

The Role of IL-6 in Cancer Cell Invasiveness and Metastasis—Overview and Therapeutic Opportunities

 ,
,  ,
,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
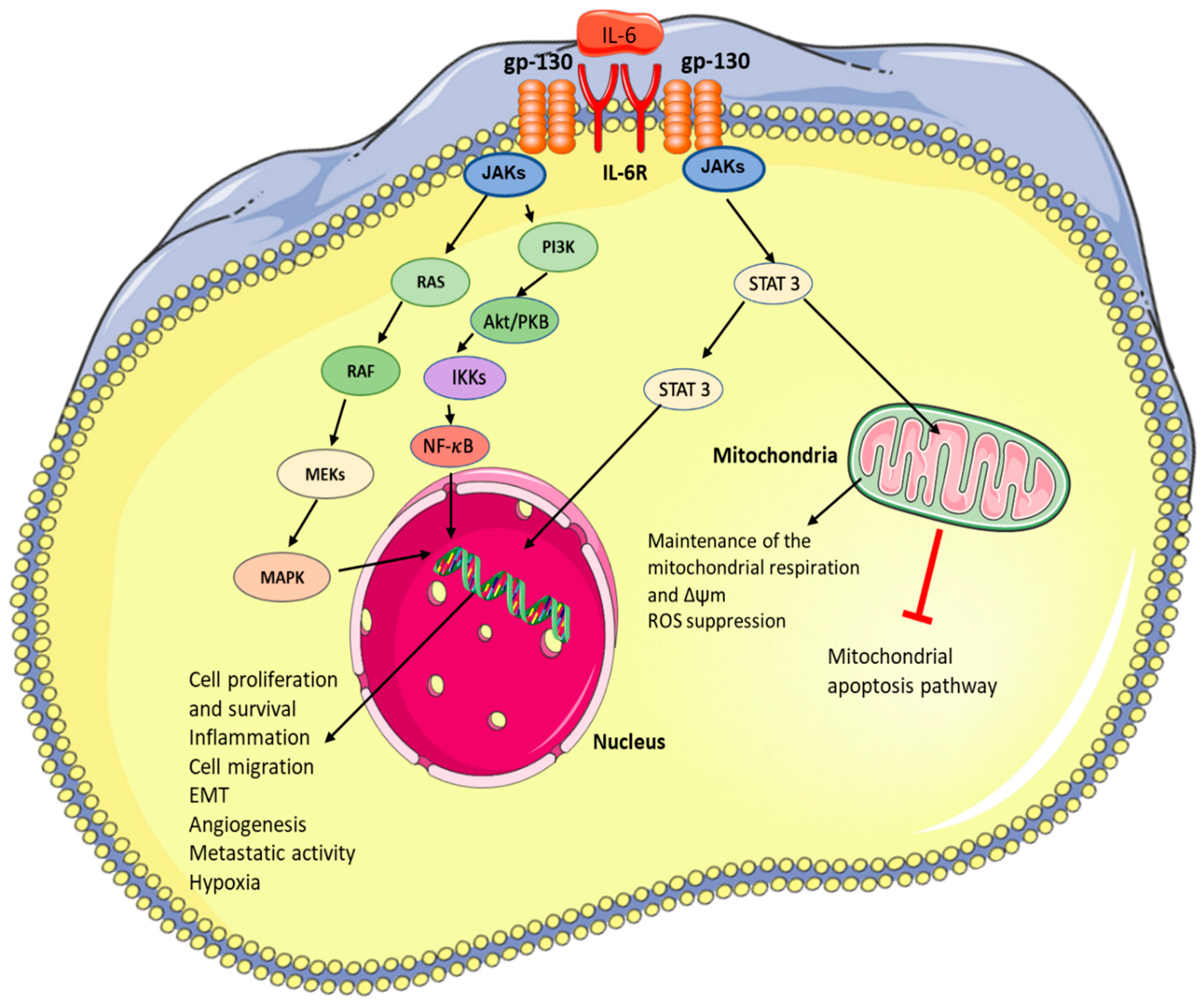
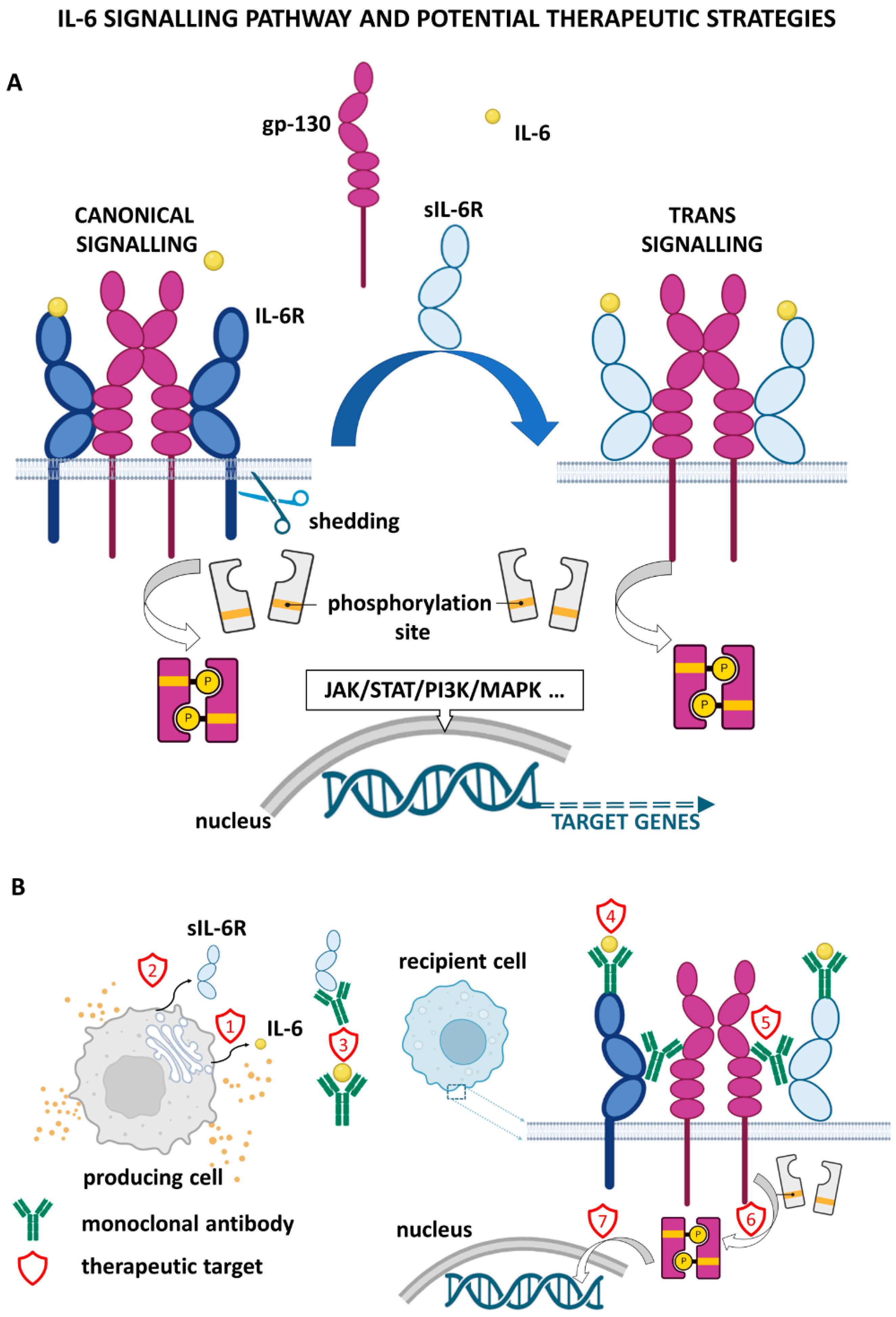
2. IL-6 Signalling and Downstream Effects
3. IL-6 Signalling in Promoting Tumorigenesis, Invasiveness, and Metastasis in Cancer
4. IL-6 Contributes to the Formation of the Pre-Metastatic Niche
5. Metastasis Repression by Targeting IL-6 Signalling
6. Inhibition of IL-6 Signalling in Combination Therapy
7. IL-6 Signalling in Selected Cancer Types
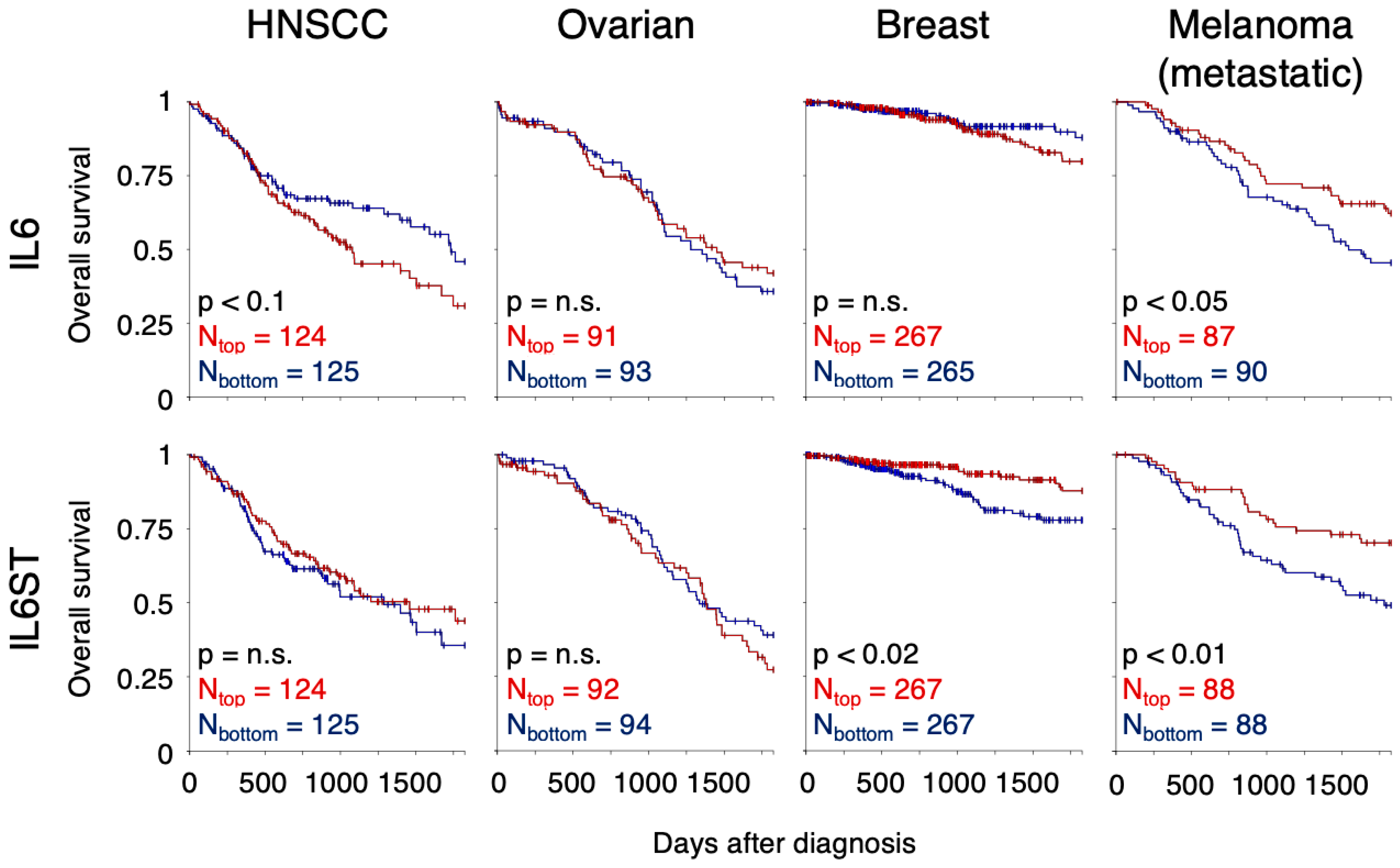
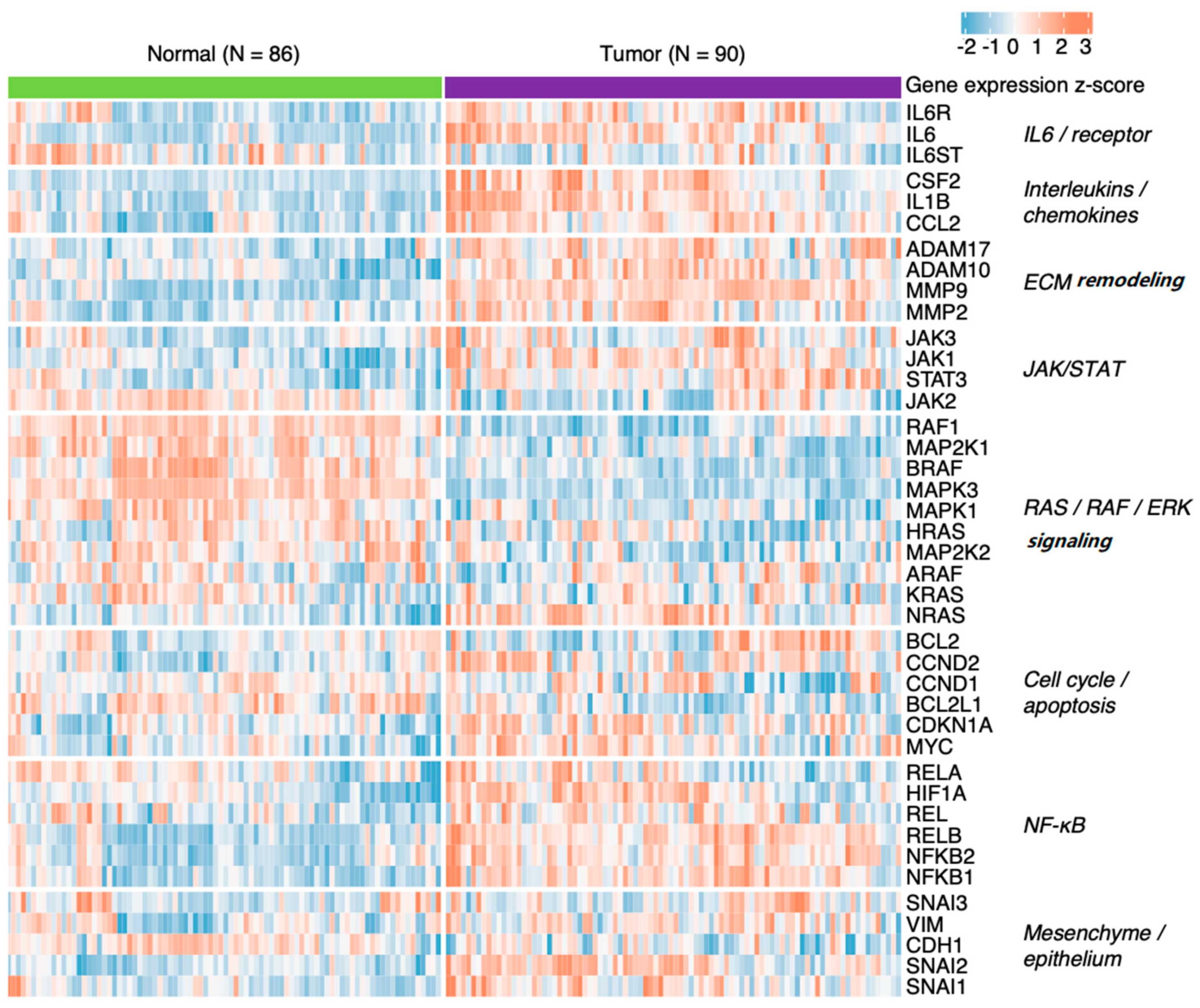
7.1. Head and Neck Squamous Cell Carcinoma
7.2. Ovarian Cancer
7.3. Breast Cancer
7.4. Melanoma and Cutaneous Squamous Cell Carcinoma
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gál, P.; Brábek, J.; Holub, M.; Jakubek, M.; Šedo, A.; Lacina, L.; Strnadová, K.; Dubový, P.; Hornychová, H.; Ryška, A.; et al. Autoimmunity, cancer and COVID-19 abnormally activate wound healing pathways: Critical role of inflammation. Histochem. Cell Biol. 2022, 158, 415–434. [Google Scholar] [CrossRef]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Brábek, J.; Jakubek, M.; Vellieux, F.; Novotný, J.; Kolář, M.; Lacina, L.; Szabo, P.; Strnadová, K.; Rösel, D.; Dvořánková, B.; et al. Interleukin-6: Molecule in the Intersection of Cancer, Ageing and COVID-19. Int. J. Mol. Sci 2020, 21, 7937. [Google Scholar] [CrossRef]
- Rose-John, S.; Scheller, J.; Elson, G.; Jones, S.A. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J. Leukoc. Biol. 2006, 80, 227–236. [Google Scholar] [CrossRef]
- Hirano, T.; Yasukawa, K.; Harada, H.; Taga, T.; Watanabe, Y.; Matsuda, T.; Kashiwamura, S.; Nakajima, K.; Koyama, K.; Iwamatsu, A.; et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 1986, 324, 73–76. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669. [Google Scholar] [CrossRef]
- Novák, Š.; Kolář, M.; Szabó, A.; Vernerová, Z.; Lacina, L.; Strnad, H.; Šáchová, J.; Hradilová, M.; Havránek, J.; Španko, M.; et al. Desmoplastic Crosstalk in Pancreatic Ductal Adenocarcinoma Is Reflected by Different Responses of Panc-1, MIAPaCa-2, PaTu-8902, and CAPAN-2 Cell Lines to Cancer-associated/Normal Fibroblasts. Cancer Genom. Proteom. 2021, 18, 221–243. [Google Scholar] [CrossRef]
- Španko, M.; Strnadová, K.; Pavlíček, A.J.; Szabo, P.; Kodet, O.; Valach, J.; Dvořánková, B.; Smetana, K.; Lacina, L. IL-6 in the Ecosystem of Head and Neck Cancer: Possible Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 11027. [Google Scholar] [CrossRef]
- Magidey-Klein, K.; Cooper, T.J.; Kveler, K.; Normand, R.; Zhang, T.; Timaner, M.; Raviv, Z.; James, B.P.; Gazit, R.; Ronai, Z.A.; et al. IL-6 contributes to metastatic switch via the differentiation of monocytic-dendritic progenitors into prometastatic immune cells. J. Immunother. Cancer 2021, 9, e002856. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Lacina, L.; Brábek, J.; Král, V.; Kodet, O.; Smetana, K., Jr. Interleukin-6: A molecule with complex biological impact in cancer. Histol. Histopathol. 2019, 34, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Novick, D.; Engelmann, H.; Wallach, D.; Rubinstein, M. Soluble cytokine receptors are present in normal human urine. J. Exp. Med. 1989, 170, 1409–1414. [Google Scholar] [CrossRef] [PubMed]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front. Oncol. 2022, 12, 866014. [Google Scholar] [CrossRef]
