
Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes


 , ,
, , 
 and
and 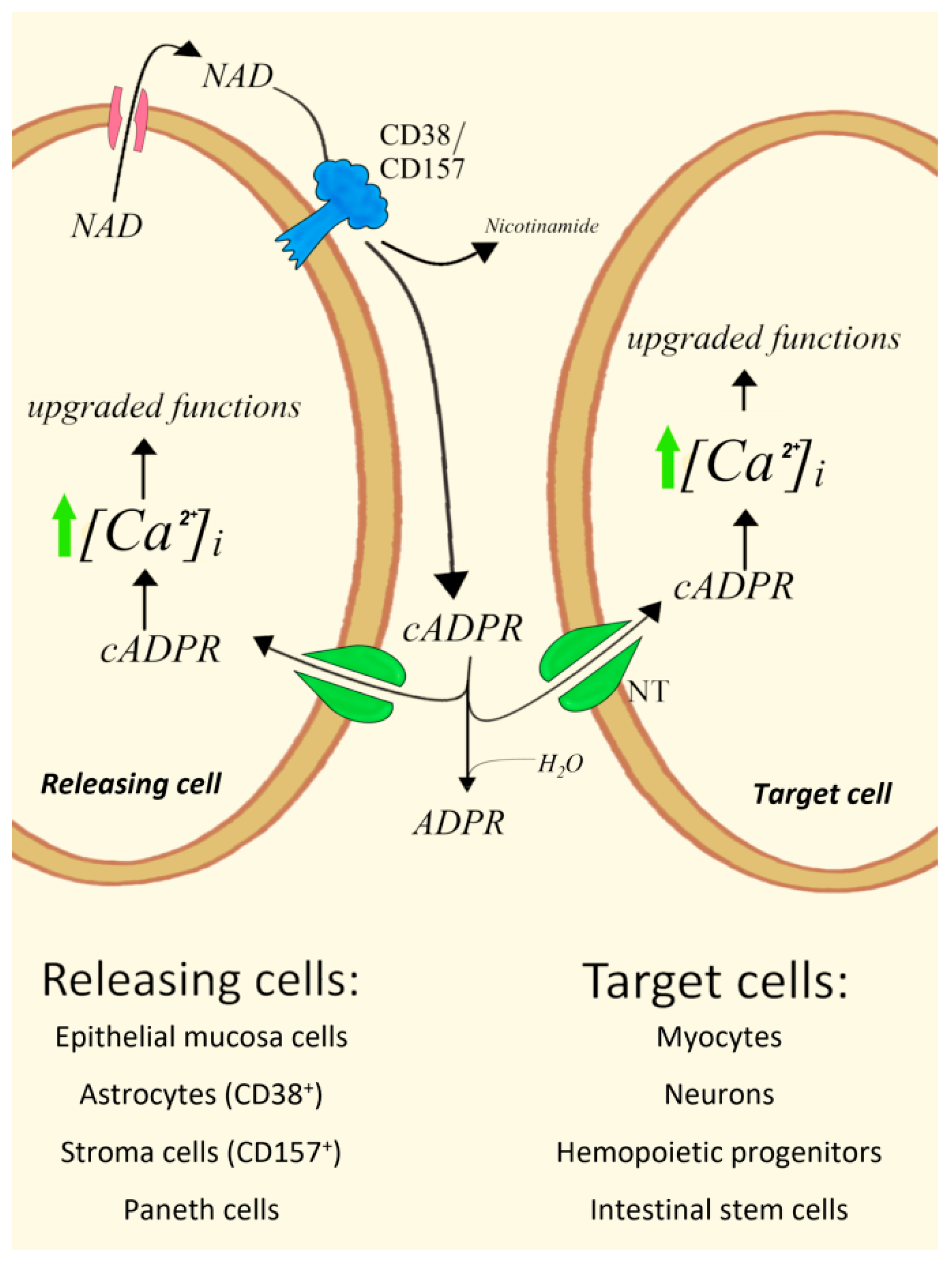{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Type II CD38 and NAD+ and cADPR Transporters
3. Type III CD38
- (a)
- CD38 catalyzes the intracellular synthesis of both cADPR and NAADP+.
- (b)
- A cGMP-PKG cascade triggered by the [Ca2+]i release from the ER through IP3 induces the internalization of type III CD38 from the plasma membrane into early endosomes with the generation of cADPR at their outer surface and the release of cADPR in the cytosol.
- (c)
