
Structure and Function of the ABCD1 Variant Database: 20 Years, 940 Pathogenic Variants, and 3400 Cases of Adrenoleukodystrophy


Abstract
:1. Introduction
2. ABCD1 Variant Database
2.1. ABCD1 Variant Analysis, 2001: Initial Results
2.2. Evolution of the Database: Experimental Data and Variant Reclassification
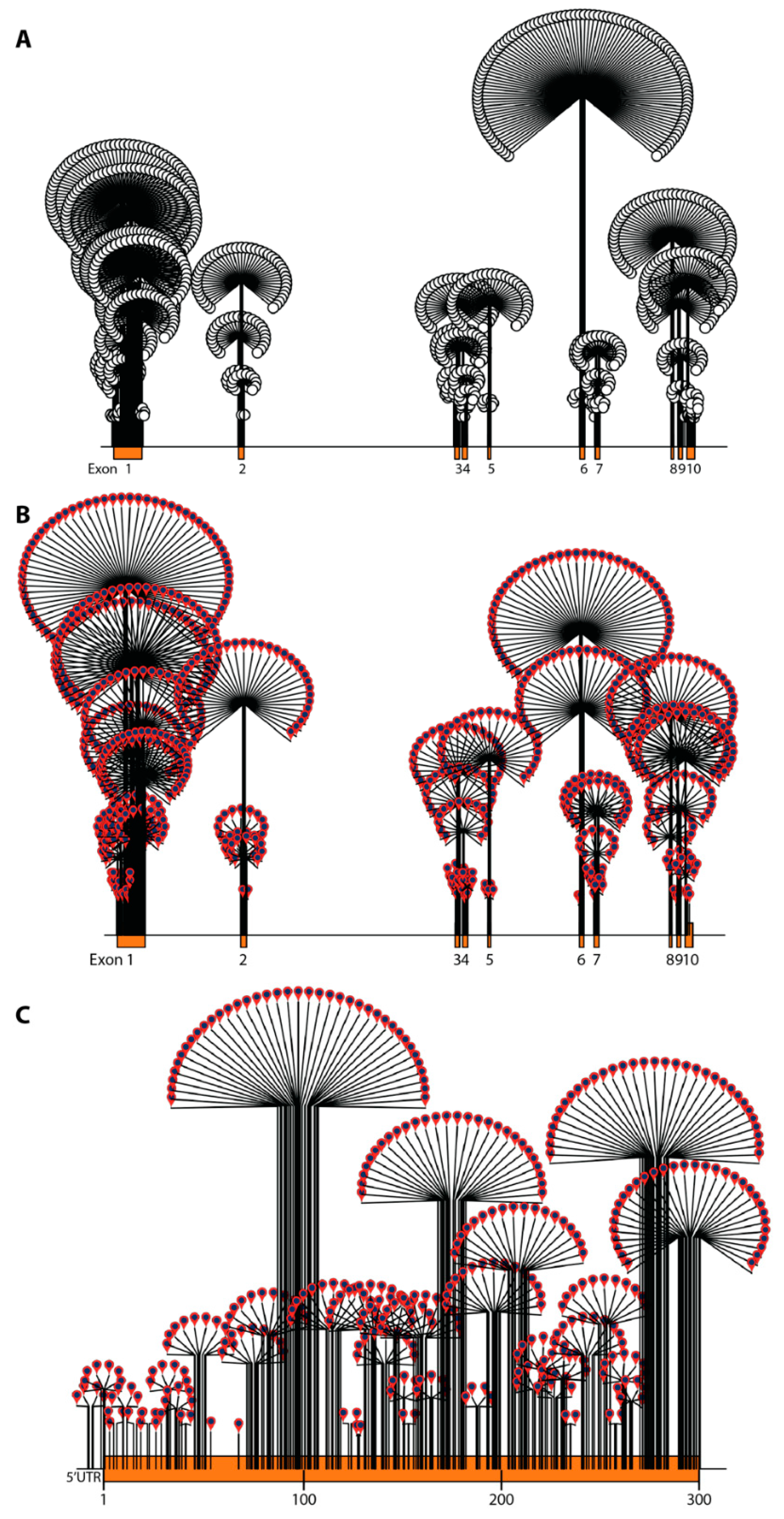
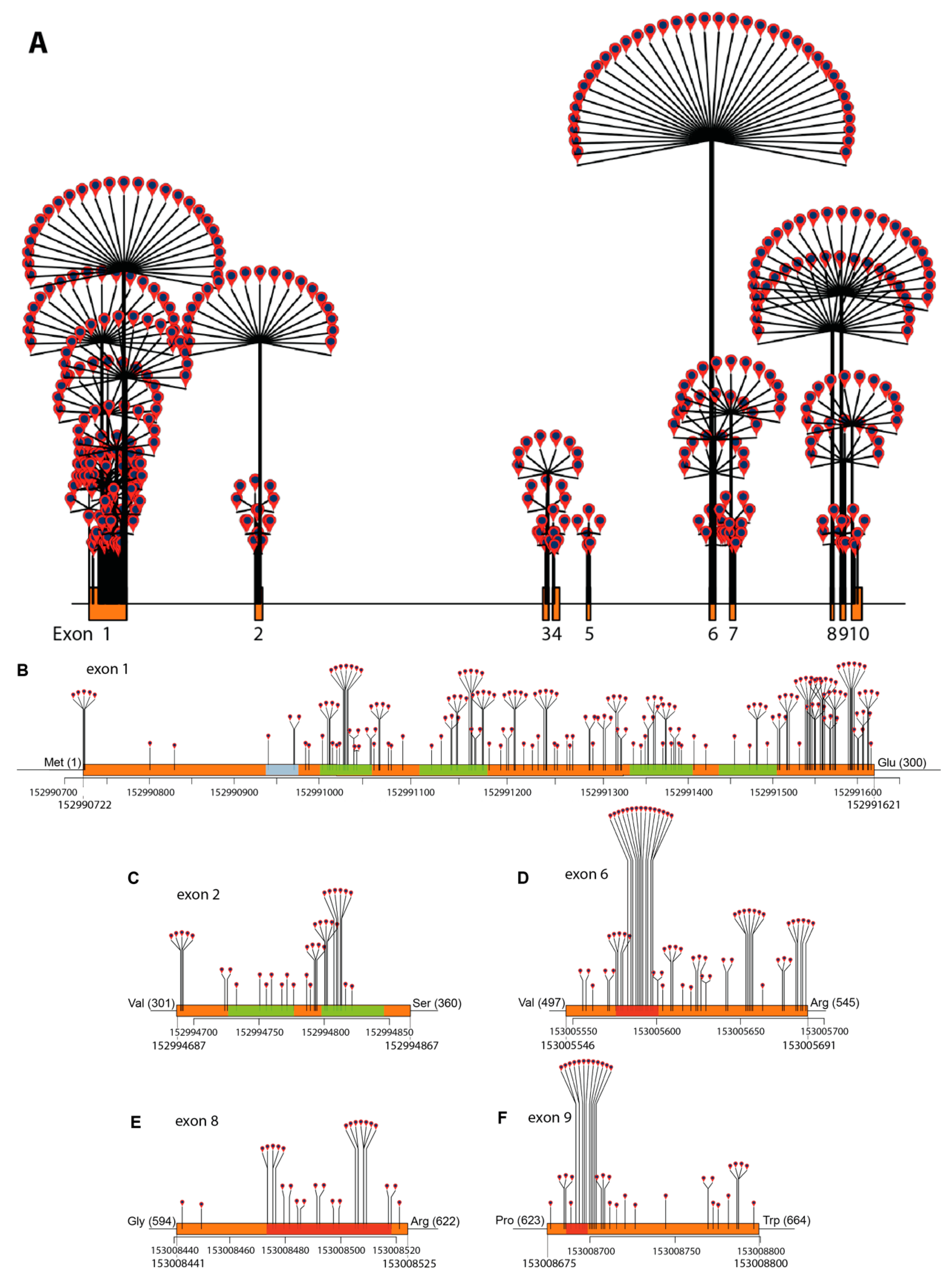
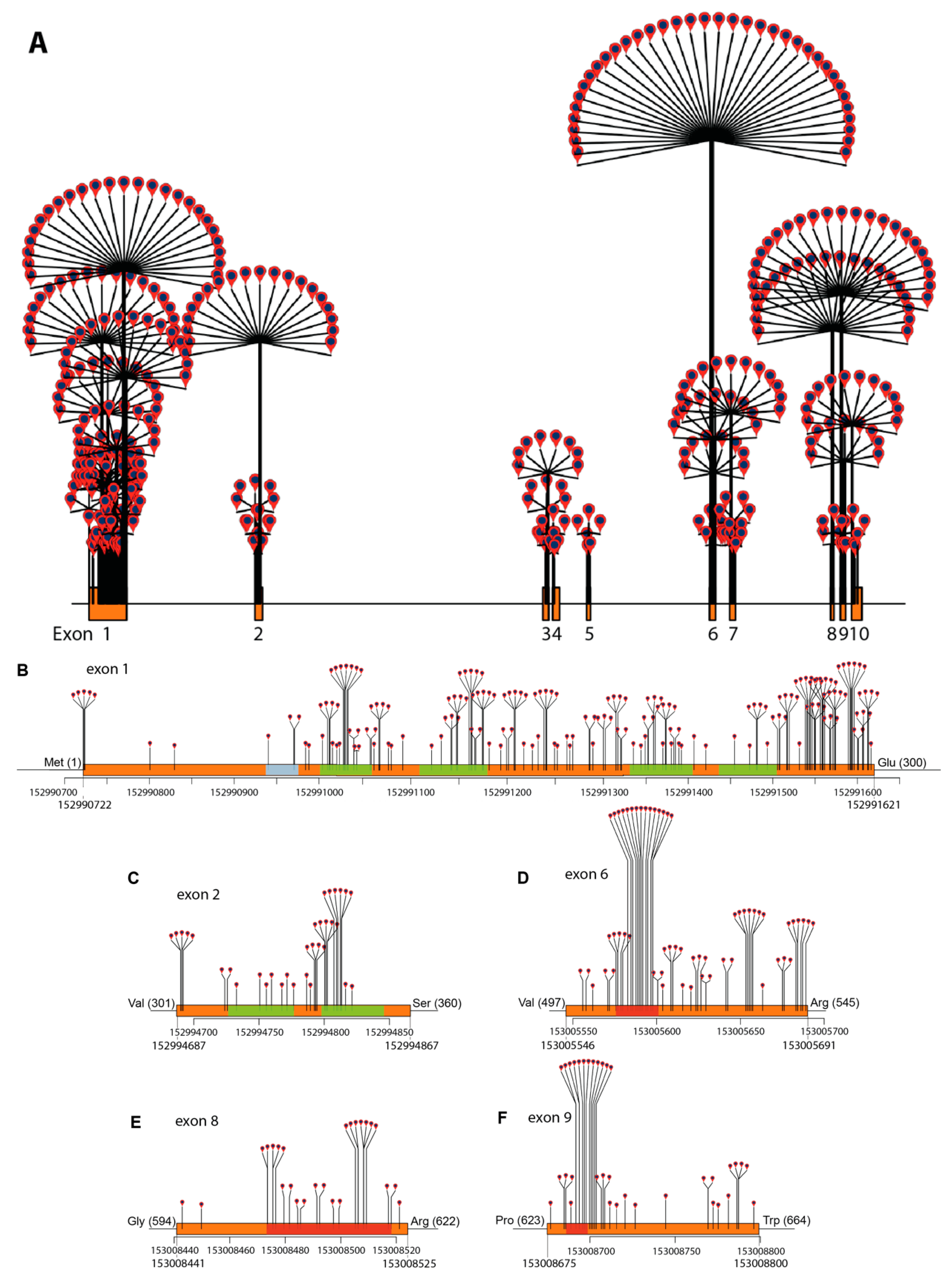
2.3. ABCD1 Variant Analysis, 2021: Materials and Methods
2.4. ABCD1 Variant Analysis, 2021: Results
3. ABCD1 Variant Interpretation: 2001 vs. 2021
4. ABCD1 Variant Interpretation in the Era of ALD Newborn Screening
4.1. Newborn Screening for ALD
4.2. Asymptomatic Diagnosis Requires a Platform to Resolve Variants of Uncertain Significance
4.3. The Clinical and Collaborative Importance of the ABCD1 Variant Database
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mosser, J.; Douar, A.M.; Sarde, C.O.; Kioschis, P.; Feil, R.; Moser, H.; Poustka, A.M.; Mandel, J.L.; Aubourg, P. Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters. Nature 1993, 361, 726–730. [Google Scholar] [CrossRef]
- Sarde, C.O.; Mosser, J.; Kioschis, P.; Kretz, C.; Vicaire, S.; Aubourg, P.; Poustka, A.; Mandel, J.L. Genomic organization of the adrenoleukodystrophy gene. Genomics 1994, 22, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Mosser, J.; Lutz, Y.; Stoeckel, M.E.; Sarde, C.O.; Kretz, C.; Douar, A.M.; Lopez, J.; Aubourg, P.; Mandel, J.L. The gene responsible for adrenoleukodystrophy encodes a peroxisomal membrane protein. Hum. Mol. Genet. 1994, 3, 265–271. [Google Scholar] [CrossRef]
- Kemp, S.; Theodoulou, F.L.; Wanders, R.J. Mammalian peroxisomal ABC transporters: From endogenous substrates to pathology and clinical significance Correspondence. Br. J. Pharmacol. 2011, 164, 1753–1766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, A.B.; Kreiter, N.; Bezman, L.; Lu, S.; Raymond, G.V.; Naidu, S.; Moser, H.W. Plasma very long chain fatty acids in 3,000 peroxisome disease patients and 29,000 controls. Ann. Neurol. 1999, 45, 100–110. [Google Scholar] [CrossRef]
- Bezman, L.; Moser, H.W. Incidence of X-linked adrenoleukodystrophy and the relative frequency of its phenotypes. Am. J. Med. Genet. 1998, 76, 415–419. [Google Scholar] [CrossRef]
