

Smoking-Associated Exposure of Human Primary Bronchial Epithelial Cells to Aldehydes: Impact on Molecular Mechanisms Controlling Mitochondrial Content and Function

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. PBEC Isolation
2.2. PBEC Proliferation and Differentiation
2.3. Validation and Characterization of Differentiation of PBEC at ALI
2.4. Monitoring PBEC Differentiation and Monolayer Integrity
2.5. Dosimetry and Exposure Regime
2.6. PBEC Exposure to CS or Mixture of Aldehydes
2.7. RNA Isolation and Real-Time Quantitative PCR
2.8. DNA Isolation and Analysis of Mitochondrial DNA (mtDNA) Copy Number
2.9. Fixation for Paraffin-Embedded Section and Immunohistochemistry Staining
2.10. Cytotoxicity Assay
2.11. Inflammatory Protein Secretion
2.12. Protein Isolation and Western Blotting
2.13. Analysis of Bead Motion Induced by Ciliary Beating
2.14. Statistical Analysis
3. Results
3.1. Validation and Characterization of Differentiation of PBEC at ALI
3.2. No Impact of CS or Aldehydes Exposure on Cell Viability and Monolayer Integrity
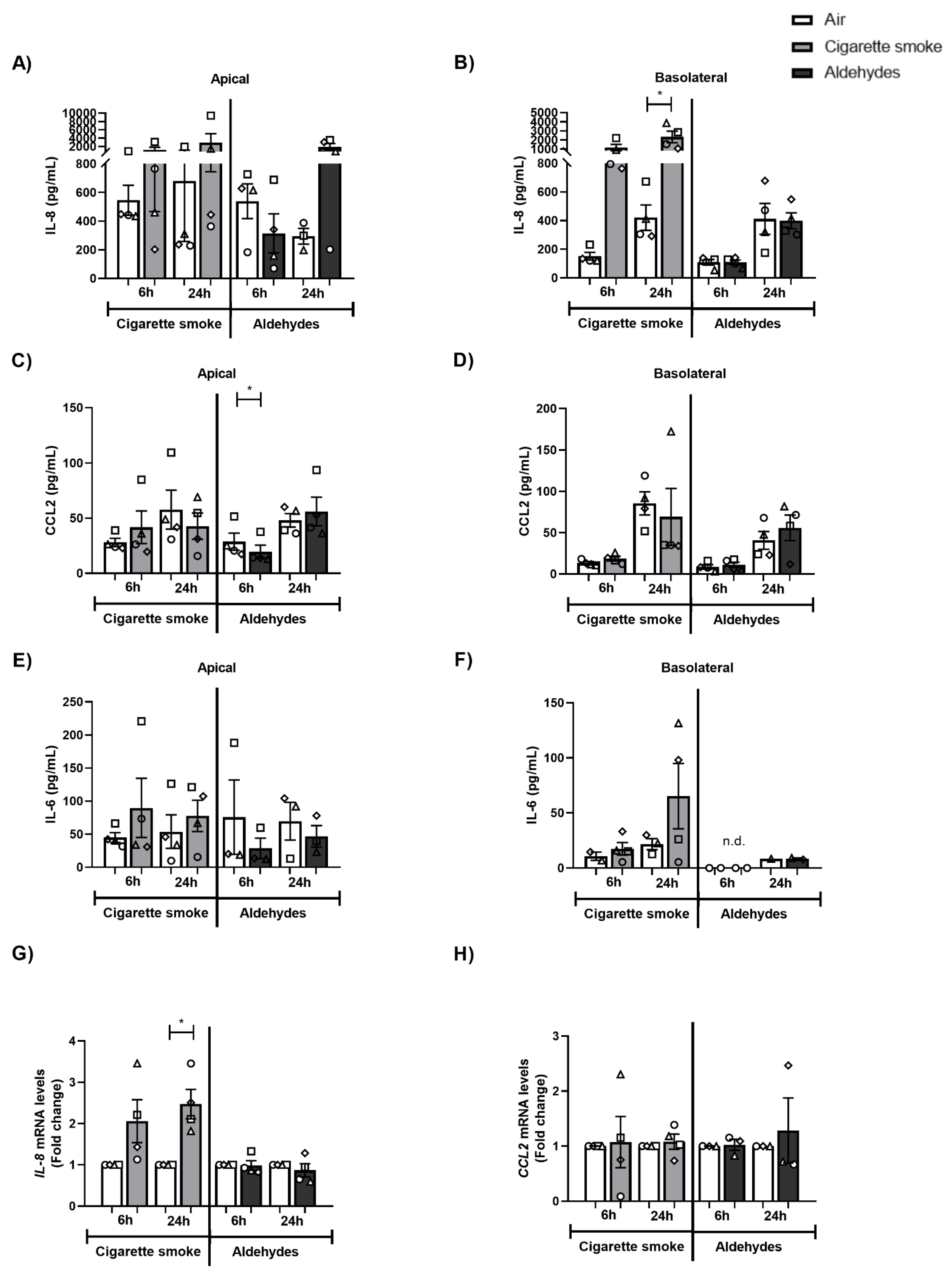
3.3. CS Exposure Affects Inflammatory Protein Production and Antioxidant Gene Expression
3.4. Altered Abundance of Molecules Associated with Autophagy Following CS or Aldehyde Exposure
3.5. Exposure to CS Decreases Expression of Components of the Mitophagy Machinery
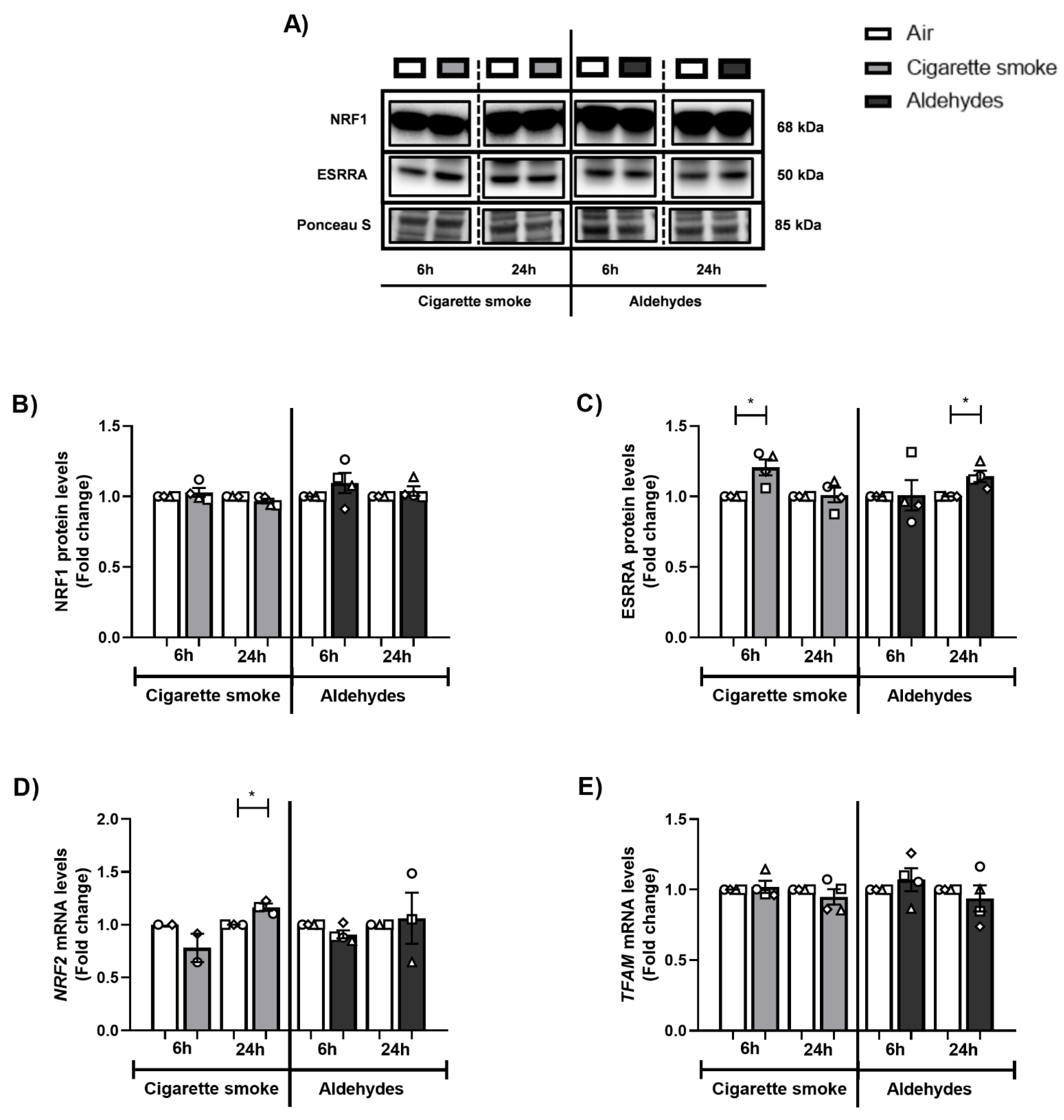
3.6. Alterations in the Expression of Proteins and Genes Involved in Mitochondrial Biogenesis in Response to CS
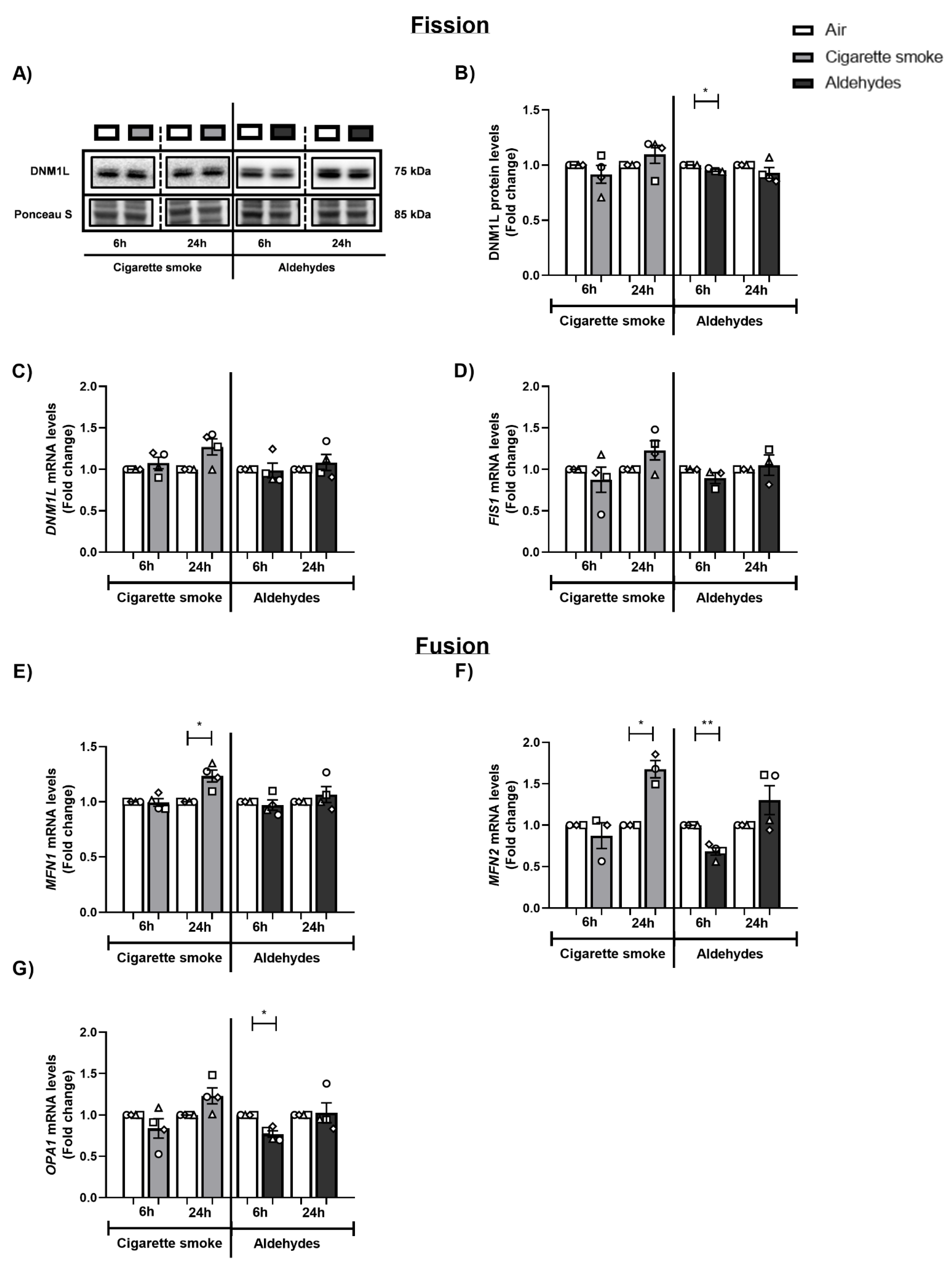
3.7. CS and Aldehydes Affect the Abundance of Mitochondrial Fission and Fusion Regulators
3.8. Minor Changes in the Abundance of Subunits of Oxidative Phosphorylation Complexes in Response to Smoking-Associated Aldehyde Exposure
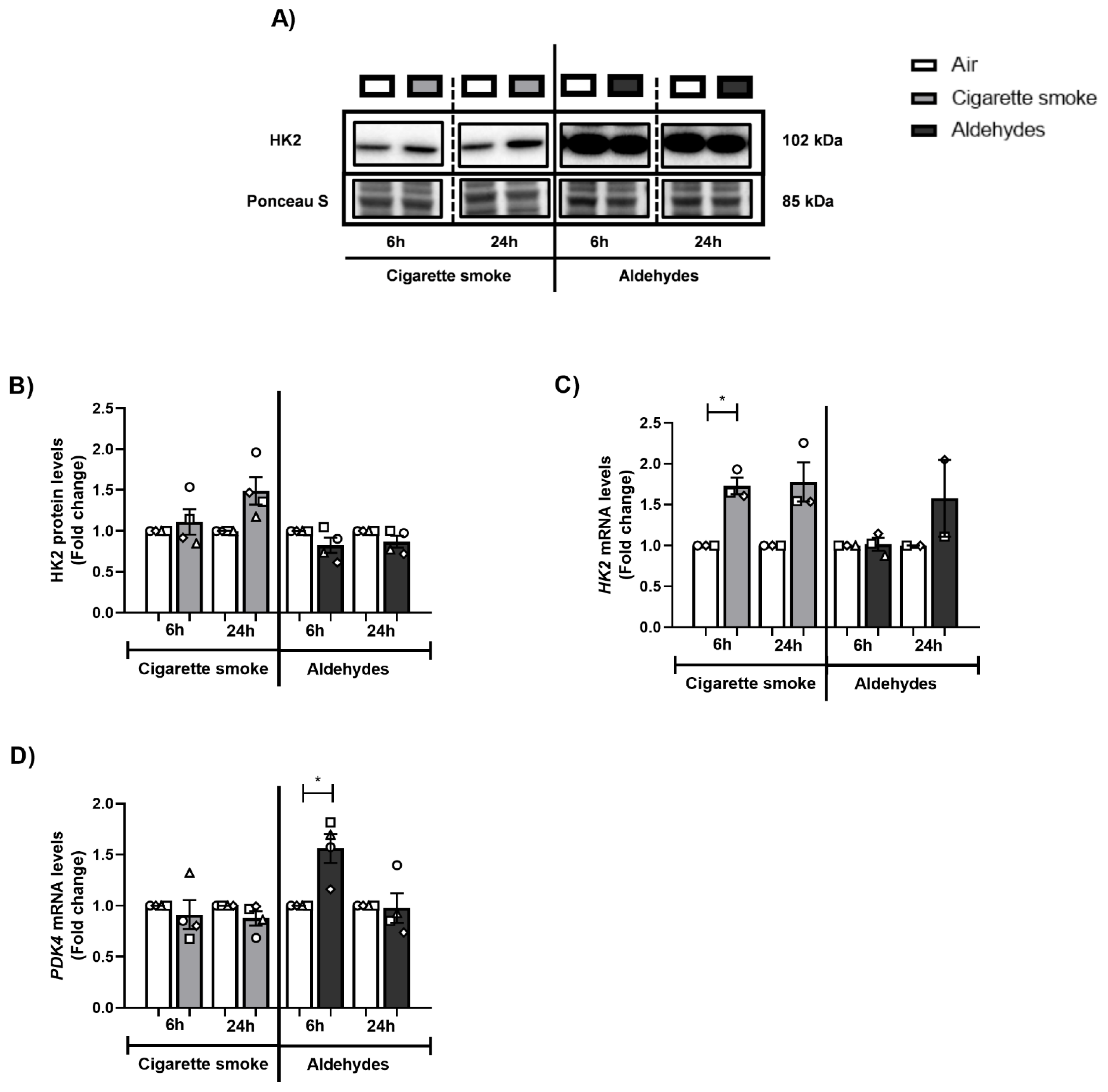
3.9. CS or Aldehyde Exposure Induced Expression of Genes Involved in Glucose Metabolism
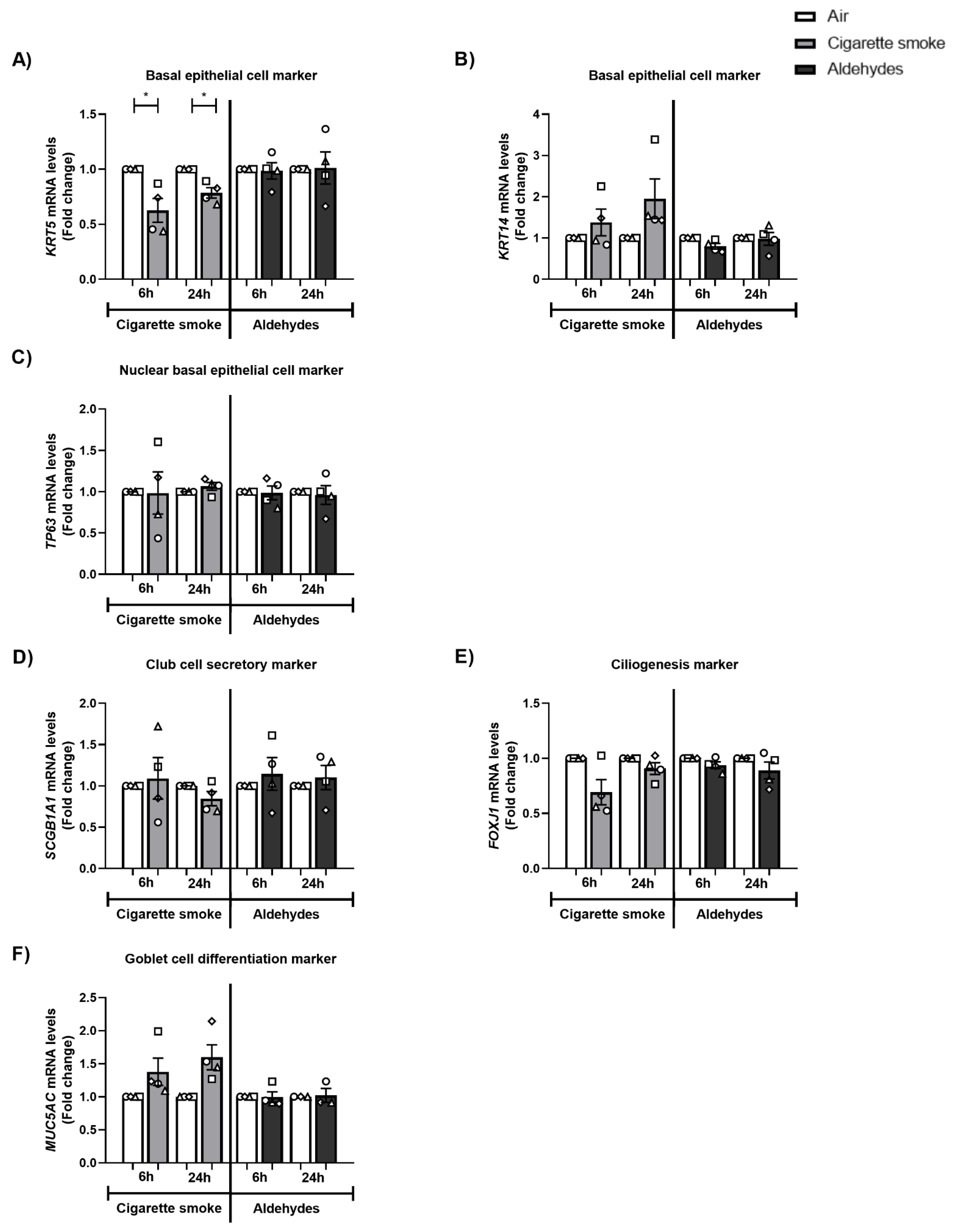
3.10. The Expression of Basal Epithelial Cell Marker KRT5 Is Decreased after CS Exposure
3.11. Impact of CS or Aldehyde Exposure on Cilia Bead Flow
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2015 Tobacco Collaborators. Smoking prevalence and attributable disease burden in 195 countries and territories, 1990–2015: A systematic analysis from the Global Burden of Disease Study 2015. Lancet 2017, 389, 1885–1906. [Google Scholar] [CrossRef]
- Celli, B.R.; Wedzicha, J.A. Update on Clinical Aspects of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Rodgman, A.; Perfetti, T.A. The Chemical Components of Tobacco and Tobacco Smoke; CRC Press: Boca Raton, FL, USA, 2013. [Google Scholar]
- Burns, D.M.; Dybing, E.; Gray, N.; Hecht, S.; Anderson, C.; Sanner, T.; O’Connor, R.; Djordjevic, M.; Dresler, C.; Hainaut, P.; et al. Mandated lowering of toxicants in cigarette smoke: A description of the World Health Organization TobReg proposal. Tob. Control 2008, 17, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Cheah, N.P. Volatile Aldehydes in Tobacco Smoke: Source Fate and Risk. Ph.D. Thesis, Maastricht University, Maastricht, The Netherlands, 2016. [Google Scholar]
- Corley, R.A.; Kabilan, S.; Kuprat, A.P.; Carson, J.P.; Jacob, R.E.; Minard, K.R.; Teeguarden, J.G.; Timchalk, C.; Pipavath, S.; Glenny, R.; et al. Comparative Risks of Aldehyde Constituents in Cigarette Smoke Using Transient Computational Fluid Dynamics/Physiologically Based Pharmacokinetic Models of the Rat and Human Respiratory Tracts. Toxicol. Sci. 2015, 146, 65–88. [Google Scholar] [CrossRef]
