
Metabolism in Cancer Stem Cells: Targets for Clinical Treatment


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Overview of Cancer Metabolism
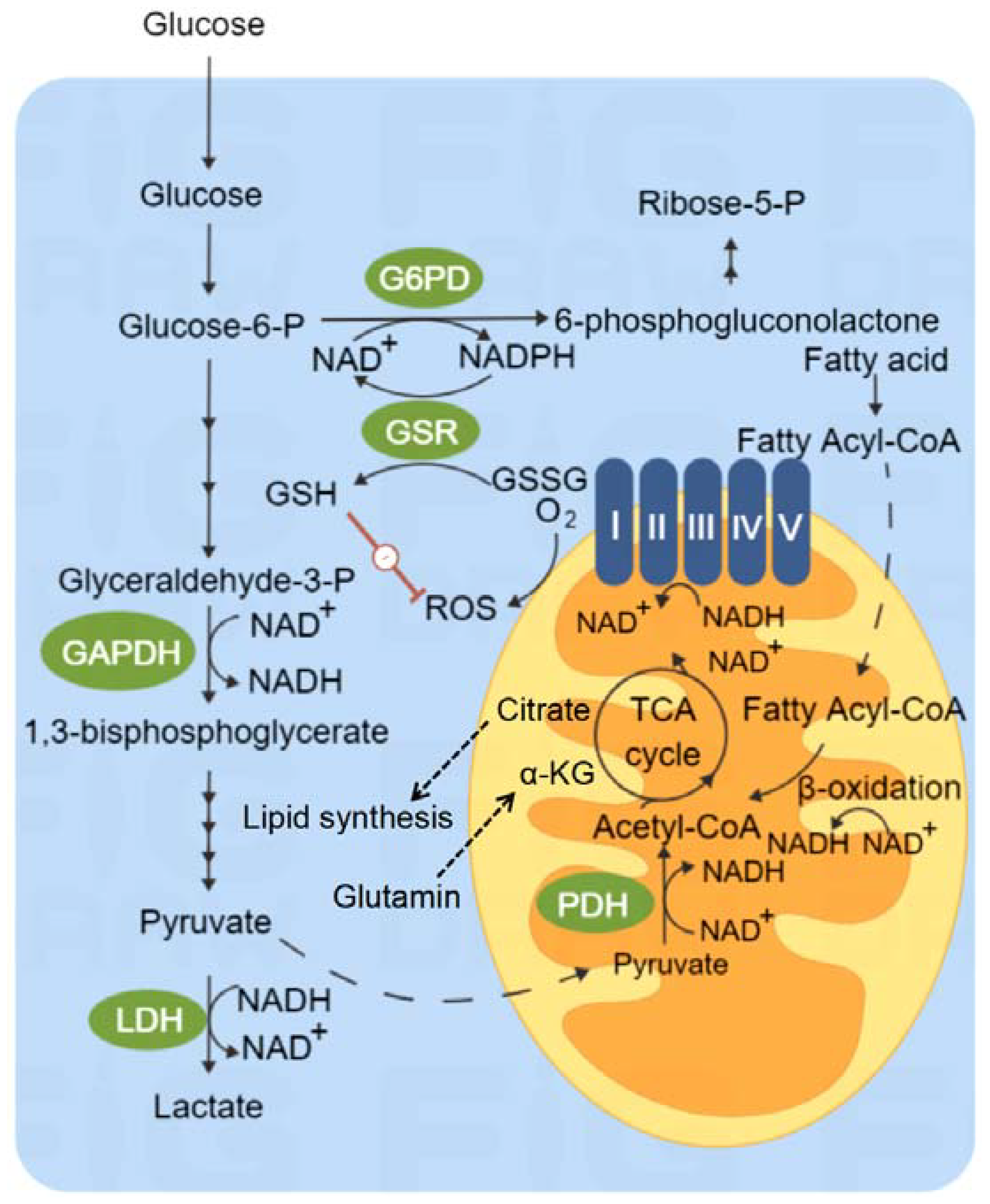
3. Glucose Metabolism
4. Mitochondrial Metabolism
5. Glutamine Metabolism
6. Lipid Metabolism
7. Iron Metabolism and Ferroptosis
8. Metabolism of ALDH1 Positive Cancer Stem Cells
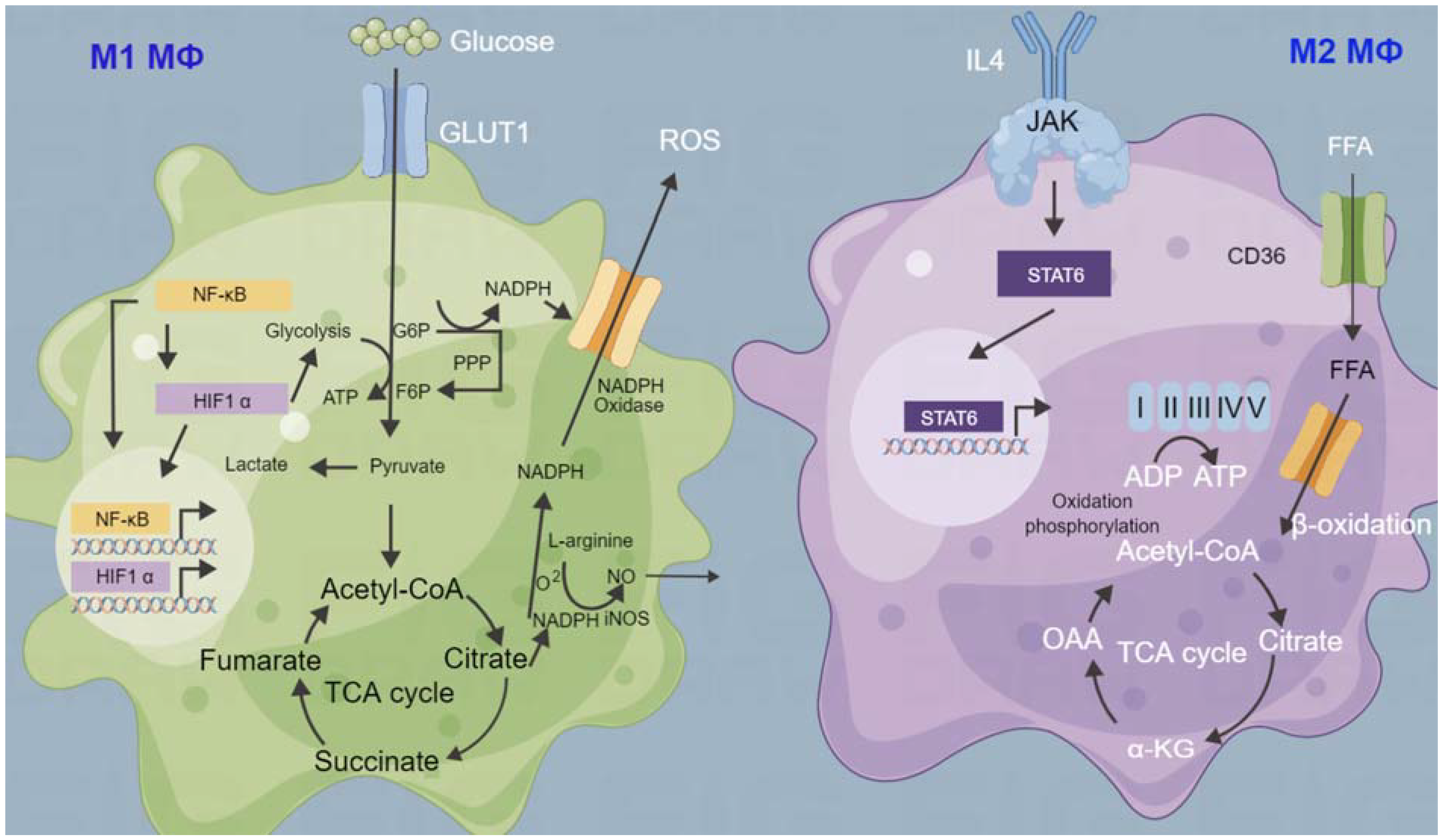
9. Tumor Microenvironment and Metabolism
10. Cancer Stem Cell Metabolism and Treatment
11. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells: Current status and evolving complexities. Cell Stem Cell 2012, 10, 717–728. [Google Scholar] [CrossRef]
- Schatton, T.; Frank, N.Y.; Frank, M.H. Identification and targeting of cancer stem cells. Bioessays 2009, 31, 1038–1049. [Google Scholar] [CrossRef]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Skvortsov, S.; Debbage, P.; Skvortsova, I. Proteomics of cancer stem cells. Int. J. Radiat. Biol. 2014, 90, 653–658. [Google Scholar] [CrossRef]
- Jagust, P.; de Luxán-Delgado, B.; Parejo-Alonso, B.; Sancho, P. Metabolism-Based Therapeutic Strategies Targeting Cancer Stem Cells. Front. Pharmacol. 2019, 10, 203. [Google Scholar] [CrossRef]
- López de Andrés, J.; Griñán-Lisón, C.; Jiménez, G.; Marchal, J.A. Cancer stem cell secretome in the tumor microenvironment: A key point for an effective personalized cancer treatment. J. Hematol. Oncol. 2020, 13, 136. [Google Scholar] [CrossRef]
- Menendez, J.A. Metabolic control of cancer cell stemness: Lessons from iPS cells. Cell Cycle 2015, 14, 3801–3811. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, C. Tracking down the origin of cancer: Metabolic reprogramming as a driver of stemness and tumorigenesis. Crit. Rev. Oncog. 2014, 19, 363–382. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The emerging hallmarks of cancer metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Fendt, S.M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Johar, D.; Elmehrath, A.O.; Khalil, R.M.; Elberry, M.H.; Zaky, S.; Shalabi, S.A.; Bernstein, L.H. Protein networks linking Warburg and reverse Warburg effects to cancer cell metabolism. Biofactors 2021, 47, 713–728. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.E.; Krebs, H. The evolution of metabolic cycles. Nature 1981, 291, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Shi, J.; Qin, X.; Zheng, Z.; Chen, M.; Lin, Z.; Ye, J.; Li, M. Hormone-Glutamine Metabolism: A Critical Regulatory Axis in Endocrine-Related Cancers. Int. J. Mol. Sci. 2022, 23, 10086. [Google Scholar] [CrossRef]
- Erin, N.; Grahovac, J.; Brozovic, A.; Efferth, T. Tumor microenvironment and epithelial mesenchymal transition as targets to overcome tumor multidrug resistance. Drug Resist. Updat. 2020, 53, 100715. [Google Scholar] [CrossRef]
- Jinesh, G.G.; Brohl, A.S. Classical epithelial-mesenchymal transition (EMT) and alternative cell death process-driven blebbishield metastatic-witch (BMW) pathways to cancer metastasis. Signal Transduct. Target. Ther. 2022, 7, 296. [Google Scholar] [CrossRef]
- Sun, H.; Yang, X.; Liang, L.; Zhang, M.; Li, Y.; Chen, J.; Wang, F.; Yang, T.; Meng, F.; Lai, X.; et al. Metabolic switch and epithelial-mesenchymal transition cooperate to regulate pluripotency. EMBO J. 2020, 39, e102961. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Li, Q.; Wu, F.; Lin, J.; Chen, J.; Zheng, H.; Guo, L. Epithelial-Mesenchymal Transition and Metabolic Switching in Cancer: Lessons From Somatic Cell Reprogramming. Front. Cell Dev. Biol. 2020, 8, 760. [Google Scholar] [CrossRef]
- Wahl, G.M.; Spike, B.T. Cell state plasticity, stem cells, EMT, and the generation of intra-tumoral heterogeneity. NPJ Breast Cancer 2017, 3, 14. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zeng, Y.; Hu, L.; Hao, J.; Chen, Y.; Yun, X.; Lin, Q.; Li, H. Based on different immune responses under the glucose metabolizing type of papillary thyroid cancer and the response to anti-PD-1 therapy. Front. Immunol. 2022, 13, 991656. [Google Scholar] [CrossRef]