- cADPR and ER-mobilized Ca2+ activate a PKA-EPAC-Rap1 cascade driven by cAMP production that stimulates NAADP+ formation by type II CD38 inside acidic lysosomes.
- (d)
- The type II-mediated generation of NAADP+ inside acidic organelles takes place through a nicotinic acid and nicotinamide base exchange between NAAD (an intermediate of NAD+ biosynthesis) and NADP+ (which is taken up from the cytosol across activated Cx43 hemichannels) [43].
- (e)
- The time sequence of second messengers is IP3-cADPR-NAADP+ and the consequent [Ca2+]i increases downstream of their binding to specific receptors (IP3, RyR and TPC, respectively) occur in a transient fashion (cADPR) followed by a long-lasting signal (NAADP+).
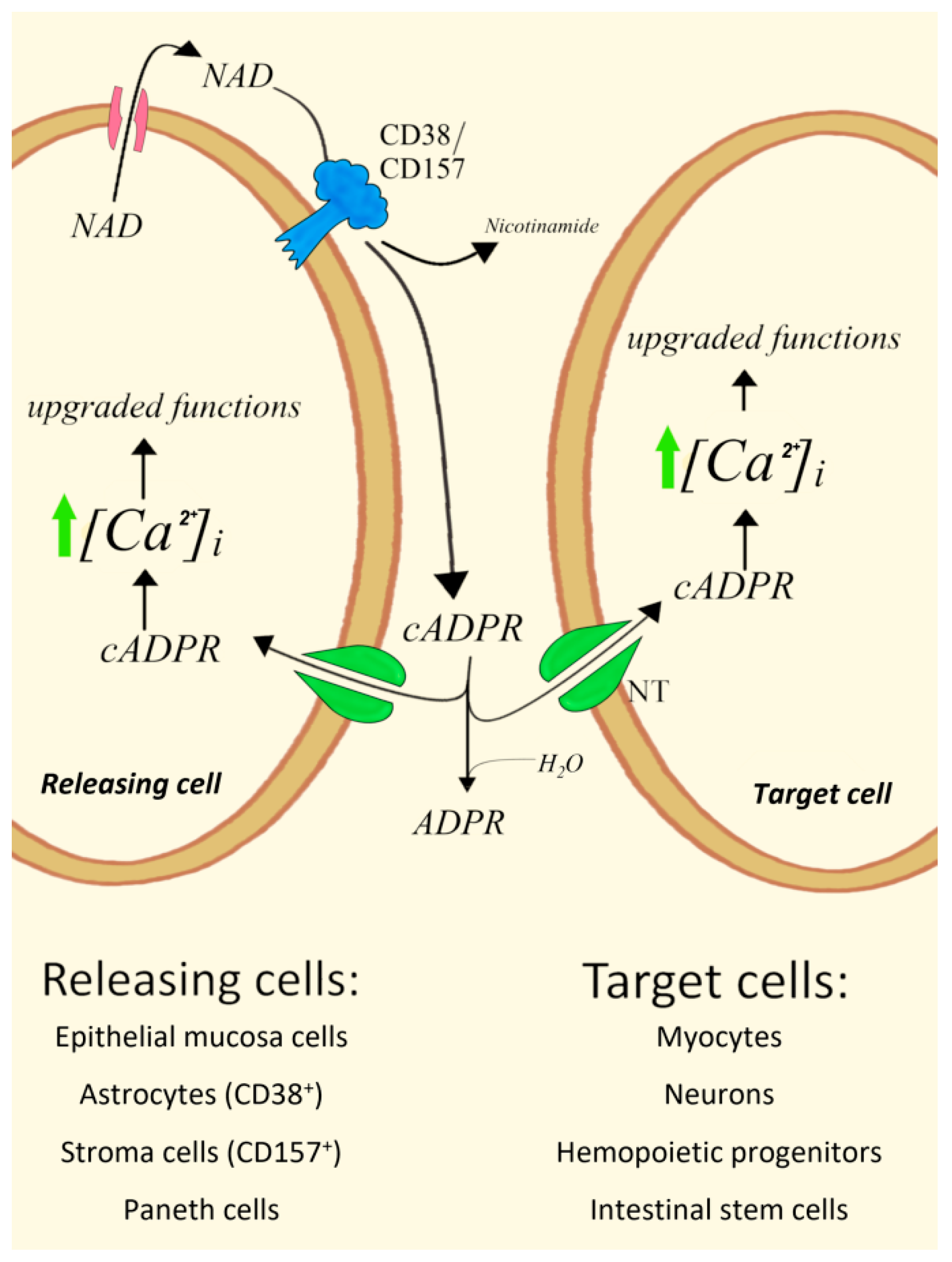
4. Type II CD38 and CD157 cADPR-Synthesizing Ectoenzymes Mediate Paracrine Mechanisms
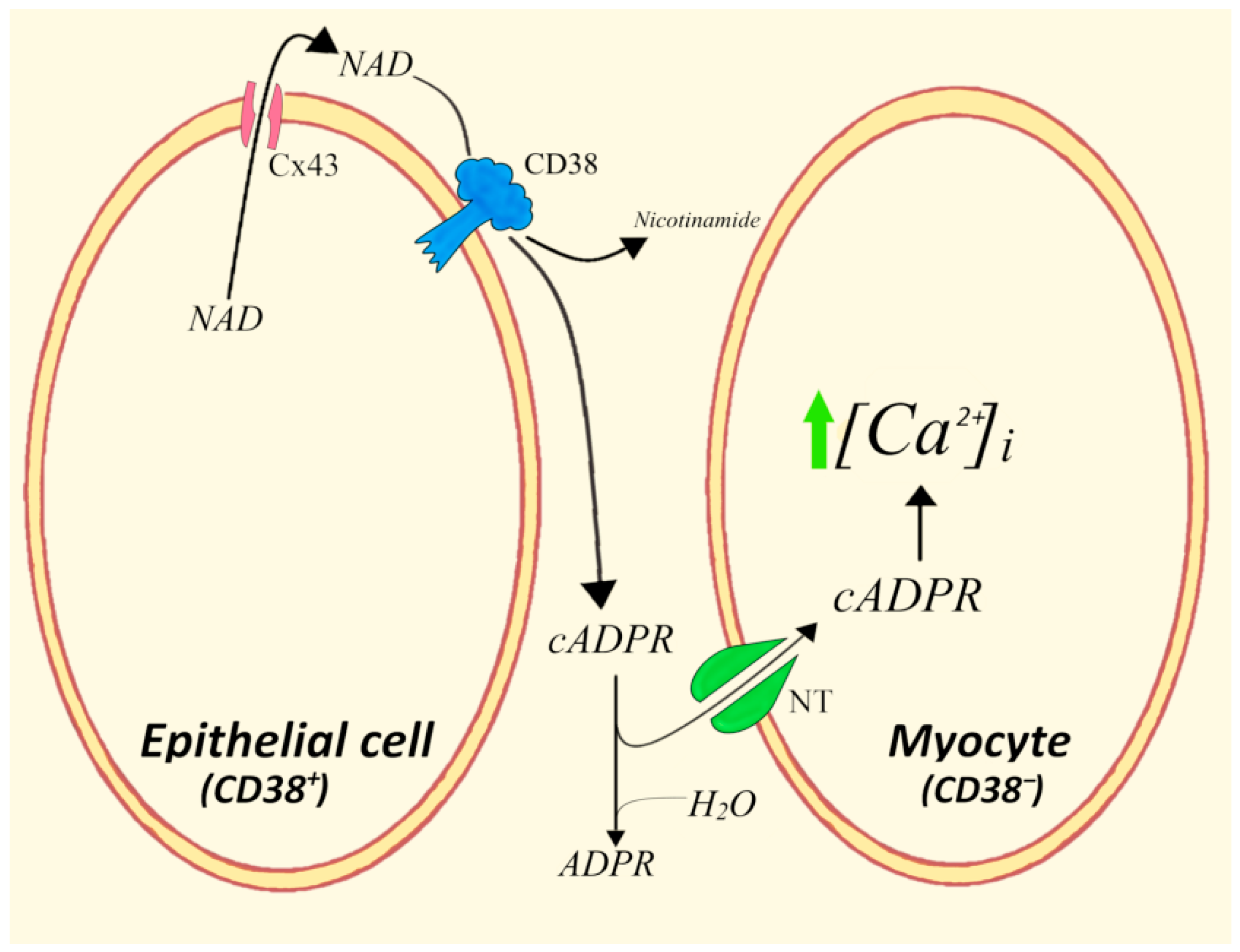
4.1. Epithelial Mucosa Cells and Smooth Myocytes in Bovine Trachea
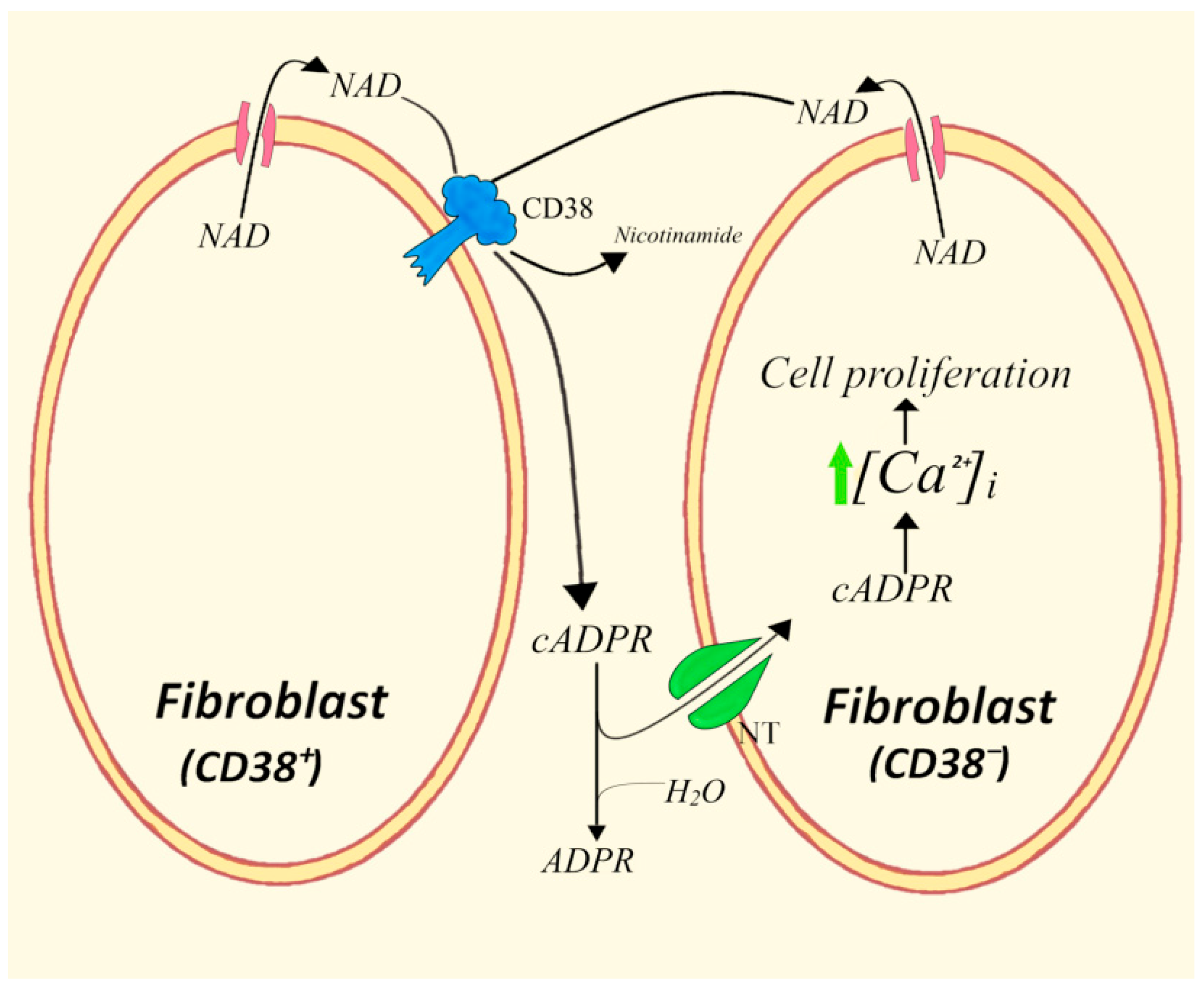
4.2. CD38+/CD38− Cells in Homotypic Co-Cultures of 3T3 Fibroblasts
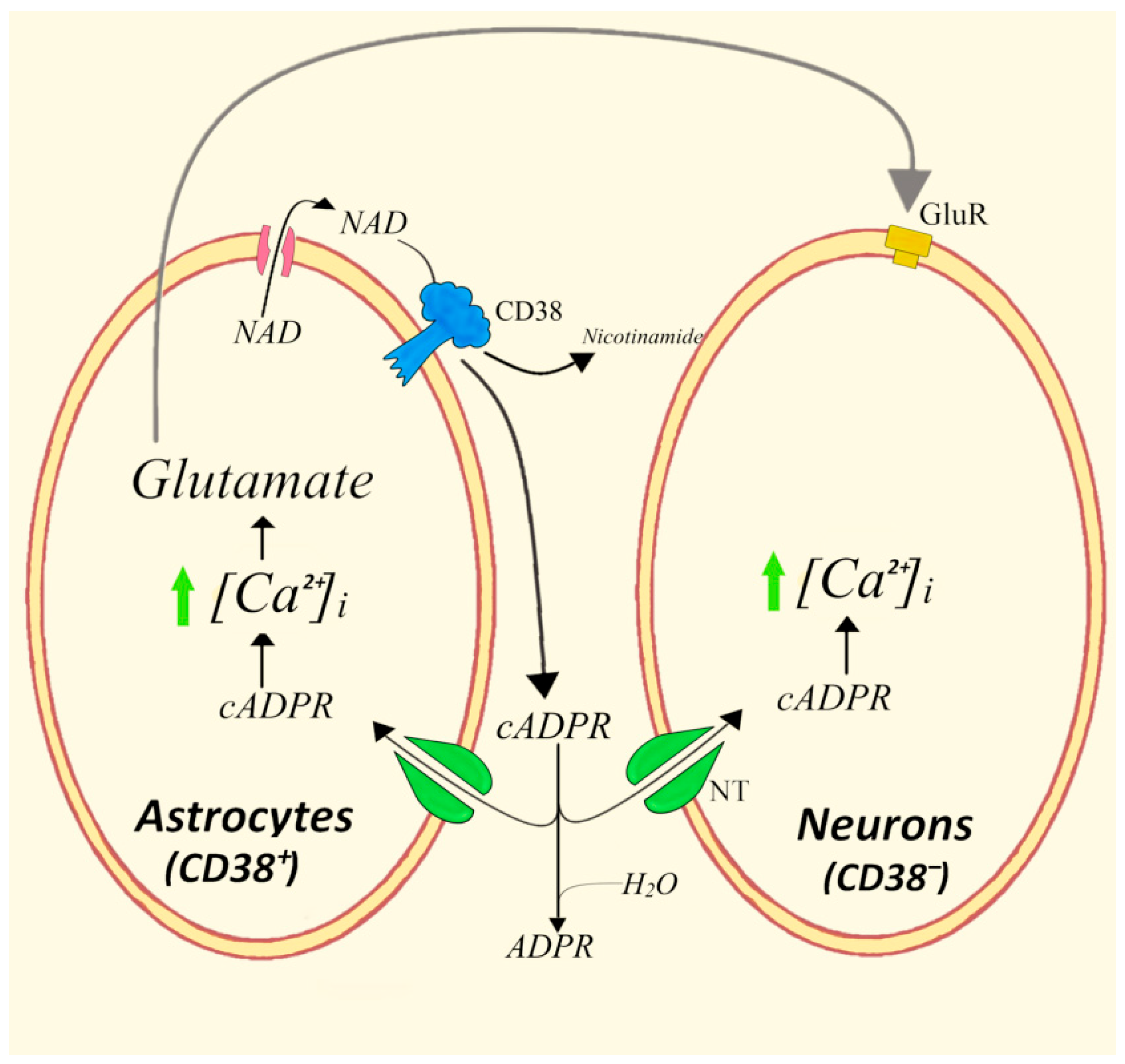
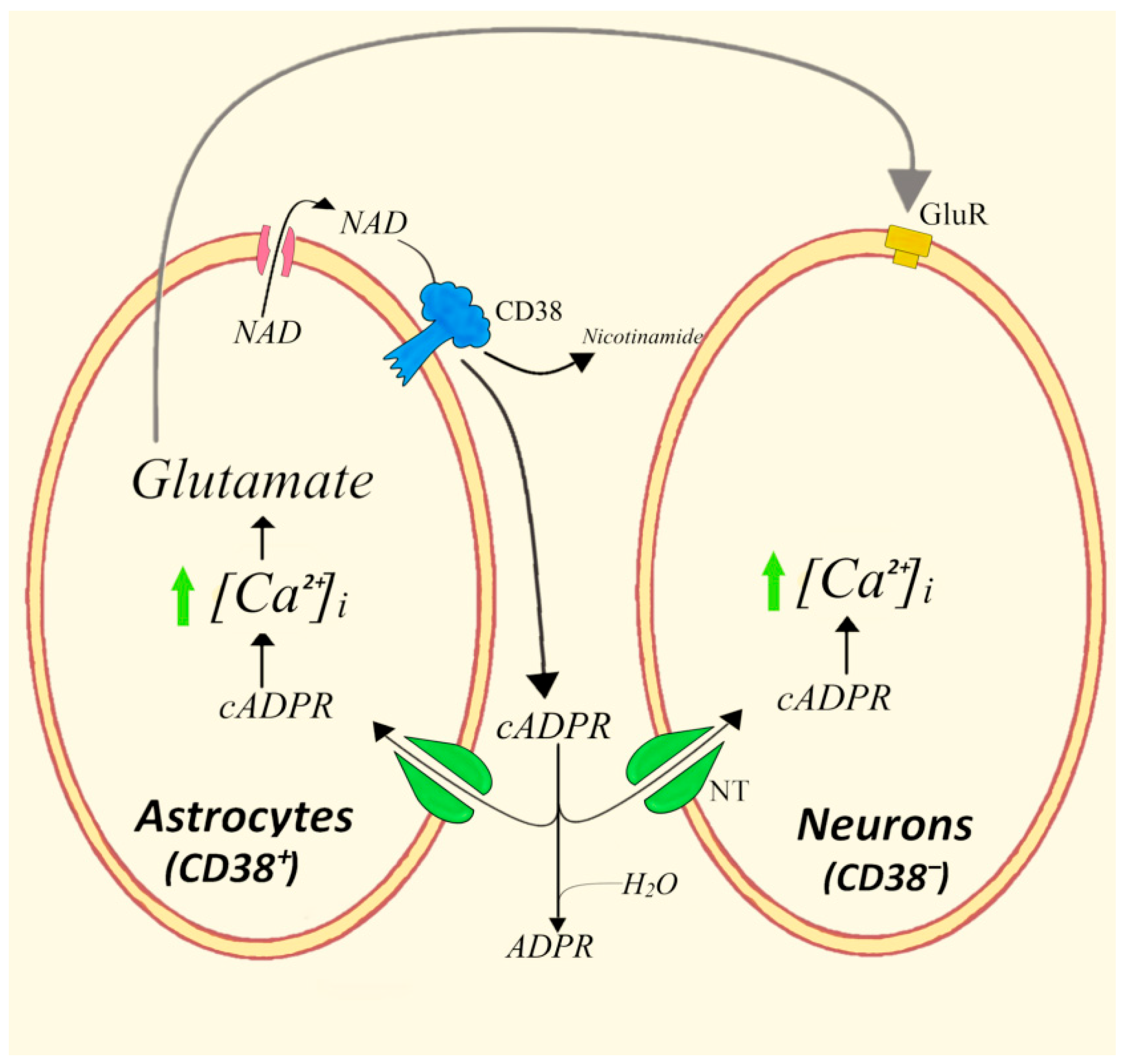