- Moser, A.B.; Jones, R.O.; Hubbard, W.C.; Tortorelli, S.; Orsini, J.; Caggana, M.; Vogel, B.H.; Raymond, G.V. Newborn screening for X-linked adrenoleukodystrophy. Int. J. Neonatal Screen. 2016, 2, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Karnebeek, C.D.M.; Richmond, P.A.; van der Kloet, F.; Wasserman, W.W.; Engelen, M.; Kemp, S. The variability conundrum in neurometabolic degenerative diseases. Mol. Genet. Metab. 2020, 131, 367–369. [Google Scholar] [CrossRef]
- Kemp, S.; Huffnagel, I.C.; Linthorst, G.E.; Wanders, R.J.A.; Engelen, M. Adrenoleukodystrophy—Neuroendocrine pathogenesis and redefinition of natural history. Nat. Rev. Endocrinol. 2016, 12, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Huffnagel, I.C.; Laheji, F.K.; Aziz-Bose, R.; Tritos, N.A.; Marino, R.; Linthorst, G.E.; Kemp, S.; Engelen, M.; Eichler, F. The Natural History of Adrenal Insufficiency in X-Linked Adrenoleukodystrophy: An International Collaboration. J. Clin. Endocrinol. Metab. 2019, 104, 118–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, H.W.; Loes, D.J.; Melhem, E.R.; Raymond, G.V.; Bezman, L.; Cox, C.S.; Lu, S.E. X-Linked adrenoleukodystrophy: Overview and prognosis as a function of age and brain magnetic resonance imaging abnormality. A study involving 372 patients. Neuropediatrics 2000, 31, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Kemp, S.; de Visser, M.; van Geel, B.M.; Wanders, R.J.; Aubourg, P.; Poll-The, B. X-linked adrenoleukodystrophy (X-ALD): Clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J. Rare Dis. 2012, 7, 51. [Google Scholar] [CrossRef]
- Huffnagel, I.C.; van Ballegoij, W.J.C.; van Geel, B.M.; Vos, J.M.B.W.; Kemp, S.; Engelen, M. Progression of myelopathy in males with adrenoleukodystrophy: Towards clinical trial readiness. Brain 2019, 142, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Engelen, M.; Barbier, M.; Dijkstra, I.M.E.; Schür, R.; de Bie, R.M.A.; Verhamme, C.; Dijkgraaf, M.G.W.; Aubourg, P.A.; Wanders, R.J.A.; van Geel, B.M.; et al. X-linked adrenoleukodystrophy in women: A cross-sectional cohort study. Brain 2014, 137, 693–706. [Google Scholar] [CrossRef] [Green Version]