- Böttcher, J.P.; Schanz, O.; Garbers, C.; Zaremba, A.; Hegenbarth, S.; Kurts, C.; Beyer, M.; Schultze, J.L.; Kastenmüller, W.; Rose-John, S.; et al. IL-6 trans-signaling-dependent rapid development of cytotoxic CD8+ T cell function. Cell Rep. 2014, 8, 1318–1327. [Google Scholar] [CrossRef]
- Chalaris, A.; Garbers, C.; Rabe, B.; Rose-John, S.; Scheller, J. The soluble Interleukin 6 receptor: Generation and role in inflammation and cancer. Eur. J. Cell Biol. 2011, 90, 484–494. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- McLoughlin, R.M.; Jenkins, B.J.; Grail, D.; Williams, A.S.; Fielding, C.A.; Parker, C.R.; Ernst, M.; Topley, N.; Jones, S.A. IL-6 trans-signaling via STAT3 directs T cell infiltration in acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9589–9594. [Google Scholar] [CrossRef]
- Szulc-Kielbik, I.; Kielbik, M.; Nowak, M.; Klink, M. The implication of IL-6 in the invasiveness and chemoresistance of ovarian cancer cells. Systematic review of its potential role as a biomarker in ovarian cancer patients. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188639. [Google Scholar] [CrossRef]
- Cressman, D.E.; Greenbaum, L.E.; DeAngelis, R.A.; Ciliberto, G.; Furth, E.E.; Poli, V.; Taub, R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science 1996, 274, 1379–1383. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Kamran, M.Z.; Patil, P.; Gude, R.P. Role of STAT3 in cancer metastasis and translational advances. BioMed Res. Int. 2013, 2013, 421821. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Stanam, A.; Love-Homan, L.; Joseph, T.S.; Espinosa-Cotton, M.; Simons, A.L. Upregulated interleukin-6 expression contributes to erlotinib resistance in head and neck squamous cell carcinoma. Mol. Oncol. 2015, 9, 1371–1383. [Google Scholar] [CrossRef]
- Krishnamurthy, S.; Warner, K.A.; Dong, Z.; Imai, A.; Nör, C.; Ward, B.B.; Helman, J.I.; Taichman, R.S.; Bellile, E.L.; McCauley, L.K.; et al. Endothelial interleukin-6 defines the tumorigenic potential of primary human cancer stem cells. Stem Cells 2014, 32, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Gwak, H.; Kim, H.S.; Kim, B.; Dhanasekaran, D.N.; Song, Y.S. Malignant ascites enhances migratory and invasive properties of ovarian cancer cells with membrane bound IL-6R in vitro. Oncotarget 2016, 7, 83148–83159. [Google Scholar] [CrossRef]
- Gasche, J.A.; Hoffmann, J.; Boland, C.R.; Goel, A. Interleukin-6 promotes tumorigenesis by altering DNA methylation in oral cancer cells. Int. J. Cancer 2011, 129, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef]
