, Y.; Chen, Y. Delayed PARP-1 inhibition alleviates post-stroke inflammation in male versus female mice: Differences and similarities. Front. Cell Neurosci. 2020, 14, 77. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, S.; Egi, Y.; Yuki, S.; Horikawa, T.; Satoh, H.; Akira, T. MP-124, a novel poly(ADP-ribose) polymerase-1 (PARP-1) inhibitor, ameliorates ischemic brain damage in a non-human primate model. Brain Res. 2011, 1410, 122–131. [Google Scholar] [CrossRef]
- Meier-Soelch, J.; Mayr-Buro, C.; Juli, J.; Leib, L.; Linne, U.; Dreute, J.; Papantonis, A.; Schmitz, M.L.; Kracht, M. Monitoring the levels of cellular NF-κB activation states. Cancers 2021, 13, 5351. [Google Scholar] [CrossRef] [PubMed]
- Lefloch, R.; Pouyssegur, J.; Lenormand, P. Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol. Cell Biol. 2008, 28, 511–527. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Zito, G.; Buscetta, M.; Cimino, M.; Dino, P.; Bucchieri, F.; Cipollina, C. Cellular models and assays to study NLRP3 inflammasome biology. Int. J. Mol. Sci. 2020, 21, 4294. [Google Scholar] [CrossRef]

























Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, V.; Santos, S.S.; Rodrigues, L.d.O.C.P.; Salomao, R.; Liaudet, L.; Szabo, C. Efficacy of Clinically Used PARP Inhibitors in a Murine Model of Acute Lung Injury. Cells 2022, 11, 3789. https://doi.org/10.3390/cells11233789
Martins V, Santos SS, Rodrigues LdOCP, Salomao R, Liaudet L, Szabo C. Efficacy of Clinically Used PARP Inhibitors in a Murine Model of Acute Lung Injury. Cells. 2022; 11(23):3789. https://doi.org/10.3390/cells11233789
Chicago/Turabian StyleMartins, Vanessa, Sidneia S. Santos, Larissa de O. C. P. Rodrigues, Reinaldo Salomao, Lucas Liaudet, and Csaba Szabo. 2022. "Efficacy of Clinically Used PARP Inhibitors in a Murine Model of Acute Lung Injury" Cells 11, no. 23: 3789. https://doi.org/10.3390/cells11233789
APA StyleMartins, V., Santos, S. S., Rodrigues, L. d. O. C. P., Salomao, R., Liaudet, L., & Szabo, C. (2022). Efficacy of Clinically Used PARP Inhibitors in a Murine Model of Acute Lung Injury. Cells, 11(23), 3789. https://doi.org/10.3390/cells11233789