

Targeting Mast Cells in Allergic Disease: Current Therapies and Drug Repurposing

 ,
,
Abstract
1. Introduction
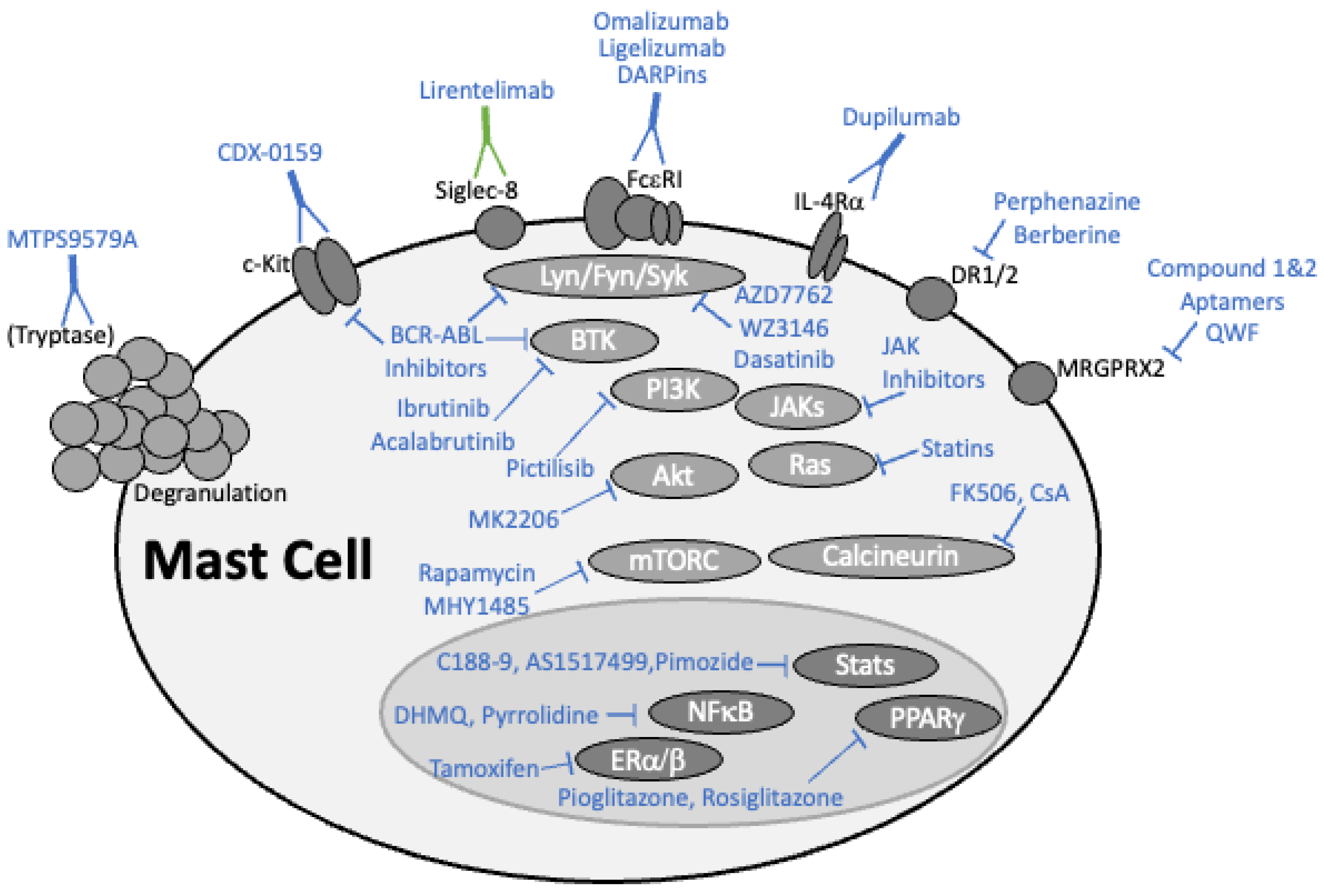
2. Targeting Mast Cells with the Latest Drugs
2.1. Monoclonal Antibody-Based Drugs
2.1.1. Omalizumab and Ligelizumab Antagonize IgE-FcεRI Binding
2.1.2. Dupilumab Suppresses IL-4 and IL-13 Signaling
2.1.3. Rituximab Reduces IgE Levels by Depleting B Cells
2.1.4. Lirentelimab Activates Siglec-8
2.1.5. MTPS9579A Disrupts Tryptase Tetramers
2.1.6. CDX-0159 Targets KIT Receptor
2.2. DARPins
2.3. Kinase Inhibitors
2.3.1. BCR-ABL Tyrosine Kinase Inhibitors
2.3.2. Bruton’s Tyrosine Kinase Inhibitors
2.3.3. Lyn, Fyn, and Syk Tyrosine Kinase Inhibitors
2.3.4. PI3K/AKT/mTOR Pathway Inhibition
2.4. Targeting Critical Transcription Factors
2.4.1. Inhibiting NFκB
2.4.2. Targeting NFAT
2.4.3. Inhibiting the JAK-STAT Pathway
2.4.4. Estrogen Receptor Signaling
2.4.5. The Complex Effects of PPAR-γ Function
2.5. Dopamine Signaling
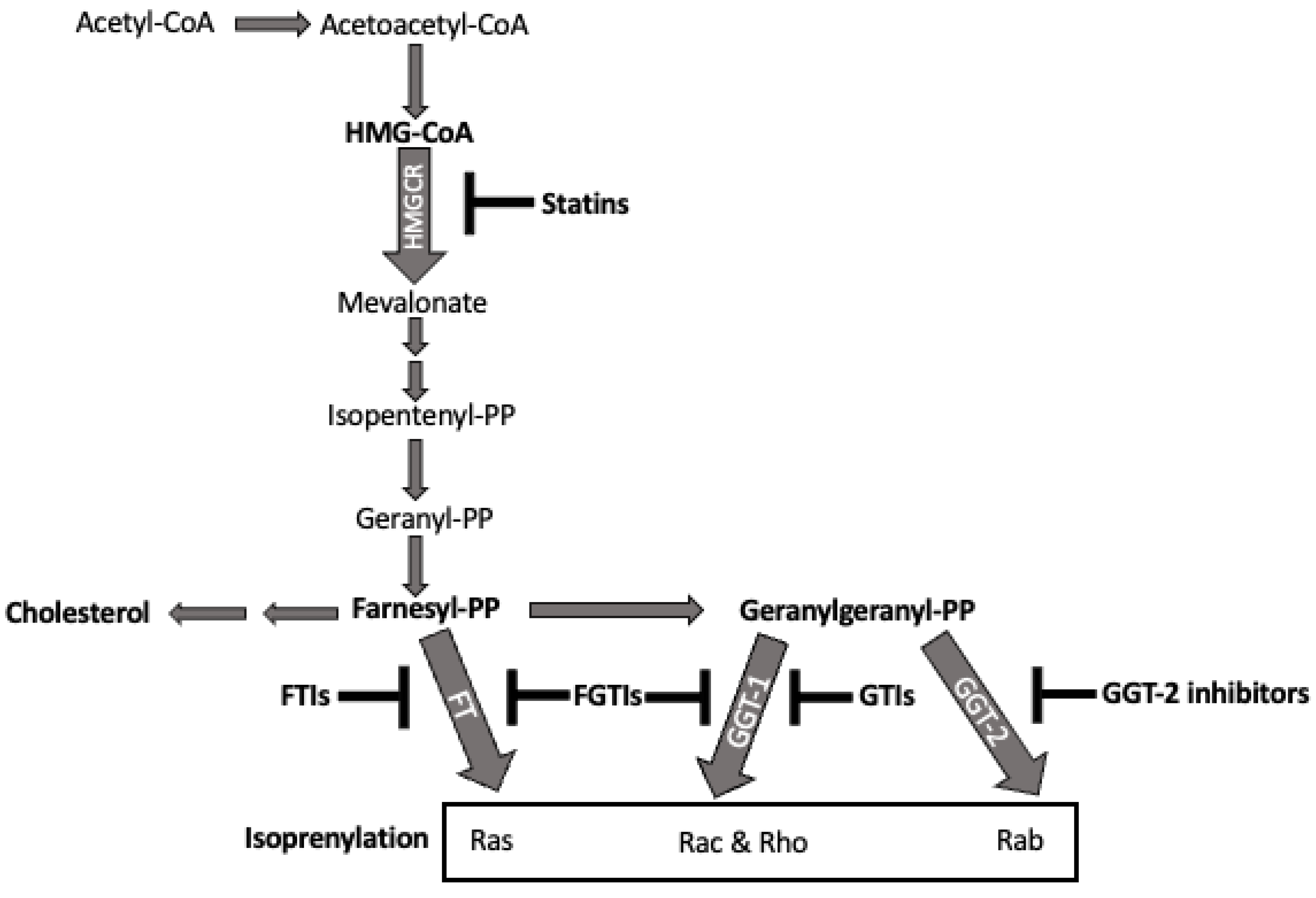
2.6. Statins and the Isoprenyl Lipid Pathway
2.7. Antidepressants as Mast Cell Inhibitors in Allergic Disease
2.8. MRGPR Proteins Driving Mast Cell Activation
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- To, T.; Stanojevic, S.; Moores, G.; Gershon, A.S.; Bateman, E.D.; Cruz, A.A.; Boulet, L.P. Global asthma prevalence in adults: Findings from the cross-sectional world health survey. BMC Public Health 2012, 12, 204. [Google Scholar] [CrossRef] [PubMed]
- Nurmagambetov, T.; Kuwahara, R.; Garbe, P. The Economic Burden of Asthma in the United States, 2008–2013. Ann. Am. Thorac. Soc. 2018, 15, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Oborne, J.; Mortimer, K.; Hubbard, R.B.; Tattersfield, A.E.; Harrison, T.W. Quadrupling the dose of inhaled corticosteroid to prevent asthma exacerbations: A randomized, double-blind, placebo-controlled, parallel-group clinical trial. Am. J. Respir. Crit. Care Med. 2009, 180, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Smits, R.A.; Leurs, R.; de Esch, I.J. Major advances in the development of histamine H4 receptor ligands. Drug Discov. Today 2009, 14, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fu, J.; Zhou, Y. Research Progress in Atopic March. Front. Immunol. 2020, 11, 1907. [Google Scholar] [CrossRef]
- Berdigaliyev, N.; Aljofan, M. An overview of drug discovery and development. Future Med. Chem. 2020, 12, 939–947. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef]
- Chakraborty, C.; Sharma, A.R.; Bhattacharya, M.; Agoramoorthy, G.; Lee, S.S. The Drug Repurposing for COVID-19 Clinical Trials Provide Very Effective Therapeutic Combinations: Lessons Learned From Major Clinical Studies. Front. Pharmacol. 2021, 12, 704205. [Google Scholar] [CrossRef]
- Broide, D.H.; Gleich, G.J.; Cuomo, A.J.; Coburn, D.A.; Federman, E.C.; Schwartz, L.B.; Wasserman, S.I. Evidence of ongoing mast cell and eosinophil degranulation in symptomatic asthma airway. J. Allergy Clin. Immunol. 1991, 88, 637–648. [Google Scholar] [CrossRef]
- Brightling, C.E.; Bradding, P.; Symon, F.A.; Holgate, S.T.; Wardlaw, A.J.; Pavord, I.D. Mast-cell infiltration of airway smooth muscle in asthma. N. Engl. J. Med. 2002, 346, 1699–1705. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Baines, K.J.; Fu, J.J.; Wood, L.G.; Simpson, J.L.; McDonald, V.M.; Cowan, D.C.; Taylor, D.R.; Cowan, J.O.; Gibson, P.G. Sputum mast cell subtypes relate to eosinophilia and corticosteroid response in asthma. Eur. Respir. J. 2016, 47, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, R.H.; Sidhu, S.S.; Raman, K.; Solon, M.; Solberg, O.D.; Caughey, G.H.; Woodruff, P.G.; Fahy, J.V. Accumulation of intraepithelial mast cells with a unique protease phenotype in T(H)2-high asthma. J. Allergy Clin. Immunol. 2010, 125, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Kanjarawi, R.; Dy, M.; Bardel, E.; Sparwasser, T.; Dubois, B.; Mecheri, S.; Kaiserlian, D. Regulatory CD4+Foxp3+ T cells control the severity of anaphylaxis. PLoS ONE 2013, 8, e69183. [Google Scholar] [CrossRef]
- Zhou, J.S.; Xing, W.; Friend, D.S.; Austen, K.F.; Katz, H.R. Mast cell deficiency in KitW-sh mice does not impair antibody-mediated arthritis. J. Exp. Med. 2007, 204, 2797–2802. [Google Scholar] [CrossRef]
- Reber, L.L.; Marichal, T.; Mukai, K.; Kita, Y.; Tokuoka, S.M.; Roers, A.; Hartmann, K.; Karasuyama, H.; Nadeau, K.C.; Tsai, M.; et al. Selective ablation of mast cells or basophils reduces peanut-induced anaphylaxis in mice. J. Allergy Clin. Immunol. 2013, 132, 881–888. [Google Scholar] [CrossRef]
- Kobayashi, T.; Miura, T.; Haba, T.; Sato, M.; Serizawa, I.; Nagai, H.; Ishizaka, K. An Essential Role of Mast Cells in the Development of Airway Hyperresponsiveness in a Murine Asthma Model. J. Immunol. 2000, 164, 3855–3861. [Google Scholar] [CrossRef]
- Williams, C.M.; Galli, S.J. Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J. Exp. Med. 2000, 192, 455–462. [Google Scholar] [CrossRef]
- Fuchs, B.; Sjöberg, L.; Westerberg, C.M.; Ekoff, M.; Swedin, L.; Dahlén, S.-E.; Adner, M.; Nilsson, G.P. Mast cell engraftment of the peripheral lung enhances airway hyperresponsiveness in a mouse asthma model. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 303, L1027–L1036. [Google Scholar] [CrossRef]
- Kawada, N.; Tanaka, H.; Takizawa, T.; Yamada, T.; Takahashi, Y.; Masuda, T.; Inagaki, N.; Nagai, H. Role of mast cells in antigen-induced airway inflammation and bronchial hyperresponsiveness in rats. Jpn. J. Pharmacol. 2001, 85, 250–259. [Google Scholar] [CrossRef]
- Kim, Y.S.; Ko, H.M.; Kang, N.I.; Song, C.H.; Zhang, X.; Chung, W.C.; Kim, J.H.; Choi, I.H.; Park, Y.M.; Kim, G.Y.; et al. Mast cells play a key role in the development of late airway hyperresponsiveness through TNF-alpha in a murine model of asthma. Eur. J. Immunol. 2007, 37, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.S. The role of the mast cell in asthma: Induction of airway hyperresponsiveness by interaction with smooth muscle? J. Allergy Clin. Immunol. 2004, 114, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Quan, P.L.; Sabaté-Brescó, M.; Guo, Y.; Martín, M.; Gastaminza, G. The Multifaceted Mas-Related G Protein-Coupled Receptor Member X2 in Allergic Diseases and Beyond. Int. J. Mol. Sci. 2021, 22, 4421. [Google Scholar] [CrossRef] [PubMed]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021, 42, 1143–1158. [Google Scholar] [CrossRef]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Kishiro, Y.; Kagawa, M.; Naito, I.; Sado, Y. A novel method of preparing rat-monoclonal antibody-producing hybridomas by using rat medial iliac lymph node cells. Cell Struct. Funct. 1995, 20, 151–156. [Google Scholar] [CrossRef]
- Spieker-Polet, H.; Sethupathi, P.; Yam, P.C.; Knight, K.L. Rabbit monoclonal antibodies: Generating a fusion partner to produce rabbit-rabbit hybridomas. Proc. Natl. Acad. Sci. USA 1995, 92, 9348–9352. [Google Scholar] [CrossRef]
- Duvall, M.; Bradley, N.; Fiorini, R.N. A novel platform to produce human monoclonal antibodies: The next generation of therapeutic human monoclonal antibodies discovery. MAbs 2011, 3, 203–208. [Google Scholar] [CrossRef][Green Version]
- Molderings, G.J.; Dumoulin, F.L.; Homann, J.; Sido, B.; Textor, J.; Mücke, M.; Qagish, G.J.; Barion, R.; Raithel, M.; Klingmüller, D.; et al. Adrenal insufficiency is a contraindication for omalizumab therapy in mast cell activation disease: Risk for serum sickness. Naunyn Schmiedebergs Arch. Pharmacol. 2020, 393, 1573–1580. [Google Scholar] [CrossRef]
- Belliveau, P.P. Omalizumab: A monoclonal anti-IgE antibody. MedGenMed 2005, 7, 27. [Google Scholar]
- Gericke, J.; Ohanyan, T.; Church, M.K.; Maurer, M.; Metz, M. Omalizumab may not inhibit mast cell and basophil activation in vitro. J. Eur. Acad. Dermatol. Venereol. 2015, 29, 1832–1836. [Google Scholar] [CrossRef] [PubMed]
- Çildağ, S.; Şentürk, T. The effect of omalizumab treatment on IgE and other immunoglobulin levels in patients with chronic spontaneous urticaria and its association with treatment response. Postepy Dermatol. Alergol. 2018, 35, 516–519. [Google Scholar] [CrossRef]