- Huffnagel, I.C.; Dijkgraaf, M.G.W.; Janssens, G.E.; van Weeghel, M.; van Geel, B.M.; Poll-The, B.T.; Kemp, S.; Engelen, M. Disease progression in women with X-linked adrenoleukodystrophy is slow. Orphanet J. Rare Dis. 2019, 14, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemp, S.; Pujol, A.; Waterham, H.R.; van Geel, B.M.; Boehm, C.D.; Raymond, G.V.; Cutting, G.R.; Wanders, R.J.A.; Moser, H.W. ABCD1 mutations and the X-linked adrenoleukodystrophy mutation database: Role in diagnosis and clinical correlations. Hum. Mutat. 2001, 18, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Boehm, C.D.; Cutting, G.R.; Lachtermacher, M.B.; Moser, H.W.; Chong, S.S. Accurate DNA-based diagnostic and carrier testing for X-linked adrenoleukodystrophy. Mol. Genet. Metab. 1999, 66, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.; Ligtenberg, M.J.; van Geel, B.M.; Barth, P.G.; Wolterman, R.A.; Schoute, F.; Sarde, C.O.; Mandel, J.L.; Van Oost, B.A.; Bolhuis, P.A. Identification of a two base pair deletion in five unrelated families with adrenoleukodystrophy: A possible hot spot for mutations. Biochem. Biophys. Res. Commun. 1994, 202, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Ligtenberg, M.J.; Kemp, S.; Sarde, C.O.; van Geel, B.M.; Kleijer, W.J.; Barth, P.G.; Mandel, J.L.; van Oost, B.A.; Bolhuis, P.A. Spectrum of mutations in the gene encoding the adrenoleukodystrophy protein. Am. J. Hum. Genet. 1995, 56, 44–50. [Google Scholar]
- Higgins, J.; Dalgleish, R.; den Dunnen, J.T.; Barsh, G.; Freeman, P.J.; Cooper, D.N.; Cullinan, S.; Davies, K.E.; Dorkins, H.; Gong, L.; et al. Verifying nomenclature of DNA variants in submitted manuscripts: Guidance for journals. Hum. Mutat. 2021, 42, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; De Marcos Lousa, C.; Schutte-Lensink, N.; Ofman, R.; Wanders, R.J.; Baldwin, S.A.; Baker, A.; Kemp, S.; Theodoulou, F.L. Conservation of targeting but divergence in function and quality control of peroxisomal ABC transporters: An analysis using cross-kingdom expression. Biochem. J. 2011, 436, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Gärtner, J.; Dehmel, T.; Klusmann, A.; Roerig, P. Functional characterization of the adrenoleukodystrophy protein (ALDP) and disease pathogenesis. Endocr. Res. 2002, 28, 741–748. [Google Scholar] [CrossRef]