- Abulaiti, A.; Shintani, Y.; Funaki, S.; Nakagiri, T.; Inoue, M.; Sawabata, N.; Minami, M.; Okumura, M. Interaction between non-small-cell lung cancer cells and fibroblasts via enhancement of TGF-β signaling by IL-6. Lung Cancer 2013, 82, 204–213. [Google Scholar] [CrossRef]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Browning, L.; Patel, M.R.; Horvath, E.B.; Tawara, K.; Jorcyk, C.L. IL-6 and ovarian cancer: Inflammatory cytokines in promotion of metastasis. Cancer Manag. Res. 2018, 10, 6685–6693. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Dragulev, B.; Zigrino, P.; Mauch, C.; Fox, J.W. The invasive potential of human melanoma cell lines correlates with their ability to alter fibroblast gene expression in vitro and the stromal microenvironment in vivo. Int. J. Cancer 2009, 125, 1796–1804. [Google Scholar] [CrossRef]
- Jobe, N.P.; Rösel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef]
- Gandalovičová, A.; Rosel, D.; Fernandes, M.; Veselý, P.; Heneberg, P.; Čermák, V.; Petruželka, L.; Kumar, S.; Sanz-Moreno, V.; Brábek, J. Migrastatics-Anti-metastatic and Anti-invasion Drugs: Promises and Challenges. Trends Cancer 2017, 3, 391–406. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Meirson, T.; Gil-Henn, H.; Samson, A.O. Invasion and metastasis: The elusive hallmark of cancer. Oncogene 2020, 39, 2024–2026. [Google Scholar] [CrossRef]
- van Zijl, F.; Krupitza, G.; Mikulits, W. Initial steps of metastasis: Cell invasion and endothelial transmigration. Mutat. Res. 2011, 728, 23–34. [Google Scholar] [CrossRef]
- Paget, S. THE DISTRIBUTION OF SECONDARY GROWTHS IN CANCER OF THE BREAST. Lancet 1889, 133, 571–573. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Y.; Guo, N.; Wang, S. MDSCs: Key Criminals of Tumor Pre-metastatic Niche Formation. Front. Immunol. 2019, 10, 172. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Giles, A.J.; Reid, C.M.; Evans, J.D.; Murgai, M.; Vicioso, Y.; Highfill, S.L.; Kasai, M.; Vahdat, L.; Mackall, C.L.; Lyden, D.; et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre-metastatic Niche. Cancer Res. 2016, 76, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Han, Z.J.; Li, Y.B.; Yang, L.X.; Cheng, H.J.; Liu, X.; Chen, H. Roles of the CXCL8-CXCR1/2 Axis in the Tumor Microenvironment and Immunotherapy. Molecules 2021, 27, 137. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, K.; Yang, D.; Oppenheim, J.J. Interleukin-8: An evolving chemokine. Cytokine 2022, 153, 155828. [Google Scholar] [CrossRef] [PubMed]
- Biggioggero, M.; Crotti, C.; Becciolini, A.; Favalli, E.G. Tocilizumab in the treatment of rheumatoid arthritis: An evidence-based review and patient selection. Drug Des. Devel. 2019, 13, 57–70. [Google Scholar] [CrossRef]
- Ando, K.; Takahashi, F.; Motojima, S.; Nakashima, K.; Kaneko, N.; Hoshi, K.; Takahashi, K. Possible Role for Tocilizumab, an Anti–Interleukin-6 Receptor Antibody, in Treating Cancer Cachexia. J. Clin. Oncol. 2013, 31, e69–e72. [Google Scholar] [CrossRef]
- KEVZARA (Sarilumab) [Prescribing Information]. 2017. Available online: https://www.ema.europa.eu/en/documents/product-information/kevzara-epar-product-information_en.pdf (accessed on 20 October 2022).