- Balakrishnan, K. Hepatocellular carcinoma stage: An almost loss of fatty acid metabolism and gain of glucose metabolic pathways dysregulation. Med. Oncol. 2022, 39, 247. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Coles, G.; Bröer, A.; McLeod, M.D.; George, A.J.; Hannan, R.D.; Bröer, S. Identification and characterization of a novel SNAT2 (SLC38A2) inhibitor reveals synergy with glucose transport inhibition in cancer cells. Front. Pharmacol. 2022, 13, 963066. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Hu, C.M.; Hsu, Y.S.; Lee, W.H. Interplays of glucose metabolism and KRAS mutation in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022, 13, 817. [Google Scholar] [CrossRef]
- Vinaik, R.; Barayan, D.; Auger, C.; Abdullahi, A.; Jeschke, M.G. Regulation of glycolysis and the Warburg effect in wound healing. JCI Insight 2020, 5, e138949. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg effect: How does it benefit cancer cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Abad, E.; Samino, S.; Yanes, O.; Potesil, D.; Zdrahal, Z.; Lyakhovich, A. Activation of glycogenolysis and glycolysis in breast cancer stem cell models. Biochim. Biophys. Acta. Mol. Basis Dis. 2020, 1866, 165886. [Google Scholar] [CrossRef]
- Mulukutla, B.C.; Yongky, A.; Le, T.; Mashek, D.G.; Hu, W.S. Regulation of Glucose Metabolism—A Perspective From Cell Bioprocessing. Trends Biotechnol. 2016, 34, 638–651. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol. Life Sci. 2016, 73, 377–392. [Google Scholar] [CrossRef]
- Hopp, A.K.; Grüter, P.; Hottiger, M.O. Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells 2019, 8, 890. [Google Scholar] [CrossRef]
- Walsh, H.R.; Cruickshank, B.M.; Brown, J.M.; Marcato, P. The Flick of a Switch: Conferring Survival Advantage to Breast Cancer Stem Cells Through Metabolic Plasticity. Front. Oncol. 2019, 9, 753. [Google Scholar] [CrossRef] [PubMed]
- Nozawa-Suzuki, N.; Nagasawa, H.; Ohnishi, K.; Morishige, K. The inhibitory effect of hypoxic cytotoxin on the expansion of cancer stem cells in ovarian cancer. Biochem. Biophys. Res. Commun. 2015, 457, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.H.; Lai, A.G. Aberrations in Notch-Hedgehog signalling reveal cancer stem cells harbouring conserved oncogenic properties associated with hypoxia and immunoevasion. Br. J. Cancer 2019, 121, 666–678. [Google Scholar] [CrossRef]
- Tirinato, L.; Pagliari, F.; Di Franco, S.; Sogne, E.; Marafioti, M.G.; Jansen, J.; Falqui, A.; Todaro, M.; Candeloro, P.; Liberale, C.; et al. ROS and Lipid Droplet accumulation induced by high glucose exposure in healthy colon and Colorectal Cancer Stem Cells. Genes Dis. 2019, 7, 620–635. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, M.; Inoue, T.; Shirai, T.; Takamatsu, K.; Kunihiro, S.; Ishii, H.; Nishikata, T. Simultaneous expression of cancer stem cell-like properties and cancer-associated fibroblast-like properties in a primary culture of breast cancer cells. Cancers 2014, 6, 1570–1578. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, I.G.; Choi, B.H.; Ku, S.K.; Kwak, M.K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef]
- Icard, P.; Simula, L.; Wu, Z.; Berzan, D.; Sogni, P.; Dohan, A.; Dautry, R.; Coquerel, A.; Lincet, H.; Loi, M.; et al. Why may citrate sodium significantly increase the effectiveness of transarterial chemoembolization in hepatocellular carcinoma? Drug Resist. Updat. 2021, 59, 100790. [Google Scholar] [CrossRef]
- Shen, Y.A.; Wang, C.Y.; Hsieh, Y.T.; Chen, Y.J.; Wei, Y.H. Metabolic reprogramming orchestrates cancer stem cell properties in nasopharyngeal carcinoma. Cell Cycle 2015, 14, 86–98. [Google Scholar] [CrossRef]
- Ji, C.C.; Hu, Y.Y.; Cheng, G.; Liang, L.; Gao, B.; Ren, Y.P.; Liu, J.T.; Cao, X.L.; Zheng, M.H.; Li, S.Z.; et al. A ketogenic diet attenuates proliferation and stemness of glioma stem-like cells by altering metabolism resulting in increased ROS production. Int. J. Oncol. 2020, 56, 606–617. [Google Scholar] [CrossRef]
- Birk, A.V.; Chao, W.M.; Bracken, C.; Warren, J.D.; Szeto, H.H. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br. J. Pharmacol. 2014, 171, 2017–2028. [Google Scholar] [CrossRef]