- Bos, P.M.J.; Soeteman-Hernández, L.G.; Talhout, R. Risk assessment of components in tobacco smoke and e-cigarette aerosols: A pragmatic choice of dose metrics. Inhal. Toxicol. 2021, 33, 81–95. [Google Scholar] [CrossRef]
- Yeager, R.P.; Kushman, M.; Chemerynski, S.; Weil, R.; Fu, X.; White, M.; Callahan-Lyon, P.; Rosenfeldt, H. Proposed Mode of Action for Acrolein Respiratory Toxicity Associated with Inhaled Tobacco Smoke. Toxicol. Sci. 2016, 151, 347–364. [Google Scholar] [CrossRef]
- Bernardini, L.; Barbosa, E.; Charão, M.F.; Brucker, N. Formaldehyde toxicity reports from in vitro and in vivo studies: A review and updated data. Drug Chem. Toxicol. 2020, 45, 1–13. [Google Scholar] [CrossRef]
- National Research Council (US) Committee. Emergency and Continuous Exposure Guidance Levels for Selected Submarine Contaminants; National Academies Press (US): Washington, DC, USA, 2009; Volume 3. [Google Scholar]
- Pauwels, C.; Klerx, W.N.M.; Pennings, J.L.A.; Boots, A.W.; van Schooten, F.J.; Opperhuizen, A.; Talhout, R. Cigarette Filter Ventilation and Smoking Protocol Influence Aldehyde Smoke Yields. Chem. Res. Toxicol. 2018, 31, 462–471. [Google Scholar] [CrossRef]
- World Health Organization Framework Convention on Tobacco Control Secretariat. Partial Guidelines for Implementation of Articles 9 and 10—Regulation of the Contents of Tobacco Products and Regulation of Tobacco Product Disclosures; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- World Health Organization; World Health Organization Tobacco Free Initiative. The Scientific Basis of Tobacco Product Regulation: Second Report of a WHO Study Group; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Hiemstra, P.S.; Grootaers, G.; van der Does, A.M.; Krul, C.A.M.; Kooter, I.M. Human lung epithelial cell cultures for analysis of inhaled toxicants: Lessons learned and future directions. Toxicol. Vitro 2018, 47, 137–146. [Google Scholar] [CrossRef]
- Crapo, J.D.; Barry, B.E.; Gehr, P.; Bachofen, M.; Weibel, E.R. Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 1982, 126, 332–337. [Google Scholar]
- Hiemstra, P.S.; McCray, P.B.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur. Respir. J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Choi, A.M. Mitochondria in lung disease. J. Clin. Investig. 2016, 126, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Aghapour, M.; Remels, A.H.V.; Pouwels, S.D.; Bruder, D.; Hiemstra, P.S.; Cloonan, S.M.; Heijink, I.H. Mitochondria: At the crossroads of regulating lung epithelial cell function in chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L149–L164. [Google Scholar] [CrossRef] [PubMed]
- MacNee, W. Pathogenesis of Chronic Obstructive Pulmonary Disease. Clin. Chest Med. 2007, 28, 479–513. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Jonker, M.R.; Brandenburg, S.M.; de Bruin, H.G.; Ten Hacken, N.H.T.; van Oosterhout, A.J.M.; Heijink, I.H. Mitochondrial dysfunction increases pro-inflammatory cytokine production and impairs repair and corticosteroid responsiveness in lung epithelium. Sci. Rep. 2019, 9, 15047. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Bouhamida, E.; Leo, S.; Pinton, P.; Rimessi, A. Mitochondrial Oxidative Stress and “Mito-Inflammation”: Actors in the Diseases. Biomedicines 2021, 9, 216. [Google Scholar] [CrossRef]
- Fritsch, L.E.; Moore, M.E.; Sarraf, S.A.; Pickrell, A.M. Ubiquitin and Receptor-Dependent Mitophagy Pathways and Their Implication in Neurodegeneration. J. Mol. Biol. 2020, 432, 2510–2524. [Google Scholar] [CrossRef]
- Lin, J.; Handschin, C.; Spiegelman, B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005, 1, 361–370. [Google Scholar] [CrossRef]
