


Cancer Stem Cells—The Insight into Non-Coding RNAs

 ,
,  ,
,
Abstract
1. Introduction
2. Biology of CSCs and Their Interaction with Tumor Microenvironment
2.1. CSCs Signaling Pathways
2.1.1. PI3K/Akt/mTOR Signaling Pathway
2.1.2. Wnt Signaling Pathway
2.1.3. JAK/STAT Signaling Pathway
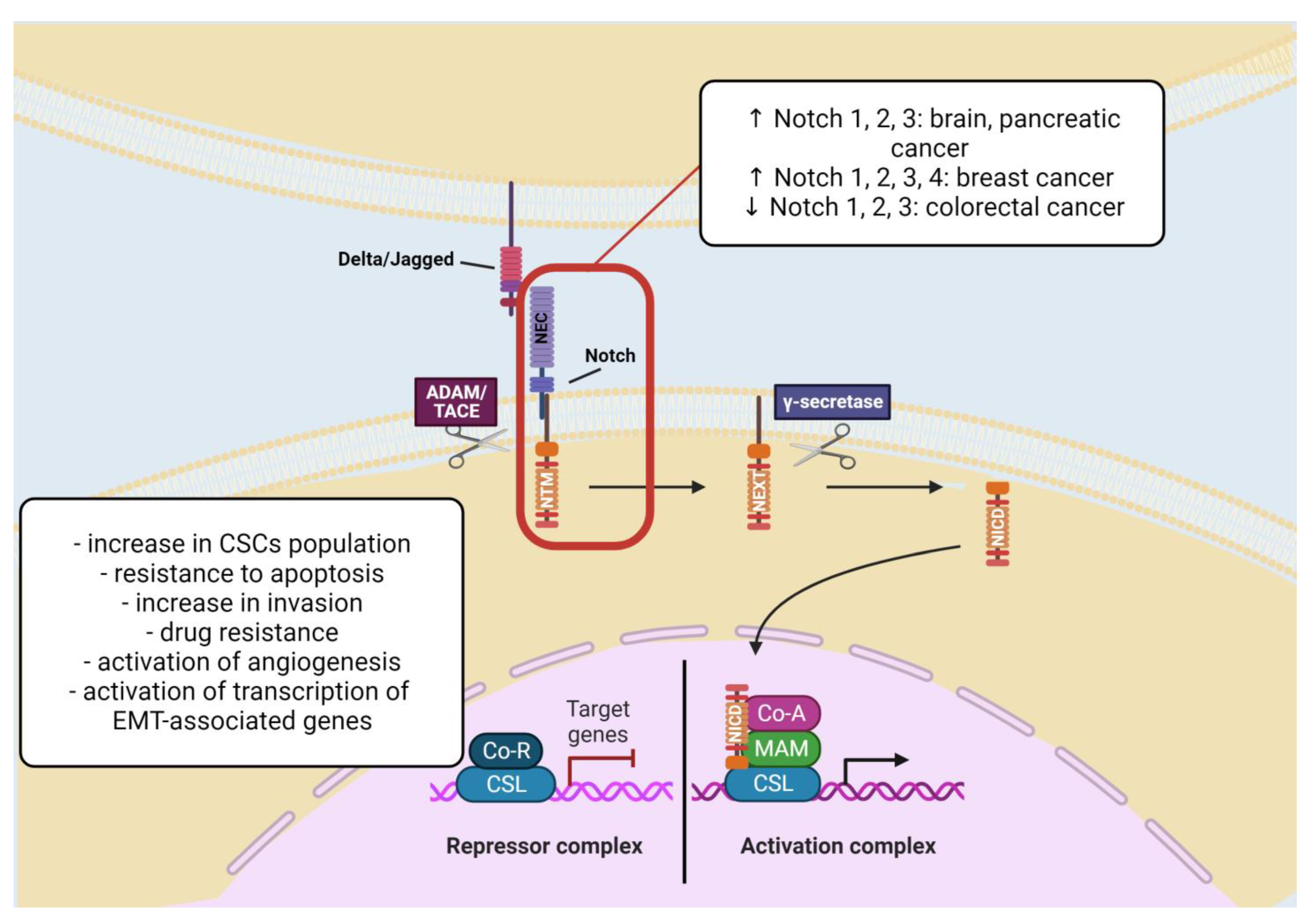
2.1.4. Notch Signaling Pathway
2.2. CSCs Plasticity, EMT and Dormancy
2.3. CSCs Metabolic Changes
2.4. Epigenetic Regulation of CSCs—The Role of microRNAs, circRNAs and lncRNAs
2.4.1. miRNAs in CSCs
miRNAs as Promoters of CSCs
miRNAs as Suppressors of CSCs
2.4.2. circRNAs in CSCs
circRNAs as Promoters of CSCs
circRNAs as Suppressors of CSCs
2.4.3. lncRNAs in CSCs
lncRNAs as Promoters of CSCs
lncRNAs as Suppressors of CSCs
3. Non-Coding RNAs in Therapeutic Anti-CSCs Strategies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lambert, A.W.; Weinberg, R.A. Linking EMT Programmes to Normal and Neoplastic Epithelial Stem Cells. Nat. Rev. Cancer 2021, 21, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A Cell Initiating Human Acute Myeloid Leukaemia after Transplantation into SCID Mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human Acute Myeloid Leukemia Is Organized as a Hierarchy That Originates from a Primitive Hematopoietic Cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of Human Brain Tumour Initiating Cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; de Maria, R. Identification and Expansion of Human Colon-Cancer-Initiating Cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Hwang-Verslues, W.W.; Kuo, W.H.; Chang, P.H.; Pan, C.C.; Wang, H.H.; Tsai, S.; Jeng, Y.M.; Shew, J.Y.; Kung, J.T.; Chen, C.H.; et al. Multiple Lineages of Human Breast Cancer Stem/Progenitor Cells Identified by Profiling with Stem Cell Markers. PLoS ONE 2009, 4, e8377. [Google Scholar] [CrossRef]
- Capp, J.P. Cancer Stem Cells: From Historical Roots to a New Perspective. J. Oncol. 2019, 2019, 5189232. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 8. [Google Scholar] [CrossRef]
- Lei, M.M.L.; Lee, T.K.W. Cancer Stem Cells: Emerging Key Players in Immune Evasion of Cancers. Front. Cell Dev. Biol. 2021, 9, 1643. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Rutella, S.; Ferrone, S.; Maccalli, C. Cancer Stem Cells: The Players of Immune Evasion from Immunotherapy. In Cancer Stem Cell Resistance to Targeted Therapy; Springer: Berlin/Heidelberg, Germany, 2019. [Google Scholar]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-Coding RNA Networks in Cancer. Nat. Rev. Cancer 2017, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-Coding RNA Regulatory Networks. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194417. [Google Scholar] [CrossRef] [PubMed]
- Romano, G.; Veneziano, D.; Acunzo, M.; Croce, C.M. Small Non-Coding RNA and Cancer. Carcinogenesis 2017, 38, 485–491. [Google Scholar] [CrossRef]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in Cancer: Opportunities and Challenges in the Field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The Role of MicroRNAs in Human Cancer. Signal Transduct. Target Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- de Angelis, M.L.; Francescangeli, F.; Zeuner, A. Breast Cancer Stem Cells as Drivers of Tumor Chemoresistance, Dormancy and Relapse: New Challenges and Therapeutic Opportunities. Cancers 2019, 11, 1569. [Google Scholar] [CrossRef]
- Lizárraga-Verdugo, E.; Avendaño-Félix, M.; Bermúdez, M.; Ramos-Payán, R.; Pérez-Plasencia, C.; Aguilar-Medina, M. Cancer Stem Cells and Its Role in Angiogenesis and Vasculogenic Mimicry in Gastrointestinal Cancers. Front. Oncol. 2020, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer Stem Cell Metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef]
- Whiteside, T.L. Exosomes and Tumor-Mediated Immune Suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef]
- Ravindran, S.; Rasool, S.; Maccalli, C. The Cross Talk between Cancer Stem Cells/Cancer Initiating Cells and Tumor Microenvironment: The Missing Piece of the Puzzle for the Efficient Targeting of These Cells with Immunotherapy. Cancer Microenviron. 2019, 12, 133–148. [Google Scholar] [CrossRef]
- Shidal, C.; Singh, N.P.; Nagarkatti, P.; Nagarkatti, M. MicroRNA-92 Expression in CD133þ Melanoma Stem Cells Regulates Immunosuppression in the Tumor Microenvironment via Integrin-Dependent Activation of TGFb. Cancer Res. 2019, 79, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and Drug Resistance: The Mechanistic Link and Clinical Implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Klepinina, L.; Klepinin, A.; Truu, L.; Chekulayev, V.; Vija, H.; Kuus, K.; Teino, I.; Pook, M.; Maimets, T.; Kaambre, T. Colon Cancer Cell Differentiation by Sodium Butyrate Modulates Metabolic Plasticity of Caco-2 Cells via Alteration of Phosphotransfer Network. PLoS ONE 2021, 16, e0245348. [Google Scholar] [CrossRef]
- Zhang, R.; Tu, J.; Liu, S. Novel Molecular Regulators of Breast Cancer Stem Cell Plasticity and Heterogeneity. Semin. Cancer Biol. 2022, 82, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Francis, J.C.; Capper, A.; Ning, J.; Knight, E.; de Bono, J.; Swain, A. SOX9 Is a Driver of Aggressive Prostate Cancer by Promoting Invasion, Cell Fate and Cytoskeleton Alterations and Epithelial to Mesenchymal Transition. Oncotarget 2018, 9, 7604–7615. [Google Scholar] [CrossRef]
- Qian, J.; Rankin, E.B. Hypoxia-Induced Phenotypes That Mediate Tumor Heterogeneity. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 1136. [Google Scholar]