- Chang, T.W.; Chen, C.; Lin, C.J.; Metz, M.; Church, M.K.; Maurer, M. The potential pharmacologic mechanisms of omalizumab in patients with chronic spontaneous urticaria. J. Allergy Clin. Immunol. 2015, 135, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Gasser, P.; Tarchevskaya, S.S.; Guntern, P.; Brigger, D.; Ruppli, R.; Zbären, N.; Kleinboelting, S.; Heusser, C.; Jardetzky, T.S.; Eggel, A. The mechanistic and functional profile of the therapeutic anti-IgE antibody ligelizumab differs from omalizumab. Nat. Commun. 2020, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Arm, J.P.; Bottoli, I.; Skerjanec, A.; Floch, D.; Groenewegen, A.; Maahs, S.; Owen, C.E.; Jones, I.; Lowe, P.J. Pharmacokinetics, pharmacodynamics and safety of QGE031 (ligelizumab), a novel high-affinity anti-IgE antibody, in atopic subjects. Clin. Exp. Allergy 2014, 44, 1371–1385. [Google Scholar] [CrossRef] [PubMed]
- Advani, R.H.; Buggy, J.J.; Sharman, J.P.; Smith, S.M.; Boyd, T.E.; Grant, B.; Kolibaba, K.S.; Furman, R.R.; Rodriguez, S.; Chang, B.Y.; et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J. Clin. Oncol. 2013, 31, 88–94. [Google Scholar] [CrossRef]
- Keegan, A.D.; Zamorano, J.; Keselman, A.; Heller, N.M. IL-4 and IL-13 Receptor Signaling From 4PS to Insulin Receptor Substrate 2: There and Back Again, a Historical View. Front. Immunol. 2018, 9, 1037. [Google Scholar] [CrossRef]
- Karo-Atar, D.; Bitton, A.; Benhar, I.; Munitz, A. Therapeutic Targeting of the Interleukin-4/Interleukin-13 Signaling Pathway: In Allergy and Beyond. BioDrugs 2018, 32, 201–220. [Google Scholar] [CrossRef]
- Gandhi, N.A.; Pirozzi, G.; Graham, N.M.H. Commonality of the IL-4/IL-13 pathway in atopic diseases. Expert Rev. Clin. Immunol. 2017, 13, 425–437. [Google Scholar] [CrossRef]
- Walsh, G.M. Biologics for asthma and allergy. Curr. Opin. Otolaryngol. Head Neck Surg. 2017, 25, 231–234. [Google Scholar] [CrossRef]
- Ma, L.L.; O’Byrne, P.M. The pharmacological modulation of allergen-induced asthma. Inflammopharmacology 2013, 21, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Bissonnette, R.; Ungar, B.; Suárez-Fariñas, M.; Ardeleanu, M.; Esaki, H.; Suprun, M.; Estrada, Y.; Xu, H.; Peng, X.; et al. Dupilumab progressively improves systemic and cutaneous abnormalities in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2019, 143, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Le Floc’h, A.; Allinne, J.; Nagashima, K.; Scott, G.; Birchard, D.; Asrat, S.; Bai, Y.; Lim, W.K.; Martin, J.; Huang, T.; et al. Dual blockade of IL-4 and IL-13 with dupilumab, an IL-4Rα antibody, is required to broadly inhibit type 2 inflammation. Allergy 2020, 75, 1188–1204. [Google Scholar] [CrossRef] [PubMed]
- Conde, E.; Bertrand, R.; Balbino, B.; Bonnefoy, J.; Stackowicz, J.; Caillot, N.; Colaone, F.; Hamdi, S.; Houmadi, R.; Loste, A.; et al. Dual vaccination against IL-4 and IL-13 protects against chronic allergic asthma in mice. Nat. Commun. 2021, 12, 2574. [Google Scholar] [CrossRef] [PubMed]
- Barmettler, S.; Ong, M.S.; Farmer, J.R.; Choi, H.; Walter, J. Association of Immunoglobulin Levels, Infectious Risk, and Mortality With Rituximab and Hypogammaglobulinemia. JAMA Netw. Open 2018, 1, e184169. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, F.; Guggino, G.; Rizzo, A.; Alessandro, R.; Carubbi, F.; Giardina, A.; Cipriani, P.; Ferrante, A.; Cannizzaro, A.; Giacomelli, R.; et al. Rituximab modulates IL-17 expression in the salivary glands of patients with primary Sjögren’s syndrome. Rheumatology 2014, 53, 1313–1320. [Google Scholar] [CrossRef]
- Dasgupta, A.; Radford, K.; Arnold, D.M.; Thabane, L.; Nair, P. The effects of rituximab on serum IgE and BAFF. Allergy Asthma Clin. Immunol. 2013, 9, 39. [Google Scholar] [CrossRef]
- Casal Moura, M.; Berti, A.; Keogh, K.A.; Volcheck, G.W.; Specks, U.; Baqir, M. Asthma control in eosinophilic granulomatosis with polyangiitis treated with rituximab. Clin. Rheumatol. 2020, 39, 1581–1590. [Google Scholar] [CrossRef]
- Teixeira, V.; Mohammad, A.J.; Jones, R.B.; Smith, R.; Jayne, D. Efficacy and safety of rituximab in the treatment of eosinophilic granulomatosis with polyangiitis. RMD Open 2019, 5, e000905. [Google Scholar] [CrossRef]
- Dellon, E.S.; Peterson, K.A.; Murray, J.A.; Falk, G.W.; Gonsalves, N.; Chehade, M.; Genta, R.M.; Leung, J.; Khoury, P.; Klion, A.D.; et al. Anti-Siglec-8 Antibody for Eosinophilic Gastritis and Duodenitis. N. Engl. J. Med. 2020, 383, 1624–1634. [Google Scholar] [CrossRef]
- Yokoi, H.; Choi, O.H.; Hubbard, W.; Lee, H.S.; Canning, B.J.; Lee, H.H.; Ryu, S.D.; von Gunten, S.; Bickel, C.A.; Hudson, S.A.; et al. Inhibition of FcepsilonRI-dependent mediator release and calcium flux from human mast cells by sialic acid-binding immunoglobulin-like lectin 8 engagement. J. Allergy Clin. Immunol. 2008, 121, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.A.; Brock, E.C.; Leung, J.; Falahati, R.; Bryce, P.J.; Bright, J.; Williams, J.; Shultz, L.D.; Greiner, D.L.; Brehm, M.A.; et al. AK002, a Humanized Sialic Acid-Binding Immunoglobulin-Like Lectin-8 Antibody that Induces Antibody-Dependent Cell-Mediated Cytotoxicity against Human Eosinophils and Inhibits Mast Cell-Mediated Anaphylaxis in Mice. Int. Arch. Allergy Immunol. 2019, 180, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.A.; Brock, E.C.; Leung, J.; Falahati, R.; Bochner, B.S.; Rasmussen, H.S.; Peterson, K.; Bebbington, C.; Tomasevic, N. Siglec-8 antibody reduces eosinophils and mast cells in a transgenic mouse model of eosinophilic gastroenteritis. JCI Insight 2019, 4, e126219. [Google Scholar] [CrossRef] [PubMed]
- Dispenza, M.C.; Bochner, B.S.; MacGlashan, D.W., Jr. Targeting the FcεRI Pathway as a Potential Strategy to Prevent Food-Induced Anaphylaxis. Front. Immunol. 2020, 11, 614402. [Google Scholar] [CrossRef] [PubMed]
- Anesi, S.D.; Tauber, J.; Nguyen, Q.D.; Chang, P.; Berdy, G.J.; Lin, C.C.; Chu, D.S.; Levine, H.T.; Fernandez, A.D.; Roy, N.; et al. Lirentelimab for severe and chronic forms of allergic conjunctivitis. J. Allergy Clin. Immunol. 2022, 150, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Rymut, S.M.; Sukumaran, S.; Sperinde, G.; Bremer, M.; Galanter, J.; Yoshida, K.; Smith, J.; Banerjee, P.; Sverkos, V.; Cai, F.; et al. Dose-dependent inactivation of airway tryptase with a novel dissociating anti-tryptase antibody (MTPS9579A) in healthy participants: A randomized trial. Clin. Transl. Sci. 2022, 15, 451–463. [Google Scholar] [CrossRef]
- Hallgren, J.; Lindahl, S.; Pejler, G. Structural requirements and mechanism for heparin-dependent activation and tetramerization of human betaI- and betaII-tryptase. J. Mol. Biol. 2005, 345, 129–139. [Google Scholar] [CrossRef]
- Schwartz, L.B.; Bradford, T.R. Regulation of tryptase from human lung mast cells by heparin. Stabilization of the active tetramer. J. Biol. Chem. 1986, 261, 7372–7379. [Google Scholar] [CrossRef]
- Sommerhoff, C.P. Mast cell tryptases and airway remodeling. Am. J. Respir. Crit. Care Med. 2001, 164, S52–S58. [Google Scholar] [CrossRef]
- Maun, H.R.; Jackman, J.K.; Choy, D.F.; Loyet, K.M.; Staton, T.L.; Jia, G.; Dressen, A.; Hackney, J.A.; Bremer, M.; Walters, B.T.; et al. An Allosteric Anti-tryptase Antibody for the Treatment of Mast Cell-Mediated Severe Asthma. Cell 2019, 179, 417–431. [Google Scholar] [CrossRef]
- Rymut, S.; Yoshida, K.; Sukumaran, S.; Cai, F.; Sperinde, G.; Sverkos, V.; Banerjee, P.; Belloni, P.; Lin, J. Local Airway Concentration of Anti-Tryptase Antibody (MTPS9579A) Predicts Extent of Tryptase Disruption. J. Allergy Clin. Immunol. 2020, 145, AB172. [Google Scholar] [CrossRef]