- Ferreros, P.; Trapero, I. Interleukin Inhibitors in Cytokine Release Syndrome and Neurotoxicity Secondary to CAR-T Therapy. Diseases 2022, 10, 41. [Google Scholar] [CrossRef]
- Zafar, E.; Maqbool, M.F.; Iqbal, A.; Maryam, A.; Shakir, H.A.; Irfan, M.; Khan, M.; Li, Y.; Ma, T. A comprehensive review on anticancer mechanism of bazedoxifene. Biotechnol. Appl. Biochem. 2022, 69, 767–782. [Google Scholar] [CrossRef]
- Kim, L.; Park, S.A.; Park, H.; Kim, H.; Heo, T.H. Bazedoxifene, a GP130 Inhibitor, Modulates EMT Signaling and Exhibits Antitumor Effects in HPV-Positive Cervical Cancer. Int. J. Mol. Sci. 2021, 22, 8693. [Google Scholar] [CrossRef]
- Song, W.; Lv, Y.; Tang, Z.; Nie, F.; Huang, P.; Pei, Q.; Guo, R. Bazedoxifene Plays a Protective Role against Inflammatory Injury of Endothelial Cells by Targeting CD40. Cardiovasc. Ther. 2020, 2020, 1795853. [Google Scholar] [CrossRef] [PubMed]
- Taneja, V.; El-Dallal, M.; Haq, Z.; Tripathi, K.; Systrom, H.K.; Wang, L.F.; Said, H.; Bain, P.A.; Zhou, Y.; Feuerstein, J.D. Effectiveness and Safety of Tofacitinib for Ulcerative Colitis: Systematic Review and Meta-analysis. J. Clin. Gastroenterol. 2022, 56, e323–e333. [Google Scholar] [CrossRef] [PubMed]
- Curtis, J.R.; Lee, E.B.; Kaplan, I.V.; Kwok, K.; Geier, J.; Benda, B.; Soma, K.; Wang, L.; Riese, R. Tofacitinib, an oral Janus kinase inhibitor: Analysis of malignancies across the rheumatoid arthritis clinical development programme. Ann. Rheum. Dis. 2016, 75, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Huo, W.X.; Yang, Y.; Shen, M.Z.; Mo, X.D. Efficacy and safety of ruxolitinib in steroid-refractory graft-versus-host disease: A meta-analysis. Front. Immunol. 2022, 13, 954268. [Google Scholar] [CrossRef]
- Passamonti, F.; Palandri, F.; Saydam, G.; Callum, J.; Devos, T.; Guglielmelli, P.; Vannucchi, A.M.; Zor, E.; Zuurman, M.; Gilotti, G.; et al. Ruxolitinib versus best available therapy in inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): 5-year follow up of a randomised, phase 3b study. Lancet Haematol. 2022, 9, e480–e492. [Google Scholar] [CrossRef]
- Mesa, R.; Harrison, C.; Oh, S.T.; Gerds, A.T.; Gupta, V.; Catalano, J.; Cervantes, F.; Devos, T.; Hus, M.; Kiladjian, J.J.; et al. Overall survival in the SIMPLIFY-1 and SIMPLIFY-2 phase 3 trials of momelotinib in patients with myelofibrosis. Leukemia 2022, 36, 2261–2268. [Google Scholar] [CrossRef]
- Hayashi, M.; Kim, Y.P.; Takamatsu, S.; Enomoto, A.; Shinose, M.; Takahashi, Y.; Tanaka, H.; Komiyama, K.; Omura, S. Madindoline, a novel inhibitor of IL-6 activity from Streptomyces sp. K93-0711. I. Taxonomy, fermentation, isolation and biological activities. J. Antibiot. 1996, 49, 1091–1095. [Google Scholar] [CrossRef]
- Hayashi, M.; Rho, M.C.; Enomoto, A.; Fukami, A.; Kim, Y.P.; Kikuchi, Y.; Sunazuka, T.; Hirose, T.; Komiyama, K.; Omura, S. Suppression of bone resorption by madindoline A, a novel nonpeptide antagonist to gp130. Proc. Natl. Acad. Sci. USA 2002, 99, 14728–14733. [Google Scholar] [CrossRef]
- Kino, T.; Boos, T.L.; Sulima, A.; Siegel, E.M.; Gold, P.W.; Rice, K.C.; Chrousos, G.P. 3-O-Formyl-20R,21-epoxyresibufogenin suppresses IL-6-type cytokine actions by targeting the glycoprotein 130 subunit: Potential clinical implications. J. Allergy Clin. Immunol. 2007, 120, 437–444. [Google Scholar] [CrossRef]
- Hayashi, M.; Rho, M.C.; Fukami, A.; Enomoto, A.; Nonaka, S.; Sekiguchi, Y.; Yanagisawa, T.; Yamashita, A.; Nogawa, T.; Kamano, Y.; et al. Biological activity of a novel nonpeptide antagonist to the interleukin-6 receptor 20S,21-epoxy-resibufogenin-3-formate. J. Pharm. Exp. 2002, 303, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Rho, M.C.; Komiyama, K.; Hayashi, M. Inhibitory effects of bufadienolides on interleukin-6 in MH-60 cells. J. Nat. Prod. 2004, 67, 2070–2072. [Google Scholar] [CrossRef]
- Adachi, M.; Cui, C.; Dodge, C.T.; Bhayani, M.K.; Lai, S.Y. Targeting STAT3 inhibits growth and enhances radiosensitivity in head and neck squamous cell carcinoma. Oral Oncol. 2012, 48, 1220–1226. [Google Scholar] [CrossRef]
- Bendell, J.C.; Hong, D.S.; Burris, H.A., 3rd; Naing, A.; Jones, S.F.; Falchook, G.; Bricmont, P.; Elekes, A.; Rock, E.P.; Kurzrock, R. Phase 1, open-label, dose-escalation, and pharmacokinetic study of STAT3 inhibitor OPB-31121 in subjects with advanced solid tumors. Cancer Chemother. Pharm. 2014, 74, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; Lee, S.H.; Han, S.W.; Kim, M.J.; Kim, T.M.; Kim, T.Y.; Heo, D.S.; Yuasa, M.; Yanagihara, Y.; Bang, Y.J. Phase I Study of OPB-31121, an Oral STAT3 Inhibitor, in Patients with Advanced Solid Tumors. Cancer Res. Treat. 2015, 47, 607–615. [Google Scholar] [CrossRef]
- Handle, F.; Puhr, M.; Schaefer, G.; Lorito, N.; Hoefer, J.; Gruber, M.; Guggenberger, F.; Santer, F.R.; Marques, R.B.; van Weerden, W.M.; et al. The STAT3 Inhibitor Galiellalactone Reduces IL6-Mediated AR Activity in Benign and Malignant Prostate Models. Mol. Cancer Ther. 2018, 17, 2722–2731. [Google Scholar] [CrossRef] [PubMed]
- Escobar, Z.; Bjartell, A.; Canesin, G.; Evans-Axelsson, S.; Sterner, O.; Hellsten, R.; Johansson, M.H. Preclinical Characterization of 3β-(N-Acetyl l-cysteine methyl ester)-2aβ,3-dihydrogaliellalactone (GPA512), a Prodrug of a Direct STAT3 Inhibitor for the Treatment of Prostate Cancer. J. Med. Chem. 2016, 59, 4551–4562. [Google Scholar] [CrossRef]
- Witt, K.; Evans-Axelsson, S.; Lundqvist, A.; Johansson, M.; Bjartell, A.; Hellsten, R. Inhibition of STAT3 augments antitumor efficacy of anti-CTLA-4 treatment against prostate cancer. Cancer Immunol. Immunother. 2021, 70, 3155–3166. [Google Scholar] [CrossRef]
- Ogura, M.; Uchida, T.; Terui, Y.; Hayakawa, F.; Kobayashi, Y.; Taniwaki, M.; Takamatsu, Y.; Naoe, T.; Tobinai, K.; Munakata, W.; et al. Phase I study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015, 106, 896–901. [Google Scholar] [CrossRef]
- Boonstra, P.S.; Polk, A.; Brown, N.; Hristov, A.C.; Bailey, N.G.; Kaminski, M.S.; Phillips, T.; Devata, S.; Mayer, T.; Wilcox, R.A. A single center phase II study of ixazomib in patients with relapsed or refractory cutaneous or peripheral T-cell lymphomas. Am. J. Hematol. 2017, 92, 1287–1294. [Google Scholar] [CrossRef]
- Shuai, T.; Zhang, C.; Zhang, M.; Wang, Y.; Xiong, H.; Huang, Q.; Liu, J. Low-dose theophylline in addition to ICS therapy in COPD patients: A systematic review and meta-analysis. PLoS ONE 2021, 16, e0251348. [Google Scholar] [CrossRef] [PubMed]
- Montaño, L.M.; Sommer, B.; Gomez-Verjan, J.C.; Morales-Paoli, G.S.; Ramírez-Salinas, G.L.; Solís-Chagoyán, H.; Sanchez-Florentino, Z.A.; Calixto, E.; Pérez-Figueroa, G.E.; Carter, R.; et al. Theophylline: Old Drug in a New Light, Application in COVID-19 through Computational Studies. Int. J. Mol. Sci. 2022, 23, 4167. [Google Scholar] [CrossRef] [PubMed]