- Sun, X.; Lv, X.; Yan, Y.; Zhao, Y.; Ma, R.; He, M.; Wei, M. Hypoxia-Mediated Cancer Stem Cell Resistance and Targeted Therapy. Biomed. Pharmacother. 2020, 130, 110623. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, Z.; Zhu, Y.; Chen, J.; Li, W. Role of Hypoxia Inducible Factor-1 in Cancer Stem Cells (Review). Mol. Med. Rep. 2021, 23, 17. [Google Scholar] [CrossRef]
- Olmeda, D.; Montes, A.; Flores, J.M.; Portillo, F.; Cano, A. Snai1 and Snai2 Collaborate on Tumor Growth and Metastasis Properties of Mouse Skin Carcinoma Cell Lines. Oncogene 2008, 27, 4690–4701. [Google Scholar] [CrossRef]
- Li, Y.; Wu, Y.; Abbatiello, T.C.; Wu, W.L.; Kim, J.R.; Sarkissyan, M.; Sarkissyan, S.; Chung, S.S. Slug Contributes to Cancer Progression by Direct Regulation of ER α Signaling Pathway. Int. J. Oncol. 2015, 46, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The Roles of ZEB1 in Tumorigenic Progression and Epigenetic Modi Fi Cations. Biomed. Pharmacother. 2019, 110, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Hulea, L.; Gravel, S.P.; Morita, M.; Cargnello, M.; Uchenunu, O.; Im, Y.K.; Lehuédé, C.; Ma, E.H.; Leibovitch, M.; McLaughlan, S.; et al. Translational and HIF-1α-Dependent Metabolic Reprogramming Underpin Metabolic Plasticity and Responses to Kinase Inhibitors and Biguanides. Cell Metab. 2018, 28, 817–832.e8. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Kim, J.S.; Chae, C.W.; Han, H.J. Role of HIF1α Regulatory Factors in Stem Cells. Int. J. Stem Cells 2019, 12, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-A.; Wang, C.-Y.; Hsieh, Y.-T.; Chen, Y.-J.; Wei, Y.-H. Metabolic Reprogramming Orchestrates Cancer Stem Cell Properties in Nasopharyngeal Carcinoma. Cell Cycle 2015, 14, 86–98. [Google Scholar] [CrossRef]
- Smigiel, J.M.; Parameswaran, N.; Jackson, M.W. Targeting Pancreatic Cancer Cell Plasticity: The Latest in Therapeutics. Cancers 2018, 10, 14. [Google Scholar] [CrossRef]
- Yadav, U.P.; Singh, T.; Kumar, P.; Sharma, P.; Kaur, H.; Sharma, S.; Singh, S.; Kumar, S.; Mehta, K. Metabolic Adaptations in Cancer Stem Cells. Front Oncol 2020, 10, 1010. [Google Scholar] [CrossRef]
- O’Brien-Ball, C.; Biddle, A. Reprogramming to Developmental Plasticity in Cancer Stem Cells. Dev. Biol. 2017, 430, 266–274. [Google Scholar] [CrossRef]
- Garcia-Heredia, J.M.; Lucena-Cacace, A.; Verdugo-Sivianes, E.M.; Perez, M.; Carnero, A. The Cargo Protein MAP17 (PDZK1IP1) Regulates the Cancer Stem Cell Pool Activating the Notch Pathway by Abducting NUMB. Clin. Cancer Res. 2017, 23, 3871–3883. [Google Scholar] [CrossRef]
- Murgai, M.; Ju, W.; Eason, M.; Kline, J.; Beury, D.W.; Kaczanowska, S.; Miettinen, M.M.; Kruhlak, M.; Lei, H.; Shern, J.F.; et al. KLF4-Dependent Perivascular Cell Plasticity Mediates Pre-Metastatic Niche Formation and Metastasis. Nat. Med. 2017, 23, 1176–1190. [Google Scholar] [CrossRef]
- Joseph, D.; Gonsky, J.P.; Blain, S.W. Macrophage Inhibitory Factor-1 (MIF-1) Controls the Plasticity of Multiple Myeloma Tumor Cells. PLoS ONE 2018, 1, e0206368. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Giorgi, C.; Lebiedzinska, M.; Esposito, G.; Bartoli, A.; Gough, D.J.; Turkson, J.; Levy, D.E.; Christine, J.; Angeli, D.; et al. STAT3-Mediated Metabolic Switch Is Involved in Tumour Transformation and STAT3 Addiction. Aging 2010, 2, 823–842. [Google Scholar] [CrossRef] [PubMed]
- Turdo, A.; Veschi, V.; Gaggianesi, M.; Chinnici, A.; Bianca, P.; Todaro, M.; Stassi, G. Meeting the Challenge of Targeting Cancer Stem Cells. Front. Cell Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol Cancer 2019, 18, 26. [Google Scholar] [CrossRef]
- Deng, J.; Bai, X.; Feng, X.; Ni, J.; Beretov, J.; Graham, P.; Li, Y. Inhibition of PI3K/Akt/MTOR Signaling Pathway Alleviates Ovarian Cancer Chemoresistance through Reversing Epithelial-Mesenchymal Transition and Decreasing Cancer Stem Cell Marker Expression. BMC Cancer 2019, 19, 618. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Buján, J.; García-Honduvilla, N.; Coca, S. Signal Transduction Pathways in Breast Cancer: The Important Role of PI3K/Akt/MTOR. J. Oncol. 2020, 2020, 9258396. [Google Scholar] [CrossRef]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-MTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef]
- Nanta, R.; Shrivastava, A.; Sharma, J.; Shankar, S.; Srivastava, R.K. Inhibition of Sonic Hedgehog and PI3K/Akt/MTOR Pathways Cooperate in Suppressing Survival, Self-Renewal and Tumorigenic Potential of Glioblastoma-Initiating Cells. Mol. Cell Biochem. 2019, 454, 11–23. [Google Scholar] [CrossRef]
- Han, C.Y.; Patten, D.A.; Richardson, R.B.; Harper, M.E.; Tsang, B.K. Tumor Metabolism Regulating Chemosensitivity in Ovarian Cancer. Genes Cancer 2018, 9, 155–175. [Google Scholar] [CrossRef]
- Paplomata, E.; O’regan, R. The PI3K/AKT/MTOR Pathway in Breast Cancer: Targets, Trials and Biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef]
- Madsen, R.R. PI3K in Stemness Regulation: From Development to Cancer. Biochem. Soc. Trans. 2020, 48, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting Signalling Pathways and the Immune Microenvironment of Cancer Stem Cells—A Clinical Update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. Canonical and Non-Canonical WNT Signaling in Cancer Stem Cells and Their Niches: Cellular Heterogeneity, Omics Reprogramming, Targeted Therapy and Tumor Plasticity (Review). Int. J. Oncol. 2017, 51, 1357–1369. [Google Scholar] [CrossRef]