- Alvarado, D.; Maurer, M.; Gedrich, R.; Seibel, S.B.; Murphy, M.B.; Crew, L.; Goldstein, J.; Crocker, A.; Vitale, L.A.; Morani, P.A.; et al. Anti-KIT monoclonal antibody CDX-0159 induces profound and durable mast cell suppression in a healthy volunteer study. Allergy 2022, 77, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Caputi, A.P.; Navarra, P. Beyond antibodies: Ankyrins and DARPins. From basic research to drug approval. Curr. Opin. Pharmacol. 2020, 51, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Eggel, A.; Baravalle, G.; Hobi, G.; Kim, B.; Buschor, P.; Forrer, P.; Shin, J.S.; Vogel, M.; Stadler, B.M.; Dahinden, C.A.; et al. Accelerated dissociation of IgE-FcεRI complexes by disruptive inhibitors actively desensitizes allergic effector cells. J. Allergy Clin. Immunol. 2014, 133, 1709–1719. [Google Scholar] [CrossRef]
- Zellweger, F.; Gasser, P.; Brigger, D.; Buschor, P.; Vogel, M.; Eggel, A. A novel bispecific DARPin targeting FcγRIIB and FcεRI-bound IgE inhibits allergic responses. Allergy 2017, 72, 1174–1183. [Google Scholar] [CrossRef]
- Fellmann, M.; Buschor, P.; Röthlisberger, S.; Zellweger, F.; Vogel, M. High affinity targeting of CD23 inhibits IgE synthesis in human B cells. Immun. Inflamm. Dis. 2015, 3, 339–349. [Google Scholar] [CrossRef]
- Gilfillan, A.M.; Rivera, J. The tyrosine kinase network regulating mast cell activation. Immunol. Rev. 2009, 228, 149–169. [Google Scholar] [CrossRef]
- Rossari, F.; Minutolo, F.; Orciuolo, E. Past, present, and future of Bcr-Abl inhibitors: From chemical development to clinical efficacy. J. Hematol. Oncol. 2018, 11, 84. [Google Scholar] [CrossRef]
- Hantschel, O.; Rix, U.; Schmidt, U.; Bürckstümmer, T.; Kneidinger, M.; Schütze, G.; Colinge, J.; Bennett, K.L.; Ellmeier, W.; Valent, P.; et al. The Btk tyrosine kinase is a major target of the Bcr-Abl inhibitor dasatinib. Proc. Natl. Acad. Sci. USA 2007, 104, 13283–13288. [Google Scholar] [CrossRef]
- El-Agamy, D.S. Anti-allergic effects of nilotinib on mast cell-mediated anaphylaxis like reactions. Eur. J. Pharmacol. 2012, 680, 115–121. [Google Scholar] [CrossRef]
- Cahill, K.N.; Katz, H.R.; Cui, J.; Lai, J.; Kazani, S.; Crosby-Thompson, A.; Garofalo, D.; Castro, M.; Jarjour, N.; DiMango, E.; et al. KIT Inhibition by Imatinib in Patients with Severe Refractory Asthma. N. Engl. J. Med. 2017, 376, 1911–1920. [Google Scholar] [CrossRef]
- Cerny-Reiterer, S.; Rabenhorst, A.; Stefanzl, G.; Herndlhofer, S.; Hoermann, G.; Müllauer, L.; Baumgartner, S.; Beham-Schmid, C.; Sperr, W.R.; Mannhalter, C.; et al. Long-term treatment with imatinib results in profound mast cell deficiency in Ph+ chronic myeloid leukemia. Oncotarget 2015, 6, 3071–3084. [Google Scholar] [CrossRef]
- Hata, D.; Kawakami, Y.; Inagaki, N.; Lantz, C.S.; Kitamura, T.; Khan, W.N.; Maeda-Yamamoto, M.; Miura, T.; Han, W.; Hartman, S.E.; et al. Involvement of Bruton’s tyrosine kinase in FcepsilonRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 1998, 187, 1235–1247. [Google Scholar] [CrossRef] [PubMed]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Dispenza, M.C.; Krier-Burris, R.A.; Chhiba, K.D.; Undem, B.J.; Robida, P.A.; Bochner, B.S. Bruton’s tyrosine kinase inhibition effectively protects against human IgE-mediated anaphylaxis. J. Clin. Investig. 2020, 130, 4759–4770. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.N. Regulation of B lymphocyte development and activation by Bruton’s tyrosine kinase. Immunol. Res. 2001, 23, 147–156. [Google Scholar] [CrossRef]
- Chang, B.Y.; Huang, M.M.; Francesco, M.; Chen, J.; Sokolove, J.; Magadala, P.; Robinson, W.H.; Buggy, J.J. The Bruton tyrosine kinase inhibitor PCI-32765 ameliorates autoimmune arthritis by inhibition of multiple effector cells. Arthritis Res. Ther. 2011, 13, R115. [Google Scholar] [CrossRef]
- Smiljkovic, D.; Blatt, K.; Stefanzl, G.; Dorofeeva, Y.; Skrabs, C.; Focke-Tejkl, M.; Sperr, W.R.; Jaeger, U.; Valenta, R.; Valent, P. BTK inhibition is a potent approach to block IgE-mediated histamine release in human basophils. Allergy 2017, 72, 1666–1676. [Google Scholar] [CrossRef]
- Gamperl, S.; Stefanzl, G.; Peter, B.; Smiljkovic, D.; Bauer, K.; Willmann, M.; Valent, P.; Hadzijusufovic, E. Effects of ibrutinib on proliferation and histamine release in canine neoplastic mast cells. Vet. Comp. Oncol. 2019, 17, 553–561. [Google Scholar] [CrossRef]
- Dubovsky, J.A.; Beckwith, K.A.; Natarajan, G.; Woyach, J.A.; Jaglowski, S.; Zhong, Y.; Hessler, J.D.; Liu, T.M.; Chang, B.Y.; Larkin, K.M.; et al. Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 2013, 122, 2539–2549. [Google Scholar] [CrossRef]
- Regan, J.A.; Cao, Y.; Dispenza, M.C.; Ma, S.; Gordon, L.I.; Petrich, A.M.; Bochner, B.S. Ibrutinib, a Bruton’s tyrosine kinase inhibitor used for treatment of lymphoproliferative disorders, eliminates both aeroallergen skin test and basophil activation test reactivity. J. Allergy Clin. Immunol. 2017, 140, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Dispenza, M.C.; Pongracic, J.A.; Singh, A.M.; Bochner, B.S. Short-term ibrutinib therapy suppresses skin test responses and eliminates IgE-mediated basophil activation in adults with peanut or tree nut allergy. J. Allergy Clin. Immunol. 2018, 141, 1914–1916. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Yamasaki, S.; Hasegawa, A.; Iwamatsu, A.; Koseki, H.; Hirano, T. Gab2, via PI-3K, regulates ARF1 in FcεRI-mediated granule translocation and mast cell degranulation. J. Immunol. 2011, 187, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, V.; Gadina, M.; Kovarova, M.; Odom, S.; Gonzalez-Espinosa, C.; Furumoto, Y.; Saitoh, S.; Samelson, L.E.; O’Shea, J.J.; Rivera, J. Fyn kinase initiates complementary signals required for IgE-dependent mast cell degranulation. Nat. Immunol. 2002, 3, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Kim, D.K.; Kim, H.W.; Kim, H.S.; Lee, D.; Lee, M.B.; Min, K.Y.; Koo, J.; Kim, S.J.; Kang, C.; et al. Repositioning of anti-cancer drug candidate, AZD7762, to an anti-allergic drug suppressing IgE-mediated mast cells and allergic responses via the inhibition of Lyn and Fyn. Biochem. Pharmacol. 2018, 154, 270–277. [Google Scholar] [CrossRef]
- Park, Y.H.; Kim, D.K.; Kim, H.S.; Lee, D.; Lee, M.B.; Min, K.Y.; Jo, M.G.; Lee, J.E.; Kim, Y.M.; Choi, W.S. WZ3146 inhibits mast cell Lyn and Fyn to reduce IgE-mediated allergic responses in vitro and in vivo. Toxicol. Appl. Pharmacol. 2019, 383, 114763. [Google Scholar] [CrossRef]
- Lee, D.; Park, Y.H.; Lee, J.E.; Kim, H.S.; Min, K.Y.; Jo, M.G.; Kim, H.S.; Choi, W.S.; Kim, Y.M. Dasatinib Inhibits Lyn and Fyn Src-Family Kinases in Mast Cells to Suppress Type I Hypersensitivity in Mice. Biomol. Ther. (Seoul) 2020, 28, 456–464. [Google Scholar] [CrossRef]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef]
- Kim, M.S.; Radinger, M.; Gilfillan, A.M. The multiple roles of phosphoinositide 3-kinase in mast cell biology. Trends Immunol. 2008, 29, 493–501. [Google Scholar] [CrossRef]
- Weichhart, T.; Säemann, M.D. The PI3K/Akt/mTOR pathway in innate immune cells: Emerging therapeutic applications. Ann. Rheum. Dis. 2008, 67 (Suppl. 3), iii70–iii74. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Perry, M.W.D.; Brown, J.R.; André, F.; Okkenhaug, K. PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 2021, 20, 741–769. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Franke, K.; Bal, G.; Li, Z.; Zuberbier, T.; Babina, M. MRGPRX2-Mediated Degranulation of Human Skin Mast Cells Requires the Operation of G(αi), G(αq), Ca++ Channels, ERK1/2 and PI3K-Interconnection between Early and Late Signaling. Cells 2022, 11, 953–970. [Google Scholar] [CrossRef] [PubMed]
- Schwartzberg, P.L.; Finkelstein, L.D.; Readinger, J.A. TEC-family kinases: Regulators of T-helper-cell differentiation. Nat. Rev. Immunol. 2005, 5, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Cho, H.; Thorvaldsen, J.L.; Chu, Q.; Feng, F.; Birnbaum, M.J. Akt1/PKBalpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J. Biol. Chem. 2001, 276, 38349–38352. [Google Scholar] [CrossRef]
- Gonzalez, E.; McGraw, T.E. The Akt kinases: Isoform specificity in metabolism and cancer. Cell Cycle 2009, 8, 2502–2508. [Google Scholar] [CrossRef]
- Downward, J. PI 3-kinase, Akt and cell survival. Semin. Cell Dev. Biol. 2004, 15, 177–182. [Google Scholar] [CrossRef]