- Scarpulla, R.C. Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim. Biophys. Acta 2011, 1813, 1269–1278. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef]
- Hara, H.; Kuwano, K.; Araya, J. Mitochondrial Quality Control in COPD and IPF. Cells 2018, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Ryter, S.W.; Rosas, I.O.; Owen, C.A.; Martinez, F.J.; Choi, M.E.; Lee, C.G.; Elias, J.A.; Choi, A.M.K. Mitochondrial Dysfunction as a Pathogenic Mediator of Chronic Obstructive Pulmonary Disease and Idiopathic Pulmonary Fibrosis. Ann. Am. Thorac. Soc. 2018, 15, S266–S272. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.R.; Yin, F.; Cadenas, E. Short-term cigarette smoke exposure leads to metabolic alterations in lung alveolar cells. Am. J. Respir. Cell Mol. Biol. 2014, 51, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Cloonan, S.M.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabon, M.A.; Konrad, C.; Polverino, F.; Siempos, I.I.; Perez, E.; et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat. Med. 2016, 22, 163–174. [Google Scholar] [CrossRef]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef]
- Malinska, D.; Szymanski, J.; Patalas-Krawczyk, P.; Michalska, B.; Wojtala, A.; Prill, M.; Partyka, M.; Drabik, K.; Walczak, J.; Sewer, A.; et al. Assessment of mitochondrial function following short- and long-term exposure of human bronchial epithelial cells to total particulate matter from a candidate modified-risk tobacco product and reference cigarettes. Food Chem. Toxicol. 2018, 115, 1–12. [Google Scholar] [CrossRef]
- Mizumura, K.; Cloonan, S.M.; Nakahira, K.; Bhashyam, A.R.; Cervo, M.; Kitada, T.; Glass, K.; Owen, C.A.; Mahmood, A.; Washko, G.R.; et al. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Investig. 2014, 124, 3987–4003. [Google Scholar] [CrossRef]
- Sundar, I.K.; Maremanda, K.P.; Rahman, I. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol. Lett. 2019, 317, 92–101. [Google Scholar] [CrossRef]
- Valdivieso, Á.G.; Dugour, A.V.; Sotomayor, V.; Clauzure, M.; Figueroa, J.M.; Santa-Coloma, T.A. N-acetyl cysteine reverts the proinflammatory state induced by cigarette smoke extract in lung Calu-3 cells. Redox Biol. 2018, 16, 294–302. [Google Scholar] [CrossRef]
- Van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Luan, G.; Xu, Y.; Shen, S.; Qian, S.; Zhu, Z.; Zhang, X.; Yin, S.; Ye, J. Cigarette smoke extract increases mitochondrial membrane permeability through activation of adenine nucleotide translocator (ANT) in lung epithelial cells. Biochem. Biophys. Res. Commun. 2020, 525, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, T.; Sundar, I.K.; Lerner, C.A.; Gerloff, J.; Tormos, A.M.; Yao, H.; Rahman, I. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease. FASEB J. 2015, 29, 2912–2929. [Google Scholar] [CrossRef]
- Chen, Z.H.; Kim, H.P.; Sciurba, F.C.; Lee, S.J.; Feghali-Bostwick, C.; Stolz, D.B.; Dhir, R.; Landreneau, R.J.; Schuchert, M.J.; Yousem, S.A.; et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS ONE 2008, 3, e3316. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Araya, J.; Kurita, Y.; Kobayashi, K.; Takasaka, N.; Yoshida, M.; Hara, H.; Minagawa, S.; Wakui, H.; Fujii, S.; et al. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 2015, 11, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Kyung, S.Y.; Kim, Y.J.; Son, E.S.; Jeong, S.H.; Park, J.W. The Phosphodiesterase 4 Inhibitor Roflumilast Protects against Cigarette Smoke Extract-Induced Mitophagy-Dependent Cell Death in Epithelial Cells. Tuberc. Respir. Dis. 2018, 81, 138–147. [Google Scholar] [CrossRef]