- Roerig, P.; Mayerhofer, P.; Holzinger, A.; Gärtner, J. Characterization and functional analysis of the nucleotide binding fold in human peroxisomal ATP binding cassette transporters. FEBS Lett. 2001, 492, 66–72. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, N.; Morita, M.; Maeda, T.; Harayama, Y.; Shimozawa, N.; Suzuki, Y.; Furuya, H.; Sato, R.; Kashiwayama, Y.; Imanaka, T. Adrenoleukodystrophy: Subcellular localization and degradation of adrenoleukodystrophy protein (ALDP/ABCD1) with naturally occurring missense mutations. J. Neurochem. 2007, 101, 1632–1643. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Dvorakova, L.; Storkanova, G.; Unterrainer, G.; Hujova, J.; Kmoch, S.; Zeman, J.; Hrebicek, M.; Berger, J. Eight novel ABCD1 gene mutations and three polymorphisms in patients with X-linked adrenoleukodystrophy: The first polymorphism causing an amino acid exchange. Hum. Mutat. 2001, 18, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.; Gentleman, R.; Carey, V. rtracklayer: An R package for interfacing with genome browsers. Bioinformatics 2009, 25, 1841–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.; Zhu, L.J. trackViewer: A Bioconductor package for interactive and integrative visualization of multi-omics data. Nat. Methods 2019, 16, 453–454. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, J.; Sarde, C.O.; Vicaire, S.; Yates, J.R.; Mandel, J.L. A new human gene (DXS1357E) with ubiquitous expression, located in Xq28 adjacent to the adrenoleukodystrophy gene. Genomics 1994, 22, 469–471. [Google Scholar] [CrossRef]
- Corzo, D.; Gibson, W.; Johnson, K.; Mitchell, G.; LePage, G.; Cox, G.F.; Casey, R.; Zeiss, C.; Tyson, H.; Cutting, G.R.; et al. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: A novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am. J. Hum. Genet. 2002, 70, 1520–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Kurisu, M.; Kashiwayama, Y.; Yokota, S.; Imanaka, T. ATP-binding and -hydrolysis activities of ALDP (ABCD1) and ALDRP (ABCD2), human peroxisomal ABC proteins, overexpressed in Sf21 cells. Biol. Pharm. Bull. 2006, 29, 1836–1842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwayama, Y.; Morita, M.; Kamijo, K.; Imanaka, T. Nucleotide-induced conformational changes of PMP70, an ATP binding cassette transporter on rat liver peroxisomal membranes. Biochem. Biophys. Res. Commun. 