- Fernández-Vizarra, E.; López-Calcerrada, S.; Sierra-Magro, A.; Pérez-Pérez, R.; Formosa, L.E.; Hock, D.H.; Illescas, M.; Peñas, A.; Brischigliaro, M.; Ding, S.; et al. Two independent respiratory chains adapt OXPHOS performance to glycolytic switch. Cell Metab. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Karp, I.; Lyakhovich, A. Targeting cancer stem cells with antibiotics inducing mitochondrial dysfunction as an alternative anticancer therapy. Biochem. Pharmacol. 2022, 198, 114966. [Google Scholar] [CrossRef]
- De Luca, A.; Fiorillo, M.; Peiris-Pagès, M.; Ozsvari, B.; Smith, D.L.; Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Cappello, A.R.; Pezzi, V.; Lisanti, M.P.; et al. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget 2015, 6, 14777–14795. [Google Scholar] [CrossRef]
- Vikramdeo, K.S.; Sudan, S.K.; Singh, A.P.; Singh, S.; Dasgupta, S. Mitochondrial respiratory complexes: Significance in human mitochondrial disorders and cancers. J. Cell Physiol. 2022. Online ahead of print. [Google Scholar] [CrossRef]
- Lavorato, M.; Nakamaru-Ogiso, E.; Mathew, N.D.; Herman, E.; Shah, N.; Haroon, S.; Xiao, R.; Seiler, C.; Falk, M.J. Dichloroacetate improves mitochondrial function, physiology, and morphology in FBXL4 disease models. JCI Insight 2022, 7, e156346. [Google Scholar] [CrossRef]
- Shen, H.; Decollogne, S.; Dilda, P.J.; Hau, E.; Chung, S.A.; Luk, P.P.; Hogg, P.J.; McDonald, K.L. Dual-targeting of aberrant glucose metabolism in glioblastoma. J. Exp. Clin. Cancer Res. 2015, 34, 14. [Google Scholar] [CrossRef] [PubMed]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Pinho, S.A.; Afonso, G.J.M.; Oliveira, P.J.; Cunha-Oliveira, T.; Saso, L. NRF2 and Mitochondrial Function in Cancer and Cancer Stem Cells. Cells 2022, 11, 2401. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Qin, S.; Townsend, D.; Schulte, B.A.; Tew, K.D.; Wang, G.Y. Oxidative stress induces senescence in breast cancer stem cells. Biochem. Biophys. Res. Commun. 2019, 514, 1204–1209. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Metabolic and mind shifts: From glucose to glutamine and acetate addictions in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 346–353. [Google Scholar] [CrossRef]
- Dringen, R.; Brandmann, M.; Hohnholt, M.C.; Blumrich, E.M. Glutathione-Dependent Detoxification Processes in Astrocytes. Neurochem. Res. 2015, 40, 2570–2582. [Google Scholar] [CrossRef] [PubMed]
- Claiborne, M.D.; Leone, R. Differential glutamine metabolism in the tumor microenvironment—Studies in diversity and heterogeneity: A mini-review. Front. Oncol. 2022, 12, 1011191. [Google Scholar] [CrossRef] [PubMed]
- Jogo, T.; Oki, E.; Nakanishi, R.; Ando, K.; Nakashima, Y.; Kimura, Y.; Saeki, H.; Oda, Y.; Maehara, Y.; Mori, M. Expression of CD44 variant 9 induces chemoresistance of gastric cancer by controlling intracellular reactive oxygen spices accumulation. Gastric. Cancer 2021, 24, 1089–1099. [Google Scholar] [CrossRef]
- Ishimoto, T.; Nagano, O.; Yae, T.; Tamada, M.; Motohara, T.; Oshima, H.; Oshima, M.; Ikeda, T.; Asaba, R.; Yagi, H.; et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell 2011, 19, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Vidal, M.; Gasol, E.; Zorzano, A.; Nunes, V.; Palacín, M.; Chillarón, J. Thiol modification of cysteine 327 in the eighth transmembrane domain of the light subunit xCT of the heteromeric cystine/glutamate antiporter suggests close proximity to the substrate binding site/permeation pathway. J. Biol. Chem. 2004, 279, 11214–11221. [Google Scholar] [CrossRef]
- Wada, F.; Koga, H.; Akiba, J.; Niizeki, T.; Iwamoto, H.; Ikezono, Y.; Nakamura, T.; Abe, M.; Masuda, A.; Sakaue, T.; et al. High expression of CD44v9 and xCT in chemoresistant hepatocellular carcinoma: Potential targets by sulfasalazine. Cancer Sci. 2018, 109, 2801–2810. [Google Scholar] [CrossRef]
- Sugano, K.; Maeda, K.; Ohtani, H.; Nagahara, H.; Shibutani, M.; Hirakawa, K. Expression of xCT as a predictor of disease recurrence in patients with colorectal cancer. Anticancer Res. 2015, 35, 677–682. [Google Scholar] [PubMed]
- Lanzardo, S.; Conti, L.; Rooke, R.; Ruiu, R.; Accart, N.; Bolli, E.; Arigoni, M.; Macagno, M.; Barrera, G.; Pizzimenti, S.; et al. Immunotargeting of Antigen xCT Attenuates Stem-like Cell Behavior and Metastatic Progression in Breast Cancer. Cancer Res. 2016, 76, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Qian, J.; Rahman, S.M.J.; Siska, P.J.; Zou, Y.; Harris, B.K.; Hoeksema, M.D.; Trenary, I.A.; Heidi, C.; Eisenberg, R.; et al. xCT (SLC7A11)-mediated metabolic reprogramming promotes non-small cell lung cancer progression. Oncogene 2018, 37, 5007–5019. [Google Scholar] [CrossRef]