- Kim, J.H.; Park, S.Y.; Jun, Y.; Kim, J.Y.; Nam, J.S. Roles of Wnt Target Genes in the Journey of Cancer Stem Cells. Int. J. Mol. Sci. 2017, 18, 1604. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The Role of JAK/STAT Signalling in the Pathogenesis, Prognosis and Treatment of Solid Tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef]
- Kim, S.L.; Choi, H.S.; Kim, J.H.; Jeong, D.K.; Kim, K.S.; Lee, D.S. Dihydrotanshinone-Induced Nox5 Activation Inhibits Breast Cancer Stem Cell through the ROS/STAT3 Signaling Pathway. Oxid. Med. Cell Longev. 2019, 2019, 9296439. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Z.; Yang, X.; Cheng, W. OCT4 Accelerates Tumorigenesis through Activating JAK/STAT Signaling in Ovarian Cancer Side Population Cells. Cancer Manag. Res. 2019, 11, 389–399. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, C.J.; Choi, J.H.; Kim, J.H.; Kim, J.W.; Kim, J.Y.; Nam, J.S. The JAK2/STAT3/CCND2 Axis Promotes Colorectal Cancer Stem Cell Persistence and Radioresistance. J. Exp. Clin. Cancer Res. 2019, 38, 399. [Google Scholar] [CrossRef]
- Li, X.; Dong, Y.; Yin, H.; Qi, Z.; Wang, D.; Ren, S. Mesenchymal Stem Cells Induced Regulatory Dendritic Cells from Hemopoietic Progenitor Cells through Notch Pathway and TGF-β Synergistically. Immunol. Lett. 2020, 222, 49–57. [Google Scholar] [CrossRef]
- Meisel, C.T.; Porcheri, C.; Mitsiadis, T.A. Cancer Stem Cells, Quo Vadis? The Notch Signaling Pathway in Tumor Initiation and Progression. Cells 2020, 9, 1879. [Google Scholar] [CrossRef] [PubMed]
- BeLow, M.; Osipo, C. Notch Signaling in Breast Cancer: A Role in Drug Resistance. Cells 2020, 9, 2204. [Google Scholar] [CrossRef] [PubMed]
- Leontovich, A.A.; Jalalirad, M.; Salisbury, J.L.; Mills, L.; Haddox, C.; Schroeder, M.; Tuma, A.; Guicciardi, M.E.; Zammataro, L.; Gambino, M.W.; et al. NOTCH3 Expression Is Linked to Breast Cancer Seeding and Distant Metastasis. Breast Cancer Res. 2018, 20, 105. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of Breast Cancer Stem Cell Activity by Signaling through the Notch4 Receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef]
- Islam, S.S.; Aboussekhra, A. Sequential Combination of Cisplatin with Eugenol Targets Ovarian Cancer Stem Cells through the Notch-Hes1 Signalling Pathway. J. Exp. Clin. Cancer Res. 2019, 38, 382. [Google Scholar] [CrossRef]
- Venkatesh, V.; Nataraj, R.; Thangaraj, G.S.; Karthikeyan, M.; Gnanasekaran, A.; Kaginelli, S.B.; Kuppanna, G.; Kallappa, C.G.; Basalingappa, K.M. Targeting Notch Signalling Pathway of Cancer Stem Cells. Stem Cell Investig. 2018, 5, 5. [Google Scholar] [CrossRef]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic Plasticity, Bet-Hedging, and Androgen Independence in Prostate Cancer: Role of Non-Genetic Heterogeneity. Front. Oncol. 2018, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.G.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J.C. Vascular Channel Formation by Human Melanoma Cells in Vivo and in Vitro: Vasculogenic Mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef]
- Knopik-Skrocka, A.; Krȩplewska, P.; Jarmołowska-Jurczyszyn, D. Tumor Blood Vessels and Vasculogenic Mimicry-Current Knowledge and Searching for New Cellular/Molecular Targets of Anti-Angiogenic Therapy. Adv. Cell Biol. 2017, 5, 50–71. [Google Scholar] [CrossRef]
- Ayala-Domínguez, L.; Olmedo-Nieva, L.; Muñoz-Bello, J.O.; Contreras-Paredes, A.; Manzo-Merino, J.; Martínez-Ramírez, I.; Lizano, M. Mechanisms of Vasculogenic Mimicry in Ovarian Cancer. Front. Oncol. 2019, 9, 998. [Google Scholar] [CrossRef]
- Sun, H.; Yao, N.; Cheng, S.; Li, L.; Liu, S.; Yang, Z.; Shang, G.; Zhang, D.; Yao, Z. Cancer Stem-like Cells Directly Participate in Vasculogenic Mimicry Channels in Triple-Negative Breast Cancer. Cancer Biol. Med. 2019, 16, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Fazioli, F.; Colella, G.; Miceli, R.; di Salvatore, M.G.; Gallo, M.; Boccella, S.; de Chiara, A.; Ruosi, C.; de Nigris, F. Post-Surgery Fluids Promote Transition of Cancer Stem Cellto- Endothelial and AKT/MTOR Activity, Contributing to Relapse of Giant Cell Tumors of Bone. Oncotarget 2017, 8, 85040–85053. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.P.; Liao, Y.D.; Mai, D.M.; Xie, P.; Qiang, Y.Y.; Zheng, L.S.; Wang, M.Y.; Mei, Y.; Meng, D.F.; Xu, L.; et al. Tumor Vasculogenic Mimicry Predicts Poor Prognosis in Cancer Patients: A Meta-Analysis. Angiogenesis 2016, 19, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Soundararajan, R.; Paranjape, A.N.; Maity, S.; Aparicio, A.; Mani, S.A. EMT, Stemness and Tumor Plasticity in Aggressive Variant Neuroendocrine Prostate Cancers. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 229–238. [Google Scholar] [CrossRef]
- Wang, H.; Unternaehrer, J.J. Epithelial-Mesenchymal Transition and Cancer Stem Cells: At the Crossroads of Differentiation and Dedifferentiation. Dev. Dyn. 2019, 248, 10–20. [Google Scholar] [CrossRef]
- Garg, M. Epithelial Plasticity and Cancer Stem Cells: Major Mechanisms of Cancer Pathogenesis and Therapy Resistance. World J Stem Cells 2017, 9, 118–126. [Google Scholar] [CrossRef]
- Phi, L.T.H.; Sari, I.N.; Yang, Y.G.; Lee, S.H.; Jun, N.; Kim, K.S.; Lee, Y.K.; Kwon, H.Y. Cancer Stem Cells (CSCs) in Drug Resistance and Their Therapeutic Implications in Cancer Treatment. Stem Cells Int. 2018, 2018, 5416923. [Google Scholar] [CrossRef]