- Hers, I.; Vincent, E.E.; Tavaré, J.M. Akt signalling in health and disease. Cell Signal. 2011, 23, 1515–1527. [Google Scholar] [CrossRef]
- Ma, B.; Athari, S.S.; Mehrabi Nasab, E.; Zhao, L. PI3K/AKT/mTOR and TLR4/MyD88/NF-κB Signaling Inhibitors Attenuate Pathological Mechanisms of Allergic Asthma. Inflammation 2021, 44, 1895–1907. [Google Scholar] [CrossRef]
- Kitaura, J.; Asai, K.; Maeda-Yamamoto, M.; Kawakami, Y.; Kikkawa, U.; Kawakami, T. Akt-dependent cytokine production in mast cells. J. Exp. Med. 2000, 192, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Jabbarzadeh Kaboli, P.; Salimian, F.; Aghapour, S.; Xiang, S.; Zhao, Q.; Li, M.; Wu, X.; Du, F.; Zhao, Y.; Shen, J.; et al. Akt-targeted therapy as a promising strategy to overcome drug resistance in breast cancer—A comprehensive review from chemotherapy to immunotherapy. Pharmacol. Res. 2020, 156, 104806. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Sootome, H.; Nakatsuru, Y.; Miyama, K.; Taguchi, S.; Tsujioka, K.; Ueno, Y.; Hatch, H.; Majumder, P.K.; Pan, B.S.; et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol. Cancer Ther. 2010, 9, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef]
- Cui, H.; Cheng, Y.; He, Y.; Cheng, W.; Zhao, W.; Zhao, H.; Zhou, F.H.; Wang, L.; Dong, J.; Cai, S. The AKT inhibitor MK2206 suppresses airway inflammation and the pro-remodeling pathway in a TDI-induced asthma mouse model. Mol. Med. Rep. 2020, 22, 3723–3734. [Google Scholar] [CrossRef]
- Qian, Q.Q.; Zhang, X.; Wang, Y.W.; Xu, J.W.; Dong, H.Q.; Li, N.N.; Qian, Y.N.; Gui, B. Pro-inflammatory role of high-mobility group box-1 on brain mast cells via the RAGE/NF-κB pathway. J. Neurochem. 2019, 151, 595–607. [Google Scholar] [CrossRef]
- Honda, T.; Nishio, Y.; Sakai, H.; Asagiri, M.; Yoshimura, K.; Inui, M.; Kuramasu, A. Calcium/calmodulin-dependent regulation of Rac GTPases and Akt in histamine-induced chemotaxis of mast cells. Cell Signal. 2021, 83, 109973. [Google Scholar] [CrossRef]
- Morita, M.; Gravel, S.P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef]
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. mTOR: A Cellular Regulator Interface in Health and Disease. Cells 2019, 8, 18–40. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Yamaki, K.; Yoshino, S. Preventive and therapeutic effects of rapamycin, a mammalian target of rapamycin inhibitor, on food allergy in mice. Allergy 2012, 67, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.B.; Theoharides, T.C. Methoxyluteolin Inhibits Neuropeptide-stimulated Proinflammatory Mediator Release via mTOR Activation from Human Mast Cells. J. Pharmacol. Exp. Ther. 2017, 361, 462–471. [Google Scholar] [CrossRef]
- Rakhmanova, V.; Jin, M.; Shin, J. Inhibition of Mast Cell Function and Proliferation by mTOR Activator MHY1485. Immune Netw. 2018, 18, e18. [Google Scholar] [CrossRef] [PubMed]
- Rakhmanova, V.; Park, S.; Lee, S.; Kim, Y.H.; Shin, J. 3-Benzyl-5-((2-nitrophenoxy) methyl)-dihydrofuran-2(3H)-one suppresses FcεRI-mediated mast cell degranulation via the inhibition of mTORC2-Akt signaling. Biochem. Biophys. Res. Commun. 2020, 521, 72–76. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Srivastava, M.; Kaplan, M.H. Transcription Factors in the Development and Pro-Allergic Function of Mast Cells. Front. Allergy 2021, 2, 679121. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Gaynor, R.B. IkappaB kinases: Key regulators of the NF-kappaB pathway. Trends Biochem. Sci. 2004, 29, 72–79. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, W.; Zeng, X.; Hu, G.; Zhang, H.; He, S.; Zhang, S. Histamine induces upregulated expression of histamine receptors and increases release of inflammatory mediators from microglia. Mol. Neurobiol. 2014, 49, 1487–1500. [Google Scholar] [CrossRef]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of NF-kappaB signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164–5205. [Google Scholar] [CrossRef]
- Wilhelm, T.; Bick, F.; Peters, K.; Mohta, V.; Tirosh, B.; Patterson, J.B.; Kharabi-Masouleh, B.; Huber, M. Infliction of proteotoxic stresses by impairment of the unfolded protein response or proteasomal inhibition as a therapeutic strategy for mast cell leukemia. Oncotarget 2018, 9, 2984–3000. [Google Scholar] [CrossRef]
- Lyberg, K.; Ali, H.A.; Grootens, J.; Kjellander, M.; Tirfing, M.; Arock, M.; Hägglund, H.; Nilsson, G.; Ungerstedt, J. Histone deacetylase inhibitor SAHA mediates mast cell death and epigenetic silencing of constitutively active D816V KIT in systemic mastocytosis. Oncotarget 2017, 8, 9647–9659. [Google Scholar] [CrossRef]
- Matsumoto, N.; Ariga, A.; To-e, S.; Nakamura, H.; Agata, N.; Hirano, S.; Inoue, J.; Umezawa, K. Synthesis of NF-kappaB activation inhibitors derived from epoxyquinomicin C. Bioorg. Med. Chem. Lett. 2000, 10, 865–869. [Google Scholar] [CrossRef]
- Noma, N.; Asagiri, M.; Takeiri, M.; Ohmae, S.; Takemoto, K.; Iwaisako, K.; Minato, N.; Maeda-Yamamoto, M.; Simizu, S.; Umezawa, K. Inhibition of MMP-2-Mediated Mast Cell Invasion by NF-κB Inhibitor DHMEQ in Mast Cells. Int. Arch. Allergy Immunol. 2015, 166, 84–90. [Google Scholar] [CrossRef]
- Kwon, O.; Lee, E.; Moon, T.C.; Jung, H.; Lin, C.X.; Nam, K.S.; Baek, S.H.; Min, H.K.; Chang, H.W. Expression of cyclooxygenase-2 and pro-inflammatory cytokines induced by 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB 153) in human mast cells requires NF-kappa B activation. Biol. Pharm. Bull. 2002, 25, 1165–1168. [Google Scholar] [CrossRef][Green Version]
- Ulleras, E.; Karlberg, M.; Moller Westerberg, C.; Alfredsson, J.; Gerondakis, S.; Strasser, A.; Nilsson, G. NFAT but not NF-kappaB is critical for transcriptional induction of the prosurvival gene A1 after IgE receptor activation in mast cells. Blood 2008, 111, 3081–3089. [Google Scholar] [CrossRef]
- Lee, J.U.; Kim, L.K.; Choi, J.M. Revisiting the Concept of Targeting NFAT to Control T Cell Immunity and Autoimmune Diseases. Front. Immunol. 2018, 9, 2747. [Google Scholar] [CrossRef]
- Takeuchi, M.; Ohno, K.; Takata, K.; Gion, Y.; Tachibana, T.; Orita, Y.; Yoshino, T.; Sato, Y. Interleukin 13-positive mast cells are increased in immunoglobulin G4-related sialadenitis. Sci. Rep. 2015, 5, 7696. [Google Scholar] [CrossRef]
- de Paulis, A.; Stellato, C.; Cirillo, R.; Ciccarelli, A.; Oriente, A.; Marone, G. Anti-inflammatory effect of FK-506 on human skin mast cells. J. Investig. Dermatol. 1992, 99, 723–728. [Google Scholar] [CrossRef]
- Wershil, B.K.; Furuta, G.T.; Lavigne, J.A.; Choudhury, A.R.; Wang, Z.S.; Galli, S.J. Dexamethasone or cyclosporin A suppress mast cell-leukocyte cytokine cascades. Multiple mechanisms of inhibition of IgE- and mast cell-dependent cutaneous inflammation in the mouse. J. Immunol. 1995, 154, 1391–1398. [Google Scholar]
- Hatfield, S.M.; Roehm, N.W. Cyclosporine and FK506 inhibition of murine mast cell cytokine production. J. Pharmacol. Exp. Ther. 1992, 260, 680–688. [Google Scholar]
- Warbrick, E.V.; Thomas, A.L.; Williams, C.M. The effects of cyclosporin A, dexamethasone and other immunomodulatory drugs on induced expression of IL-3, IL-4 and IL-8 mRNA in a human mast cell line. Toxicology 1997, 116, 211–218. [Google Scholar] [CrossRef]
- Ito, F.; Toyota, N.; Sakai, H.; Takahashi, H.; Iizuka, H. FK506 and cyclosporin A inhibit stem cell factor-dependent cell proliferation/survival, while inducing upregulation of c-kit expression in cells of the mast cell line MC/9. Arch. Dermatol. Res. 1999, 291, 275–283. [Google Scholar] [CrossRef]
- McLauchlan, P.E.; Roberts, H.C.; Loxton, N.J.; Wastling, J.M.; Newlands, G.F.; Chappell, L.H. Mucosal mast cell responses and release of mast cell protease-I in infections of mice with Hymenolepis diminuta and H. microstoma: Modulation by cyclosporin A. Parasite Immunol. 1999, 21, 151–161. [Google Scholar] [CrossRef]
- Harrison, C.A.; Bastan, R.; Peirce, M.J.; Munday, M.R.; Peachell, P.T. Role of calcineurin in the regulation of human lung mast cell and basophil function by cyclosporine and FK506. Br. J. Pharmacol. 2007, 150, 509–518. [Google Scholar] [CrossRef]