- Zhong, W.; Weiss, H.L.; Jayswal, R.D.; Hensley, P.J.; Downes, L.M.; St Clair, D.K.; Chaiswing, L. Extracellular redox state shift: A novel approach to target prostate cancer invasion. Free Radic. Biol. Med. 2018, 117, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Polewski, M.D.; Reveron-Thornton, R.F.; Cherryholmes, G.A.; Marinov, G.K.; Aboody, K.S. SLC7A11 Overexpression in Glioblastoma Is Associated with Increased Cancer Stem Cell-Like Properties. Stem Cells Dev. 2017, 26, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Samanta, D.; Xiang, L.; Zhang, H.; Hu, H.; Chen, I.; Bullen, J.W.; Semenza, G.L. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. USA 2015, 112, E4600–E4609. [Google Scholar] [CrossRef]
- Nagarajan, S.R.; Butler, L.M.; Hoy, A.J. The diversity and breadth of cancer cell fatty acid metabolism. Cancer Metab. 2021, 9, 2. [Google Scholar] [CrossRef]
- Günenc, A.N.; Graf, B.; Stark, H.; Chari, A. Fatty Acid Synthase: Structure, Function, and Regulation. Subcell Biochem. 2022, 99, 1–33. [Google Scholar]
- Qu, Q.; Zeng, F.; Liu, X.; Wang, Q.J.; Deng, F. Fatty acid oxidation and carnitine palmitoyltransferase I: Emerging therapeutic targets in cancer. Cell Death Dis. 2016, 7, e2226. [Google Scholar] [CrossRef]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef]
- Sun, M.; Yang, Z. Metabolomic Studies of Live Single Cancer Stem Cells Using Mass Spectrometry. Anal. Chem. 2019, 91, 2384–2391. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Z.; Song, L.; Gao, J.; Liu, Y. Lipid metabolism of cancer stem cells. Oncol. Lett. 2022, 23, 119. [Google Scholar] [CrossRef]
- Bort, A.; Sánchez, B.G.; de Miguel, I.; Mateos-Gómez, P.A.; Diaz-Laviada, I. Dysregulated lipid metabolism in hepatocellular carcinoma cancer stem cells. Mol. Biol. Rep. 2020, 47, 2635–2647. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Park, J.H.; Park, J.; Son, D.M.; Baek, J.Y.; Jang, H.J.; Jung, W.K.; Byun, Y.; Kim, S.K.; Park, S.K. Stereospecific inhibition of AMPK by (R)-crizotinib induced changes to the morphology and properties of cancer and cancer stem cell-like cells. Eur. J. Pharmacol. 2021, 911, 174525. [Google Scholar] [CrossRef]
- Yeh, S.C.; Wang, P.Y.; Lou, Y.W.; Khoo, K.H.; Hsiao, M.; Hsu, T.L.; Wong, C.H. Glycolipid GD3 and GD3 synthase are key drivers for glioblastoma stem cells and tumorigenicity. Proc. Natl. Acad. Sci. USA 2016, 113, 5592–5597. [Google Scholar] [CrossRef]
- Juarez, D.; Fruman, D.A. Targeting the Mevalonate Pathway in Cancer. Trends Cancer 2021, 7, 525–540. [Google Scholar] [CrossRef]
- Guerra, B.; Recio, C.; Aranda-Tavío, H.; Guerra-Rodríguez, M.; García-Castellano, J.M.; Fernández-Pérez, L. The Mevalonate Pathway, a Metabolic Target in Cancer Therapy. Front. Oncol. 2021, 11, 626971. [Google Scholar] [CrossRef]
- Trub, A.G.; Wagner, G.R.; Anderson, K.A.; Crown, S.B.; Zhang, G.F.; Thompson, J.W.; Ilkayeva, O.R.; Stevens, R.D.; Grimsrud, P.A.; Kulkarni, R.A.; et al. Statin therapy inhibits fatty acid synthase via dynamic protein modifications. Nat. Commun. 2022, 13, 2542. [Google Scholar] [CrossRef]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef]
- Mancini, R.; Noto, A.; Pisanu, M.E.; De Vitis, C.; Maugeri-Saccà, M.; Ciliberto, G. Metabolic features of cancer stem cells: The emerging role of lipid metabolism. Oncogene 2018, 37, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Iannelli, F.; Roca, M.S.; Lombardi, R.; Ciardiello, C.; Grumetti, L.; De Rienzo, S.; Moccia, T.; Vitagliano, C.; Sorice, A.; Costantini, S.; et al. Synergistic antitumor interaction of valproic acid and simvastatin sensitizes prostate cancer to docetaxel by targeting CSCs compartment via YAP inhibition. J. Exp. Clin. Cancer Res. 2020, 39, 213. [Google Scholar] [CrossRef] [PubMed]