4.3. Bidirectional Astrocytes/Neurons Communications Involving the CD38/cADPR System and Glutamate
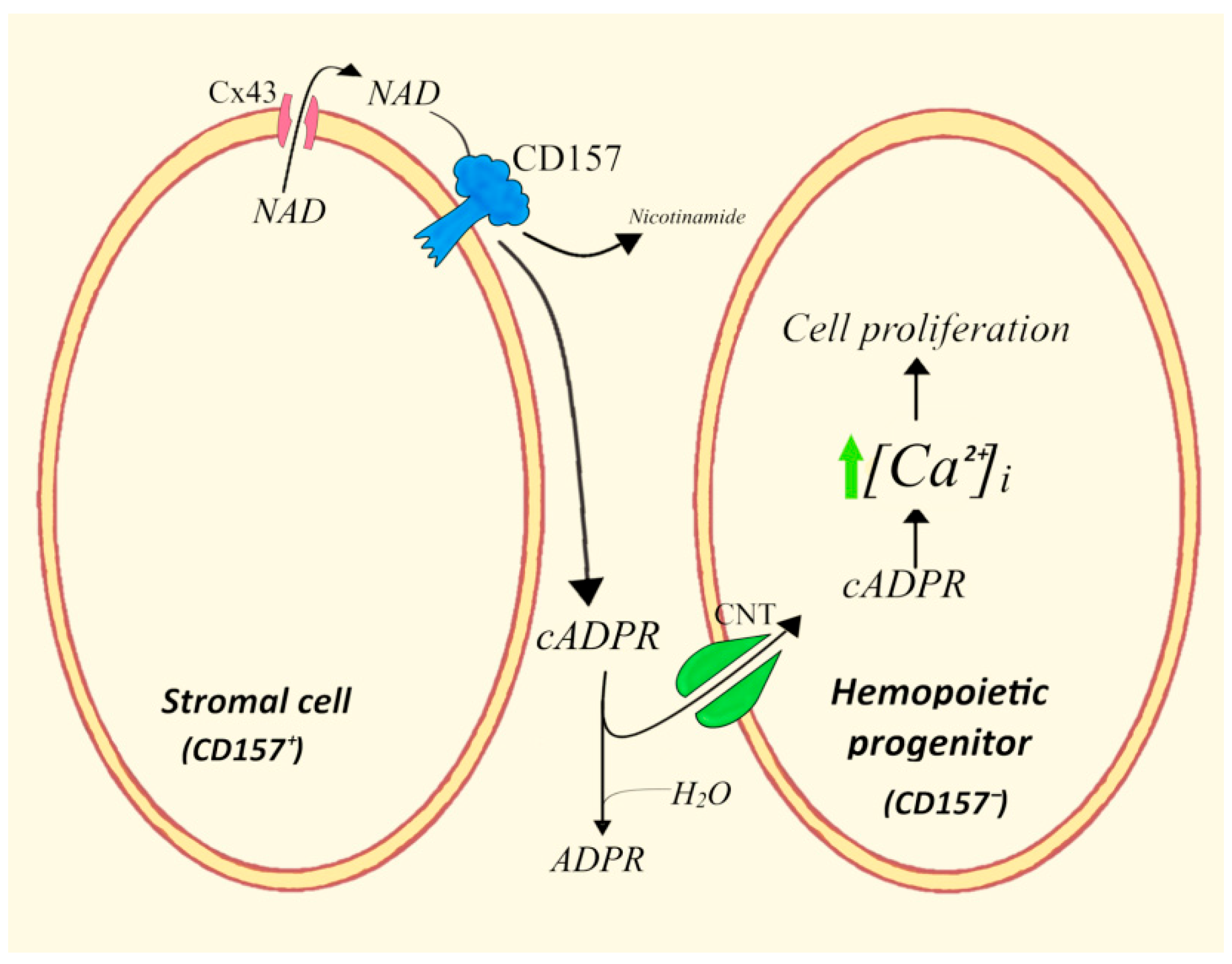
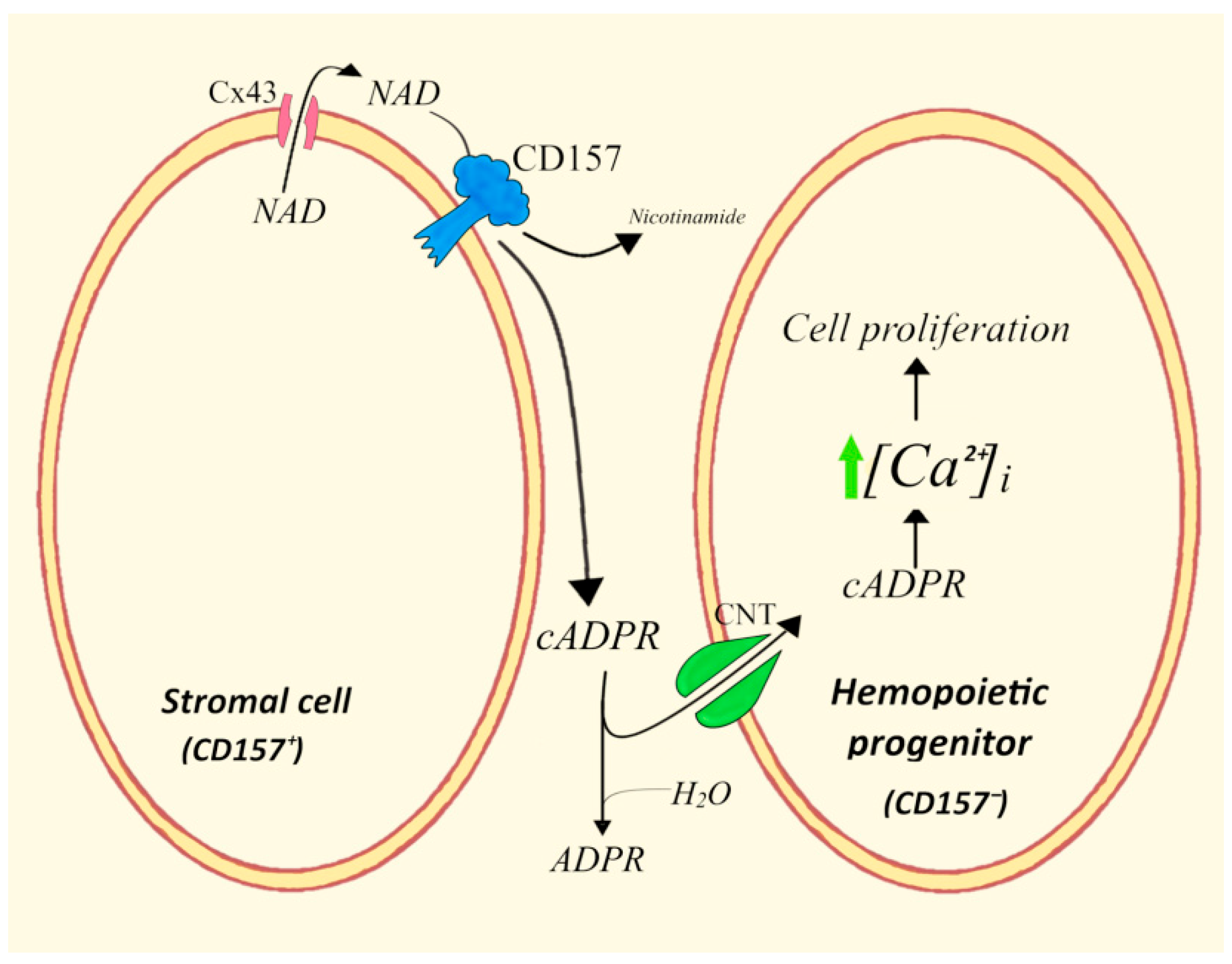
4.4. ADPRC+ Stroma Cells/Human Hemopoietic Progenitors (HPs)
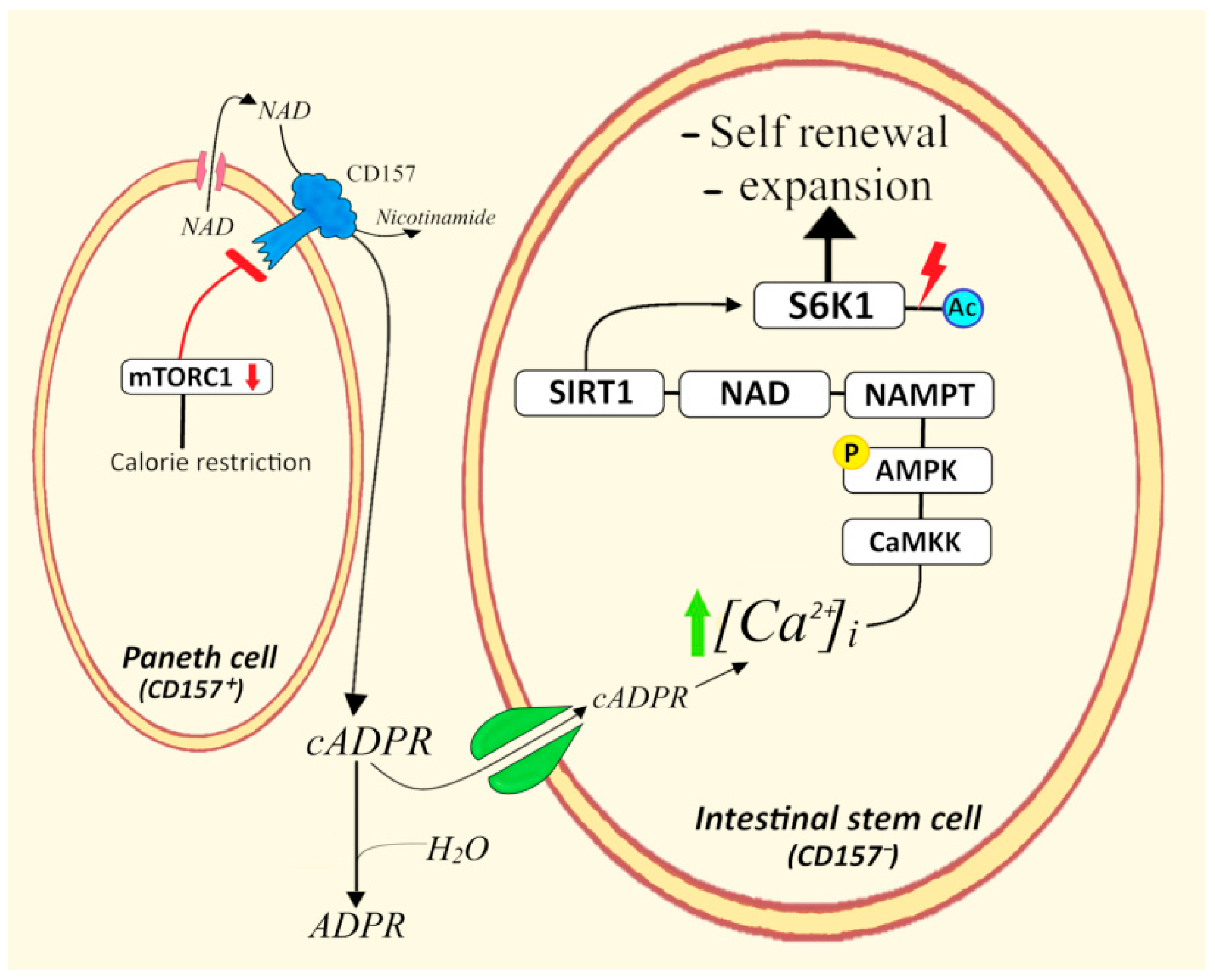
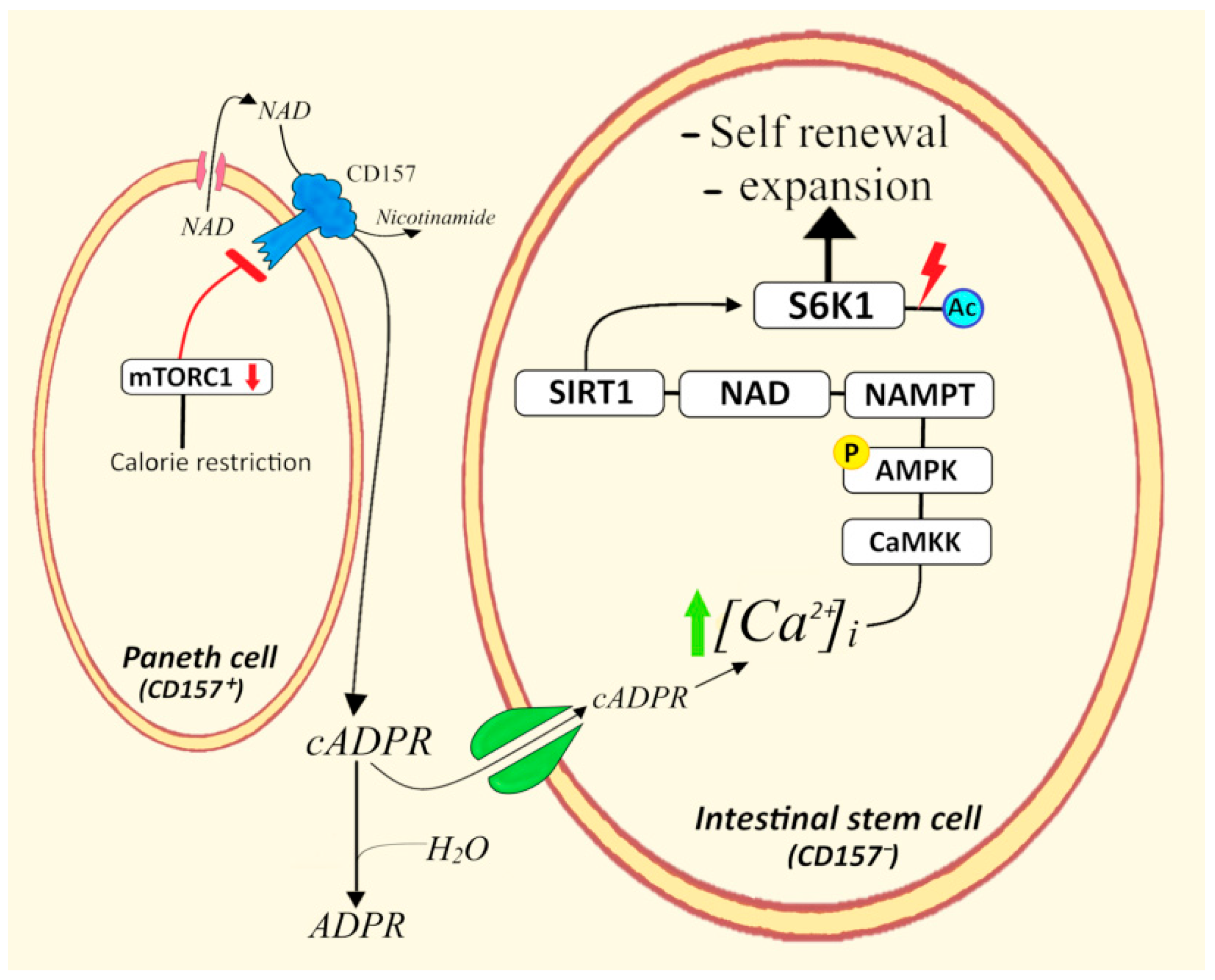
4.5. CD157+ Paneth Cells/Intestinal Stem Cells
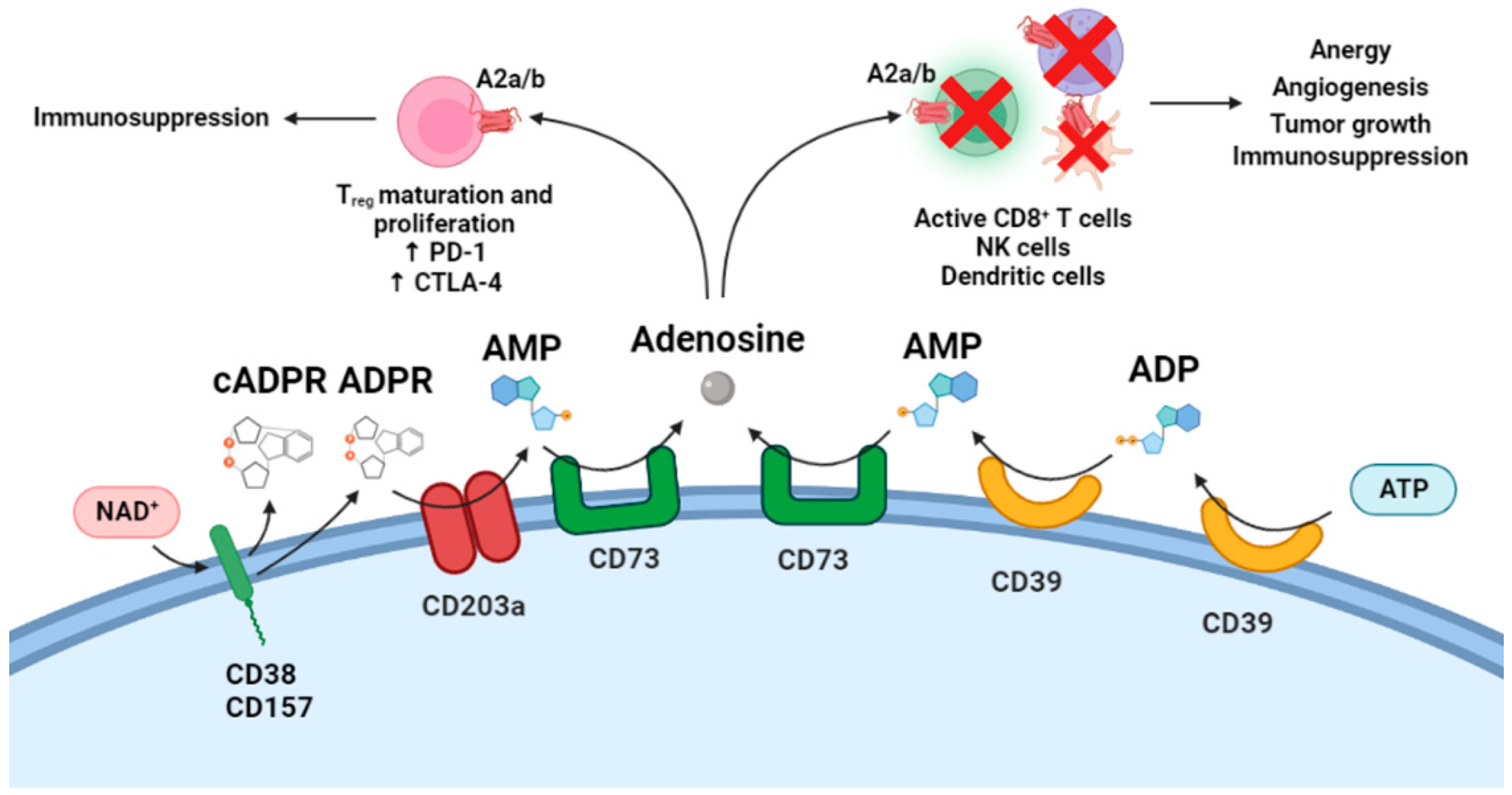
5. Cytotoxic T Lymphocytes and CD38-Mediated Adenosine Generation in Tumor Cells
6. Conclusions and Perspectives
- -
- NAD+e as a hormone.
- -
- cADPRe/i as a second messenger.
- -
- [Ca2+]i as a third messenger.