- Mizumura, K.; Justice, M.J.; Schweitzer, K.S.; Krishnan, S.; Bronova, I.; Berdyshev, E.V.; Hubbard, W.C.; Pewzner-Jung, Y.; Futerman, A.H.; Choi, A.M.K.; et al. Sphingolipid regulation of lung epithelial cell mitophagy and necroptosis during cigarette smoke exposure. FASEB J. 2018, 32, 1880–1890. [Google Scholar] [CrossRef]
- Park, E.J.; Park, Y.J.; Lee, S.J.; Lee, K.; Yoon, C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol. Lett. 2019, 303, 55–66. [Google Scholar] [CrossRef]
- Son, E.S.; Kim, S.H.; Ryter, S.W.; Yeo, E.J.; Kyung, S.Y.; Kim, Y.J.; Jeong, S.H.; Lee, C.S.; Park, J.W. Quercetogetin protects against cigarette smoke extract-induced apoptosis in epithelial cells by inhibiting mitophagy. Toxicol. Vitro 2018, 48, 170–178. [Google Scholar] [CrossRef]
- Song, C.; Luo, B.; Gong, L. Resveratrol reduces the apoptosis induced by cigarette smoke extract by upregulating MFN2. PLoS ONE 2017, 12, e0175009. [Google Scholar] [CrossRef]
- Zhang, M.; Shi, R.; Zhang, Y.; Shan, H.; Zhang, Q.; Yang, X.; Li, Y.; Zhang, J. Nix/BNIP3L-dependent mitophagy accounts for airway epithelial cell injury induced by cigarette smoke. J. Cell Physiol. 2019, 234, 14210–14220. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Song, X.; Wei, L.; Liu, Q.; Li, C.; Wang, J. Role of mitophagy in cigarette smoke-induced lung epithelial cell injury in vitro. Curr. Mol. Med. 2022; ahead of print. [Google Scholar] [CrossRef]
- Morita, K.; Masuda, N.; Oniki, K.; Saruwatari, J.; Kajiwara, A.; Otake, K.; Ogata, Y.; Nakagawa, K. Association between the aldehyde dehydrogenase 2*2 allele and smoking-related chronic airway obstruction in a Japanese general population: A pilot study. Toxicol. Lett. 2015, 236, 117–122. [Google Scholar] [CrossRef]
- Costa, D.L.; Kutzman, R.S.; Lehmann, J.R.; Drew, R.T. Altered lung function and structure in the rat after subchronic exposure to acrolein. Am. Rev. Respir. Dis. 1986, 133, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Tulen, C.B.M.; Wang, Y.; Beentjes, D.; Jessen, P.J.J.; Ninaber, D.K.; Reynaert, N.L.; van Schooten, F.J.; Opperhuizen, A.; Hiemstra, P.S.; Remels, A.H.V. Dysregulated mitochondrial metabolism upon cigarette smoke exposure in various human bronchial epithelial cell models. Dis. Models Mech. 2022, 15, dmm049247. [Google Scholar] [CrossRef] [PubMed]
- Van der Does, A.M.; Mahbub, R.M.; Ninaber, D.K.; Rathnayake, S.N.H.; Timens, W.; van den Berge, M.; Aliee, H.; Theis, F.J.; Nawijn, M.C.; Hiemstra, P.S.; et al. Early transcriptional responses of bronchial epithelial cells to whole cigarette smoke mirror those of in-vivo exposed human bronchial mucosa. Respir. Res. 2022, 23, 227. [Google Scholar] [CrossRef]
- Van Wetering, S.; van der Linden, A.C.; van Sterkenburg, M.A.; de Boer, W.I.; Kuijpers, A.L.; Schalkwijk, J.; Hiemstra, P.S. Regulation of SLPI and elafin release from bronchial epithelial cells by neutrophil defensins. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L51–L58. [Google Scholar] [CrossRef]
- Van Wetering, S.; Zuyderduyn, S.; Ninaber, D.K.; van Sterkenburg, M.A.J.A.; Rabe, K.F.; Hiemstra, P.S. Epithelial differentiation is a determinant in the production of eotaxin-2 and -3 by bronchial epithelial cells in response to IL-4 and IL-13. Mol. Immunol. 2007, 44, 803–811. [Google Scholar] [CrossRef]
- World Health Organization; TobLabNet. SOP 1—Standard Operating Procedure for Intense Smoking of Cigarettes; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- World Health Organization; TobLabNet. SOP 8—Standard Operating Procedure for Determination of Aldehydes in Mainstream Cigarette Smoke under ISO and Intense Smoking Conditions; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Rasband, W.S. ImageJ, 1997–2018; National Institutes of Health: Bethesda, MD, USA, 2019. [Google Scholar]