- Sharon, C.; Baranwal, S.; Patel, N.J.; Rodriguez-Agudo, D.; Pandak, W.M.; Majumdar, A.P.; Krystal, G.; Patel, B.B. Inhibition of insulin-like growth factor receptor/AKT/mammalian target of rapamycin axis targets colorectal cancer stem cells by attenuating mevalonate-isoprenoid pathway in vitro and in vivo. Oncotarget 2015, 6, 15332–15347. [Google Scholar] [CrossRef]
- Brandi, J.; Dando, I.; Pozza, E.D.; Biondani, G.; Jenkins, R.; Elliott, V.; Park, K.; Fanelli, G.; Zolla, L.; Costello, E.; et al. Proteomic analysis of pancreatic cancer stem cells: Functional role of fatty acid synthesis and mevalonate pathways. J. Proteom. 2017, 150, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Ehmsen, S.; Pedersen, M.H.; Wang, G.; Terp, M.G.; Arslanagic, A.; Hood, B.L.; Conrads, T.P.; Leth-Larsen, R.; Ditzel, H.J. Increased Cholesterol Biosynthesis Is a Key Characteristic of Breast Cancer Stem Cells Influencing Patient Outcome. Cell Rep. 2019, 27, 3927–3938.e6. [Google Scholar] [CrossRef] [PubMed]
- Richardson, D.R.; Kalinowski, D.S.; Lau, S.; Jansson, P.J.; Lovejoy, D.B. Cancer cell iron metabolism and the development of potent iron chelators as anti-tumour agents. Biochim. Biophys. Acta 2009, 1790, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Torti, S.V.; Torti, F.M. Iron and cancer: More ore to be mined. Nat. Rev. Cancer 2013, 13, 342–355. [Google Scholar] [CrossRef] [PubMed]
- Levina, A.; Chetcuti, A.R.M.; Lay, P.A. Controversial Role of Transferrin in the Transport of Ruthenium Anticancer Drugs. Biomolecules 2022, 12, 1319. [Google Scholar] [CrossRef]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Cui, C.; Cheng, X.; Yan, L.; Ding, H.; Guan, X.; Zhang, W.; Tian, X.; Hao, C. Downregulation of TfR1 promotes progression of colorectal cancer via the JAK/STAT pathway. Cancer Manag. Res. 2019, 11, 6323–6341. [Google Scholar] [CrossRef]
- Zhu, B.; Zhi, Q.; Xie, Q.; Wu, X.; Gao, Y.; Chen, X.; Shi, L. Reduced expression of ferroportin1 and ceruloplasmin predicts poor prognosis in adrenocortical carcinoma. J. Trace Elem. Med. Biol. 2019, 56, 52–59. [Google Scholar] [CrossRef]
- Tseng, H.H.; Chang, J.G.; Hwang, Y.H.; Yeh, K.T.; Chen, Y.L.; Yu, H.S. Expression of hepcidin and other iron-regulatory genes in human hepatocellular carcinoma and its clinical implications. J. Cancer Res. Clin. Oncol. 2009, 135, 1413–1420. [Google Scholar] [CrossRef]
- Khiroya, H.; Moore, J.S.; Ahmad, N.; Kay, J.; Woolnough, K.; Langman, G.; Ismail, I.; Naidu, B.; Tselepis, C.; Turner, A.M. IRP2 as a potential modulator of cell proliferation, apoptosis and prognosis in nonsmall cell lung cancer. Eur. Respir. J. 2017, 49, 1600711. [Google Scholar] [CrossRef]
- Mu, L.M.; Bu, Y.Z.; Liu, L.; Xie, H.J.; Ju, R.J.; Wu, J.S.; Zeng, F.; Zhao, Y.; Zhang, J.Y.; Lu, W.L. Lipid vesicles containing transferrin receptor binding peptide TfR-T12 and octa-arginine conjugate stearyl-R8 efficiently treat brain glioma along with glioma stem cells. Sci. Rep. 2017, 7, 3487. [Google Scholar] [CrossRef]
- Abuzaid, H.; Abdelrazig, S.; Ferreira, L.; Collins, H.M.; Kim, D.H.; Lim, K.H.; Kam, T.S.; Turyanska, L.; Bradshaw, T.D. Apoferritin-Encapsulated Jerantinine A for Transferrin Receptor Targeting and Enhanced Selectivity in Breast Cancer Therapy. ACS Omega 2022, 7, 21473–21482. [Google Scholar] [CrossRef] [PubMed]
- Chanvorachote, P.; Luanpitpong, S. Iron induces cancer stem cells and aggressive phenotypes in human lung cancer cells. Am. J. Physiol. Cell Physiol. 2016, 310, C728–C739. [Google Scholar] [CrossRef] [PubMed]
- Ozer, U. The role of Iron on breast cancer stem-like cells. Cell Mol. Biol. 2016, 62, 25–30. [Google Scholar] [PubMed]
- Mirhadi, S.; Zhang, W.; Pham, N.A.; Karimzadeh, F.; Pintilie, M.; Tong, J.; Taylor, P.; Krieger, J.; Pitcher, B.; Sykes, J.; et al. Mitochondrial aconitase ACO2 links iron homeostasis with tumorigenicity in non-small cell lung cancer. Mol. Cancer Res. 2022. MCR-22-0163. [Google Scholar] [CrossRef]
- Raggi, C.; Gammella, E.; Correnti, M.; Buratti, P.; Forti, E.; Andersen, J.B.; Alpini, G.; Glaser, S.; Alvaro, D.; Invernizzi, P.; et al. Dysregulation of Iron Metabolism in Cholangiocarcinoma Stem-like Cells. Sci. Rep. 2017, 7, 17667. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Mu, Y.; Lu, C.; Tang, S.; Lu, K.; Qiu, X.; Wei, A.; Cheng, Y.; Wei, W. The iron chelator desferrioxamine synergizes with chemotherapy for cancer treatment. J. Trace Elem. Med. Biol. 2019, 56, 131–138. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Zhu, Y.; Wu, Z.; Cui, C.; Cai, F. Anticancer Mechanisms of Salinomycin in Breast Cancer and Its Clinical Applications. Front. Oncol. 2021, 11, 654428. [Google Scholar] [CrossRef]