- Erdinest, N.; Ben-Eli, H.; Solomon, A. Topical tacrolimus for allergic eye diseases. Curr. Opin. Allergy Clin. Immunol. 2019, 19, 535–543. [Google Scholar] [CrossRef]
- Rustin, M.H. The safety of tacrolimus ointment for the treatment of atopic dermatitis: A review. Br. J. Dermatol. 2007, 157, 861–873. [Google Scholar] [CrossRef]
- Morales, J.K.; Falanga, Y.T.; Depcrynski, A.; Fernando, J.; Ryan, J.J. Mast cell homeostasis and the JAK-STAT pathway. Genes Immun. 2010, 11, 599–608. [Google Scholar] [CrossRef]
- Siegel, A.M.; Stone, K.D.; Cruse, G.; Lawrence, M.G.; Olivera, A.; Jung, M.Y.; Barber, J.S.; Freeman, A.F.; Holland, S.M.; O’Brien, M.; et al. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. J. Allergy Clin. Immunol. 2013, 132, 1388–1396. [Google Scholar] [CrossRef]
- Erlich, T.H.; Yagil, Z.; Kay, G.; Peretz, A.; Migalovich-Sheikhet, H.; Tshori, S.; Nechushtan, H.; Levi-Schaffer, F.; Saada, A.; Razin, E. Mitochondrial STAT3 plays a major role in IgE-antigen-mediated mast cell exocytosis. J. Allergy Clin. Immunol. 2014, 134, 460–469. [Google Scholar] [CrossRef]
- Pullen, N.A.; Barnstein, B.O.; Falanga, Y.T.; Wang, Z.; Suzuki, R.; Tamang, T.D.; Khurana, M.C.; Harry, E.A.; Draber, P.; Bunting, K.D.; et al. Novel mechanism for Fc{epsilon}RI-mediated signal transducer and activator of transcription 5 (STAT5) tyrosine phosphorylation and the selective influence of STAT5B over mast cell cytokine production. J. Biol. Chem. 2012, 287, 2045–2054. [Google Scholar] [CrossRef]
- Harir, N.; Boudot, C.; Friedbichler, K.; Sonneck, K.; Kondo, R.; Martin-Lannerée, S.; Kenner, L.; Kerenyi, M.; Yahiaoui, S.; Gouilleux-Gruart, V.; et al. Oncogenic Kit controls neoplastic mast cell growth through a Stat5/PI3-kinase signaling cascade. Blood 2008, 112, 2463–2473. [Google Scholar] [CrossRef] [PubMed]
- Barnstein, B.O.; Li, G.; Wang, Z.; Kennedy, S.; Chalfant, C.; Nakajima, H.; Bunting, K.D.; Ryan, J.J. Stat5 expression is required for IgE-mediated mast cell function. J. Immunol. 2006, 177, 3421–3426. [Google Scholar] [CrossRef] [PubMed]
- Shelburne, C.P.; McCoy, M.E.; Piekorz, R.; Sexl, V.; Roh, K.H.; Jacobs-Helber, S.M.; Gillespie, S.R.; Bailey, D.P.; Mirmonsef, P.; Mann, M.N.; et al. Stat5 expression is critical for mast cell development and survival. Blood 2003, 102, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Ando, T.; Xiao, W.; Gao, P.; Namiranian, S.; Matsumoto, K.; Tomimori, Y.; Hong, H.; Yamashita, H.; Kimura, M.; Kashiwakura, J.; et al. Critical role for mast cell Stat5 activity in skin inflammation. Cell Rep. 2014, 6, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.A.; Xi, L.; Cauff, B.; DeZure, A.; Freeman, A.F.; Hambleton, S.; Kleiner, G.; Leahy, T.R.; O’Sullivan, M.; Makiya, M.; et al. Somatic STAT5b gain-of-function mutations in early onset nonclonal eosinophilia, urticaria, dermatitis, and diarrhea. Blood 2017, 129, 650–653. [Google Scholar] [CrossRef]
- Masuda, A.; Matsuguchi, T.; Yamaki, K.; Hayakawa, T.; Yoshikai, Y. Interleukin-15 prevents mouse mast cell apoptosis through STAT6-mediated Bcl-xL expression. J. Biol. Chem. 2001, 276, 26107–26113. [Google Scholar] [CrossRef]
- Sherman, M.A. The role of STAT6 in mast cell IL-4 production. Immunol. Rev. 2001, 179, 48–56. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Xiang, M.; Weisberg, E.; Bar-Natan, M.; Barrett, R.; Liu, S.; Kharbanda, S.; Christie, A.L.; Nicolais, M.; et al. The STAT5 Inhibitor Pimozide Displays Efficacy in Models of Acute Myelogenous Leukemia Driven by FLT3 Mutations. Genes Cancer 2012, 3, 503–511. [Google Scholar] [CrossRef]
- Douglas, W.W.; Nemeth, E.F. On the calcium receptor activating exocytosis: Inhibitory effects of calmodulin-interacting drugs on rat mast cells. J. Physiol. 1982, 323, 229–244. [Google Scholar] [CrossRef]
- Hou, Y.B.; Zhang, L.N.; Wang, H.N.; Zhao, Z.F.; Sun, Y.T.; Ji, K.; Chen, J.J. The antipsychotic drug pimozide inhibits IgE-mediated mast cell degranulation and migration. Int. Immunopharmacol. 2020, 84, 106500. [Google Scholar] [CrossRef]
- Hox, V.; O’Connell, M.P.; Lyons, J.J.; Sackstein, P.; Dimaggio, T.; Jones, N.; Nelson, C.; Boehm, M.; Holland, S.M.; Freeman, A.F.; et al. Diminution of signal transducer and activator of transcription 3 signaling inhibits vascular permeability and anaphylaxis. J. Allergy Clin. Immunol. 2016, 138, 187–199. [Google Scholar] [CrossRef]
- Nagashima, S.; Hondo, T.; Nagata, H.; Ogiyama, T.; Maeda, J.; Hoshii, H.; Kontani, T.; Kuromitsu, S.; Ohga, K.; Orita, M.; et al. Novel 7H-pyrrolo[2,3-d]pyrimidine derivatives as potent and orally active STAT6 inhibitors. Bioorg. Med. Chem. 2009, 17, 6926–6936. [Google Scholar] [CrossRef]
- Li, X.; Han, Z.; Wang, F.; Qiao, J. The STAT6 inhibitor AS1517499 reduces the risk of asthma in mice with 2,4-dinitrochlorobenzene-induced atopic dermatitis by blocking the STAT6 signaling pathway. Allergy Asthma Clin. Immunol. 2022, 18, 12. [Google Scholar] [CrossRef]
- Yamaki, K.; Yoshino, S. Remission of food allergy by the Janus kinase inhibitor ruxolitinib in mice. Int. Immunopharmacol. 2014, 18, 217–224. [Google Scholar] [CrossRef]
- Yacoub, A.; Prochaska, L. Ruxolitinib improves symptoms and quality of life in a patient with systemic mastocytosis. Biomark Res. 2016, 4, 2. [Google Scholar] [CrossRef][Green Version]
- Li, Y.; Liu, X.; Yu, J.; Li, Z.; Chen, Y.; Li, H.; Chen, X.; Su, W.; Liang, D. Tofacitinib suppresses mast cell degranulation and attenuates experimental allergic conjunctivitis. Int. Immunopharmacol. 2020, 86, 106737. [Google Scholar] [CrossRef]
- de Mello Souza, C.H.; Shiomitsu, K.; Hwang, B. Cytokine production and the effects of oclacitinib in three canine mast cell tumour cell lines. Vet. Dermatol. 2022, 33, 159-e146. [Google Scholar] [CrossRef]
- Fukuyama, T.; Ehling, S.; Cook, E.; Bäumer, W. Topically Administered Janus-Kinase Inhibitors Tofacitinib and Oclacitinib Display Impressive Antipruritic and Anti-Inflammatory Responses in a Model of Allergic Dermatitis. J. Pharmacol. Exp. Ther. 2015, 354, 394–405. [Google Scholar] [CrossRef]
- Jin, W.; Huang, W.; Chen, L.; Jin, M.; Wang, Q.; Gao, Z.; Jin, Z. Topical Application of JAK1/JAK2 Inhibitor Momelotinib Exhibits Significant Anti-Inflammatory Responses in DNCB-Induced Atopic Dermatitis Model Mice. Int. J. Mol. Sci. 2018, 19, 3973–3986. [Google Scholar] [CrossRef]
- Levy, L.L.; Urban, J.; King, B.A. Treatment of recalcitrant atopic dermatitis with the oral Janus kinase inhibitor tofacitinib citrate. J. Am. Acad. Dermatol. 2015, 73, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Bissonnette, R.; Papp, K.A.; Poulin, Y.; Gooderham, M.; Raman, M.; Mallbris, L.; Wang, C.; Purohit, V.; Mamolo, C.; Papacharalambous, J.; et al. Topical tofacitinib for atopic dermatitis: A phase IIa randomized trial. Br. J. Dermatol. 2016, 175, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors: A 2022 update. Pharmacol. Res. 2022, 175, 106037. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Newcomb, D.C. Sex Bias in Asthma Prevalence and Pathogenesis. Front. Immunol. 2018, 9, 2997. [Google Scholar] [CrossRef]
- Yasar, P.; Ayaz, G.; User, S.D.; Gupur, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef]
- Zhu, T.H.; Ding, S.J.; Li, T.T.; Zhu, L.B.; Huang, X.F.; Zhang, X.M. Estrogen is an important mediator of mast cell activation in ovarian endometriomas. Reproduction 2018, 155, 73–83. [Google Scholar] [CrossRef]
- Zaitsu, M.; Narita, S.; Lambert, K.C.; Grady, J.J.; Estes, D.M.; Curran, E.M.; Brooks, E.G.; Watson, C.S.; Goldblum, R.M.; Midoro-Horiuti, T. Estradiol activates mast cells via a non-genomic estrogen receptor-alpha and calcium influx. Mol. Immunol. 2007, 44, 1977–1985. [Google Scholar] [CrossRef]
- Zhang, J.J.; Jacob, T.J.; Valverde, M.A.; Hardy, S.P.; Mintenig, G.M.; Sepúlveda, F.V.; Gill, D.R.; Hyde, S.C.; Trezise, A.E.; Higgins, C.F. Tamoxifen blocks chloride channels. A possible mechanism for cataract formation. J. Clin. Investig. 1994, 94, 1690–1697. [Google Scholar] [CrossRef]