Funding
Data Availability Statement
Conflicts of Interest
References
- Jackson, D.G.; Bell, J.I. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J. Immunol. 1990, 144, 2811–2815. [Google Scholar] [PubMed]
- Lee, H.C.; Aarhus, R. ADP-ribosyl cyclase: An enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Rusinko, N.; Lee, H.C. Widespread occurrence in animal tissues of an enzyme catalyzing the conversion of NAD+ into a cyclic metabolite with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 11725–11731. [Google Scholar] [CrossRef]
- States, D.J.; Walseth, T.F.; Lee, H.C. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem. Sci. 1992, 17, 495. [Google Scholar] [CrossRef]
- Lee, H.C.; Zocchi, E.; Guida, L.; Franco, L.; Benatti, U.; De Flora, A. Production and hydrolysis of cyclic ADP-ribose at the outer surface of human erythrocytes. Biochem. Biophys. Res. Commun. 1993, 191, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Grimaldi, J.C.; Bazan, J.F.; Lund, F.E.; Santos-Argumedo, L.; Parkhouse, R.M.; Walseth, T.F.; Lee, H.C. Formation and hydrolysis of cyclic ADP-ribose catalyzed by lymphocyte antigen CD38. Science 1993, 262, 1056–1059. [Google Scholar] [CrossRef]
- Takasawa, S.; Tohgo, A.; Noguchi, N.; Koguma, T.; Nata, K.; Sugimoto, T.; Yonekura, H.; Okamoto, H. Synthesis and hydrolysis of cyclic ADP-ribose by human leukocyte antigen CD38 and inhibition of the hydrolysis by ATP. J. Biol. Chem. 1993, 268, 26052–26054. [Google Scholar] [CrossRef]
- Zocchi, E.; Franco, L.; Guida, L.; Benatti, U.; Bargellesi, A.; Malavasi, F.; Lee, H.C.; De Flora, A. A single protein immunologically identified as CD38 displays NAD+ glycohydrolase, ADP-ribosyl cyclase and cyclic ADP-ribose hydrolase activities at the outer surface of human erythrocytes. Biochem. Biophys. Res. Commun. 1993, 196, 1459–1465. [Google Scholar] [CrossRef]
- Summerhill, R.J.; Jackson, D.G.; Galione, A. Human lymphocyte antigen CD38 catalyzes the production of cyclic ADP-ribose. FEBS Lett. 1993, 335, 231–233. [Google Scholar] [CrossRef]
- Galione, A. Cyclic ADP-ribose, the ADP-ribosyl cyclase pathway and calcium signalling. Mol. Cell Endocrinol. 1994, 98, 125–131. [Google Scholar] [CrossRef]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Structure and enzymatic functions of human CD38. Mol. Med. 2006, 12, 317–323. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012, 5, ra67. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Zhu, W.J.; Wang, X.W.; Zhang, L.H.; Lee, H.C. Determinants of the membrane orientation of a calcium signaling enzyme CD38. Biochim. Biophys. Acta. 2015, 1853, 2095–2103. [Google Scholar] [CrossRef]
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar] [CrossRef]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [CrossRef]
- Kim, H.; Jacobson, E.L.; Jacobson, M.K. Position of cyclization in cyclic ADP-ribose. Biochem. Biophys. Res. Commun. 1993, 194, 1143–1147. [Google Scholar] [CrossRef]
- Galione, A.; Lee, H.C.; Busa, W.B. Ca(2+)-induced Ca2+ release in sea urchin egg homogenates: Modulation by cyclic ADP-ribose. Science 1991, 253, 1143–1146. [Google Scholar] [CrossRef]
- Churchill, G.C.; Okada, Y.; Thomas, J.M.; Genazzani, A.A.; Patel, S.; Galione, A. NAADP mobilizes Ca(2+) from reserve granules, lysosome-related organelles, in sea urchin eggs. Cell 2002, 111, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Dammermann, W.; Guse, A.H. Functional ryanodine receptor expression is required for NAADP-mediated local Ca2+ signaling in T-lymphocytes. J. Biol. Chem. 2005, 280, 21394–21399. [Google Scholar] [CrossRef]
- Guse, A.H.; Lee, H.C. NAADP: A universal Ca2+ trigger. Sci. Signal. 2008, 1, re10. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Zhao, Y.J. Resolving the topological enigma in Ca2+ signaling by cyclic ADP-ribose and NAADP. J. Biol. Chem. 2019, 294, 19831–19843. [Google Scholar] [CrossRef]
- Lee, H.C.; Deng, Q.W.; Zhao, Y.J. The calcium signaling enzyme CD38− a paradigm for membrane topology defining distinct protein functions. Cell Calcium 2022, 101, 102514. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Kimura, N.; Sato, K.; Ohsugi, Y.; Takasawa, S.; Okamoto, H.; Ishikawa, J.; Kaisho, T.; Ishihara, K.; Hirano, T. ADP ribosyl cyclase activity of a novel bone marrow stromal cell surface molecule, BST-1. FEBS Lett. 1994, 356, 244–248. [Google Scholar] [CrossRef]
- Itoh, M.; Ishihara, K.; Tomizawa, H.; Tanaka, H.; Kobune, Y.; Ishikawa, J.; Kaisho, T.; Hirano, T. Molecular cloning of murine BST-1 having homology with CD38 and Aplysia ADP-ribosyl cyclase. Biochem. Biophys. Res. Commun. 1994, 203, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.Y.; Xie, X.J.; Li, W.H.; Liu, J.; Chen, Z.; Zhang, B.; Li, T.; Li, S.L.; Lu, J.G.; Zhang, L.; et al. A Cell-Permeant Mimetic of NMN Activates SARM1 to Produce Cyclic ADP-Ribose and Induce Non-apoptotic Cell Death. iScience 2019, 15, 452–466. [Google Scholar] [CrossRef] [PubMed]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD+ Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e5. [Google Scholar]
- Li, W.H.; Huang, K.; Cai, Y.; Wang, Q.W.; Zhu, W.J.; Hou, Y.N.; Wang, S.; Cao, S.; Zhao, Z.Y.; Xie, X.J.; et al. Permeant fluorescent probes visualize the activation of SARM1 and uncover an anti-neurodegenerative drug candidate. Elife 2021, 10, e67381. [Google Scholar] [CrossRef] [PubMed]
- De Flora, A.; Franco, L.; Guida, L.; Bruzzone, S.; Usai, C.; Zocchi, E. Topology of CD38. Chem. Immunol. 2000, 75, 79–98. [Google Scholar]
- De Flora, A.; Guida, L.; Franco, L.; Zocchi, E. The CD38/cyclic ADP-ribose system: A topological paradox. Int. J. Biochem. Cell Biol. 1997, 29, 1149–1166. [Google Scholar] [CrossRef]
- De Flora, A.; Zocchi, E.; Guida, L.; Franco, L.; Bruzzone, S. Autocrine and Paracrine Calcium Signaling by the CD38/NAD+/Cyclic ADP-Ribose System. Ann. N. Y. Acad. Sci. 2004, 1028, 176–191. [Google Scholar] [PubMed]
- Billington, R.A.; Bruzzone, S.; De Flora, A.; Genazzani, A.A.; Koch-Nolte, F.; Ziegler, M.; Zocchi, E. Emerging functions of extracellular pyridine nucleotides. Mol. Med. 2006, 12, 324–327. [Google Scholar] [PubMed]
- Bruzzone, S.; Guida, L.; Sturla, L.; Usai, C.; Zocchi, E.; De Flora, A. Subcellular and Intercellular Traffic of NAD+, NAD+ Precursors and NAD+-Derived Signal Metabolites and Second Messengers: Old and New Topological Paradoxes. Messenger 2012, 1, 34–52. [Google Scholar] [CrossRef]
- Bruzzone, S.; Kunerth, S.; Zocchi, E.; De Flora, A.; Guse, A.H. Spatio-temporal propagation of Ca2+ signals by cyclic ADP-ribose in 3T3 cells stimulated via purinergic P2Y receptors. J. Cell Biol. 2003, 163, 837–845. [Google Scholar] [CrossRef]