- Ichiyama, T.; Hasegawa, S.; Matsubara, T.; Hayashi, T.; Furukawa, S. Theophylline inhibits NF-κB activation and IκBα degradation in human pulmonary epithelial cells. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2001, 364, 558–561. [Google Scholar] [CrossRef] [PubMed]
- Viana, S.D.; Reis, F.; Alves, R. Therapeutic Use of mTOR Inhibitors in Renal Diseases: Advances, Drawbacks, and Challenges. Oxid. Med. Cell. Longev. 2018, 2018, 3693625. [Google Scholar] [CrossRef] [PubMed]
- Liesveld, J.L.; Baran, A.; Azadniv, M.; Misch, H.; Nedrow, K.; Becker, M.; Loh, K.P.; O’Dwyer, K.M.; Mendler, J.H. A phase II study of sequential decitabine and rapamycin in acute myelogenous leukemia. Leuk. Res. 2022, 112, 106749. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, N.; Li, J.; Zhang, D.; Lv, X. Rapamycin alleviates proinflammatory cytokines and nociceptive behavior induced by chemotherapeutic paclitaxel. Neurol. Res. 2019, 41, 52–59. [Google Scholar] [CrossRef]
- Wu, C.-F.; Wu, C.-Y.; Chiou, R.Y.Y.; Yang, W.-C.; Lin, C.-F.; Wang, C.-M.; Hou, P.-H.; Lin, T.-C.; Kuo, C.-Y.; Chang, G.-R. The Anti-Cancer Effects of a Zotarolimus and 5-Fluorouracil Combination Treatment on A549 Cell-Derived Tumors in BALB/c Nude Mice. Int. J. Mol. Sci. 2021, 22, 4562. [Google Scholar] [CrossRef]
- Chen, J.S.; Alfajaro, M.M.; Chow, R.D.; Wei, J.; Filler, R.B.; Eisenbarth, S.C.; Wilen, C.B. Non-steroidal anti-inflammatory drugs dampen the cytokine and antibody response to SARS-CoV-2 infection. J. Virol. 2021, 95, e00014-21. [Google Scholar] [CrossRef]
- Mao, J.T.; Roth, M.D.; Fishbein, M.C.; Aberle, D.R.; Zhang, Z.F.; Rao, J.Y.; Tashkin, D.P.; Goodglick, L.; Holmes, E.C.; Cameron, R.B.; et al. Lung cancer chemoprevention with celecoxib in former smokers. Cancer Prev. Res. 2011, 4, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Panahi, Y.; Saadat, A.; Beiraghdar, F.; Sahebkar, A. Adjuvant Therapy with Bioavailability-Boosted Curcuminoids Suppresses Systemic Inflammation and Improves Quality of Life in Patients with Solid Tumors: A Randomized Double-Blind Placebo-Controlled Trial. Phytother. Res. 2014, 28, 1461–1467. [Google Scholar] [CrossRef]
- Liao, F.; Liu, L.; Luo, E.; Hu, J. Curcumin enhances anti-tumor immune response in tongue squamous cell carcinoma. Arch. Oral Biol. 2018, 92, 32–37. [Google Scholar] [CrossRef]
- Dhillon, N.; Aggarwal, B.B.; Newman, R.A.; Wolff, R.A.; Kunnumakkara, A.B.; Abbruzzese, J.L.; Ng, C.S.; Badmaev, V.; Kurzrock, R. Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin. Cancer Res. 2008, 14, 4491–4499. [Google Scholar] [CrossRef]
- Du, Y.; Long, Q.; Zhang, L.; Shi, Y.; Liu, X.; Li, X.; Guan, B.; Tian, Y.; Wang, X.; Li, L.; et al. Curcumin inhibits cancer-associated fibroblast-driven prostate cancer invasion through MAOA/mTOR/HIF-1α signaling. Int. J. Oncol. 2015, 47, 2064–2072. [Google Scholar] [CrossRef]
- Choi, H.; Chun, Y.-S.; Kim, S.-W.; Kim, M.-S.; Park, J.-W. Curcumin Inhibits Hypoxia-Inducible Factor-1 by Degrading Aryl Hydrocarbon Receptor Nuclear Translocator: A Mechanism of Tumor Growth Inhibition. Mol. Pharmacol. 2006, 70, 1664–1671. [Google Scholar] [CrossRef]
- Hu, Y.; McIntosh, G.H.; Le Leu, R.K.; Somashekar, R.; Meng, X.Q.; Gopalsamy, G.; Bambaca, L.; McKinnon, R.A.; Young, G.P. Supplementation with Brazil nuts and green tea extract regulates targeted biomarkers related to colorectal cancer risk in humans. Br. J. Nutr. 2016, 116, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ren, X.; Deng, C.; Yang, L.; Yan, E.; Guo, T.; Li, Y.; Xu, M.X. Mechanism of the inhibition of the STAT3 signaling pathway by EGCG. Oncol. Rep. 2013, 30, 2691–2696. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Santegoets, S.J.; Reyners, A.K.; Goedemans, R.; Wouters, M.C.; Kenter, G.G.; van Erkel, A.R.; van Poelgeest, M.I.; Nijman, H.W.; van der Hoeven, J.J.; et al. A phase I trial combining carboplatin/doxorubicin with tocilizumab, an anti-IL-6R monoclonal antibody, and interferon-α2b in patients with recurrent epithelial ovarian cancer. Ann. Oncol. 2015, 26, 2141–2149. [Google Scholar] [CrossRef]
- Dijkgraaf, E.M.; Heusinkveld, M.; Tummers, B.; Vogelpoel, L.T.; Goedemans, R.; Jha, V.; Nortier, J.W.; Welters, M.J.; Kroep, J.R.; van der Burg, S.H. Chemotherapy alters monocyte differentiation to favor generation of cancer-supporting M2 macrophages in the tumor microenvironment. Cancer Res. 2013, 73, 2480–2492. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.C.; Maurer, M.J.; Oberg, A.L.; Visscher, D.W.; Kalli, K.R.; Hartmann, L.C.; Goode, E.L.; Knutson, K.L. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PLoS ONE 2013, 8, e80063. [Google Scholar] [CrossRef]
- Kennedy, G.A.; Tey, S.K.; Buizen, L.; Varelias, A.; Gartlan, K.H.; Curley, C.; Olver, S.D.; Chang, K.; Butler, J.P.; Misra, A.; et al. A phase 3 double-blind study of the addition of tocilizumab vs placebo to cyclosporin/methotrexate GVHD prophylaxis. Blood 2021, 137, 1970–1979. [Google Scholar] [CrossRef]
- Nguyen, M.L.T.; Bui, K.C.; Scholta, T.; Xing, J.; Bhuria, V.; Sipos, B.; Wilkens, L.; Nguyen Linh, T.; Velavan, T.P.; Bozko, P.; et al. Targeting interleukin 6 signaling by monoclonal antibody siltuximab on cholangiocarcinoma. J. Gastroenterol. Hepatol. 2021, 36, 1334–1345. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.Z.; Greenman, K.L.; Billings, S.; Van Vranken, D.L.; Krolewski, J.J. Binding of madindoline A to the extracellular domain of gp130. Biochemistry 2005, 44, 10822–10827. [Google Scholar] [CrossRef]
- Aigner, P.; Just, V.; Stoiber, D. STAT3 isoforms: Alternative fates in cancer? Cytokine 2019, 118, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.X.; Yang, P.L.; Li, E.M.; Xu, L.Y. STAT3beta, a distinct isoform from STAT3. Int. J. Biochem. Cell Biol. 2019, 110, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Lavecchia, A.; Di Giovanni, C.; Cerchia, C. Novel inhibitors of signal transducer and activator of transcription 3 signaling pathway: An update on the recent patent literature. Expert Opin. Ther. Pat. 2014, 25, 1305–1317. [Google Scholar] [CrossRef]
- Rah, B.; Rather, R.A.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front. Pharmacol. 2022, 13, 821344. [Google Scholar] [CrossRef]