- Greenburg, G.; Hay, E.D. Epithelia Suspended in Collagen Gels Can Lose Polarity and Express Characteristics of Migrating Mesenchymal Cells. J. Cell Biol. 1982, 95, 333–339. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Bornes, L.; Belthier, G.; van Rheenen, J. Epithelial-to-Mesenchymal Transition in the Light of Plasticity and Hybrid e/m States. J. Clin. Med. 2021, 10, 2403. [Google Scholar] [CrossRef]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and Definitions for Research on Epithelial–Mesenchymal Transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Kröger, C.; Afeyan, A.; Mraz, J.; Eaton, E.N.; Reinhardt, F.; Khodor, Y.L.; Thiru, P.; Bierie, B.; Ye, X.; Burge, C.B.; et al. Acquisition of a Hybrid E/M State Is Essential for Tumorigenicity of Basal Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2019, 116, 7353–7362. [Google Scholar] [CrossRef]
- Thankamony, A.P.; Saxena, K.; Murali, R.; Jolly, M.K.; Nair, R. Cancer Stem Cell Plasticity–A Deadly Deal. Front. Mol. Biosci. 2020, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.J.; Ho, C.C.; Chang, Y.L.; Chen, H.Y.; Lin, C.A.; Ling, T.Y.; Yu, S.L.; Yuan, S.S.; Louisa Chen, Y.J.; Lin, C.Y.; et al. Cancer-Associated Fibroblasts Regulate the Plasticity of Lung Cancer Stemness via Paracrine Signalling. Nat. Commun. 2014, 5, 3472. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.T.; Lo, J.; Cheng, B.Y.L.; Ma, M.K.F.; Lee, J.M.F.; Ng, J.K.Y.; Chai, S.; Lin, C.H.; Tsang, S.Y.; Ma, S.; et al. Cancer-Associated Fibroblasts Regulate Tumor-Initiating Cell Plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 Signaling. Cell Rep. 2016, 15, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Kim, J.K.; Shiozawa, Y.; Wang, J.; Mishra, A.; Joseph, J.; Berry, J.E.; McGee, S.; Lee, E.; Sun, H.; et al. Recruitment of Mesenchymal Stem Cells into Prostate Tumours Promotes Metastasis. Nat. Commun. 2013, 4, 1795. [Google Scholar] [CrossRef]
- Vaziri, N.; Shariati, L.; Zarrabi, A.; Farazmand, A.; Javanmard, S.H. Cancer-Associated Fibroblasts Regulate the Plasticity of Breast Cancer Stemness through the Production of Leukemia Inhibitory Factor. Life 2021, 11, 1298. [Google Scholar] [CrossRef]
- Doherty, M.R.; Parvani, J.G.; Tamagno, I.; Junk, D.J.; Bryson, B.L.; Cheon, H.J.; Stark, G.R.; Jackson, M.W. The Opposing Effects of Interferon-Beta and Oncostatin-M as Regulators of Cancer Stem Cell Plasticity in Triple-Negative Breast Cancer. Breast Cancer Res. 2019, 21, 54. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, B.E.; Leong, K.G.; Yue, P.; Li, L.; Jhunjhunwala, S.; Chen, D.; Seo, K.; Modrusan, Z.; Gao, W.Q.; et al. Androgen Deprivation Causes Epithelial-Mesenchymal Transition in the Prostate: Implications for Androgen- Deprivation Therapy. Cancer Res. 2012, 72, 527–536. [Google Scholar] [CrossRef]
- Guo, Y.; Zhang, K.; Cheng, C.; Ji, Z.; Wang, X.; Wang, M.; Chu, M.; Tang, D.G.; Zhu, H.H.; Gao, W.Q. Numb-/Low Enriches a Castration-Resistant Prostate Cancer Cell Subpopulation Associated with Enhanced Notch and Hedgehog Signaling. Clin. Cancer Res. 2017, 23, 6744–6756. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and Cancer Stem Cells: An Enigma for Cancer Therapeutic Targeting. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 2019; Volume 141, pp. 43–84. [Google Scholar]
- Crea, F.; Nur Saidy, N.R.; Collins, C.C.; Wang, Y. The Epigenetic/Noncoding Origin of Tumor Dormancy. Trends Mol. Med. 2015, 21, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.L.; Zhang, M.; Tang, Y.L.; Liang, X.H. Cancer Cell Dormancy: Mechanisms and Implications of Cancer Recurrence and Metastasis. OncoTargets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Wang, L.; Tang, J.; Cao, P.; Luo, Z.; Sun, J.; Kiflu, A.; Sai, B.; Zhang, M.; Wang, F.; et al. Activation of Anaphase-Promoting Complex by P53 Induces a State of Dormancy in Cancer Cells against Chemotherapeutic Stress. Oncotarget 2016, 7, 25478–25492. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Zhang, C.; Ling, S.; Wei, R.; Wang, J.; Xu, X. The Metabolic Flexibility of Quiescent CSC: Implications for Chemotherapy Resistance. Cell Death Dis. 2021, 12, 835. [Google Scholar] [CrossRef] [PubMed]
- Szlasa, W.; Wala, K.; Kiełbik, A.; Zalesińska, A.; Saczko, J.; Kulbacka, J. Connection between Warburg Effect and Oncometabolites Biosynthesis with Connection between Warburg Effect and Oncometabolites Biosynthesis with Its Clinical Implications. Org. Med. Chem. 2020, 9, 103–111. [Google Scholar] [CrossRef]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef]
- Elgendy, M.; Cirò, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of Hypoglycemia and Metformin Impairs Tumor Metabolic Plasticity and Growth by Modulating the PP2A-GSK3β-MCL-1 Axis. Cancer Cell 2019, 35, 798–815.e5. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in Cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Matzke, M.A. Epigenetics: Regulation through Repression. Science 1999, 286, 481–486. [Google Scholar] [CrossRef]
- Kara, G.; Calin, G.A.; Ozpolat, B. RNAi-Based Therapeutics and Tumor Targeted Delivery in Cancer. Adv. Drug Deliv. Rev. 2022, 182, 114113. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.J. Therapeutic SiRNA: State of the Art. Signal Transduct. Target Ther. 2020, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Hattab, D.; Gazzali, A.M.; Bakhtiar, A. Clinical Advances of Sirna-Based Nanotherapeutics for Cancer Treatment. Pharmaceutics 2021, 13, 1009. [Google Scholar] [CrossRef] [PubMed]