- Mai, T.T.; Hamaï, A.; Hienzsch, A.; Cañeque, T.; Müller, S.; Wicinski, J.; Cabaud, O.; Leroy, C.; David, A.; Acevedo, V.; et al. Salinomycin kills cancer stem cells by sequestering iron in lysosomes. Nat. Chem. 2017, 9, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.R.; Fedorka, S.R.; Gad, I.; Shah, R.; Alqahtani, H.D.; Koranne, R.; Kuganesan, N.; Dlamini, S.; Rogers, T.; Al-Hamashi, A.; et al. Small-Molecule Ferroptotic Agents with Potential to Selectively Target Cancer Stem Cells. Sci. Rep. 2019, 9, 5926. [Google Scholar] [CrossRef]
- Liu, M.R.; Zhu, W.T.; Pei, D.S. System Xc−: A key regulatory target of ferroptosis in cancer. Investig. New Drugs 2021, 39, 1123–1131. [Google Scholar] [CrossRef]
- Basuli, D.; Tesfay, L.; Deng, Z.; Paul, B.; Yamamoto, Y.; Ning, G.; Xian, W.; McKeon, F.; Lynch, M.; Crum, C.P.; et al. Iron addiction: A novel therapeutic target in ovarian cancer. Oncogene 2017, 36, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Shepherd, T.G.; Hoskin, D.W. Contribution of reactive oxygen species to ovarian cancer cell growth arrest and killing by the anti-malarial drug artesunate. Mol. Carcinog. 2017, 56, 75–93. [Google Scholar] [CrossRef] [PubMed]
- Shitara, K.; Doi, T.; Nagano, O.; Imamura, C.K.; Ozeki, T.; Ishii, Y.; Tsuchihashi, K.; Takahashi, S.; Nakajima, T.E.; Hironaka, S.; et al. Dose-escalation study for the targeting of CD44v+ cancer stem cells by sulfasalazine in patients with advanced gastric cancer (EPOC1205). Gastric. Cancer 2017, 20, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cui, D.; Ye, L.; Li, Y.; Zhu, L.; Yang, L.; Bai, B.; Nie, Z.; Gao, J.; Cao, Y. Codelivery of salinomycin and docetaxel using poly(D,L-lactic-co-glycolic acid)-poly(ethylene glycol) nanoparticles to target both gastric cancer cells and cancer stem cells. Anticancer Drugs 2017, 28, 989–1001. [Google Scholar] [CrossRef]
- Ju, H.Q.; Lu, Y.X.; Chen, D.L.; Tian, T.; Mo, H.Y.; Wei, X.L.; Liao, J.W.; Wang, F.; Zeng, Z.L.; Pelicano, H.; et al. Redox Regulation of Stem-like Cells Though the CD44v-xCT Axis in Colorectal Cancer: Mechanisms and Therapeutic Implications. Theranostics 2016, 6, 1160–1175. [Google Scholar] [CrossRef]
- Buccarelli, M.; Marconi, M.; Pacioni, S.; De Pascalis, I.; D’Alessandris, Q.G.; Martini, M.; Ascione, B.; Malorni, W.; Larocca, L.M.; Pallini, R.; et al. Inhibition of autophagy increases susceptibility of glioblastoma stem cells to temozolomide by igniting ferroptosis. Cell Death Dis. 2018, 9, 841. [Google Scholar] [CrossRef]
- Monteleone, L.; Speciale, A.; Valenti, G.E.; Traverso, N.; Ravera, S.; Garbarino, O.; Leardi, R.; Farinini, E.; Roveri, A.; Ursini, F.; et al. PKCα Inhibition as a Strategy to Sensitize Neuroblastoma Stem Cells to Etoposide by Stimulating Ferroptosis. Antioxidants 2021, 10, 691. [Google Scholar] [CrossRef]
- Jackson, B.; Brocker, C.; Thompson, D.C.; Black, W.; Vasiliou, K.; Nebert, D.W.; Vasiliou, V. Update on the aldehyde dehydrogenase gene (ALDH) superfamily. Hum. Genom. 2011, 5, 283–303. [Google Scholar] [CrossRef]
- Muzio, G.; Maggiora, M.; Paiuzzi, E.; Oraldi, M.; Canuto, R.A. Aldehyde dehydrogenases and cell proliferation. Free Radic. Biol. Med. 2012, 52, 735–746. [Google Scholar] [CrossRef]
- Napoli, J.L. Retinoic acid: Its biosynthesis and metabolism. Prog. Nucleic Acid Res. Mol. Biol. 1999, 63, 139–188. [Google Scholar]
- Huo, W.; Du, M.; Pan, X.; Zhu, X.; Li, Z. Prognostic value of ALDH1 expression in lung cancer: A meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 2045–2051. [Google Scholar] [PubMed]
- Guan, X.; Dong, Y.; Fan, Z.; Zhan, Y.; Xie, X.; Xu, G.; Zhang, Y.; Guo, G.; Shi, A. Aldehyde dehydrogenase 1 (ALDH1) immunostaining in axillary lymph node metastases is an independent prognostic factor in ALDH1-positive breast cancer. J. Int. Med. Res. 2021, 49, 3000605211047279. [Google Scholar] [CrossRef]