- Meijering, E.; Dzyubachyk, O.; Smal, I. Methods for cell and particle tracking. Methods Enzymol. 2012, 504, 183–200. [Google Scholar] [CrossRef]
- Mio, T.; Romberger, D.J.; Thompson, A.B.; Robbins, R.A.; Heires, A.; Rennard, S.I. Cigarette smoke induces interleukin-8 release from human bronchial epithelial cells. Am. J. Respir. Crit. Care Med. 1997, 155, 1770–1776. [Google Scholar] [CrossRef]
- Moretto, N.; Facchinetti, F.; Southworth, T.; Civelli, M.; Singh, D.; Patacchini, R. alpha, beta-Unsaturated aldehydes contained in cigarette smoke elicit IL-8 release in pulmonary cells through mitogen-activated protein kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L839–L848. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, A.M.; Upadhyay, S.; Johanson, G.; Ernstgard, L.; Palmberg, L. Inflammatory effects of acrolein, crotonaldehyde and hexanal vapors on human primary bronchial epithelial cells cultured at air-liquid interface. Toxicol. Vitro 2018, 46, 219–228. [Google Scholar] [CrossRef]
- Nyunoya, T.; Mebratu, Y.; Contreras, A.; Delgado, M.; Chand, H.S.; Tesfaigzi, Y. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am. J. Respir. Cell Mol. Biol. 2014, 50, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Janocha, A.J.; Leahy, R.A.; Klatte, R.; Dudzinski, D.; Mavrakis, L.A.; Comhair, S.A.; Lauer, M.E.; Cotton, C.U.; Erzurum, S.C. A novel method for pulmonary research: Assessment of bioenergetic function at the air-liquid interface. Redox Biol. 2014, 2, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Mavin, E.; Verdon, B.; Carrie, S.; Saint-Criq, V.; Powell, J.; Kuttruff, C.A.; Ward, C.; Garnett, J.P.; Miwa, S. Real-time measurement of cellular bioenergetics in fully differentiated human nasal epithelial cells grown at air-liquid-interface. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L1158–L1164. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Cloonan, S.; Choi, M.E.; Hashimoto, S.; Nakahira, K.; Ryter, S.W.; Choi, A.M. Autophagy: Friend or Foe in Lung Disease? Ann. Am. Thorac. Soc. 2016, 13 (Suppl. S1), S40–S47. [Google Scholar] [CrossRef]
- Jiang, S.; Sun, J.; Mohammadtursun, N.; Hu, Z.; Li, Q.; Zhao, Z.; Zhang, H.; Dong, J. Dual role of autophagy/mitophagy in chronic obstructive pulmonary disease. Pulm. Pharmacol. Ther. 2019, 56, 116–125. [Google Scholar] [CrossRef]
- Monick, M.M.; Powers, L.S.; Walters, K.; Lovan, N.; Zhang, M.; Gerke, A.; Hansdottir, S.; Hunninghake, G.W. Identification of an autophagy defect in smokers’ alveolar macrophages. J. Immunol. 2010, 185, 5425–5435. [Google Scholar] [CrossRef]
- Fujii, S.; Hara, H.; Araya, J.; Takasaka, N.; Kojima, J.; Ito, S.; Minagawa, S.; Yumino, Y.; Ishikawa, T.; Numata, T.; et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology 2012, 1, 630–641. [Google Scholar] [CrossRef]
- Barnes, P.J.; Baker, J.; Donnelly, L.E. Autophagy in asthma and chronic obstructive pulmonary disease. Clin. Sci. 2022, 136, 733–746. [Google Scholar] [CrossRef]
- Leonard, A.P.; Cameron, R.B.; Speiser, J.L.; Wolf, B.J.; Peterson, Y.K.; Schnellmann, R.G.; Beeson, C.C.; Rohrer, B. Quantitative analysis of mitochondrial morphology and membrane potential in living cells using high-content imaging, machine learning, and morphological binning. Biochim. Biophys. Acta 2015, 1853, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.T.; Lin, J.H.; Yang, C.H.; Haung, C.H.; Weng, C.W.; Maan-Yuh Lin, A.; Lo, Y.L.; Chen, W.S.; Tang, M.S. Acrolein induces mtDNA damages, mitochondrial fission and mitophagy in human lung cells. Oncotarget 2017, 8, 70406–70421. [Google Scholar] [CrossRef] [PubMed]