- Rupaimoole, R.; Slack, F.J. MicroRNA Therapeutics: Towards a New Era for the Management of Cancer and Other Diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.H.; Burge, C.B.; Bartel, D.P. Most Mammalian MRNAs Are Conserved Targets of MicroRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grässer, F.A.; Lenhof, H.P.; et al. An Estimate of the Total Number of True Human MiRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Jonas, S.; Izaurralde, E. Towards a Molecular Understanding of MicroRNA-Mediated Gene Silencing. Nat. Rev. Genet. 2015, 16, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ahmed, E.; Elareer, N.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of MiRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells 2019, 8, 840. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Bu, P. Non-Coding RNAs in Cancer Stem Cells. Cancer Lett. 2018, 421, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Al-Sowayan, B.S.; Al-Shareeda, A.T.; Alrfaei, B.M. Cancer Stem Cell-Exosomes, Unexposed Player in Tumorigenicity. Front. Pharmacol. 2020, 11, 384. [Google Scholar] [CrossRef]
- Lv, C.; Li, F.; Li, X.; Tian, Y.; Zhang, Y.; Sheng, X.; Song, Y.; Meng, Q.; Yuan, S.; Luan, L.; et al. MiR-31 Promotes Mammary Stem Cell Expansion and Breast Tumorigenesis by Suppressing Wnt Signaling Antagonists. Nat. Commun. 2017, 8, 1036. [Google Scholar] [CrossRef]
- Zhang, L.; Cai, J.; Fang, L.; Huang, Y.; Li, R.; Xu, X.; Hu, Z.; Zhang, L.; Yang, Y.; Zhu, X.; et al. Simultaneous Overactivation of Wnt/β-Catenin and TGFβ Signalling by MiR-128-3p Confers Chemoresistance-Associated Metastasis in NSCLC. Nat. Commun. 2017, 8, 15870. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, C.; Becker, S.A.; Hurst, K.; Nogueira, L.M.; Findlay, V.J.; Camp, E.R. MiR-145 Antagonizes SNAI1-Mediated Stemness and Radiation Resistance in Colorectal Cancer. Mol. Ther. 2018, 26, 744–754. [Google Scholar] [CrossRef]
- Wang, T.W.; Chern, E.; Hsu, C.W.; Tseng, K.C.; Chao, H.M. SIRT1-Mediated Expression of CD24 and Epigenetic Suppression of Novel Tumor Suppressor MiR-1185-1 Increases Colorectal Cancer Stemness. Cancer Res. 2020, 80, 5257–5269. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Zhang, D.; Deng, Q.; Liu, B.; Chao, H.P.; Rycaj, K.; Takata, Y.; Lin, K.; Lu, Y.; et al. MicroRNA-141 Suppresses Prostate Cancer Stem Cells and Metastasis by Targeting a Cohort of pro-Metastasis Genes. Nat. Commun. 2017, 8, 14270. [Google Scholar] [CrossRef]
- Bhatia, V.; Yadav, A.; Tiwari, R.; Nigam, S.; Goel, S.; Carskadon, S.; Gupta, N.; Goel, A.; Palanisamy, N.; Ateeq, B. Epigenetic Silencing of MiRNA-338-5p and MiRNA-421 Drives SPINK1-Positive Prostate Cancer. Clin. Cancer Res. 2019, 25, 2755–2768. [Google Scholar] [CrossRef]
- Jiang, L.; Hermeking, H. MiR-34a and MiR-34b/c Suppress Intestinal Tumorigenesis. Cancer Res. 2017, 77, 2746–2758. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, C.; Liu, X.; Tang, D.G.; Wang, J. The MicroRNA MiR-34a Inhibits Non-Small Cell Lung Cancer (NSCLC) Growth and the CD44hi Stem-like NSCLC Cells. PLoS ONE 2014, 9, e90022. [Google Scholar] [CrossRef] [PubMed]
- Park, E.Y.; Chang, E.S.; Lee, E.J.; Lee, H.W.; Kang, H.G.; Chun, K.H.; Woo, Y.M.; Kong, H.K.; Ko, J.Y.; Suzuki, H.; et al. Targeting of MiR34a-NOTCH1 Axis Reduced Breast Cancer Stemness and Chemoresistance. Cancer Res. 2014, 74, 7573–7582. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.; Climent, M.; Panebianco, F.; Tordonato, C.; Santoro, A.; Marzi, M.J.; Pelicci, P.G.; Ventura, A.; Nicassio, F. Dual Role for MiR-34a in the Control of Early Progenitor Proliferation and Commitment in the Mammary Gland and in Breast Cancer. Oncogene 2019, 38, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Wang, L.; Chen, K.Y.; Srinivasan, T.; Murthy, P.K.L.; Tung, K.L.; Varanko, A.K.; Chen, H.J.; Ai, Y.; King, S.; et al. A MiR-34a-Numb Feedforward Loop Triggered by Inflammation Regulates Asymmetric Stem Cell Division in Intestine and Colon Cancer. Cell Stem Cell 2016, 18, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Chen, K.Y.; Chen, J.H.; Wang, L.; Walters, J.; Shin, Y.J.; Goerger, J.P.; Sun, J.; Witherspoon, M.; Rakhilin, N.; et al. A MicroRNA MiR-34a-Regulated Bimodal Switch Targets Notch in Colon Cancer Stem Cells. Cell Stem Cell 2013, 12, 602–615. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The MicroRNA MiR-34a Inhibits Prostate Cancer Stem Cells and Metastasis by Directly Repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef]
- Holdt, L.M.; Kohlmaier, A.; Teupser, D. Molecular Roles and Function of Circular RNAs in Eukaryotic Cells. Cell. Mol. Life Sci. 2018, 75, 1071–1098. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Erratum: Circular RNAs Are Abundant, Conserved, and Associated with ALU Repeats (RNA (156)). RNA 2013, 19, 426. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs Are a Large Class of Animal RNAs with Regulatory Potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs Are the Predominant Transcript Isoform from Hundreds of Human Genes in Diverse Cell Types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA Biogenesis Competes with Pre-MRNA Splicing. Mol Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Agarwal, V.; Guo, H.; Bartel, D.P. Expanded Identification and Characterization of Mammalian Circular RNAs. Genome Biol. 2014, 15, 409. [Google Scholar] [CrossRef] [PubMed]
- Starke, S.; Jost, I.; Rossbach, O.; Schneider, T.; Schreiner, S.; Hung, L.H.; Bindereif, A. Exon Circularization Requires Canonical Splice Signals. Cell Rep. 2015, 10, 103–111. [Google Scholar] [CrossRef]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA Circles Function as Efficient MicroRNA Sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Piwecka, M.; Glažar, P.; Hernandez-Miranda, L.R.; Memczak, S.; Wolf, S.A.; Rybak-Wolf, A.; Filipchyk, A.; Klironomos, F.; Jara, C.A.C.; Fenske, P.; et al. Loss of a Mammalian Circular RNA Locus Causes MiRNA Deregulation and Affects Brain Function. Science 2017, 357, eaam8526. [Google Scholar] [CrossRef]