2002, 291, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Morita, M. ABC Transporter Subfamily D: Distinct Differences in Behavior between ABCD1-3 and ABCD4 in Subcellular Localization, Function, and Human Disease. Biomed. Res. Int. 2016, 2016, 6786245. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.C.; Karpowich, N.; Millen, L.; Moody, J.E.; Rosen, J.; Thomas, P.J.; Hunt, J.F. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer. Mol. Cell 2002, 10, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Raymond, G.V.; Aubourg, P.; Paker, A.; Escolar, M.; Fischer, A.; Blanche, S.; Baruchel, A.; Dalle, J.H.; Michel, G.; Prasad, V.; et al. Survival and Functional Outcomes in Boys with Cerebral Adrenoleukodystrophy with and without Hematopoietic Stem Cell Transplantation. Biol. Blood Marrow Transpl. 2019, 25, 538–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckmann, N.B.; Miller, W.P.; Dietrich, M.S.; Orchard, P.J. Quality of life among boys with adrenoleukodystrophy following hematopoietic stem cell transplant. Child Neuropsychol. 2018, 24, 986–998. [Google Scholar] [CrossRef]
- Ashwal, S.; Michelson, D.; Plawner, L.; Dobyns, W.B. Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society Practice parameter: Evaluation of the child with microcephaly (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2009, 73, 887–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahmood, A.; Raymond, G.V.; Dubey, P.; Peters, C.; Moser, H.W. Survival analysis of haematopoietic cell transplantation for childhood cerebral X-linked adrenoleukodystrophy: A comparison study. Lancet Neurol. 2007, 6, 687–692. [Google Scholar] [CrossRef]
- Pierpont, E.I.; Nascene, D.R.; Shanley, R.; Kenney-Jung, D.L.; Ziegler, R.S.; Miller, W.P.; Gupta, A.O.; Lund, T.C.; Orchard, P.J.; Eisengart, J.B. Neurocognitive benchmarks following transplant for emerging cerebral adrenoleukodystrophy. Neurology 2020, 95, e591–e600. [Google Scholar] [CrossRef]
- Peters, C.; Charnas, L.R.; Tan, Y.; Ziegler, R.S.; Shapiro, E.G.; DeFor, T.; Grewal, S.S.; Orchard, P.J.; Abel, S.L.; Goldman, A.I.; et al. Cerebral X-linked adrenoleukodystrophy: The international hematopoietic cell transplantation experience from 1982 to 1999. Blood 2004, 104, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Moser, H.W.; Moser, A.B.; Smith, K.D.; Bergin, A.; Borel, J.; Shankroff, J.; Stine, O.C.; Merette, C.; Ott, J.; Krivit, W.; et al. Adrenoleukodystrophy: Phenotypic variability and implications for therapy. J. Inherit. Metab. Dis. 1992, 15, 645–664. [Google Scholar] [CrossRef]