- Kong, R.; Kang, O.H.; Seo, Y.S.; Zhou, T.; Kim, S.A.; Shin, D.W.; Kwon, D.Y. MAPKs and NF-κB pathway inhibitory effect of bisdemethoxycurcumin on phorbol-12-myristate-13-acetate and A23187-induced inflammation in human mast cells. Mol. Med. Rep. 2018, 17, 630–635. [Google Scholar] [CrossRef]
- Duffy, S.M.; Lawley, W.J.; Kaur, D.; Yang, W.; Bradding, P. Inhibition of human mast cell proliferation and survival by tamoxifen in association with ion channel modulation. J. Allergy Clin. Immunol. 2003, 112, 965–972. [Google Scholar] [CrossRef]
- Vliagoftis, H.; Dimitriadou, V.; Boucher, W.; Rozniecki, J.J.; Correia, I.; Raam, S.; Theoharides, T.C. Estradiol augments while tamoxifen inhibits rat mast cell secretion. Int. Arch. Allergy Immunol. 1992, 98, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.M.; Coquet, J.M.; Tibbitt, C.A. The Role of PPAR-gamma in Allergic Disease. Curr. Allergy Asthma Rep. 2021, 21, 45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Fang, S.; Zhu, Z.; Yao, M.; Ying, L.; Zhu, L.; Ma, Z.; Wang, W. Peroxisome proliferator-activated receptor gamma agonist suppresses mast cell maturation and induces apoptosis. Mol. Med. Rep. 2017, 16, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Kasakura, K.; Miura, R.; Yashiro, T.; Nishiyama, C. Suppressive role of PPARγ in the IgE-dependent activation of mast cells. Int. Immunol. 2020, 32, 143–150. [Google Scholar] [CrossRef]
- Narala, V.R.; Ranga, R.; Smith, M.R.; Berlin, A.A.; Standiford, T.J.; Lukacs, N.W.; Reddy, R.C. Pioglitazone is as effective as dexamethasone in a cockroach allergen-induced murine model of asthma. Respir. Res. 2007, 8, 90. [Google Scholar] [CrossRef]
- Yao, L.; Gu, Y.; Jiang, T.; Che, H. Inhibition effect of PPAR-γ signaling on mast cell-mediated allergic inflammation through down-regulation of PAK1/ NF-κB activation. Int. Immunopharmacol. 2022, 108, 108692. [Google Scholar] [CrossRef]
- Dong, H.; Zhang, X.; Qian, Y. Mast cells and neuroinflammation. Med. Sci. Monit. Basic Res. 2014, 20, 200–206. [Google Scholar] [CrossRef]
- Mori, T.; Kabashima, K.; Fukamachi, S.; Kuroda, E.; Sakabe, J.; Kobayashi, M.; Nakajima, S.; Nakano, K.; Tanaka, Y.; Matsushita, S.; et al. D1-like dopamine receptors antagonist inhibits cutaneous immune reactions mediated by Th2 and mast cells. J. Dermatol. Sci. 2013, 71, 37–44. [Google Scholar] [CrossRef]
- Heo, M.J.; Choi, S.Y.; Lee, C.; Choi, Y.M.; An, I.S.; Bae, S.; An, S.; Jung, J.H. Perphenazine Attenuates the Pro-Inflammatory Responses in Mouse Models of Th2-Type Allergic Dermatitis. Int. J. Mol. Sci. 2020, 21, 3241–3251. [Google Scholar] [CrossRef]
- Li, W.; Liu, F.; Wang, J.; Long, M.; Wang, Z. MicroRNA-21-Mediated Inhibition of Mast Cell Degranulation Involved in the Protective Effect of Berberine on 2,4-Dinitrofluorobenzene-Induced Allergic Contact Dermatitis in Rats via p38 Pathway. Inflammation 2018, 41, 689–699. [Google Scholar] [CrossRef]
- Kim, B.Y.; Park, H.R.; Jeong, H.G.; Kim, S.W. Berberine reduce allergic inflammation in a house dust mite allergic rhinitis mouse model. Rhinology 2015, 53, 353–358. [Google Scholar] [CrossRef]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef]
- Endo, A.; Kuroda, M.; Tsujita, Y. ML-236A, ML-236B, and ML-236C, new inhibitors of cholesterogensis produced by Penicillium citrinum. J. Antibiot. 1976, 29, 1346–1348. [Google Scholar] [CrossRef]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B 2010, 86, 484–493. [Google Scholar] [CrossRef]
- Hoffman, W.F.; Alberts, A.W.; Anderson, P.S.; Chen, J.S.; Smith, R.L.; Willard, A.K. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. 4. Side-chain ester derivatives of mevinolin. J. Med. Chem. 1986, 29, 849–852. [Google Scholar] [CrossRef]
- Lee, T.J.; Holtz, W.J.; Smith, R.L.; Alberts, A.W.; Gilfillan, J.L. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors. 8. Side chain ether analogs of lovastatin. J. Med. Chem. 1991, 34, 2474–2477. [Google Scholar] [CrossRef]
- Tulbah, A.S. The potential of Atorvastatin for chronic lung diseases therapy. Saudi Pharm. J. 2020, 28, 1353–1363. [Google Scholar] [CrossRef]
- Ramkumar, S.; Raghunath, A.; Raghunath, S. Statin Therapy: Review of Safety and Potential Side Effects. Acta Cardiol. Sin. 2016, 32, 631–639. [Google Scholar] [CrossRef]
- Greenwood, J.; Steinman, L.; Zamvil, S.S. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat. Rev. Immunol. 2006, 6, 358–370. [Google Scholar] [CrossRef]
- Montecucco, F.; Burger, F.; Pelli, G.; Poku, N.K.; Berlier, C.; Steffens, S.; Mach, F. Statins inhibit C-reactive protein-induced chemokine secretion, ICAM-1 upregulation and chemotaxis in adherent human monocytes. Rheumatology 2009, 48, 233–242. [Google Scholar] [CrossRef]
- Diamantis, E.; Kyriakos, G.; Quiles-Sanchez, L.V.; Farmaki, P.; Troupis, T. The Anti-Inflammatory Effects of Statins on Coronary Artery Disease: An Updated Review of the Literature. Curr. Cardiol. Rev. 2017, 13, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Janda, S.; Young, A.; FitzGerald, J.M.; Etminan, M.; Swiston, J. The effect of statins on mortality from severe infections and sepsis: A systematic review and meta-analysis. J. Crit. Care 2010, 25, 656.e7–e22. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Y.; Yao, T.-C.; Tsai, Y.-T.; Wu, A.C.; Tsai, H.-J. Increased Dose and Duration of Statin Use Is Associated with Decreased Asthma-Related Emergency Department Visits and Hospitalizations. J. Allergy Clin. Immunol. Pract. 2018, 6, 1588–1595. [Google Scholar] [CrossRef]
- Zeki, A.A.; Franzi, L.; Last, J.; Kenyon, N.J. Simvastatin Inhibits Airway Hyperreactivity: Implications for the Mevalonate Pathway and Beyond. Am. J. Respir. Crit. Care Med. 2009, 180, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Quist-Paulsen, P. Statins and inflammation: An update. Curr. Opin. Cardiol. 2010, 25, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-W.; Kang, H.-J.; Jhon, M.; Kim, J.-W.; Lee, J.-Y.; Walker, A.J.; Agustini, B.; Kim, J.-M.; Berk, M. Statins and Inflammation: New Therapeutic Opportunities in Psychiatry. Front. Psychiatry 2019, 10, 103. [Google Scholar] [CrossRef]
- Link, A.; Ayadhi, T.; Bohm, M.; Nickenig, G. Rapid immunomodulation by rosuvastatin in patients with acute coronary syndrome. Eur. Heart J. 2006, 27, 2945–2955. [Google Scholar] [CrossRef]
- Kolawole, E.M.; McLeod, J.J.A.; Ndaw, V.; Abebayehu, D.; Barnstein, B.O.; Faber, T.; Spence, A.J.; Taruselli, M.; Paranjape, A.; Haque, T.T.; et al. Fluvastatin Suppresses Mast Cell and Basophil IgE Responses: Genotype-Dependent Effects. J. Immunol. 2016, 196, 1461–1470. [Google Scholar] [CrossRef]
- Paez, P.A.; Kolawole, M.; Taruselli, M.T.; Ajith, S.; Dailey, J.M.; Kee, S.A.; Haque, T.T.; Barnstein, B.O.; McLeod, J.J.A.; Caslin, H.L.; et al. Fluvastatin Induces Apoptosis in Primary and Transformed Mast Cells. J. Pharmacol. Exp. Ther. 2020, 374, 104–112. [Google Scholar] [CrossRef]
- Yilmaz, A.; Reiss, C.; Tantawi, O.; Weng, A.; Stumpf, C.; Raaz, D.; Ludwig, J.; Berger, T.; Steinkasserer, A.; Daniel, W.G.; et al. HMG-CoA reductase inhibitors suppress maturation of human dendritic cells: New implications for atherosclerosis. Atherosclerosis 2004, 172, 85–93. [Google Scholar] [CrossRef]
- Forero-Peña, D.A.; Gutierrez, F.R.S. Statins as Modulators of Regulatory T-Cell Biology. Mediat. Inflamm. 2013, 2013, 167086. [Google Scholar] [CrossRef] [PubMed]
- Healy, A.; Berus, J.M.; Christensen, J.L.; Lee, C.; Mantsounga, C.; Dong, W.; Watts, J.P.; Assali, M.; Ceneri, N.; Nilson, R.; et al. Statins Disrupt Macrophage Rac1 Regulation Leading to Increased Atherosclerotic Plaque Calcification. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 714–732. [Google Scholar] [CrossRef]
- Fujimoto, M.; Oka, T.; Murata, T.; Hori, M.; Ozaki, H. Fluvastatin inhibits mast cell degranulation without changing the cytoplasmic Ca2+ level. Eur. J. Pharmacol. 2009, 602, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Reiner, Ž. Resistance and intolerance to statins. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Ness, G.C.; Chambers, C.M.; Lopez, D. Atorvastatin action involves diminished recovery of hepatic HMG-CoA reductase activity. J. Lipid Res. 1998, 39, 75–84. [Google Scholar] [CrossRef]