- Zheng, Q.; Bao, C.; Guo, W.; Li, S.; Chen, J.; Chen, B.; Luo, Y.; Lyu, D.; Li, Y.; Shi, G.; et al. Circular RNA Profiling Reveals an Abundant CircHIPK3 That Regulates Cell Growth by Sponging Multiple MiRNAs. Nat. Commun. 2016, 7, 11215. [Google Scholar] [CrossRef]
- Chen, N.; Zhao, G.; Yan, X.; Lv, Z.; Yin, H.; Zhang, S.; Song, W.; Li, X.; Li, L.; Du, Z.; et al. A Novel FLI1 Exonic Circular RNA Promotes Metastasis in Breast Cancer by Coordinately Regulating TET1 and DNMT1. Genome Biol. 2018, 19, 218. [Google Scholar] [CrossRef]
- Holdt, L.M.; Stahringer, A.; Sass, K.; Pichler, G.; Kulak, N.A.; Wilfert, W.; Kohlmaier, A.; Herbst, A.; Northoff, B.H.; Nicolaou, A.; et al. Circular Non-Coding RNA ANRIL Modulates Ribosomal RNA Maturation and Atherosclerosis in Humans. Nat. Commun. 2016, 7, 12429. [Google Scholar] [CrossRef]
- Legnini, I.; di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 Is a Circular RNA That Can Be Translated and Functions in Myogenesis. Mol. Cell 2017, 66, 22–37.e9. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, X.; Zhang, M.; Yan, S.; Sun, C.; Xiao, F.; Huang, N.; Yang, X.; Zhao, K.; Zhou, H.; et al. Novel Role of FBXW7 Circular RNA in Repressing Glioma Tumorigenesis. J. Natl. Cancer Inst. 2018, 110, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Huang, N.; Yang, X.; Luo, J.; Yan, S.; Xiao, F.; Chen, W.; Gao, X.; Zhao, K.; Zhou, H.; et al. A Novel Protein Encoded by the Circular Form of the SHPRH Gene Suppresses Glioma Tumorigenesis. Oncogene 2018, 37, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Lei, M.; Zheng, G.; Ning, Q.; Zheng, J.; Dong, D. Translation and Functional Roles of Circular RNAs in Human Cancer. Mol. Cancer 2020, 19, 30. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Liu, Z.; Yang, X.; Zhou, J.; Yu, H.; Zhang, R.; Li, H. The Emerging Functions and Roles of Circular RNAs in Cancer. Cancer Lett. 2018, 414, 301–309. [Google Scholar] [CrossRef]
- Su, M.; Xiao, Y.; Ma, J.; Tang, Y.; Tian, B.; Zhang, Y.; Li, X.; Wu, Z.; Yang, D.; Zhou, Y.; et al. Circular RNAs in Cancer: Emerging Functions in Hallmarks, Stemness, Resistance and Roles as Potential Biomarkers. Mol. Cancer 2019, 18, 1–17. [Google Scholar] [CrossRef]
- Li, W.; Yang, X.; Shi, C.; Zhou, Z. Hsa_circ_002178 Promotes the Growth and Migration of Breast Cancer Cells and Maintains Cancer Stem-like Cell Properties Through Regulating MiR-1258/KDM7A Axis. Cell Transplant. 2020, 29, 0963689720960174. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, X.; Yan, B.; Yu, W.; Shan, L. CircAGFG1 Drives Metastasis and Stemness in Colorectal Cancer by Modulating YY1/CTNNB1. Cell Death Dis. 2020, 11, 542. [Google Scholar] [CrossRef]
- Jian, X.; He, H.; Zhu, J.; Zhang, Q.; Zheng, Z.; Liang, X.; Chen, L.; Yang, M.; Peng, K.; Zhang, Z.; et al. Hsa_circ_001680 Affects the Proliferation and Migration of CRC and Mediates Its Chemoresistance by Regulating BMI1 through MiR-340. Mol. Cancer 2020, 19, 20. [Google Scholar] [CrossRef]
- Lin, X.; Chen, W.; Wei, F.; Xie, X. TV-CircRGPD6 Nanoparticle Suppresses Breast Cancer Stem Cell-Mediated Metastasis via the MiR-26b/YAF2 Axis. Mol. Ther. 2021, 29, 244–262. [Google Scholar] [CrossRef]
- Kung, J.T.Y.; Colognori, D.; Lee, J.T. Long Noncoding RNAs: Past, Present, and Future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 96–118. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yang, L.; Chen, L.L. The Diversity of Long Noncoding RNAs and Their Generation. Trends Genet. 2017, 33, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Chekulaeva, M.; Rajewsky, N. Roles of Long Noncoding Rnas and Circular Rnas in Translation. Cold Spring Harb. Perspect. Biol. 2019, 11, a032680. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Fullwood, M.J. Roles, Functions, and Mechanisms of Long Non-Coding RNAs in Cancer. Genom. Proteom. Bioinform. 2016, 14, 42–54. [Google Scholar] [CrossRef]
- van Heesch, S.; Witte, F.; Schneider-Lunitz, V.; Schulz, J.F.; Adami, E.; Faber, A.B.; Kirchner, M.; Maatz, H.; Blachut, S.; Sandmann, C.L.; et al. The Translational Landscape of the Human Heart. Cell 2019, 178, 242–260.e29. [Google Scholar] [CrossRef]
- Peng, F.; Li, T.T.; Wang, K.L.; Xiao, G.Q.; Wang, J.H.; Zhao, H.D.; Kang, Z.J.; Fan, W.J.; Zhu, L.L.; Li, M.; et al. H19/Let-7/LIN28 Reciprocal Negative Regulatory Circuit Promotes Breast Cancer Stem Cell Maintenance. Cell Death Dis. 2017, 8, e2569. [Google Scholar] [CrossRef]
- Guo, K.; Gong, W.; Wang, Q.; Gu, G.; Zheng, T.; Li, Y.; Li, W.; Fang, M.; Xie, H.; Yue, C.; et al. LINC01106 Drives Colorectal Cancer Growth and Stemness through a Positive Feedback Loop to Regulate the Gli Family Factors. Cell Death Dis. 2020, 11, 869. [Google Scholar] [CrossRef]
- Liu, X.; Yin, Z.; Xu, L.; Liu, H.; Jiang, L.; Liu, S.; Sun, X. Upregulation of LINC01426 Promotes the Progression and Stemness in Lung Adenocarcinoma by Enhancing the Level of SHH Protein to Activate the Hedgehog Pathway. Cell Death Dis. 2021, 12, 173. [Google Scholar] [CrossRef]
- Ma, F.; Liu, X.; Zhou, S.; Li, W.; Liu, C.; Chadwick, M.; Qian, C. Long Non-Coding RNA FGF13-AS1 Inhibits Glycolysis and Stemness Properties of Breast Cancer Cells through FGF13-AS1/IGF2BPs/Myc Feedback Loop. Cancer Lett. 2019, 450, 63–75. [Google Scholar] [CrossRef]