- Kim, M.-J.; Nam, H.-J.; Kim, H.-P.; Han, S.-W.; Im, S.-A.; Kim, T.-Y.; Oh, D.-Y.; Bang, Y.-J. OPB-31121, a novel small molecular inhibitor, disrupts the JAK2/STAT3 pathway and exhibits an antitumor activity in gastric cancer cells. Cancer Lett. 2013, 335, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Hellsten, R.; Johansson, M.; Dahlman, A.; Dizeyi, N.; Sterner, O.; Bjartell, A. Galiellalactone is a novel therapeutic candidate against hormone-refractory prostate cancer expressing activated Stat3. Prostate 2008, 68, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Han, B.; Dong, X. The Androgen Receptor and Its Crosstalk With the Src Kinase During Castrate-Resistant Prostate Cancer Progression. Front. Oncol. 2022, 12, 905398. [Google Scholar] [CrossRef]
- Fialova, J.L.; Raudenska, M.; Jakubek, M.; Kejik, Z.; Martasek, P.; Babula, P.; Matkowski, A.; Filipensky, P.; Masarik, M. Novel Mitochondria-targeted Drugs for Cancer Therapy. Mini Rev. Med. Chem. 2021, 21, 816–832. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial Stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Mantel, C.; Messina-Graham, S.; Moh, A.; Cooper, S.; Hangoc, G.; Fu, X.Y.; Broxmeyer, H.E. Mouse hematopoietic cell-targeted STAT3 deletion: Stem/progenitor cell defects, mitochondrial dysfunction, ROS overproduction, and a rapid aging-like phenotype. Blood 2012, 120, 2589–2599. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef]
- Denisenko, T.V.; Gorbunova, A.S.; Zhivotovsky, B. Mitochondrial Involvement in Migration, Invasion and Metastasis. Front. Cell Dev. Biol. 2019, 7, 355. [Google Scholar] [CrossRef]
- Passaniti, A.; Kim, M.S.; Polster, B.M.; Shapiro, P. Targeting mitochondrial metabolism for metastatic cancer therapy. Mol. Carcinog. 2022, 61, 827–838. [Google Scholar] [CrossRef]
- Caino, M.C.; Altieri, D.C. Molecular Pathways: Mitochondrial Reprogramming in Tumor Progression and Therapy. Clin. Cancer Res. 2016, 22, 540–545. [Google Scholar] [CrossRef]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and Mitochondrial Respiration: A Neglected Connection. Cancer Res. 2015, 75, 3687–3691. [Google Scholar] [CrossRef]
- Jia, D.; Park, J.H.; Jung, K.H.; Levine, H.; Kaipparettu, B.A. Elucidating the Metabolic Plasticity of Cancer: Mitochondrial Reprogramming and Hybrid Metabolic States. Cells 2018, 7, 21. [Google Scholar] [CrossRef]
- Patra, S.; Elahi, N.; Armorer, A.; Arunachalam, S.; Omala, J.; Hamid, I.; Ashton, A.W.; Joyce, D.; Jiao, X.; Pestell, R.G. Mechanisms Governing Metabolic Heterogeneity in Breast Cancer and Other Tumors. Front. Oncol. 2021, 11, 700629. [Google Scholar] [CrossRef]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef]
- Mackenzie, G.G.; Huang, L.; Alston, N.; Ouyang, N.; Vrankova, K.; Mattheolabakis, G.; Constantinides, P.P.; Rigas, B. Targeting mitochondrial STAT3 with the novel phospho-valproic acid (MDC-1112) inhibits pancreatic cancer growth in mice. PLoS ONE 2013, 8, e61532. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Li, L.; Andrew, S.; Allan, D.; Li, X.; Lee, J.; Ji, G.; Yao, Z.; Gadde, S.; Figeys, D.; et al. An autocrine inflammatory forward-feedback loop after chemotherapy withdrawal facilitates the repopulation of drug-resistant breast cancer cells. Cell Death Dis. 2017, 8, e2932. [Google Scholar] [CrossRef] [PubMed]
- Filimon, A.; Preda, I.A.; Boloca, A.F.; Negroiu, G. Interleukin-8 in Melanoma Pathogenesis, Prognosis and Therapy-An Integrated View into Other Neoplasms and Chemokine Networks. Cells 2021, 11, 120. [Google Scholar] [CrossRef]
- Xia, T.; Li, J.; Ren, X.; Liu, C.; Sun, C. Research progress of phenolic compounds regulating IL-6 to exert antitumor effects. Phytother. Res. 2021, 35, 6720–6734. [Google Scholar] [CrossRef]
- Jiang, J.; Wang, G.Z.; Wang, Y.; Huang, H.Z.; Li, W.T.; Qu, X.D. Hypoxia-induced HMGB1 expression of HCC promotes tumor invasiveness and metastasis via regulating macrophage-derived IL-6. Exp. Cell Res. 2018, 367, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.J.; Thompson, A.R.; Whyte, M.K.; Walmsley, S.R. HIF-mediated innate immune responses: Cell signaling and therapeutic implications. Hypoxia 2014, 2, 47–58. [Google Scholar] [CrossRef]
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and Cycling Hypoxia: Drivers of Cancer Chronic Inflammation through HIF-1 and NF-κB Activation: A Review of the Molecular Mechanisms. Int. J. Mol. Sci. 2021, 22, 10701. [Google Scholar] [CrossRef]
- Malkov, M.I.; Lee, C.T.; Taylor, C.T. Regulation of the Hypoxia-Inducible Factor (HIF) by Pro-Inflammatory Cytokines. Cells 2021, 10, 2340. [Google Scholar] [CrossRef]
- Jayatilaka, H.; Tyle, P.; Chen, J.J.; Kwak, M.; Ju, J.; Kim, H.J.; Lee, J.S.H.; Wu, P.H.; Gilkes, D.M.; Fan, R.; et al. Synergistic IL-6 and IL-8 paracrine signalling pathway infers a strategy to inhibit tumour cell migration. Nat. Commun. 2017, 8, 15584. [Google Scholar] [CrossRef]
- Jayatilaka, H.; Umanzor, F.G.; Shah, V.; Meirson, T.; Russo, G.; Starich, B.; Tyle, P.; Lee, J.S.H.; Khatau, S.; Gil-Henn, H.; et al. Tumor cell density regulates matrix metalloproteinases for enhanced migration. Oncotarget 2018, 9, 32556–32569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Roque, D.M.; Reader, J.; Lin, J. Combined inhibition of IL-6 and IL-8 pathways suppresses ovarian cancer cell viability and migration and tumor growth. Int. J. Oncol. 2022, 60, 5340. [Google Scholar] [CrossRef] [PubMed]
- Kejík, Z.; Kaplánek, R.; Dytrych, P.; Masařík, M.; Veselá, K.; Abramenko, N.; Hoskovec, D.; Vašáková, M.; Králová, J.; Martásek, P.; et al. Circulating Tumour Cells (CTCs) in NSCLC: From Prognosis to Therapy Design. Pharmaceutics 2021, 13, 1879. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.; Zadeh, L.R.; Baradaran, B.; Molavi, O.; Ghesmati, Z.; Sabzichi, M.; Ramezani, F. Up-down regulation of HIF-1α in cancer progression. Gene 2021, 798, 145796. [Google Scholar] [CrossRef]
- Li, M.; Lin, L.; Guo, T.; Wu, Y.; Lin, J.; Liu, Y.; Yang, K.; Hu, C. Curcumin Administered in Combination with Glu-GNPs Induces Radiosensitivity in Transplanted Tumor MDA-MB-231-luc Cells in Nude Mice. BioMed Res. Int. 2021, 2021, 9262453. [Google Scholar] [CrossRef]
- Kubatka, P.; Mazurakova, A.; Samec, M.; Koklesova, L.; Zhai, K.; Al-Ishaq, R.; Kajo, K.; Biringer, K.; Vybohova, D.; Brockmueller, A.; et al. Flavonoids against non-physiologic inflammation attributed to cancer initiation, development, and progression-3PM pathways. EPMA J. 2021, 12, 559–587. [Google Scholar] [CrossRef]
- Talianová, V.; Kejík, Z.; Kaplánek, R.; Veselá, K.; Abramenko, N.; Lacina, L.; Strnadová, K.; Dvořánková, B.; Martásek, P.; Masařík, M.; et al. New-Generation Heterocyclic Bis-Pentamethinium Salts as Potential Cytostatic Drugs with Dual IL-6R and Mitochondria-Targeting Activity. Pharmaceutics 2022, 14, 1712. [Google Scholar] [CrossRef]