- Chasman, D.I. Pharmacogenetic Study of Statin Therapy and Cholesterol Reduction. JAMA 2004, 291, 2821. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Wee, J.H.; Choi, H.G.; Park, J.Y.; Hwang, Y.I.; Jang, S.H.; Jung, K.S. Association Between Statin Medication and Asthma/Asthma Exacerbation in a National Health Screening Cohort. J. Allergy Clin. Immunol. Pract 2021, 9, 2783–2791. [Google Scholar] [CrossRef]
- Tse, S.M.; Li, L.; Butler, M.G.; Fung, V.; Kharbanda, E.O.; Larkin, E.K.; Vollmer, W.M.; Miroshnik, I.; Rusinak, D.; Weiss, S.T.; et al. Statin exposure is associated with decreased asthma-related emergency department visits and oral corticosteroid use. Am. J. Respir. Crit. Care Med. 2013, 188, 1076–1082. [Google Scholar] [CrossRef]
- Ostroukhova, M.; Kouides, R.W.; Friedman, E. The effect of statin therapy on allergic patients with asthma. Ann. Allergy Asthma Immunol. 2009, 103, 463–468. [Google Scholar] [CrossRef]
- Si, X.B.; Zhang, S.; Huo, L.Y.; Dai, W.L.; Wang, H.L. Statin therapy does not improve lung function in asthma: A meta-analysis of randomized controlled trials. J. Int. Med. Res. 2013, 41, 276–283. [Google Scholar] [CrossRef]
- Zeki, A.A.; Oldham, J.; Wilson, M.; Fortenko, O.; Goyal, V.; Last, M.; Last, A.; Patel, A.; Last, J.A.; Kenyon, N.J. Statin use and asthma control in patients with severe asthma. BMJ Open 2013, 3, e003314. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Chan, W.L.; Chen, Y.C.; Chen, T.J.; Chou, K.T.; Lin, S.J.; Chen, J.W.; Leu, H.B. Statin use in patients with asthma: A nationwide population-based study. Eur J. Clin. Investig. 2011, 41, 507–512. [Google Scholar] [CrossRef]
- Yeh, J.J.; Syue, S.H.; Lin, C.L.; Hsu, C.Y.; Shae, Z.; Kao, C.H. Statin use and Vital Organ Failure in Patients With Asthma-Chronic Obstructive Pulmonary Disease Overlap: A Time-Dependent Population-Based Study. Front. Pharmacol. 2019, 10, 889. [Google Scholar] [CrossRef]
- Jeong, A.; Suazo, K.F.; Wood, W.G.; Distefano, M.D.; Li, L. Isoprenoids and protein prenylation: Implications in the pathogenesis and therapeutic intervention of Alzheimer’s disease. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 279–310. [Google Scholar] [CrossRef] [PubMed]
- Casey, P.J.; Seabra, M.C. Protein Prenyltransferases. J. Biol. Chem. 1996, 271, 5289–5292. [Google Scholar] [CrossRef] [PubMed]
- Kalinin, A.; Thomä, N.H.; Iakovenko, A.; Heinemann, I.; Rostkova, E.; Constantinescu, A.T.; Alexandrov, K. Expression of Mammalian Geranylgeranyltransferase Type-II in Escherichia coli and Its Application for in Vitro Prenylation of Rab Proteins. Protein Expr. Purif. 2001, 22, 84–91. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef]
- Wang, M.; Casey, P.J. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Inagaki-Katashiba, N.; Ito, T.; Inaba, M.; Azuma, Y.; Tanaka, A.; Phan, V.; Kibata, K.; Satake, A.; Nomura, S. Statins can suppress DC-mediated Th2 responses through the repression of OX40-ligand and CCL17 expression. Eur. J. Immunol. 2019, 49, 2051–2062. [Google Scholar] [CrossRef]
- Takeda, N.; Kondo, M.; Ito, S.; Ito, Y.; Shimokata, K.; Kume, H. Role of RhoA Inactivation in Reduced Cell Proliferation of Human Airway Smooth Muscle by Simvastatin. Am. J. Respir. Cell Mol. Biol. 2006, 35, 722–729. [Google Scholar] [CrossRef]
- Srikanth, S.; Kim, K.D.; Gao, Y.; Woo, J.S.; Ghosh, S.; Calmettes, G.; Paz, A.; Abramson, J.; Jiang, M.; Gwack, Y. A large Rab GTPase encoded by CRACR2A is a component of subsynaptic vesicles that transmit T cell activation signals. Sci. Signal. 2016, 9, ra31. [Google Scholar] [CrossRef] [PubMed]
- Storck, E.M.; Morales-Sanfrutos, J.; Serwa, R.A.; Panyain, N.; Lanyon-Hogg, T.; Tolmachova, T.; Ventimiglia, L.N.; Martin-Serrano, J.; Seabra, M.C.; Wojciak-Stothard, B.; et al. Dual chemical probes enable quantitative system-wide analysis of protein prenylation and prenylation dynamics. Nat. Chem. 2019, 11, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Baier, A.; Ndoh, V.N.E.; Lacy, P.; Eitzen, G. Rac1 and Rac2 control distinct events during antigen-stimulated mast cell exocytosis. J. Leukoc. Biol. 2014, 95, 763–774. [Google Scholar] [CrossRef]
- Sheshachalam, A.; Baier, A.; Eitzen, G. The effect of Rho drugs on mast cell activation and degranulation. J. Leukoc. Biol. 2017, 102, 71–81. [Google Scholar] [CrossRef]
- Turner, H.; Cantrell, D.A. Distinct Ras Effector Pathways Are Involved in FcεR1 Regulation of the Transcriptional Activity of Elk-1 and NFAT in Mast Cells. J. Exp. Med. 1997, 185, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Mor, A.; Shefler, I.; Salamon, P.; Kloog, Y.; Mekori, Y.A. Characterization of ERK Activation in Human Mast Cells Stimulated by Contact with T Cells. Inflammation 2010, 33, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, W.F.; Yang, F.C.; Chen, S.; White, H.; Bessler, W.; Ingram, D.A.; Clapp, D.W. K-ras is critical for modulating multiple c-kit-mediated cellular functions in wild-type and Nf1+/− mast cells. J. Immunol. 2007, 178, 2527–2534. [Google Scholar] [CrossRef]
- Dwyer, D.F.; Barrett, N.A.; Austen, K.F. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 2016, 17, 878–887. [Google Scholar] [CrossRef]
- Fang, X.; Lang, Y.; Wang, Y.; Mo, W.; Wei, H.; Xie, J.; Yu, M. Shp2 Activates Fyn and Ras to Regulate RBL-2H3 Mast Cell Activation following FcεRI Aggregation. PLoS ONE 2012, 7, e40566. [Google Scholar] [CrossRef]
- Yamasaki, S.; Saito, T. Progress in Allergy Signal Research on Mast Cells: Signal Regulation of Multiple Mast Cell Responses Through FcεRI. J. Pharmacol. Sci. 2008, 106, 336–340. [Google Scholar] [CrossRef]
- Kuramasu, A.; Wakabayashi, M.; Inui, M.; Yanai, K. Distinct Roles of Small GTPases Rac1 and Rac2 in Histamine H(4) Receptor-Mediated Chemotaxis of Mast Cells. J. Pharmacol. Exp. Ther. 2018, 367, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.-C.; Kapur, R.; King, A.J.; Tao, W.; Kim, C.; Borneo, J.; Breese, R.; Marshall, M.; Dinauer, M.C.; Williams, D.A. Rac2 Stimulates Akt Activation Affecting BAD/Bcl-XL Expression while Mediating Survival and Actin Function in Primary Mast Cells. Immunity 2000, 12, 557–568. [Google Scholar] [CrossRef]
- Hong-Geller, E.; Holowka, D.; Siraganian, R.P.; Baird, B.; Cerione, R.A. Activated Cdc42/Rac reconstitutes Fcepsilon RI-mediated Ca2+ mobilization and degranulation in mutant RBL mast cells. Proc. Natl. Acad. Sci. USA 2001, 98, 1154–1159. [Google Scholar] [CrossRef]
- Riedel, D.; Antonin, W.; Fernandez-Chacon, R.; Alvarez de Toledo, G.; Jo, T.; Geppert, M.; Valentijn, J.A.; Valentijn, K.; Jamieson, J.D.; Südhof, T.C.; et al. Rab3D Is Not Required for Exocrine Exocytosis but for Maintenance of Normally Sized Secretory Granules. Mol. Cell. Biol. 2002, 22, 6487–6497. [Google Scholar] [CrossRef] [PubMed]
- Azouz, N.P.; Zur, N.; Efergan, A.; Ohbayashi, N.; Fukuda, M.; Amihai, D.; Hammel, I.; Rothenberg, M.E.; Sagi-Eisenberg, R. Rab5 Is a Novel Regulator of Mast Cell Secretory Granules: Impact on Size, Cargo, and Exocytosis. J. Immunol. 2014, 192, 4043–4053. [Google Scholar] [CrossRef] [PubMed]
- Karasic, T.B.; Chiorean, E.G.; Sebti, S.M.; O’Dwyer, P.J. A Phase I Study of GGTI-2418 (Geranylgeranyl Transferase I Inhibitor) in Patients with Advanced Solid Tumors. Target. Oncol. 2019, 14, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III Trial of Gemcitabine Plus Tipifarnib Compared With Gemcitabine Plus Placebo in Advanced Pancreatic Cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Marasine, N.R.; Sankhi, S.; Lamichhane, R.; Marasini, N.R.; Dangi, N.B. Use of Antidepressants among Patients Diagnosed with Depression: A Scoping Review. Biomed. Res. Int. 2021, 2021, 6699028. [Google Scholar] [CrossRef]
- Brody, D.J.; Gu, Q. Antidepressant Use Among Adults: United States, 2015–2018; US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Health Statistics: Washington, DC, USA, 2020.