- Zagorac, S.; de Giorgio, A.; Dabrowska, A.; Kalisz, M.; Casas-Vila, N.; Cathcart, P.; Yiu, A.; Ottaviani, S.; Degani, N.; Lombardo, Y.; et al. SCIRT LncRNA Restrains Tumorigenesis by Opposing Transcriptional Programs of Tumor-Initiating Cells. Cancer Res. 2021, 81, 580–593. [Google Scholar] [CrossRef]
- Le, P.; Romano, G.; Nana-Sinkam, P.; Acunzo, M. Non-Coding Rnas in Cancer Diagnosis and Therapy: Focus on Lung Cancer. Cancers 2021, 13, 1372. [Google Scholar] [CrossRef] [PubMed]
- Khvorova, A.; Watts, J.K. The Chemical Evolution of Oligonucleotide Therapies of Clinical Utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming Cellular Barriers for RNA Therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Rooij, E.; Kauppinen, S. Review Review Series: Small RNA Development of MicroRNA Therapeutics Is Coming of Age. EMBO Mol. Med. 2014, 6, 851–864. [Google Scholar] [CrossRef]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA Therapeutics—Challenges and Potential Solutions. Nat. Rev. Drug Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S. Short Hairpin RNAs (ShRNAs) Induce Sequence-Specific Silencing in Mammalian Cells. Genes Dev. 2002, 16, 948–958. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-Coding RNAs as Drug Targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 Study of MRX34, a Liposomal MiR-34a Mimic, in Patients with Advanced Solid Tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I Study of MRX34, a Liposomal MiR-34a Mimic, Administered Twice Weekly in Patients with Advanced Solid Tumors. Invest. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, Y.; Zhu, Y.; Sun, H.; Juguilon, C.; Li, F.; Fan, D.; Yin, L.; Zhang, Y. Macrophage MiR-34a Is a Key Regulator of Cholesterol Efflux and Atherosclerosis. Mol. Ther. 2020, 28, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.; Walch-Rückheim, B.; Friedmann, K.S.; Rheinheimer, S.; Tänzer, T.; Glombitza, B.; Sester, M.; Lenhof, H.P.; Hoth, M.; Schwarz, E.C.; et al. MiR-34a: A New Player in the Regulation of T Cell Function by Modulation of NF-ΚB Signaling. Cell Death Dis. 2019, 10, 46. [Google Scholar] [CrossRef] [PubMed]
- Diener, C.; Keller, A.; Meese, E. Emerging Concepts of MiRNA Therapeutics: From Cells to Clinic. Trends Genet. 2022, 38, 613–626. [Google Scholar] [CrossRef]
- Guo, S.; Li, H.; Ma, M.; Fu, J.; Dong, Y.; Guo, P. Size, Shape, and Sequence-Dependent Immunogenicity of RNA Nanoparticles. Mol. Ther. Nucleic Acids 2017, 9, 399–408. [Google Scholar] [CrossRef]
- Meganck, R.M.; Borchardt, E.K.; Castellanos Rivera, R.M.; Scalabrino, M.L.; Wilusz, J.E.; Marzluff, W.F.; Asokan, A. Tissue-Dependent Expression and Translation of Circular RNAs with Recombinant AAV Vectors In Vivo. Mol. Ther. Nucleic Acids 2018, 13, 89–98. [Google Scholar] [CrossRef]
- Han, D.; Wang, Y.; Wang, Y.; Dai, X.; Zhou, T.; Chen, J.; Tao, B.; Zhang, J.; Cao, F. The Tumor-Suppressive Human Circular RNA CircITCH Sponges MiR-330-5p to Ameliorate Doxorubicin-Induced Cardiotoxicity through Upregulating SIRT6, Survivin, and SERCA2a. Circ. Res. 2020, 127, e108–e125. [Google Scholar] [CrossRef]
- Rossbach, O. Artificial Circular RNA Sponges Targeting MicroRNAs as a Novel Tool in Molecular Biology. Mol. Ther. Nucleic Acids 2019, 17, 452–454. [Google Scholar] [CrossRef]
- Liu, X.; Abraham, J.M.; Cheng, Y.; Wang, Z.; Wang, Z.; Zhang, G.; Ashktorab, H.; Smoot, D.T.; Cole, R.N.; Boronina, T.N.; et al. Synthetic Circular RNA Functions as a MiR-21 Sponge to Suppress Gastric Carcinoma Cell Proliferation. Mol. Ther. Nucleic Acids 2018, 13, 312–321. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Jakobsen, T.; Hager, H.; Kjems, J. The Emerging Roles of CircRNAs in Cancer and Oncology. Nat. Rev. Clin. Oncol. 2022, 19, 188–206. [Google Scholar] [CrossRef]
- He, A.T.; Liu, J.; Li, F.; Yang, B.B. Targeting Circular RNAs as a Therapeutic Approach: Current Strategies and Challenges. Signal Transduct. Target Ther. 2021, 6, 185. [Google Scholar] [CrossRef] [PubMed]
- Mercer, T.R.; Munro, T.; Mattick, J.S. The Potential of Long Noncoding RNA Therapies. Trends Pharmacol. Sci. 2022, 43, 269–280. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Function | Reference |
|---|---|---|
| SNAI1 (SNAIL) | Promotes tumor growth, invasion, migration of cancer cells | [32] |
| SNAI2 (SLUG) | Prevents cell death and promotes cell survival | [33] |
| ZEB1 | EMT transcription factor, causes plasticity of non-CSCs into CSCs, promotes stem-like and tumorigenic phenotype | [34] |
| HIF1A | Promotes the expression of stem cell-associated transcription factors, prompt the transcription of genes involved in the metabolism | [35,36] |
| MCL1 | Promotes chemotherapy resistant CSC via the regulation of OXPHOS | [37] |
| VEGF | Invasion related to hypoxia | [38] |
| MYC | The driver of stemness and glycolysis | [39] |
| PIK3CA | Mutated PIK3CA induces reprogramming of lineage restricted progenitors to a multipotent stem-like state | [40] |
| NOTCH | Regulation of asymmetric division and cell stemness | [41] |
| SOX2 | Maintaining pluripotency of stem cells | [26] |
| SOX9 | Regulator of epithelial cell proliferation; acquiring properties of basal stem cells; induction of EMT. | [28] |
| KLF4 | Promotes production of ECM that creates pro-metastatic niche | [42] |
| MIF | Conversion of cells phenotype from CD138- to CD138+ | [43] |
| STAT3 | Causes shift to aerobic glycolysis | [44] |
| miRNA Name | Tumor Type | Target mRNAs | CSCs Promoter/Suppressor | Reference |
|---|---|---|---|---|
| miR-31 | Breast | DKK1, AXIN1, GSK3Β | Promoter | [118] |