- Fialova, J.L.; Hönigova, K.; Raudenska, M.; Miksatkova, L.; Zobalova, R.; Navratil, J.; Šmigová, J.; Moturu, T.R.; Vicar, T.; Balvan, J.; et al. Pentamethinium salts suppress key metastatic processes by regulating mitochondrial function and inhibiting dihydroorotate dehydrogenase respiration. Biomed. Pharmacother. 2022, 154, 113582. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian Pyrimidine Biosynthesis: Fresh Insights into an Ancient Pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef]
- Boukalova, S.; Hubackova, S.; Milosevic, M.; Ezrova, Z.; Neuzil, J.; Rohlena, J. Dihydroorotate dehydrogenase in oxidative phosphorylation and cancer. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165759. [Google Scholar] [CrossRef]
- Mochizuki, D.; Adams, A.; Warner, K.A.; Zhang, Z.; Pearson, A.T.; Misawa, K.; McLean, S.A.; Wolf, G.T.; Nör, J.E. Anti-tumor effect of inhibition of IL-6 signaling in mucoepidermoid carcinoma. Oncotarget 2015, 6, 22822–22835. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Nakayama, H.; Yoshida, R.; Hirosue, A.; Nagata, M.; Tanaka, T.; Kawahara, K.; Sakata, J.; Arita, H.; Nakashima, H.; et al. IL-6 controls resistance to radiation by suppressing oxidative stress via the Nrf2-antioxidant pathway in oral squamous cell carcinoma. Br. J. Cancer 2016, 115, 1234–1244. [Google Scholar] [CrossRef]
- Méndez-Clemente, A.; Bravo-Cuellar, A.; González-Ochoa, S.; Santiago-Mercado, M.; Palafox-Mariscal, L.; Jave-Suárez, L.; Solorzano-Ibarra, F.; Villaseñor-García, M.; Ortiz-Lazareno, P.; Hernández-Flores, G. Dual STAT-3 and IL-6R inhibition with stattic and tocilizumab decreases migration, invasion and proliferation of prostate cancer cells by targeting the IL-6/IL-6R/STAT-3 axis. Oncol. Rep. 2022, 48, 8349. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Prim. 2020, 6, 92. [Google Scholar] [CrossRef]
- Peitzsch, C.; Nathansen, J.; Schniewind, S.I.; Schwarz, F.; Dubrovska, A. Cancer Stem Cells in Head and Neck Squamous Cell Carcinoma: Identification, Characterization and Clinical Implications. Cancers 2019, 11, 616. [Google Scholar] [CrossRef] [PubMed]
- Kalavrezos, N.; Bhandari, R. Current trends and future perspectives in the surgical management of oral cancer. Oral Oncol. 2010, 46, 429–432. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Chen, Y.C.; Nör, F.; Warner, K.A.; Andrews, A.; Wagner, V.P.; Zhang, Z.; Zhang, Z.; Martins, M.D.; Pearson, A.T.; et al. Endothelial-derived interleukin-6 induces cancer stem cell motility by generating a chemotactic gradient towards blood vessels. Oncotarget 2017, 8, 100339–100352. [Google Scholar] [CrossRef]
- Chinn, S.B.; Darr, O.A.; Owen, J.H.; Bellile, E.; McHugh, J.B.; Spector, M.E.; Papagerakis, S.M.; Chepeha, D.B.; Bradford, C.R.; Carey, T.E.; et al. Cancer stem cells: Mediators of tumorigenesis and metastasis in head and neck squamous cell carcinoma. Head Neck 2015, 37, 317–326. [Google Scholar] [CrossRef]
- Wang, Y.F.; Chang, S.Y.; Tai, S.K.; Li, W.Y.; Wang, L.S. Clinical significance of interleukin-6 and interleukin-6 receptor expressions in oral squamous cell carcinoma. Head Neck 2002, 24, 850–858. [Google Scholar] [CrossRef]
- Novotný, J.; Bandúrová, V.; Strnad, H.; Chovanec, M.; Hradilová, M.; Šáchová, J.; Šteffl, M.; Grušanović, J.; Kodet, R.; Pačes, V.; et al. Analysis of HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinomas and Paired Normal Mucosae Reveals Cyclin D1 Deregulation and Compensatory Effect of Cyclin D2. Cancers 2020, 12, 792. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Macciò, A.; Madeddu, C. Inflammation and ovarian cancer. Cytokine 2012, 58, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Saini, U.; Naidu, S.; ElNaggar, A.C.; Bid, H.K.; Wallbillich, J.J.; Bixel, K.; Bolyard, C.; Suarez, A.A.; Kaur, B.; Kuppusamy, P.; et al. Elevated STAT3 expression in ovarian cancer ascites promotes invasion and metastasis: A potential therapeutic target. Oncogene 2017, 36, 168–181. [Google Scholar] [CrossRef]
- Chaluvally-Raghavan, P.; Jeong, K.J.; Pradeep, S.; Silva, A.M.; Yu, S.; Liu, W.; Moss, T.; Rodriguez-Aguayo, C.; Zhang, D.; Ram, P.; et al. Direct Upregulation of STAT3 by MicroRNA-551b-3p Deregulates Growth and Metastasis of Ovarian Cancer. Cell Rep. 2016, 15, 1493–1504. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Guo, X.; Jin, X.; Sun, W.; Zhang, X.; Xu, R.C. Interleukin-6 signaling regulates anchorage-independent growth, proliferation, adhesion and invasion in human ovarian cancer cells. Cytokine 2012, 59, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.P.; Yang, D.C.; Elliott, R.L.; Head, J.F. Reduction in serum IL-6 after vacination of breast cancer patients with tumour-associated antigens is related to estrogen receptor status. Cytokine 2000, 12, 458–465. [Google Scholar] [CrossRef]
- Tamm, I.; Cardinale, I.; Krueger, J.; Murphy, J.S.; May, L.T.; Sehgal, P.B. Interleukin 6 decreases cell-cell association and increases motility of ductal breast carcinoma cells. J. Exp. Med. 1989, 170, 1649–1669. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef]
- Oh, K.; Lee, O.Y.; Park, Y.; Seo, M.W.; Lee, D.S. IL-1β induces IL-6 production and increases invasiveness and estrogen-independent growth in a TG2-dependent manner in human breast cancer cells. BMC Cancer 2016, 16, 724. [Google Scholar] [CrossRef]
- Sanguinetti, A.; Santini, D.; Bonafè, M.; Taffurelli, M.; Avenia, N. Interleukin-6 and pro inflammatory status in the breast tumor microenvironment. World J. Surg. Oncol. 2015, 13, 129. [Google Scholar] [CrossRef]
- Markovic, S.N.; Erickson, L.A.; Rao, R.D.; Weenig, R.H.; Pockaj, B.A.; Bardia, A.; Vachon, C.M.; Schild, S.E.; McWilliams, R.R.; Hand, J.L.; et al. Malignant melanoma in the 21st century, part 1: Epidemiology, risk factors, screening, prevention, and diagnosis. Mayo Clin. Proc. 2007, 82, 364–380. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Weber, R.; Groth, C.; Lasser, S.; Arkhypov, I.; Petrova, V.; Altevogt, P.; Utikal, J.; Umansky, V. IL-6 as a major regulator of MDSC activity and possible target for cancer immunotherapy. Cell Immunol. 2021, 359, 104254. [Google Scholar] [CrossRef]
- Weber, R.; Riester, Z.; Hüser, L.; Sticht, C.; Siebenmorgen, A.; Groth, C.; Hu, X.; Altevogt, P.; Utikal, J.S.; Umansky, V. IL-6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J. Immunother. Cancer 2020, 8, e000949. [Google Scholar] [CrossRef] [PubMed]
- Hailemichael, Y.; Johnson, D.H.; Abdel-Wahab, N.; Foo, W.C.; Bentebibel, S.E.; Daher, M.; Haymaker, C.; Wani, K.; Saberian, C.; Ogata, D.; et al. Interleukin-6 blockade abrogates immunotherapy toxicity and promotes tumor immunity. Cancer Cell 2022, 40, 509–523. [Google Scholar] [CrossRef] [PubMed]
- Depner, S.; Lederle, W.; Gutschalk, C.; Linde, N.; Zajonz, A.; Mueller, M.M. Cell type specific interleukin-6 induced responses in tumor keratinocytes and stromal fibroblasts are essential for invasive growth. Int. J. Cancer 2014, 135, 551–562. [Google Scholar] [CrossRef]