- van Geel, B.M.; Bezman, L.; Loes, D.J.; Moser, H.W.; Raymond, G.V. Evolution of phenotypes in adult male patients with X-linked adrenoleukodystrophy. Ann. Neurol. 2001, 49, 186–194. [Google Scholar] [CrossRef]
- Shapiro, E.; Krivit, W.; Lockman, L.; Jambaque, I.; Peters, C.; Cowan, M.; Harris, R.; Blanche, S.; Bordigoni, P.; Loes, D.; et al. Long-term effect of bone-marrow transplantation for childhood-onset cerebral X-linked adrenoleukodystrophy. Lancet 2000, 356, 713–718. [Google Scholar] [CrossRef]
- Pierpont, E.I.; Eisengart, J.B.; Shanley, R.; Nascene, D.; Raymond, G.V.; Shapiro, E.G.; Ziegler, R.S.; Orchard, P.J.; Miller, W.P. Neurocognitive trajectory of boys who received a hematopoietic stem cell transplant at an early stage of childhood cerebral adrenoleukodystrophy. JAMA Neurol. 2017, 74, 710–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichler, F.; Duncan, C.; Musolino, P.L.; Orchard, P.J.; De Oliveira, S.; Thrasher, A.J.; Armant, M.; Dansereau, C.; Lund, T.C.; Miller, W.P.; et al. Hematopoietic stem-cell gene therapy for cerebral adrenoleukodystrophy. N. Engl. J. Med. 2017, 377, 1630–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moser, A.B.; Fatemi, A. Newborn Screening and Emerging Therapies for X-Linked Adrenoleukodystrophy. JAMA Neurol. 2018, 75, 1175–1176. [Google Scholar] [CrossRef]
- Cartier, N.; Hacein-Bey-Abina, S.; Bartholomae, C.C.; Veres, G.; Schmidt, M.; Kutschera, I.; Vidaud, M.; Abel, U.; Dal-Cortivo, L.; Caccavelli, L.; et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 2009, 326, 818–823. [Google Scholar] [CrossRef] [Green Version]
- Vogel, B.H.; Bradley, S.E.; Adams, D.J.; D’Aco, K.; Erbe, R.W.; Fong, C.; Iglesias, A.; Kronn, D.; Levy, P.; Morrissey, M.; et al. Newborn screening for X-linked adrenoleukodystrophy in New York State: Diagnostic protocol, surveillance protocol and treatment guidelines. Mol. Genet. Metab. 2015, 114, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Barendsen, R.W.; Dijkstra, I.M.E.; Visser, W.F.; Alders, M.; Bliek, J.; Boelen, A.; Bouva, M.J.; van der Crabben, S.N.; Elsinghorst, E.; van Gorp, A.G.M.; et al. Adrenoleukodystrophy Newborn Screening in the Netherlands (SCAN Study): The X-Factor. Front. cell Dev. Biol. 2020, 8, 499. [Google Scholar] [CrossRef] [PubMed]