| miR-128-3p | Lung | AXIN1, SFRP2, WIF1, SMURF2, PP1c | Promoter | [119] |
| miR-145 | Colorectal | SNAI1 | Suppressor | [120] |
| miR-1185-1 | Colorectal | CD24 | Suppressor | [121] |
| miR-141 | Prostate | CD44, EZH2, Rho GTPases (RAC1, CDC42, CDC42EP3 and ARPC5) | Suppressor | [122] |
| miRNA-338-5pmiRNA-421 | Prostate | SPINK1 | Suppressor | [123] |
| miRNA-34a, miRNA-34b/c | Colorectal | PDGFRA, PDGFRB, AXL, WASF1 (NGS-, reporter assay- and WB-validated) FGFR1, IGF1, STC1, CACNA2D2, COL6A2, COL4A2, INHBB (NGS- and qPCR-validated) | Suppressor | [124] |
| circRNA Name | Tumor Type | Target | CSCs Promoter/Suppressor | Reference |
|---|---|---|---|---|
| hsa_circ_002178 | Breast | miR-1258/KDM7A | Promoter | [149] |
| circAGFG1 | Colorectal | miR-4262/miR-185-5p/YY1/CTNNB1; WNT/β-catenin pathway activation | Promoter | [150] |
| hsa_circ_001680 | Colorectal | miR-340/BMI1 | Promoter | [151] |
| circRGPD6 | Breast | miR-26b/YAF2 | Suppressor | [152] |
| lncRNA Name | Tumor Type | Mode of Action; Interacting with RNA/DNA/Protein | CSCs Promoter/Suppressor | Reference |
|---|---|---|---|---|
| H19 | Breast | miRNA sponging-let-7/HIF-1α/PDK1 | Promoter | [159] |
| LINC01106 | Colorectal | Cytoplasmic: miRNA sponging-miR-449b-5p/Gli4; Nuclear: RBP binding-FUS/Gli1/Gli2 | Promoter | [160] |
| LINC01426 | Lung | Interacting with protein-USPP2/SHH/Hedgehog pathway activation | Promoter | [161] |
| FGF13-AS1 | Breast | RBP binding-IGF2BPs/Myc | Suppressor | [162] |
| SCIRT | Breast | transcription induction- recruitment of FOXM1 through interaction with EZH2; transcription inhibition—interaction with SOX2 and EZH2 to antagonize their effects | Suppressor | [163] |
| Therapeutic Name | Mode of Action | Tumor Type | Delivery System | Clinical Trial Number | Phase, Recruitment Status |
|---|---|---|---|---|---|
| MRX34 | miR-34 mimic | Melanoma; advanced and metastatic solid tumors | Intravenously/ vehicle transfer (liposomal) | NCT01829971 NCT02862145 | Phase I T (serious adverse events) |
| MesomiR 1 | miR-16 mimic | MPM and NSCLC | Intravenously/ vehicle transfer (nonliving minicells) | NCT02369198 | Phase I C |
| Cobomarsen/ MRG-106 | anti-miR-155 | CTCL, CLL, DLBCL or ATLL | Intravenously/chemical modification (LNA) | NCT02580552 NCT03837457 NCT03713320 | Phase I T (business reasons) |
| INT-1B3 | miR-193a-3p mimic | Solid tumors | Intravenously/vehicle transfer (lipid NP) | NCT04675996 | Phase I R |
| Therapeutic Name | Mode of Action | Tumor Type | Delivery System | Clinical Trial Number | Phase, Recruitment Status |
|---|---|---|---|---|---|
| Vigil ™ vaccine | Bi-shRNA-furin and GMCSF +carboplatin +bevacizumab +atezolizumab +irinotecan, temozolomide | Ewing’s sarcoma NSCLC Liver cancer Stage III/IV Ovarian cancer Ovarian, Cervical, Uterine cancer Ewing’s sarcoma | Intradermally/ (autologous tumor cells expressing Vigil plasmid) | NCT01061840 NCT01867086 NCT01551745 NCT03073525 NCT03495921 | Phase I C Phase II C Phase II C Phase II A Phase III A |
| pbi-shRNA™ EWS-FLI1 Type 1 LPX | bi-shRNA EWS/FLI1 | Ewing’s sarcoma | Intravenously/vehicle transfer (liposomal) | NCT02736565 | Phase I A |
| lentivirus vector CCR5 shRNA-TRIM5alpha-TAR decoy | CCR5 shRNA | HIV infection, Lymphomas | Intravenously/transduced autologous CD34+ hematopoietic progenitor cells transplantation/vehicle transfer (lentivirus) | NCT02797470 | Phase I/II R |
| lentivirus vector rHIV7-shI-TAR-CCR5RZ | shRNA targeted to an exon of the HIV-1 genes tat/rev | Lymphoma | Intravenously/transduced hematopoietic progenitor cells and non-bound CD34+ cells/vehicle transfer (lentivirus) | NCT00569985 | Phase I C |
| pbi-shRNA ™ STMN1 | STMN1 bi-shRNA | Advanced, metastatic Cancer, solid tumors | Intratumoral injection/transfer vehicle (bilamellar invaginated vesicle lipoplex (BIV LP)) | NCT01505153 | Phase I C |
| Atu027 | PKN3 siRNA + gemcitabine | Advanced solid tumors | Intravenously/vehicle transfer (liposomes) | NCT00938574 NCT01808638 | Phase I C Phase I/II C |
| ARO-HIF-2 | TRiM (RGD- HIF-2α siRNA conjugate) | ccRCC | Intravenously/bioconjugation (alpha-v beta3 targeting ligand) | NCT04169711 | Phase I C |
| siG12D-LODER | KRASG12D siRNA | Advanced pancreatic cancer | Intratumoral/Biodegradable polymeric matrix (PLGA) | NCT01188785 NCT01676259 | Phase I C Phase II R |
| TKM 080301 | PLK1 siRNA | Colorectal, pancreas, gastric, breast, ovarian cancer with hepatic metastase | Intravenously/vehicle transfer (LNP) | NCT01437007 | Phase I C |
| APN401 | Cbl-b siRNA | Inoperable metastatic solid tumors | Intravenously/siRNA-transfected Peripheral Blood Mononuclear Cells | NCT03087591 NCT02166255 | Phase I C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bryl, R.; Piwocka, O.; Kawka, E.; Mozdziak, P.; Kempisty, B.; Knopik-Skrocka, A. Cancer Stem Cells—The Insight into Non-Coding RNAs. Cells 2022, 11, 3699. https://doi.org/10.3390/cells11223699
Bryl R, Piwocka O, Kawka E, Mozdziak P, Kempisty B, Knopik-Skrocka A. Cancer Stem Cells—The Insight into Non-Coding RNAs. Cells. 2022; 11(22):3699. https://doi.org/10.3390/cells11223699
Chicago/Turabian StyleBryl, Rut, Oliwia Piwocka, Emilia Kawka, Paul Mozdziak, Bartosz Kempisty, and Agnieszka Knopik-Skrocka. 2022. "Cancer Stem Cells—The Insight into Non-Coding RNAs" Cells 11, no. 22: 3699. https://doi.org/10.3390/cells11223699
APA StyleBryl, R., Piwocka, O., Kawka, E., Mozdziak, P., Kempisty, B., & Knopik-Skrocka, A. (2022). Cancer Stem Cells—The Insight into Non-Coding RNAs. Cells, 11(22), 3699. https://doi.org/10.3390/cells11223699