- Guse, A.H. Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J. 2005, 272, 4590–4597. [Google Scholar] [CrossRef]
- Bruzzone, S.; Guida, L.; Zocchi, E.; Franco, L.; De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001, 15, 10–12. [Google Scholar] [CrossRef]
- Bruzzone, S.; Franco, L.; Guida, L.; Zocchi, E.; Contini, P.; Bisso, A.; Usai, C.; De Flora, A. A self-restricted CD38-connexin 43 cross-talk affects NAD+ and cyclic ADP-ribose metabolism and regulates intracellular calcium in 3T3 fibroblasts. J. Biol. Chem. 2001, 276, 48300–48308. [Google Scholar] [CrossRef]
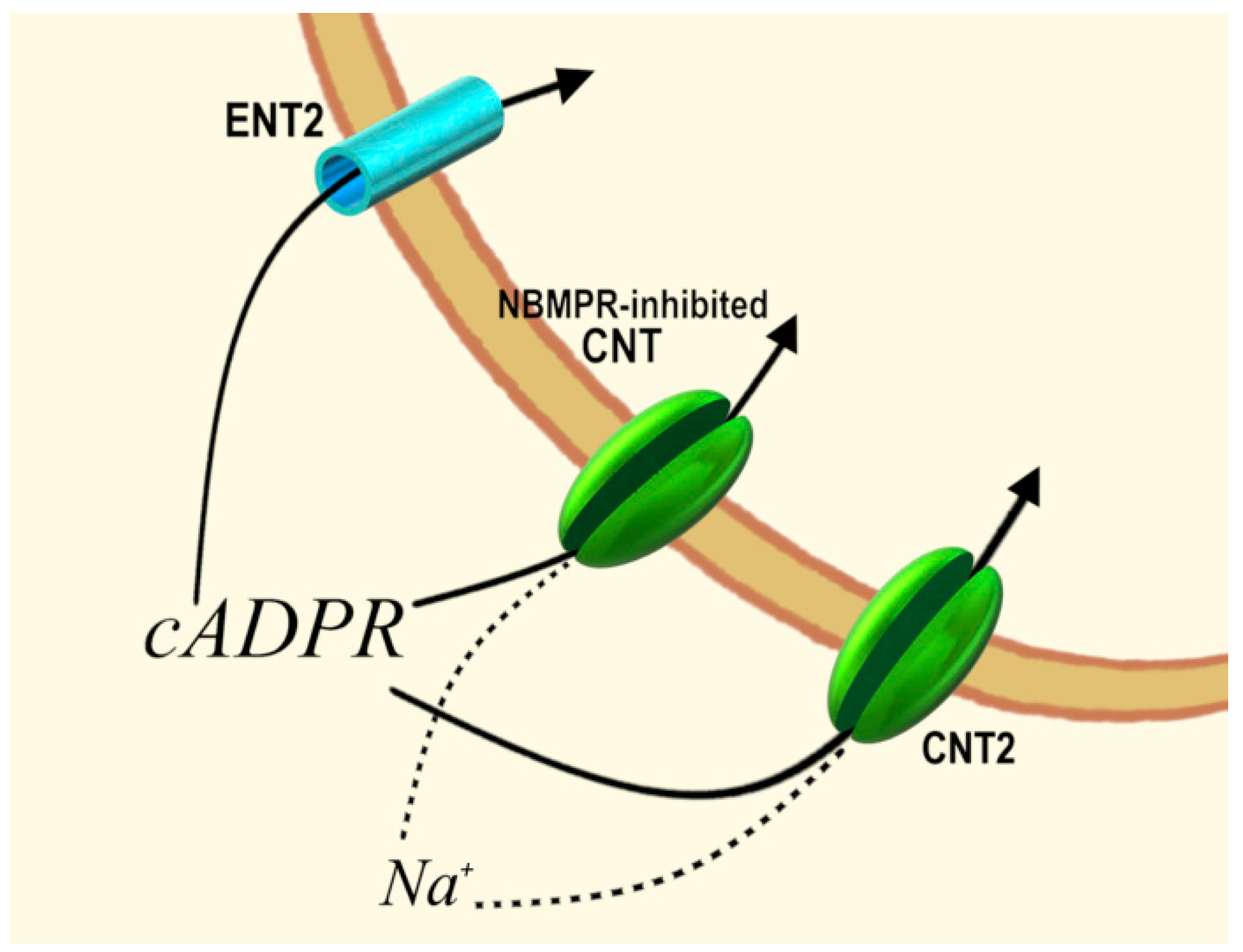
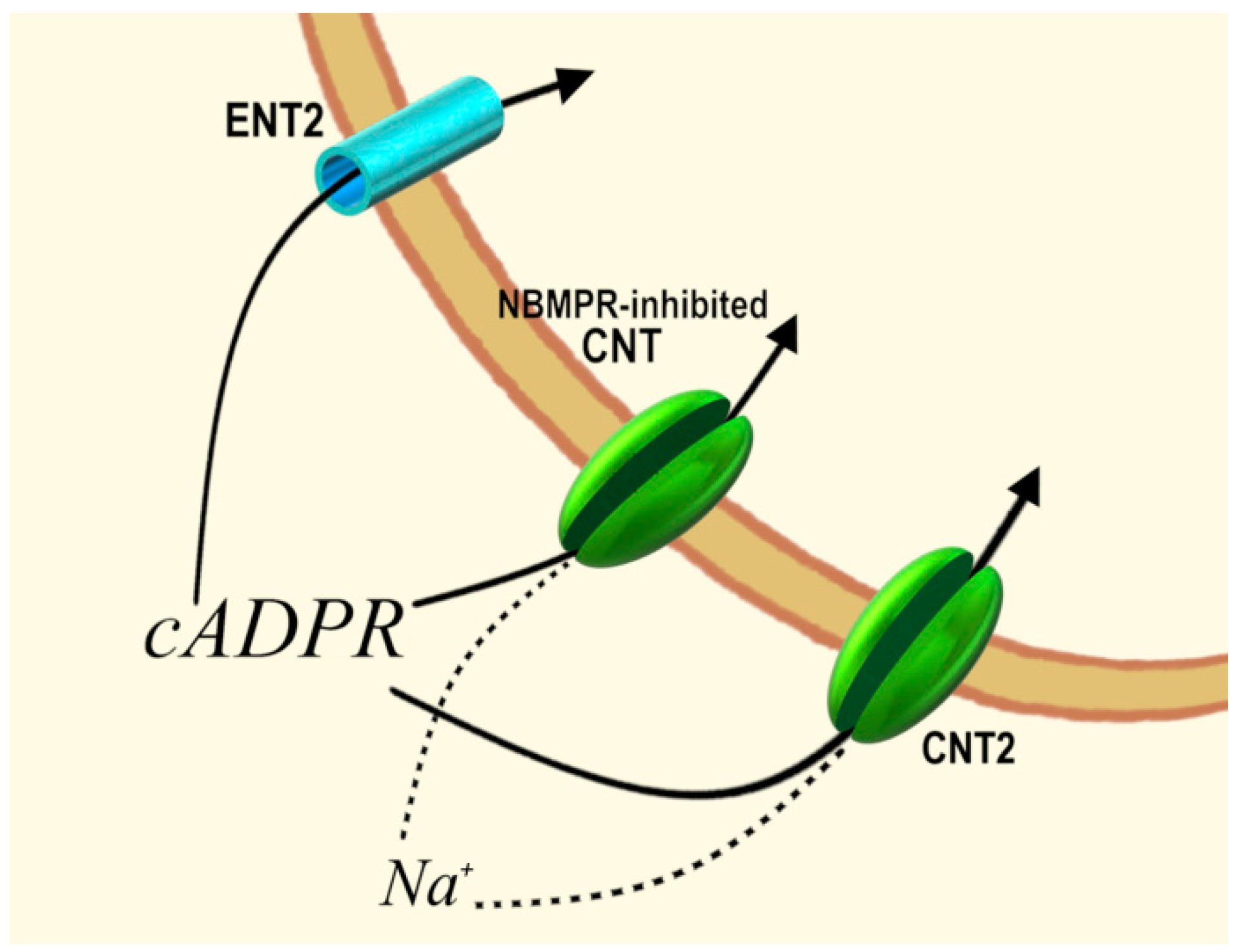
- Guida, L.; Bruzzone, S.; Sturla, L.; Franco, L.; Zocchi, E.; De Flora, A. Equilibrative and concentrative nucleoside transporters mediate influx of extracellular cyclic ADP-ribose into 3T3 murine fibroblasts. J. Biol. Chem. 2002, 277, 47097–47105. [Google Scholar] [CrossRef]
- Guida, L.; Franco, L.; Bruzzone, S.; Sturla, L.; Zocchi, E.; Basile, G.; Usai, C.; De Flora, A. Concentrative influx of functionally active cyclic ADP-ribose in dimethyl sulfoxide-differentiated HL-60 cells. J. Biol. Chem. 2004, 279, 22066–22075. [Google Scholar] [CrossRef]
- Podestà, M.; Benvenuto, F.; Pitto, A.; Figari, O.; Bacigalupo, A.; Bruzzone, S.; Guida, L.; Franco, L.; Paleari, L.; Bodrato, N.; et al. Concentrative uptake of cyclic ADP-ribose generated by BST-1+ stroma stimulates proliferation of human hematopoietic progenitors. J. Biol. Chem. 2005, 280, 5343–5349. [Google Scholar] [CrossRef]
- Liu, J.; Zhao, Y.J.; Li, W.H.; Hou, Y.N.; Li, T.; Zhao, Z.Y.; Fang, C.; Li, S.L.; Lee, H.C. Cytosolic interaction of type III human CD38 with CIB1 modulates cellular cyclic ADP-ribose levels. Proc. Natl. Acad. Sci. USA 2017, 114, 8283–8288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, U.H. Roles of cADPR and NAADP in pancreatic beta cell signaling. Cell Calcium 2022, 103, 102562. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [PubMed]
- Horenstein, A.L.; Chillemi, A.; Zini, R.; Quarona, V.; Bianchi, N.; Manfredini, R.; Gambari, R.; Malavasi, F.; Ferrari, D. Cytokine-Induced Killer Cells Express CD39, CD38, CD203a, CD73 Ectoenzymes and P1 Adenosinergic Receptors. Front. Pharmacol. 2018, 9, 196. [Google Scholar] [CrossRef]
- Ferrero, E.; Faini, A.C.; Malavasi, F. A phylogenetic view of the leukocyte ectonucleotidases. Immunol. Lett. 2019, 205, 51–58. [Google Scholar] [CrossRef]
- Quarona, V.; Ferri, V.; Chillemi, A.; Bolzoni, M.; Mancini, C.; Zaccarello, G.; Roato, I.; Morandi, F.; Marimpietri, D.; Faccani, G.; et al. Unraveling the contribution of ectoenzymes to myeloma life and survival in the bone marrow niche. Ann. N. Y. Acad. Sci. 2015, 1335, 10–22. [Google Scholar] [CrossRef]
- Morandi, F.; Morandi, B.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Zaccarello, G.; Carrega, P.; Ferlazzo, G.; Mingari, M.C.; Moretta, L.; et al. A non-canonical adenosinergic pathway led by CD38 in human melanoma cells induces suppression of T cell proliferation. Oncotarget 2015, 6, 25602–25618. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Marimpietri, D.; Horenstein, A.L.; Bolzoni, M.; Toscani, D.; Costa, F.; Castella, B.; Faini, A.C.; Massaia, M.; Pistoia, V.; et al. Microvesicles released from multiple myeloma cells are equipped with ectoenzymes belonging to canonical and non-canonical adenosinergic pathways and produce adenosine from ATP and NAD. Oncoimmunology 2018, 7, e1458809. [Google Scholar] [CrossRef]
- Vaisitti, T.; Arruga, F.; Guerra, G.; Deaglio, S. Ectonucleotidases in Blood Malignancies: A Tale of Surface Markers and Therapeutic Targets. Front. Immunol. 2019, 10, 2301. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef]
- Konen, J.M.; Fradette, J.J.; Gibbons, D.L. The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors. Cells 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittal, D.; Vijayan, D.; Smyth, M.J. Overcoming Acquired PD-1/PD-L1 Resistance with CD38 Blockade. Cancer Discov. 2018, 8, 1066–1068. [Google Scholar] [CrossRef] [PubMed]
- Zocchi, E.; Franco, L.; Guida, L.; Piccini, D.; Tacchetti, C.; De Flora, A. NAD+-dependent internalization of the transmembrane glycoprotein CD38 in human Namalwa B cells. FEBS Lett. 1996, 396, 327–332. [Google Scholar] [CrossRef]
- McCarthy, T.V.; Datar, S.; Mackrill, J.J. Activation of ryanodine receptor/Ca2+ release channels downregulates CD38 in the Namalwa B lymphoma. FEBS Lett. 2003, 554, 133–137. [Google Scholar] [CrossRef]
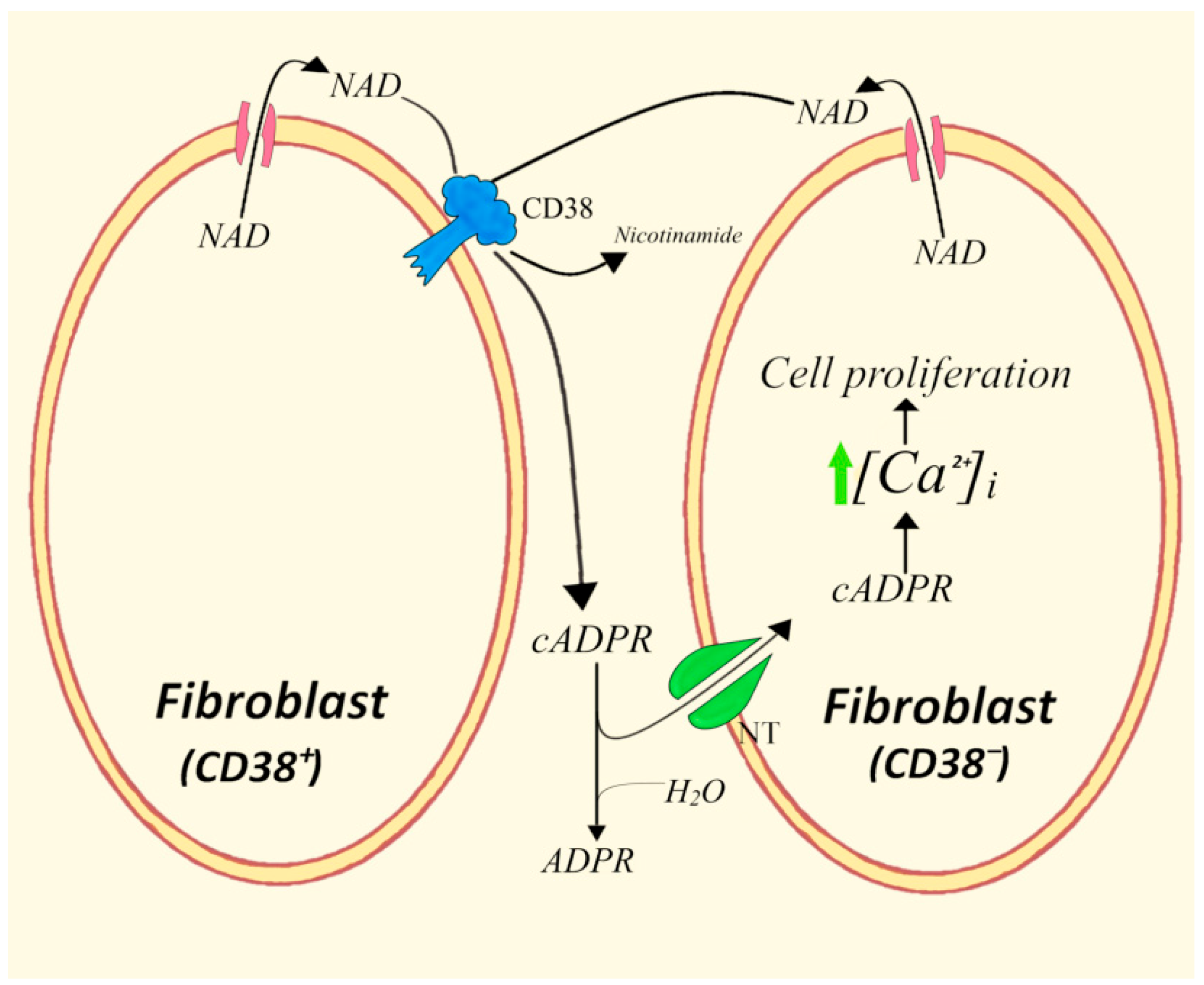
- Zocchi, E.; Daga, A.; Usai, C.; Franco, L.; Guida, L.; Bruzzone, S.; Costa, A.; Marchetti, C.; De Flora, A. Expression of CD38 increases intracellular calcium concentration and reduces doubling time in HeLa and 3T3 cells. J. Biol. Chem. 1998, 273, 8017–8024. [Google Scholar] [CrossRef]
- Axelsen, L.N.; Calloe, K.; Holstein-Rathlou, N.H.; Nielsen, M.S. Managing the complexity of communication: Regulation of gap junctions by post-translational modification. Front. Pharmacol. 2013, 4, 130. [Google Scholar] [CrossRef]
- Pogoda, K.; Kameritsch, P.; Retamal, M.A.; Vega, J.L. Regulation of gap junction channels and hemichannels by phosphorylation and redox changes: A revision. BMC Cell Biol. 2016, 17, 11. [Google Scholar] [CrossRef]
- Solan, J.L.; Lampe, P.D. Connexin43 phosphorylation: Structural changes and biological effects. Biochem. J. 2009, 419, 261–272. [Google Scholar] [CrossRef]
- Song, E.K.; Rah, S.Y.; Lee, Y.R.; Yoo, C.H.; Kim, Y.R.; Yeom, J.H.; Park, K.H.; Kim, J.S.; Kim, U.H.; Han, M.K. Connexin-43 hemichannels mediate cyclic ADP-ribose generation and its Ca2+-mobilizing activity by NAD+/cyclic ADP-ribose transport. J. Biol. Chem. 2011, 286, 44480–44490. [Google Scholar] [CrossRef]
- Vonheijne, G. Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature 1989, 341, 456–458. [Google Scholar] [CrossRef]
- Park, D.R.; Nam, T.S.; Kim, Y.W.; Bae, Y.S.; Kim, U.H. Oxidative activation of type III CD38 by NADPH oxidase-derived hydrogen peroxide in Ca2+ signaling. FASEB J. 2019, 33, 3404–3419. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, J.; Fang, L.; Lee, H.C.; Zhao, Y.J. A cytosolic chaperone complex controls folding and degradation of type III CD38. J. Biol. Chem. 2019, 294, 4247–4258. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Li, T.; Li, Y.; Xu, G.J.; Deng, Q.W.; Chen, Y.J.; Hou, Y.N.; Lee, H.C.; Zhao, Y.J. CD38 produces nicotinic acid adenosine dinucleotide phosphate in the lysosome. J. Biol. Chem. 2018, 293, 8151–8160. [Google Scholar] [CrossRef]