- Vokurka, M.; Lacina, L.; Brábek, J.; Kolář, M.; Ng, Y.Z.; Smetana, K. Cancer-Associated Fibroblasts Influence the Biological Properties of Malignant Tumours via Paracrine Secretion and Exosome Production. Int. J. Mol. Sci. 2022, 23, 964. [Google Scholar] [CrossRef]
- Strnadová, K.; Pfeiferová, L.; Přikryl, P.; Dvořánková, B.; Vlčák, E.; Frýdlová, J.; Vokurka, M.; Novotný, J.; Šáchová, J.; Hradilová, M.; et al. Exosomes produced by melanoma cells significantly influence the biological properties of normal and cancer-associated fibroblasts. Histochem. Cell Biol. 2022, 157, 153–172. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name (Trade Name) | Target: Function | Status | Ref. |
|---|---|---|---|
| Human monoclonal antibody | |||
| Tocilizumab (RoActemra) | IL-6R: receptor inhibition | Clinically used for rheumatoid arthritis | [47] |
| Case report: Reducing IL-6-mediated cachexia | [48] | ||
| Sarilumab (Kevzara) | IL-6R: receptor inhibition | Clinically used for rheumatoid arthritis | [49] |
| Under clinical trial (EMPOWER NCT04333706): triple-negative breast cancer (stage I-III, high-risk residual diseases) combination with Capecitabine | |||
| Siltuximab | IL-6: neutralization | approved for CAR-T | [50] |
| Low molecular inhibitor | |||
| Bazedoxifene (Conbriza) | Estrogen receptor modulator | Clinically used in the treatment of osteoporosis | [51] |
| gp130: inhibitor | In vivo | [52] | |
| CD40 receptor: inhibitor | In vitro | [53] | |
| Tofacitinib (Xeljanz) | JAK1/3 inhibitor | Clinically used in the treatment of moderate-severe ulcerative colitis | [54] |
| JAK pathway | Clinical trial: developed malignancies lung, breast, gastric cancer, and lymphoma; rate of malignancies by 6-month intervals of tofacitinib exposure indicates rates remained stable over time | [55] | |
| Ruxolitinib (Jakafi) | JAK1/2 inhibitor | Clinically used in the treatment of steroid refractory graft-versus-host disease | [56] |
| JAK pathway | Clinical trials: inadequately controlled polycythaemia; decrease in thromboembolic events | [57] | |
| Momelotinib | ACVR1/ALK2, JAK1 and JAK2, inhibitor | FDA accepts for the treatment of the myelofibrosis | |
| Momelotinib | Clinical trials: myelofibrosis; higher overall and leukaemia-free survival. | [58] | |
| Madindoline A and B | gp130: inhibitor | In vitro | [59,60] |
| ERBF | IL-6R: blocking interaction IL-6R with IL-6, or gp130 | In vivo | [61,62,63] |
| Stattic | STAT3: inhibition of activation | In vivo | [64] |
| OPB-31121 | STAT3: inhibition of activation | Clinical trials: advanced colon and rectal tumours; tumour shrinkage, bad pharmacokinetic (very low blood concentration) | [65,66] |
| Galiellalactone | STAT3: inhibition of DNA binding | prostatectomy samples: reduction IL-6 induced AR signalling | [67] |
| In vivo | [68] | ||
| GPB730 | STAT3: inhibition of DNA binding | In vivo | [69] |
| OPB-51602 | STAT3: activation of aggregation | Clinical trials: refractory haematological malignancies; no clear therapeutic response was observed | [70] |
| Ixazomib | NF-κB: inhibition of ubiquitin-proteasome pathway leading to loss of NF-κB activity | Clinical trials: Relapsed or Refractory Cutaneous or Peripheral T-cell Lymphomas; reduction in NF-κB activation and subsequently GATA-3 expression in the biopsy specimens | [71] |
| Theofyline (Elixophyllin, Elixophylline, Pulmophylline, Quibron-T, Theo-24, Theolair, Uniphyl) | phosphodiesterase inhibitor, adenosine receptor blocker, and histone deacetylase activator | Clinically used in chronic obstructive pulmonary disease and asthma | [72,73] |
| NF-κB: inhibition of activation | In vitro | [74] | |
| Rapamycin (Sirolimus, Rapamur) | mTOR: inhibitor | Clinically used immunosuppressive therapy | [75] |
| Clinical trials: acute myelogenous leukaemia; no effects on the composite complete remission rate | [76] | ||
| IL-6, TNF-α and IL-1β: decrease cytokine level | In vivo | [77] | |
| Zotarolimus | IL-1β, TNF-α, IL-6 and NF-κB: decrease cytokine level and NF-KB activity | In vivo | [78] |
| NSAIDs (e.g., celecoxib, aspirin, ibuprofen, naproxen, meloxicam) | cyclooxygenase inhibitors | in a broad spectrum of conditions; Analgetic, antipyretics, in rheumatic diseases | [79] |
| Celecoxib | IL-6: decrease expression by COX-2 inhibition | Clinical trials: former-smokers; bronchoscopy samples (reduction IL-6 and Ki-67 expression) | [80] |
| Food supplements | |||
| Curcumin | IL-6: decrease expression | Clinical trials: patients with solid tumour; decrease in plasma level of IL-6, TNF-a, TGF-b, substance P, hs-CRP, CGRP and MCP-1, increase patient quality life | [81] |
| STAT3: decrease activity | In vivo | [82] | |
| NF-κB: activity and expression | Clinical trials: advanced pancreatic cancer; peripheral blood mononuclear cells (decrease in expression of NF-κB, STAT-3 and COX-2) decrease in serum cytokine levels (IL-6, IL-8, IL-10, and IL-1 receptor antagonists) | [83] | |
| HIF-1α: decrease expression and activity | In vitro and In vivo | [84,85] | |
| Epigallocatechin-3-gallate | NF-κB: decrease expression | Clinical trial: subjects with a high risk of colorectal cancer; lover expression of the NF-κB and DNMT1 | [86] |
| STAT3: inhibition of activation | Molecular assay and In vitro | [87] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rašková, M.; Lacina, L.; Kejík, Z.; Venhauerová, A.; Skaličková, M.; Kolář, M.; Jakubek, M.; Rosel, D.; Smetana, K., Jr.; Brábek, J. The Role of IL-6 in Cancer Cell Invasiveness and Metastasis—Overview and Therapeutic Opportunities. Cells 2022, 11, 3698. https://doi.org/10.3390/cells11223698
Rašková M, Lacina L, Kejík Z, Venhauerová A, Skaličková M, Kolář M, Jakubek M, Rosel D, Smetana K Jr., Brábek J. The Role of IL-6 in Cancer Cell Invasiveness and Metastasis—Overview and Therapeutic Opportunities. Cells. 2022; 11(22):3698. https://doi.org/10.3390/cells11223698
Chicago/Turabian StyleRašková, Magdalena, Lukáš Lacina, Zdeněk Kejík, Anna Venhauerová, Markéta Skaličková, Michal Kolář, Milan Jakubek, Daniel Rosel, Karel Smetana, Jr., and Jan Brábek. 2022. "The Role of IL-6 in Cancer Cell Invasiveness and Metastasis—Overview and Therapeutic Opportunities" Cells 11, no. 22: 3698. https://doi.org/10.3390/cells11223698
APA StyleRašková, M., Lacina, L., Kejík, Z., Venhauerová, A., Skaličková, M., Kolář, M., Jakubek, M., Rosel, D., Smetana, K., Jr., & Brábek, J. (2022). The Role of IL-6 in Cancer Cell Invasiveness and Metastasis—Overview and Therapeutic Opportunities. Cells, 11(22), 3698. https://doi.org/10.3390/cells11223698