- Islam, F.; Gopalan, V.; Lam, A.K. Detention and Identification of Cancer Stem Cells in Esophageal Squamous Cell Carcinoma. Methods Mol. Biol. 2020, 2129, 177–191. [Google Scholar]
- Mahmood, N.A.; Abdulghany, Z.S.; Al-Sudani, I.M. Expression of Aldehyde Dehydrogenase (ALDH1) and ATP Binding Cassette Transporter G2 (ABCG2) in Iraqi Patients with Colon Cancer and the Relation with Clinicopathological Features. Int. J. Mol. Cell Med. 2018, 7, 234–240. [Google Scholar]
- Cui, J.; Li, W.; Bu, W.; Liu, J.; Chen, X.; Li, X.; Liu, C.; Meng, L.; Chen, M.; Sun, H.; et al. Folic acid-modified disulfiram/Zn-IRMOF3 nanoparticles for oral cancer therapy by inhibiting ALDH1A1+ cancer stem cells. Biomater Adv. 2022, 139, 213038. [Google Scholar] [CrossRef]
- Takebe, N.; Zhao, S.C.; Adhikari, D.; Mineishi, S.; Sadelain, M.; Hilton, J.; Colvin, M.; Banerjee, D.; Bertino, J.R. Generation of dual resistance to 4-hydroperoxycyclophosphamide and methotrexate by retroviral transfer of the human aldehyde dehydrogenase class 1 gene and a mutated dihydrofolate reductase gene. Mol. Ther. 2001, 3, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Konno, M.; Hamabe, A.; Hasegawa, S.; Kano, Y.; Ohta, K.; Fukusumi, T.; Sakai, D.; Kudo, T.; Haraguchi, N.; et al. Aldehyde dehydrogenase high gastric cancer stem cells are resistant to chemotherapy. Int. J. Oncol. 2013, 42, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Qiang, J.; Deng, Q.; Xia, J.; Deng, L.; Zhou, L.; Wang, D.; He, X.; Liu, Y.; Zhao, B.; et al. ALDH1A1 Activity in Tumor-Initiating Cells Remodels Myeloid-Derived Suppressor Cells to Promote Breast Cancer Progression. Cancer Res. 2021, 81, 5919–5934. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.; Kaufmann, A.M.; Qian, X.; Sangvatanakul, V.; Chen, C.; Kube, T.; Zhang, G.; Albers, A.E. Susceptibility to cytotoxic T cell lysis of cancer stem cells derived from cervical and head and neck tumor cell lines. J. Cancer Res. Clin. Oncol. 2013, 139, 159–170. [Google Scholar] [CrossRef]
- Fridman, W.H.; Zitvogel, L.; Sautès-Fridman, C.; Kroemer, G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Kapoor-Narula, U.; Lenka, N. Cancer stem cells and tumor heterogeneity: Deciphering the role in tumor progression and metastasis. Cytokine 2022, 157, 155968. [Google Scholar] [CrossRef] [PubMed]
- Jacobsson, H.; Harrison, H.; Hughes, É.; Persson, E.; Rhost, S.; Fitzpatrick, P.; Gustafsson, A.; Andersson, D.; Gregersson, P.; Magnusson, Y.; et al. Hypoxia-induced secretion stimulates breast cancer stem cell regulatory signalling pathways. Mol. Oncol. 2019, 13, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhao, Y.; Wu, X.; Liu, Z.; Liu, X. A novel strategy to fuel cancer immunotherapy: Targeting glucose metabolism to remodel the tumor microenvironment. Front. Oncol. 2022, 12, 931104. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Tanaka, N. IL-8-induced O-GlcNAc modification via GLUT3 and GFAT regulates cancer stem cell-like properties in colon and lung cancer cells. Oncogene 2019, 38, 1520–1533. [Google Scholar] [CrossRef]
- Wang, T.; Fahrmann, J.F.; Lee, H.; Li, Y.J.; Tripathi, S.C.; Yue, C.; Zhang, C.; Lifshitz, V.; Song, J.; Yuan, Y.; et al. JAK/STAT3-Regulated Fatty Acid β-Oxidation Is Critical for Breast Cancer Stem Cell Self-Renewal and Chemoresistance. Cell Metab. 2018, 27, 136–150.e5. [Google Scholar] [CrossRef]
- Zheng, F.; Dang, J.; Zhang, H.; Xu, F.; Ba, D.; Zhang, B.; Cheng, F.; Chang, A.E.; Wicha, M.S.; Li, Q. Cancer Stem Cell Vaccination With PD-L1 and CTLA-4 Blockades Enhances the Eradication of Melanoma Stem Cells in a Mouse Tumor Model. J. Immunother. 2018, 41, 361–368. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, X.; Wang, L.; Hong, X.; Yang, J. Metabolic reprogramming and crosstalk of cancer-related fibroblasts and immune cells in the tumor microenvironment. Front. Endocrinol. 2022, 13, 988295. [Google Scholar] [CrossRef]