- Rah, S.Y.; Mushtaq, M.; Nam, T.S.; Kim, S.H.; Kim, U.H. Generation of cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate by CD38 for Ca2+ signaling in interleukin-8-treated lymphokine-activated killer cells. J. Biol. Chem. 2010, 285, 21877–21887. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.; Bruzzone, S.; Song, P.; Guida, L.; Zocchi, E.; Walseth, T.F.; Crimi, E.; Usai, C.; De Flora, A.; Brusasco, V. Extracellular cyclic ADP-ribose potentiates ACh-induced contraction in bovine tracheal smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L98–L106. [Google Scholar] [CrossRef]
- Franco, L.; Zocchi, E.; Usai, C.; Guida, L.; Bruzzone, S.; Costa, A.; De Flora, A. Paracrine roles of NAD+ and cyclic ADP-ribose in increasing intracellular calcium and enhancing cell proliferation of 3T3 fibroblasts. J. Biol. Chem. 2001, 276, 21642–21648. [Google Scholar] [CrossRef]
- Perea, G.; Araque, A. Communication between astrocytes and neurons: A complex language. J. Physiol. Paris 2002, 96, 199–207. [Google Scholar] [CrossRef]
- Verderio, C.; Bruzzone, S.; Zocchi, E.; Fedele, E.; Schenk, U.; De Flora, A.; Matteoli, M. Evidence of a role for cyclic ADP-ribose in calcium signalling and neurotransmitter release in cultured astrocytes. J. Neurochem. 2001, 78, 646–657. [Google Scholar] [CrossRef]
- Bruzzone, S.; Verderio, C.; Schenk, U.; Fedele, E.; Zocchi, E.; Matteoli, M.; De Flora, A. Glutamate-mediated overexpression of CD38 in astrocytes cultured with neurones. J. Neurochem. 2004, 89, 264–272. [Google Scholar] [CrossRef]
- Higashida, H.; Bowden, S.E.; Yokoyama, S.; Salmina, A.; Hashii, M.; Hoshi, N.; Zhang, J.S.; Knijnik, R.; Noda, M.; Zhong, Z.G.; et al. Overexpression of human CD38/ADP-ribosyl cyclase enhances acetylcholine-induced Ca2+ signalling in rodent NG108–15 neuroblastoma cells. Neurosci. Res. 2007, 57, 339–346. [Google Scholar] [CrossRef]
- Zhang, J.S.; Jin, D.; Higashida, H. Acetylcholine stimulates cyclic ADP-ribose formation via M1 muscarinic receptors in rat superior cervical ganglion. Biochem. Biophys. Res. Commun. 2005, 335, 920–924. [Google Scholar] [CrossRef]
- Jin, D.; Liu, H.X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behavior by regulating oxytocin secretion. Nature. 2007, 446, 41–45. [Google Scholar] [CrossRef]
- Higashida, H.; Yokoyama, S.; Kikuchi, M.; Munesue, T. CD38 and its role in oxytocin secretion and social behavior. Horm. Behav. 2012, 61, 351–358. [Google Scholar] [CrossRef] [PubMed]
- Higashida, H.; Yokoyama, S.; Huang, J.J.; Liu, L.; Ma, W.J.; Akther, S.; Higashida, C.; Kikuchi, M.; Minabe, Y.; Munesue, T. Social memory, amnesia, and autism: Brain oxytocin secretion is regulated by NAD+ metabolites and single nucleotide polymorphisms of CD38. Neurochem. Int. 2012, 61, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Gerasimenko, M.; Lopatina, O.; Shabalova, A.A.; Cherepanov, S.M.; Salmina, A.B.; Yokoyama, S.; Goto, H.; Okamoto, H.; Yamamoto, Y.; Ishihara, K.; et al. Distinct physical condition and social behavior phenotypes of CD157 and CD38 knockout mice during aging. PLoS ONE 2020, 15, e0244022. [Google Scholar] [CrossRef] [PubMed]
- Higashida, H.; Hashii, M.; Tanaka, Y.; Matsukawa, S.; Higuchi, Y.; Gabata, R.; Tsubomoto, M.; Seishima, N.; Teramachi, M.; Kamijima, T.; et al. CD38, CD157, and RAGE as Molecular Determinants for Social Behavior. Cells 2019, 9, 62. [Google Scholar] [CrossRef]
- Lopatina, O.L.; Komleva, Y.K.; Malinovskaya, N.A.; Panina, Y.A.; Morgun, A.V.; Salmina, A.B. CD157 and Brain Immune System in (Patho)physiological Conditions: Focus on Brain Plasticity. Front. Immunol. 2020, 11, 585294. [Google Scholar] [CrossRef]
- Li, Y.; Pazyra-Murphy, M.F.; Avizonis, D.; de Sá Tavares Russo, M.; Tang, S.; Chen, C.Y.; Hsueh, Y.P.; Bergholz, J.S.; Jiang, T.; Zhao, J.J.; et al. Sarm1 activation produces cADPR to increase intra-axonal Ca++ and promote axon degeneration in PIPN. J. Cell Biol. 2022, 221, e202106080. [Google Scholar] [CrossRef]
- Höke, A. cADPR induced calcium influx mediates axonal degeneration caused by paclitaxel. J. Cell Biol. 2022, 221, e202112021. [Google Scholar] [CrossRef]
- Podestà, M.; Zocchi, E.; Pitto, A.; Usai, C.; Franco, L.; Bruzzone, S.; Guida, L.; Bacigalupo, A.; Scadden, D.T.; Walseth, T.F.; et al. Extracellular cyclic ADP-ribose increases intracellular free calcium concentration and stimulates proliferation of human hemopoietic progenitors. FASEB J. 2000, 14, 680–690. [Google Scholar] [CrossRef]
- Zocchi, E.; Podestà, M.; Pitto, A.; Usai, C.; Bruzzone, S.; Franco, L.; Guida, L.; Bacigalupo, A.; De Flora, A. Paracrinally stimulated expansion of early human hemopoietic progenitors by stroma-generated cyclic ADP-ribose. FASEB J. 2001, 15, 1610–1612. [Google Scholar] [CrossRef] [PubMed]
- Podestà, M.; Pitto, A.; Figari, O.; Bacigalupo, A.; Bruzzone, S.; Guida, L.; Franco, L.; De Flora, A.; Zocchi, E. Cyclic ADP-ribose generation by CD38 improves human hemopoietic stem cell engraftment into NOD/SCID mice. FASEB J. 2003, 17, 310–312. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Guarente, L. mTORC1 and SIRT1 Cooperate to Foster Expansion of Gut Adult Stem Cells during Calorie Restriction. Cell 2016, 166, 436–450. [Google Scholar] [CrossRef]
- Igarashi, M.; Guarente, L. The unexpected role of mTORC1 in intestinal stem cells during calorie restriction. Cell Cycle 2017, 16, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, Ö.H.; Katajisto, P.; Lamming, D.W.; Gültekin, Y.; Bauer-Rowe, K.E.; Sengupta, S.; Birsoy, K.; Dursun, A.; Yilmaz, V.O.; Selig, M.; et al. mTORC1 in the Paneth cell niche couples intestinal stem-cell function to calorie intake. Nature 2012, 486, 490–495. [Google Scholar] [CrossRef]
- Igarashi, M.; Miura, M.; Williams, E.; Jaksch, F.; Kadowaki, T.; Yamauchi, T.; Guarente, L. NAD+ supplementation rejuvenates aged gut adult stem cells. Aging Cell 2019, 18, e12935. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G.; Samburski, S.S.; Mortensen, S.P.; Jalkanen, S.; González-Alonso, J. Intravascular ADP and soluble nucleotidases contribute to acute prothrombotic state during vigorous exercise in humans. J. Physiol. 2007, 579, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Horenstein, A.L.; Malavasi, F. The Key Role of NAD+ in Anti-Tumor Immune Response: An Update. Front. Immunol. 2021, 12, 658263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, F.; Wang, L.; Qiu, S.; Yao, Y.; Xiong, X.; Chen, X.; Ji, Q.; Cao, J.; Li, D.; et al. Potentiating the anti-tumor response of tumor infiltrated T cells by NAD+ supplementation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Chini, E.N.; Chini, C.C.S.; Espindola Netto, J.M.; de Oliveira, G.C.; van Schooten, W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol. Sci. 2018, 39, 424–436. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Liu, Y.; Du, X.; Ma, S.; Ge, M.; Tang, H.; Han, C.; Zhao, X.; Liu, Y.; Shao, Y.; et al. The intrinsic role and mechanism of tumor expressed-CD38 on lung adenocarcinoma progression. Cell Death Dis. 2021, 12, 680. [Google Scholar] [CrossRef] [PubMed]