- Tulen, C.B.M.; Snow, S.J.; Leermakers, P.A.; Kodavanti, U.P.; van Schooten, F.J.; Opperhuizen, A.; Remels, A.H.V. Acrolein inhalation acutely affects the regulation of mitochondrial metabolism in rat lung. Toxicology 2022, 469, 153129. [Google Scholar] [CrossRef]
- Liu, Q.P.; Zhou, D.X.; Lv, M.Q.; Ge, P.; Li, Y.X.; Wang, S.J. Formaldehyde inhalation triggers autophagy in rat lung tissues. Toxicol. Ind. Health 2018, 34, 748233718796347. [Google Scholar] [CrossRef] [PubMed]
- Giordano, L.; Gregory, A.D.; Verdaguer, M.P.; Ware, S.A.; Harvey, H.; DeVallance, E.; Brzoska, T.; Sundd, P.; Zhang, Y.; Sciurba, F.C.; et al. Extracellular release of mitochondrial DNA: Triggered by cigaretee smoke and detected in COPD. Cells 2022, 11, 369. [Google Scholar] [CrossRef]
- Vanella, L.; Li Volti, G.; Distefano, A.; Raffaele, M.; Zingales, V.; Avola, R.; Tibullo, D.; Barbagallo, I. A new antioxidant formulation reduces the apoptotic and damaging effect of cigarette smoke extract on human bronchial epithelial cells. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 5478–5484. [Google Scholar] [CrossRef]
- Luo, C.; Li, Y.; Yang, L.; Feng, Z.; Li, Y.; Long, J.; Liu, J. A cigarette component acrolein induces accelerated senescence in human diploid fibroblast IMR-90 cells. Biogerontology 2013, 14, 503–511. [Google Scholar] [CrossRef]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef]
- Ballweg, K.; Mutze, K.; Königshoff, M.; Eickelberg, O.; Meiners, S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L895–L907. [Google Scholar] [CrossRef]
- Farfán Labonne, B.E.; Gutiérrez, M.; Gómez-Quiroz, L.E.; Konigsberg Fainstein, M.; Bucio, L.; Souza, V.; Flores, O.; Ortíz, V.; Hernández, E.; Kershenobich, D.; et al. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol. Toxicol. 2009, 25, 599–609. [Google Scholar] [CrossRef]
- Agarwal, A.R.; Yin, F.; Cadenas, E. Metabolic shift in lung alveolar cell mitochondria following acrolein exposure. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L764–L773. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Luo, C.; Long, J.; Wei, D.; Liu, J. Acrolein is a mitochondrial toxin: Effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion 2006, 6, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Van Buskirk, J.J.; Frisell, W.R. Inhibition by formaldehyde of energy transfer and related processes in rat liver mitochondria. II. Effects on energy-linked reactions in intact mitochondria and phosphorylating particles. Arch. Biochem. Biophys. 1969, 132, 130–138. [Google Scholar] [CrossRef]
- Zerin, T.; Kim, J.S.; Gil, H.W.; Song, H.Y.; Hong, S.Y. Effects of formaldehyde on mitochondrial dysfunction and apoptosis in SK-N-SH neuroblastoma cells. Cell Biol. Toxicol. 2015, 31, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Schamberger, A.C.; Staab-Weijnitz, C.A.; Mise-Racek, N.; Eickelberg, O. Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci. Rep. 2015, 5, 8163. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, D.; Majeed, S.; Guedj, E.; Dulize, R.; Baumer, K.; Iskandar, A.; Boue, S.; Martin, F.; Kostadinova, R.; Mathis, C.; et al. Impact assessment of repeated exposure of organotypic 3D bronchial and nasal tissue culture models to whole cigarette smoke. J. Vis. Exp. 2015, 96, e52325. [Google Scholar] [CrossRef] [PubMed]
- Aufderheide, M.; Scheffler, S.; Ito, S.; Ishikawa, S.; Emura, M. Ciliatoxicity in human primary bronchiolar epithelial cells after repeated exposure at the air–liquid interface with native mainstream smoke of K3R4F cigarettes with and without charcoal filter. Exp. Toxicol. Pathol. 2015, 67, 407–411. [Google Scholar] [CrossRef]