- Ippolito, L.; Sonveaux, P.; Chiarugi, P. Unconventional roles of lactate along the tumor and immune landscape. Trends Endocrinol. Metab. 2022, 33, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Fiaschi, T.; Marini, A.; Giannoni, E.; Taddei, M.L.; Gandellini, P.; De Donatis, A.; Lanciotti, M.; Serni, S.; Cirri, P.; Chiarugi, P. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012, 72, 5130–5140. [Google Scholar] [CrossRef] [PubMed]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Carmeliet, P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013, 18, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Parra-Bonilla, G.; Alvarez, D.F.; Al-Mehdi, A.B.; Alexeyev, M.; Stevens, T. Critical role for lactate dehydrogenase A in aerobic glycolysis that sustains pulmonary microvascular endothelial cell proliferation. Am. J. Physiol. Lung Cell Mol. Physiol. 2010, 299, L513–L522. [Google Scholar] [CrossRef] [PubMed]
- Peng, F.; Li, Q.; Sun, J.Y.; Luo, Y.; Chen, M.; Bao, Y. PFKFB3 is involved in breast cancer proliferation, migration, invasion and angiogenesis. Int. J. Oncol. 2018, 52, 945–954. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy. J. Immunol. Res. 2016, 2016, 9720912. [Google Scholar] [CrossRef]
- Luo, S.; Yang, G.; Ye, P.; Cao, N.; Chi, X.; Yang, W.H.; Yan, X. Macrophages Are a Double-Edged Sword: Molecular Crosstalk between Tumor-Associated Macrophages and Cancer Stem Cells. Biomolecules 2022, 12, 850. [Google Scholar] [CrossRef]
- Wang, S.; Liu, G.; Li, Y.; Pan, Y. Metabolic Reprogramming Induces Macrophage Polarization in the Tumor Microenvironment. Front. Immunol. 2022, 13, 840029. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Zhao, Q.; Yang, T.; Ding, W.; Zhao, Y. Cellular metabolism and macrophage functional polarization. Int. Rev. Immunol. 2015, 34, 82–100. [Google Scholar] [CrossRef]
- Nusblat, L.M.; Carroll, M.J.; Roth, C.M. Crosstalk between M2 macrophages and glioma stem cells. Cell. Oncol. 2017, 40, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gong, X.; Li, J.; Wang, H.; Xu, X.; Wu, Y.; Wang, J.; Wang, S.; Li, Y.; Zhang, Z. M2 macrophage microvesicle-inspired nanovehicles improve accessibility to cancer cells and cancer stem cells in tumors. J. Nanobiotechnol. 2021, 19, 397. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Oft, M. Immune regulation and cytotoxic T cell activation of IL-10 agonists—Preclinical and clinical experience. Semin. Immunol. 2019, 44, 101325. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Ye, H.; et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018, 34, 724–740. [Google Scholar] [CrossRef]
- Stevens, B.M.; Jones, C.L.; Pollyea, D.A.; Culp-Hill, R.; D’Alessandro, A.; Winters, A.; Krug, A.; Abbott, D.; Goosman, M.; Pei, S.; et al. Fatty acid metabolism underlies venetoclax resistance in acute myeloid leukemia stem cells. Nat. Cancer 2020, 1, 1176–1187. [Google Scholar] [CrossRef]
- Jones, C.L.; Stevens, B.M.; Pollyea, D.A.; Culp-Hill, R.; Reisz, J.A.; Nemkov, T.; Gehrke, S.; Gamboni, F.; Krug, A.; Winters, A.; et al. Nicotinamide metabolism mediates resistance to venetoclax in relapsed acute myeloid leukemia stem cells. Cell Stem Cell 2020, 27, 748–764.e4. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef]
- He, W.; Liang, B.; Wang, C.; Li, S.; Zhao, Y.; Huang, Q.; Liu, Z.; Yao, Z.; Wu, Q.; Liao, W.; et al. MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene 2019, 38, 4637–4654. [Google Scholar] [CrossRef]
- Zhang, Z.; Han, H.; Rong, Y.; Zhu, K.; Zhu, Z.; Tang, Z.; Xiong, C.; Tao, J. Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J. Exp. Clin. Cancer Res. 2018, 37, 291. [Google Scholar] [CrossRef]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Dejure, F.R.; Eilers, M. MYC and tumor metabolism: Chicken and egg. EMBO J. 2017, 36, 3409–3420. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wen, G.-M.; Xu, X.-Y.; Xia, P. Metabolism in Cancer Stem Cells: Targets for Clinical Treatment. Cells 2022, 11, 3790. https://doi.org/10.3390/cells11233790
Wen G-M, Xu X-Y, Xia P. Metabolism in Cancer Stem Cells: Targets for Clinical Treatment. Cells. 2022; 11(23):3790. https://doi.org/10.3390/cells11233790
Chicago/Turabian StyleWen, Gui-Min, Xiao-Yan Xu, and Pu Xia. 2022. "Metabolism in Cancer Stem Cells: Targets for Clinical Treatment" Cells 11, no. 23: 3790. https://doi.org/10.3390/cells11233790
APA StyleWen, G.-M., Xu, X.-Y., & Xia, P. (2022). Metabolism in Cancer Stem Cells: Targets for Clinical Treatment. Cells, 11(23), 3790. https://doi.org/10.3390/cells11233790