- Wiens, K.; Berry, S.A.; Choi, H.; Gaviglio, A.; Gupta, A.; Hietala, A.; Kenney-Jung, D.; Lund, T.; Miller, W.; Pierpont, E.I.; et al. A report on state-wide implementation of newborn screening for X-linked Adrenoleukodystrophy. Am. J. Med. Genet. A 2019, 179, 1205–1213. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Clinard, K.; Young, S.P.; Rehder, C.W.; Fan, Z.; Calikoglu, A.S.; Bali, D.S.; Bailey, D.B.; Gehtland, L.M.; Millington, D.S.; et al. Evaluation of X-Linked Adrenoleukodystrophy Newborn Screening in North Carolina. JAMA Netw. Open 2020, 3, e1920356. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.L.; Li, H.; Hagar, A.F.; Jerris, S.C.; Wittenauer, A.; Wilcox, W. Newborn Screening for X-Linked Adrenoleukodystrophy in Georgia: Experiences from a Pilot Study Screening of 51,081 Newborns. Int. J. Neonatal Screen. 2020, 6, 81. [Google Scholar] [CrossRef]
- Matteson, J.; Sciortino, S.; Feuchtbaum, L.; Bishop, T.; Olney, R.S.; Tang, H. Adrenoleukodystrophy Newborn Screening in California Since 2016: Programmatic Outcomes and Follow-Up. Int. J. Neonatal Screen. 2021, 7, 22. [Google Scholar] [CrossRef]
- Regelmann, M.O.; Kamboj, M.K.; Miller, B.S.; Nakamoto, J.M.; Sarafoglou, K.; Shah, S.; Stanley, T.L.; Marino, R. Adrenoleukodystrophy: Guidance for adrenal surveillance in males identified by newborn screen. J. Clin. Endocrinol. Metab. 2018, 103, 4324–4331. [Google Scholar] [CrossRef] [Green Version]
- Mallack, E.J.; Turk, B.R.; Yan, H.; Price, C.; Demetres, M.; Moser, A.B.; Becker, C.; Hollandsworth, K.; Adang, L.; Vanderver, A.; et al. MRI surveillance of boys with X-linked adrenoleukodystrophy identified by newborn screening: Meta-analysis and consensus guidelines. J. Inherit. Metab. Dis. 2021, 44, 728–739. [Google Scholar] [CrossRef]
- van de Stadt, S.I.W.; Mooyer, P.A.W.; Dijkstra, I.M.E.; Dekker, C.J.M.; Vats, D.; Vera, M.; Ruzhnikov, M.R.Z.; van Haren, K.; Tang, N.; Koop, K.; et al. Biochemical Studies in Fibroblasts to Interpret Variants of Unknown Significance in the ABCD1 Gene. Genes 2021, 12, 1930. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Liberato, A.P.; Mallack, E.J.; Aziz-Bose, R.; Hayden, D.; Lauer, A.; Caruso, P.A.; Musolino, P.L.; Eichler, F.S. MRI brain lesions in asymptomatic boys with X-linked adrenoleukodystrophy. Neurology 2019, 92, e1698–e1708. [Google Scholar] [CrossRef]