- Tatsuta, M.; Kan-o, K.; Ishii, Y.; Yamamoto, N.; Ogawa, T.; Fukuyama, S.; Ogawa, A.; Fujita, A.; Nakanishi, Y.; Matsumoto, K. Effects of cigarette smoke on barrier function and tight junction proteins in the bronchial epithelium: Protective role of cathelicidin LL-37. Respir. Res. 2019, 20, 251. [Google Scholar] [CrossRef]
- Yu, Q.; Chen, X.; Fang, X.; Chen, Q.; Hu, C. Caveolin-1 aggravates cigarette smoke extract-induced MUC5AC secretion in human airway epithelial cells. Int. J. Mol. Med. 2015, 35, 1435–1442. [Google Scholar] [CrossRef]
- Brekman, A.; Walters, M.S.; Tilley, A.E.; Crystal, R.G. FOXJ1 prevents cilia growth inhibition by cigarette smoke in human airway epithelium in vitro. Am. J. Respir. Cell Mol. Biol. 2014, 51, 688–700. [Google Scholar] [CrossRef]
- Gindele, J.A.; Kiechle, T.; Benediktus, K.; Birk, G.; Brendel, M.; Heinemann, F.; Wohnhaas, C.T.; LeBlanc, M.; Zhang, H.; Strulovici-Barel, Y.; et al. Intermittent exposure to whole cigarette smoke alters the differentiation of primary small airway epithelial cells in the air-liquid interface culture. Sci. Rep. 2020, 10, 6257. [Google Scholar] [CrossRef] [PubMed]
- Rigden, H.M.; Alias, A.; Havelock, T.; O’Donnell, R.; Djukanovic, R.; Davies, D.E.; Wilson, S.J. Squamous Metaplasia Is Increased in the Bronchial Epithelium of Smokers with Chronic Obstructive Pulmonary Disease. PLoS ONE 2016, 11, e0156009. [Google Scholar] [CrossRef] [PubMed]
- Crystal, R.G. Airway basal cells. The “smoking gun” of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2014, 190, 1355–1362. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K. Comparison of the structural and inflammatory features of COPD and asthma Giles F. Filley Lecture. Chest 2000, 117, 251S–260S. [Google Scholar] [CrossRef]
- Saetta, M.; Turato, G.; Baraldo, S.; Zanin, A.; Braccioni, F.; Mapp, C.E.; Maestrelli, P.; Cavallesco, G.; Papi, A.; Fabbri, L.M. Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers with both symptoms of chronic bronchitis and chronic airflow limitation. Am. J. Respir. Crit. Care Med. 2000, 161, 1016–1021. [Google Scholar] [CrossRef]
- Xiong, R.; Wu, Q.; Muskhelishvili, L.; Davis, K.; Shemansky, J.M.; Bryant, M.; Rosenfeldt, H.; Healy, S.M.; Cao, X. Evaluating Mode of Action of Acrolein Toxicity in an In Vitro Human Airway Tissue Model. Toxicol. Sci. 2018, 166, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, J.; Chen, H.; Wang, A.; Liu, Y.; Hou, H.; Hu, Q. Combined cytotoxicity of co-exposure to aldehyde mixtures on human bronchial epithelial BEAS-2B cells. Environ. Pollut. 2019, 250, 650–661. [Google Scholar] [CrossRef]
- LoPachin, R.M.; Gavin, T. Molecular mechanisms of aldehyde toxicity: A chemical perspective. Chem. Res. Toxicol. 2014, 27, 1081–1091. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, H.; Wang, A.; Liu, Y.; Hou, H.; Hu, Q. Combined effects of co-exposure to formaldehyde and acrolein mixtures on cytotoxicity and genotoxicity in vitro. Environ. Sci. Pollut. Res. Int. 2018, 25, 25306–25314. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, J.; Cheng, W.; Chen, H.; Wang, A.; Liu, Y.; Hou, H.; Hu, Q. Combined cell death of co-exposure to aldehyde mixtures on human bronchial epithelial BEAS-2B cells: Molecular insights into the joint action. Chemosphere 2020, 244, 125482. [Google Scholar] [CrossRef]