- Wo, Y.J.; Gan, A.S.P.; Lim, X.; Tay, I.S.Y.; Lim, S.; Lim, J.C.T.; Yeong, J.P.S. The Roles of CD38 and CD157 in the Solid Tumor Microenvironment and Cancer Immunotherapy. Cells 2019, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Ben Baruch, B.; Levy, A.; Geva, N.; Green, K.D.; Garneau-Tsodikova, S.; Fridman, M.; Stein, R. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int. J. Cancer. 2015, 136, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Ben Baruch, B.; Blacher, E.; Mantsur, E.; Schwartz, H.; Vaknine, H.; Erez, N.; Stein, R. Stromal CD38 regulates outgrowth of primary melanoma and generation of spontaneous metastasis. Oncotarget. 2018, 9, 31797–31811. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Munari, E.; Mariotti, F.R.; Quatrini, L.; Bertoglio, P.; Tumino, N.; Vacca, P.; Eccher, A.; Ciompi, F.; Brunelli, M.; Martignoni, G.; et al. PD-1/PD-L1 in Cancer: Pathophysiological, Diagnostic and Therapeutic Aspects. Int. J. Mol. Sci. 2021, 22, 5123. [Google Scholar] [CrossRef]
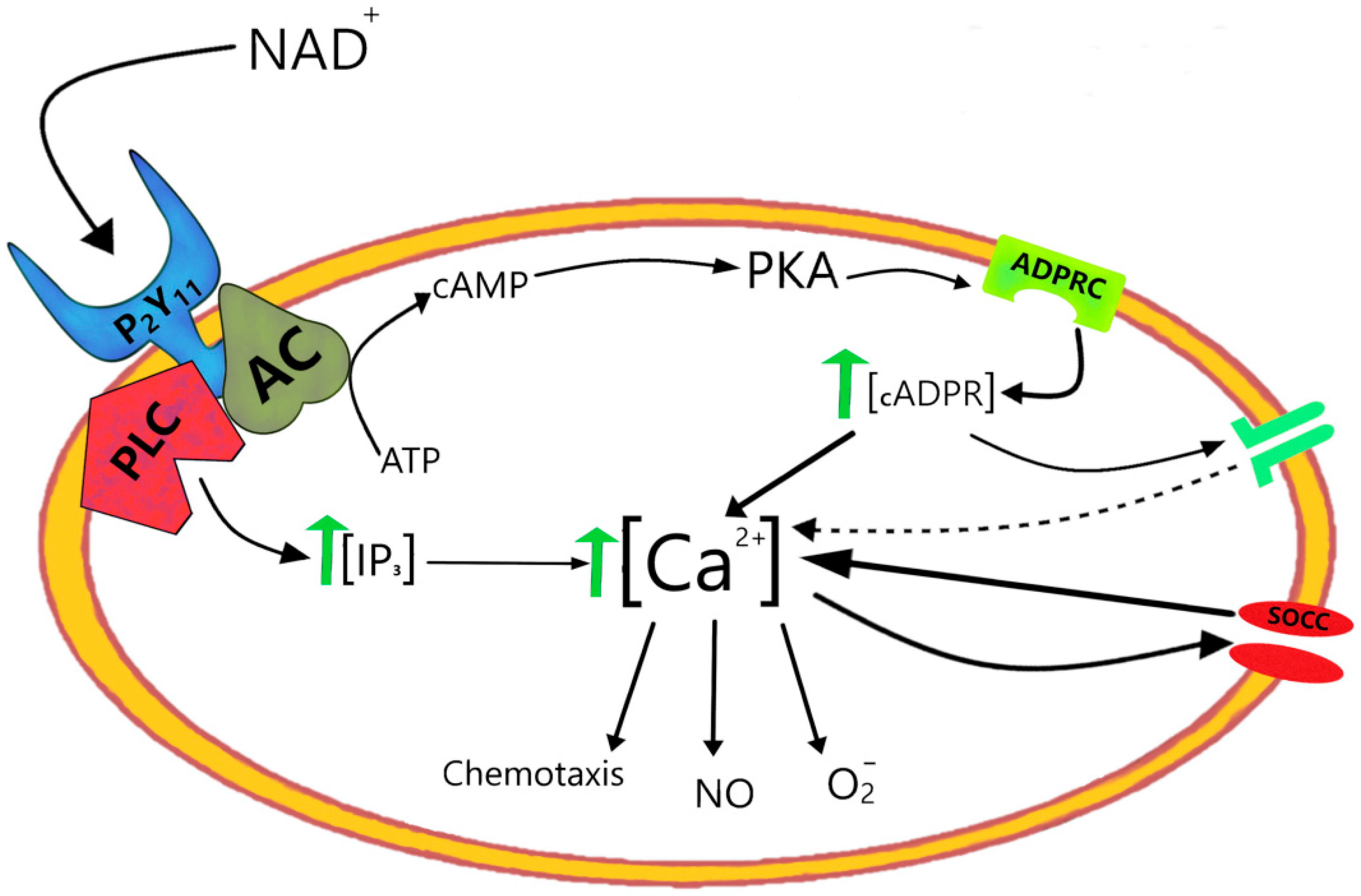
- Moreschi, I.; Bruzzone, S.; Nicholas, R.A.; Fruscione, F.; Sturla, L.; Benvenuto, F.; Usai, C.; Meis, S.; Kassack, M.U.; Zocchi, E.; et al. Extracellular NAD+ is an agonist of the human P2Y11 purinergic receptor in human granulocytes. J. Biol. Chem. 2006, 281, 31419–31429. [Google Scholar] [CrossRef]
- Moresch, I.; Bruzzone, S.; Bodrato, N.; Usai, C.; Guida, L.; Nicholas, R.A.; Kassack, M.U.; Zocchi, E.; De Flora, A. NAADP+ is an agonist of the human P2Y11 purinergic receptor. Cell Calcium 2008, 43, 344–355. [Google Scholar] [CrossRef]
- Bruzzone, S.; Moreschi, I.; Guida, L.; Usai, C.; Zocchi, E.; De Flora, A. Extracellular NAD+ regulates intracellular calcium levels and induces activation of human granulocytes. Biochem. J. 2006, 393, 697–704. [Google Scholar] [CrossRef]
- Krebs, C.; Adriouch, S.; Braasch, F.; Koestner, W.; Leiter, E.H.; Seman, M.; Lund, F.E.; Oppenheimer, N.; Haag, F.; Koch-Nolte, F. CD38 controls ADP-ribosyltransferase-2-catalyzed ADP-ribosylation of T cell surface proteins. J. Immunol. 2005, 174, 3298–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aswad, F.; Kawamura, H.; Dennert, G. High sensitivity of CD4+CD25+ regulatory T cells to extracellular metabolites nicotinamide adenine dinucleotide and ATP: A role for P2X7 receptors. J. Immunol. 2005, 175, 3075–3083. [Google Scholar] [CrossRef]
- Park, K.H.; Kim, B.J.; Shawl, A.I.; Han, M.K.; Lee, H.C.; Kim, U.H. Autocrine/paracrine function of nicotinic acid adenine dinucleotide phosphate (NAADP) for glucose homeostasis in pancreatic β-cells and adipocytes. J. Biol. Chem. 2013, 288, 35548–35558. [Google Scholar] [CrossRef] [PubMed]
- Walseth, T.F.; Lin-Moshier, Y.; Jain, P.; Ruas, M.; Parrington, J.; Galione, A.; Marchant, J.S.; Slama, J.T. Photoaffinity labeling of high affinity nicotinic acid adenine dinucleotide phosphate (NAADP)-binding proteins in sea urchin egg. J. Biol. Chem. 2012, 287, 2308–2315. [Google Scholar] [CrossRef] [PubMed]
- Roggenkamp, H.G.; Khansahib, I.; Hernandez, C.L.C.; Zhang, Y.; Lodygin, D.; Krüger, A.; Gu, F.; Möckl, F.; Löhndorf, A.; Wolters, V.; et al. HN1L/JPT2: A signaling protein that connects NAADP generation to Ca2+ microdomain formation. Sci. Signal. 2021, 14, eabd5647. [Google Scholar] [CrossRef] [PubMed]
- Gunaratne, G.S.; Brailoiu, E.; He, S.; Unterwald, E.M.; Patel, S.; Slama, J.T.; Walseth, T.F.; Marchant, J.S. Essential requirement for JPT2 in NAADP-evoked Ca2+ signaling. Sci. Signal. 2021, 14, eabd5605. [Google Scholar] [CrossRef]
- Zhang, J.; Guan, X.; Shah, K.; Yan, J. Lsm12 is an NAADP receptor and a two-pore channel regulatory protein required for calcium mobilization from acidic organelles. Nat. Commun. 2021, 12, 4739. [Google Scholar] [CrossRef]
- Zhang, K.; Sun, W.; Huang, L.; Zhu, K.; Pei, F.; Zhu, L.; Wang, Q.; Lu, Y.; Zhang, H.; Jin, H.; et al. Identifying Glyceraldehyde 3-Phosphate Dehydrogenase as a Cyclic Adenosine Diphosphoribose Binding Protein by Photoaffinity Protein-Ligand Labeling Approach. J. Am. Chem. Soc. 2017, 139, 156–170. [Google Scholar] [CrossRef]
- Noguchi, N.; Takasawa, S.; Nata, K.; Tohgo, A.; Kato, I.; Ikehata, F.; Yonekura, H.; Okamoto, H. Cyclic ADP-ribose binds to FK506-binding protein 12.6 to release Ca2+ from islet microsomes. J. Biol. Chem. 1997, 272, 3133–3136. [Google Scholar] [CrossRef]
- Higashida, H.; Liang, M.; Yoshihara, T.; Akther, S.; Fakhrul, A.; Stanislav, C.; Nam, T.S.; Kim, U.H.; Kasai, S.; Nishimura, T.; et al. An immunohistochemical, enzymatic, and behavioral study of CD157/BST-1 as a neuroregulator. BMC Neurosci. 2017, 18, 35. [Google Scholar] [CrossRef]
- Ma, K.; Sun, L.; Shen, M.; Zhang, X.; Xiao, Z.; Wang, J.; Liu, X.; Jiang, K.; Xiao-Feng Qin, F.; Guo, F.; et al. Functional assessment of the cell-autonomous role of NADase CD38 in regulating CD8+ T cell exhaustion. iScience 2022, 25, 104347. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Astigiano, C.; Benzi, A.; Laugieri, M.E.; Piacente, F.; Sturla, L.; Guida, L.; Bruzzone, S.; De Flora, A. Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes. Cells 2022, 11, 2637. https://doi.org/10.3390/cells11172637
Astigiano C, Benzi A, Laugieri ME, Piacente F, Sturla L, Guida L, Bruzzone S, De Flora A. Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes. Cells. 2022; 11(17):2637. https://doi.org/10.3390/cells11172637
Chicago/Turabian StyleAstigiano, Cecilia, Andrea Benzi, Maria Elena Laugieri, Francesco Piacente, Laura Sturla, Lucrezia Guida, Santina Bruzzone, and Antonio De Flora. 2022. "Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes" Cells 11, no. 17: 2637. https://doi.org/10.3390/cells11172637
APA StyleAstigiano, C., Benzi, A., Laugieri, M. E., Piacente, F., Sturla, L., Guida, L., Bruzzone, S., & De Flora, A. (2022). Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes. Cells, 11(17), 2637. https://doi.org/10.3390/cells11172637