- Henig, I.; Zuckerman, T. Hematopoietic stem cell transplantation-50 years of evolution and future perspectives. Rambam Maimonides Med. J. 2014, 5, e0028. [Google Scholar] [CrossRef] [PubMed]
- Corre, C.S.; Grant, N.; Sadjadi, R.; Hayden, D.; Becker, C.; Gomery, P.; Eichler, F.S. Beyond gait and balance: Urinary and bowel dysfunction in X-linked adrenoleukodystrophy. Orphanet J. Rare Dis. 2021, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Eugster, A.; Vingerhoets, A.J. Psychological aspects of in vitro fertilization: A review. Soc. Sci. Med. 1999, 48, 575–589. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Total | Unique | |||
|---|---|---|---|---|
| n | % | n | % | |
| All ABCD1 variants in the database | 3401 | 948 | 28% | |
| Missense pathogenic variants | 2087 | 61.4% | 411 | 43.4% |
| Nonsense pathogenic variants | 336 | 9.9% | 116 | 12.3% |
| Frame shift pathogenic variants | 585 | 17.2% | 262 | 27.6% |
| Amino acid insertions/deletions | 119 | 3.5% | 52 | 5.5% |
| Splice site pathogenic variants | 145 | 4.3% | 43 | 4.5% |
| One or more exons deleted | 89 | 2.6% | 24 | 2.5% |
| Benign variants | 40 | 1.2% | 40 | 4.2% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallack, E.J.; Gao, K.; Engelen, M.; Kemp, S. Structure and Function of the ABCD1 Variant Database: 20 Years, 940 Pathogenic Variants, and 3400 Cases of Adrenoleukodystrophy. Cells 2022, 11, 283. https://doi.org/10.3390/cells11020283
Mallack EJ, Gao K, Engelen M, Kemp S. Structure and Function of the ABCD1 Variant Database: 20 Years, 940 Pathogenic Variants, and 3400 Cases of Adrenoleukodystrophy. Cells. 2022; 11(2):283. https://doi.org/10.3390/cells11020283
Chicago/Turabian StyleMallack, Eric J., Kerry Gao, Marc Engelen, and Stephan Kemp. 2022. "Structure and Function of the ABCD1 Variant Database: 20 Years, 940 Pathogenic Variants, and 3400 Cases of Adrenoleukodystrophy" Cells 11, no. 2: 283. https://doi.org/10.3390/cells11020283
APA StyleMallack, E. J., Gao, K., Engelen, M., & Kemp, S. (2022). Structure and Function of the ABCD1 Variant Database: 20 Years, 940 Pathogenic Variants, and 3400 Cases of Adrenoleukodystrophy. Cells, 11(2), 283. https://doi.org/10.3390/cells11020283