- Iskandar, A.R.; Xiang, Y.; Frentzel, S.; Talikka, M.; Leroy, P.; Kuehn, D.; Guedj, E.; Martin, F.; Mathis, C.; Ivanov, N.V.; et al. Impact Assessment of Cigarette Smoke Exposure on Organotypic Bronchial Epithelial Tissue Cultures: A Comparison of Mono-Culture and Coculture Model Containing Fibroblasts. Toxicol. Sci. 2015, 147, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Van Riet, S.; van Schadewijk, A.; de Vos, S.; Vandeghinste, N.; Rottier, R.J.; Stolk, J.; Hiemstra, P.S.; Khedoe, P. Modulation of Airway Epithelial Innate Immunity and Wound Repair by M(GM-CSF) and M(M-CSF) Macrophages. J. Innate Immun. 2020, 12, 410–421. [Google Scholar] [CrossRef]
- Ishikawa, S.; Matsumura, K.; Kitamura, N.; Takanami, Y.; Ito, S. Multi-omics analysis: Repeated exposure of a 3D bronchial tissue culture to whole-cigarette smoke. Toxicol. Vitro 2019, 54, 251–262. [Google Scholar] [CrossRef]
- Chen, Z.H.; Lam, H.C.; Jin, Y.; Kim, H.P.; Cao, J.; Lee, S.J.; Ifedigbo, E.; Parameswaran, H.; Ryter, S.W.; Choi, A.M. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc. Natl. Acad. Sci. USA 2010, 107, 18880–18885. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cheng, Y.; Tang, Z.; Mei, X.; Cao, X.; Liu, J. Toxicity mechanism of acrolein on DNA damage and apoptosis in BEAS-2B cells: Insights from cell biology and molecular docking analyses. Toxicology 2022, 466, 153083. [Google Scholar] [CrossRef]
- Peterson, L.A.; Oram, M.K.; Flavin, M.; Seabloom, D.; Smith, W.E.; O’Sullivan, M.G.; Vevang, K.R.; Upadhyaya, P.; Stornetta, A.; Floeder, A.C.; et al. Coexposure to Inhaled Aldehydes or Carbon Dioxide Enhances the Carcinogenic Properties of the Tobacco-Specific Nitrosamine 4-Methylnitrosamino-1-(3-pyridyl)-1-butanone in the A/J Mouse Lung. Chem. Res. Toxicol. 2021, 34, 723–732. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment | Primary Bronchial Epithelial Cell Donors |
|---|---|
| N | 4 |
| Male/female | 3/1 |
| Age (years) | 69.75 ± 2.02 |
| Body mass index | 28.00 ± 4.38 |
| Pack years (years) | 35.00 ± 15.00 $ |
| FEV1 absolute (L) | 2.73 ± 0.19 |
| Tiffeneau Index | 76.06 ± 2.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tulen, C.B.M.; Duistermaat, E.; Cremers, J.W.J.M.; Klerx, W.N.M.; Fokkens, P.H.B.; Weibolt, N.; Kloosterboer, N.; Dentener, M.A.; Gremmer, E.R.; Jessen, P.J.J.; et al. Smoking-Associated Exposure of Human Primary Bronchial Epithelial Cells to Aldehydes: Impact on Molecular Mechanisms Controlling Mitochondrial Content and Function. Cells 2022, 11, 3481. https://doi.org/10.3390/cells11213481
Tulen CBM, Duistermaat E, Cremers JWJM, Klerx WNM, Fokkens PHB, Weibolt N, Kloosterboer N, Dentener MA, Gremmer ER, Jessen PJJ, et al. Smoking-Associated Exposure of Human Primary Bronchial Epithelial Cells to Aldehydes: Impact on Molecular Mechanisms Controlling Mitochondrial Content and Function. Cells. 2022; 11(21):3481. https://doi.org/10.3390/cells11213481
Chicago/Turabian StyleTulen, Christy B. M., Evert Duistermaat, Johannes W. J. M. Cremers, Walther N. M. Klerx, Paul H. B. Fokkens, Naömi Weibolt, Nico Kloosterboer, Mieke A. Dentener, Eric R. Gremmer, Phyllis J. J. Jessen, and et al. 2022. "Smoking-Associated Exposure of Human Primary Bronchial Epithelial Cells to Aldehydes: Impact on Molecular Mechanisms Controlling Mitochondrial Content and Function" Cells 11, no. 21: 3481. https://doi.org/10.3390/cells11213481
APA StyleTulen, C. B. M., Duistermaat, E., Cremers, J. W. J. M., Klerx, W. N. M., Fokkens, P. H. B., Weibolt, N., Kloosterboer, N., Dentener, M. A., Gremmer, E. R., Jessen, P. J. J., Koene, E. J. C., Maas, L., Opperhuizen, A., van Schooten, F.-J., Staal, Y. C. M., & Remels, A. H. V. (2022). Smoking-Associated Exposure of Human Primary Bronchial Epithelial Cells to Aldehydes: Impact on Molecular Mechanisms Controlling Mitochondrial Content and Function. Cells, 11(21), 3481. https://doi.org/10.3390/cells11213481