- Dregan, A.; Matcham, F.; Harber-Aschan, L.; Rayner, L.; Brailean, A.; Davis, K.; Hatch, S.; Pariante, C.; Armstrong, D.; Stewart, R.; et al. Common mental disorders within chronic inflammatory disorders: A primary care database prospective investigation. Ann. Rheum. Dis. 2019, 78, 688–695. [Google Scholar] [CrossRef]
- Felger, J.C. The Role of Dopamine in Inflammation-Associated Depression: Mechanisms and Therapeutic Implications. Curr Top Behav. Neurosci. 2017, 31, 199–219. [Google Scholar] [CrossRef]
- Koutsouraki, E.; Hatzifilipou, E.; Michmizos, D.; Cotsavasiloglou, C.; Costa, V.; Baloyannis, S. Increase in interleukin-6 levels is related to depressive phenomena in the acute (relapsing) phase of multiple sclerosis. J. Neuropsychiatry Clin. Neurosci. 2011, 23, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, H.; Kato, T.A.; Mizoguchi, Y.; Monji, A.; Seki, Y.; Ohkuri, T.; Gotoh, L.; Yonaha, M.; Ueda, T.; Hashioka, S.; et al. Inhibitory effects of SSRIs on IFN-gamma induced microglial activation through the regulation of intracellular calcium. Prog. Neuropsychopharmacol. Biol. Psychiatry 2010, 34, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Kohler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: A systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Sukhatme, V.P.; Reiersen, A.M.; Vayttaden, S.J.; Sukhatme, V.V. Fluvoxamine: A Review of Its Mechanism of Action and Its Role in COVID-19. Front. Pharmacol. 2021, 12, 652688. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, D.; Dhabhar, F.S.; James, S.J.; Hough, C.M.; Jain, F.A.; Bersani, F.S.; Reus, V.I.; Verhoeven, J.E.; Epel, E.S.; Mahan, L.; et al. Oxidative stress, inflammation and treatment response in major depression. Psychoneuroendocrinology 2017, 76, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Deshwal, S.; Forkink, M.; Hu, C.H.; Buonincontri, G.; Antonucci, S.; Di Sante, M.; Murphy, M.P.; Paolocci, N.; Mochly-Rosen, D.; Krieg, T.; et al. Monoamine oxidase-dependent endoplasmic reticulum-mitochondria dysfunction and mast cell degranulation lead to adverse cardiac remodeling in diabetes. Cell Death Differ. 2018, 25, 1671–1685. [Google Scholar] [CrossRef]
- Sitges, M.; Gomez, C.D.; Aldana, B.I. Sertraline reduces IL-1beta and TNF-alpha mRNA expression and overcomes their rise induced by seizures in the rat hippocampus. PLoS ONE 2014, 9, e111665. [Google Scholar] [CrossRef]
- Rossi, A.; Barraco, A.; Donda, P. Fluoxetine: A review on evidence based medicine. Ann. Gen. Psychiatry 2004, 3, 2. [Google Scholar] [CrossRef]
- Sherkawy, M.M.; Abo-Youssef, A.M.; Salama, A.A.A.; Ismaiel, I.E. Fluoxetine protects against OVA induced bronchial asthma and depression in rats. Eur. J. Pharmacol. 2018, 837, 25–32. [Google Scholar] [CrossRef]
- Kubera, M.; Simbirtsev, A.; Mathison, R.; Maes, M. Effects of repeated fluoxetine and citalopram administration on cytokine release in C57BL/6 mice. Psychiatry Res. 2000, 96, 255–266. [Google Scholar] [CrossRef]
- Sharbaf Shoar, N.; Fariba, K.A.; Padhy, R.K. Citalopram. [Updated 11 December 2021]. In StatPearls; [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2021. Available online: https://www.ncbi.nlm.nih.gov/books/NBK482222/ (accessed on 19 September 2022).
- Brown, E.S.; Sayed, N.; Van Enkevort, E.; Kulikova, A.; Nakamura, A.; Khan, D.A.; Ivleva, E.I.; Sunderajan, P.; Bender, B.G.; Holmes, T. A Randomized, Double-Blind, Placebo-Controlled Trial of Escitalopram in Patients with Asthma and Major Depressive Disorder. J. Allergy Clin. Immunol. Pract. 2018, 6, 1604–1612. [Google Scholar] [CrossRef] [PubMed]
- Akulova, M.N.; Ovcharenko, S.I.; Ishchenko, E.N. Is it possible to decrease asthma severity by use of antidepressants? Eur. Respir. J. 2011, 38, 523. [Google Scholar]
- Håkansson, K.E.J.; Renzi-Lomholt, M.; Backer, V.; Ulrik, C.S. High Use of Antidepressant Medication in Both Mild-to-Modelate and Possible Severe Asthma—A Nationwide Cohort Study. J. Asthma Allergy 2022, 15, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhen, H.; Yao, W.; Bian, F.; Mao, X.; Yang, X.; Jin, S. Antidepressant drug, desipramine, alleviates allergic rhinitis by regulating Treg and Th17 cells. Int. J. Immunopathol. Pharmacol. 2013, 26, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Nazimek, K.; Kozlowski, M.; Bryniarski, P.; Strobel, S.; Bryk, A.; Myszka, M.; Tyszka, A.; Kuszmiersz, P.; Nowakowski, J.; Filipczak-Bryniarska, I. Repeatedly administered antidepressant drugs modulate humoral and cellular immune response in mice through action on macrophages. Exp. Biol. Med. (Maywood) 2016, 241, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, P.; Nessler, S.; Kalluri, S.R.; Hartung, H.P.; Hemmer, B. The antidepressant venlafaxine ameliorates murine experimental autoimmune encephalomyelitis by suppression of pro-inflammatory cytokines. Int. J. Neuropsychopharmacol. 2009, 12, 525–536. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Kempuraj, D. Important role of mast cells in multiple sclerosis. Mult. Scler. Relat. Disord. 2016, 5, 77–80. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.O.; Pullen, N.A. Beyond IgE: Alternative Mast Cell Activation Across Different Disease States. Int. J. Mol. Sci. 2020, 21, 1498. [Google Scholar] [CrossRef]
- Cianferoni, A. Non-IgE-mediated anaphylaxis. J. Allergy Clin. Immunol. 2021, 147, 1123–1131. [Google Scholar] [CrossRef]
- Falduto, G.H.; Pfeiffer, A.; Luker, A.; Metcalfe, D.D.; Olivera, A. Emerging mechanisms contributing to mast cell-mediated pathophysiology with therapeutic implications. Pharmacol. Ther. 2021, 220, 107718. [Google Scholar] [CrossRef]
- Kumar, M.; Duraisamy, K.; Chow, B.K. Unlocking the Non-IgE-Mediated Pseudo-Allergic Reaction Puzzle with Mas-Related G-Protein Coupled Receptor Member X2 (MRGPRX2). Cells 2021, 10, 1033. [Google Scholar] [CrossRef] [PubMed]
- Tatemoto, K.; Nozaki, Y.; Tsuda, R.; Konno, S.; Tomura, K.; Furuno, M.; Ogasawara, H.; Edamura, K.; Takagi, H.; Iwamura, H.; et al. Immunoglobulin E-independent activation of mast cell is mediated by Mrg receptors. Biochem. Biophys. Res. Commun. 2006, 349, 1322–1328. [Google Scholar] [CrossRef] [PubMed]
- McNeil, B.D.; Pundir, P.; Meeker, S.; Han, L.; Undem, B.J.; Kulka, M.; Dong, X. Identification of a mast-cell-specific receptor crucial for pseudo-allergic drug reactions. Nature 2015, 519, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, H.; Noguchi, M. Therapeutic Potential of MRGPRX2 Inhibitors on Mast Cells. Cells 2021, 10, 2906. [Google Scholar] [CrossRef] [PubMed]
- Tsvilovskyy, V.; Solis-Lopez, A.; Almering, J.; Richter, C.; Birnbaumer, L.; Dietrich, A.; Freichel, M. Analysis of Mrgprb2 Receptor-Evoked Ca2+ Signaling in Bone Marrow Derived (BMMC) and Peritoneal (PMC) Mast Cells of TRPC-Deficient Mice. Front. Immunol. 2020, 11, 564. [Google Scholar] [CrossRef]
- Subramanian, H.; Gupta, K.; Ali, H. Roles of Mas-related G protein-coupled receptor X2 on mast cell-mediated host defense, pseudoallergic drug reactions, and chronic inflammatory diseases. J. Allergy Clin. Immunol. 2016, 138, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Wang, J.; Liu, R.; Zhang, G.; Dong, K.; Zhang, T. Paeoniflorin inhibits MRGPRX2-mediated pseudo-allergic reaction via calcium signaling pathway. Phytother. Res. 2020, 34, 401–408. [Google Scholar] [CrossRef]
- Ding, Y.; Che, D.; Li, C.; Cao, J.; Wang, J.; Ma, P.; Zhao, T.; An, H.; Zhang, T. Quercetin inhibits Mrgprx2-induced pseudo-allergic reaction via PLCγ-IP3R related Ca2+ fluctuations. Int. Immunopharmacol. 2019, 66, 185–197. [Google Scholar] [CrossRef]
- Azimi, E.; Reddy, V.B.; Shade, K.C.; Anthony, R.M.; Talbot, S.; Pereira, P.J.S.; Lerner, E.A. Dual action of neurokinin-1 antagonists on Mas-related GPCRs. JCI Insight 2016, 1, e89362. [Google Scholar] [CrossRef]
- Suzuki, Y.; Liu, S.; Ogasawara, T.; Sawasaki, T.; Takasaki, Y.; Yorozuya, T.; Mogi, M. A novel MRGPRX2-targeting antagonistic DNA aptamer inhibits histamine release and prevents mast cell-mediated anaphylaxis. Eur. J. Pharmacol. 2020, 878, 173104. [Google Scholar] [CrossRef]
- Ogasawara, H.; Furuno, M.; Edamura, K.; Noguchi, M. Novel MRGPRX2 antagonists inhibit IgE-independent activation of human umbilical cord blood-derived mast cells. J. Leukoc. Biol. 2019, 106, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Centers for Disease Control and Prevention 2022; Most Recent National Asthma Data Centers for Disease Control and Prevention: Atlanta, GA, USA, 2022. Available online: https://www.cdc.gov/asthma/most_recent_national_asthma_data.htm (accessed on 19 September 2022).
- Gupta, R.S.; Warren, C.M.; Smith, B.M.; Blumenstock, J.A.; Jiang, J.; Davis, M.M.; Nadeau, K.C. The Public Health Impact of Parent-Reported Childhood Food Allergies in the United States. Pediatrics 2018, 142, e20181235. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Warren, C.M.; Smith, B.M.; Jiang, J.; Blumenstock, J.A.; Davis, M.M.; Schleimer, R.P.; Nadeau, K.C. Prevalence and Severity of Food Allergies Among US Adults. JAMA Netw. Open 2019, 2, e185630. [Google Scholar] [CrossRef]
- Warren, C.M.; Brewer, A.G.; Grobman, B.; Jiang, J.; Gupta, R.S. Racial/Ethnic Differences in Food Allergy. Immunol. Allergy Clin. N. Am. 2021, 41, 189–203. [Google Scholar] [CrossRef] [PubMed]
- MacGlashan, D.W., Jr.; Bochner, B.S.; Adelman, D.C.; Jardieu, P.M.; Togias, A.; McKenzie-White, J.; Sterbinsky, S.A.; Hamilton, R.G.; Lichtenstein, L.M. Down-regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients with anti-IgE antibody. J. Immunol. 1997, 158, 1438–1445. [Google Scholar]
- Holgate, S.; Casale, T.; Wenzel, S.; Bousquet, J.; Deniz, Y.; Reisner, C. The anti-inflammatory effects of omalizumab confirm the central role of IgE in allergic inflammation. J. Allergy Clin. Immunol. 2005, 115, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Buendia, J.A.; Guerrero Patiño, D.; Cossio-Giraldo, Y.E. Cost-effectiveness of tiotropium versus omalizumab for uncontrolled allergic asthma. J. Asthma 2021, 16, 1–8. [Google Scholar] [CrossRef]
- Jourdan, J.P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Drug | Target | FDA Status |
|---|---|---|
| Omalizumab | IgE | Approved for some allergic disease |
| Ligelizumab | IgE | Phase 3 trials for allergic disease |
| Dupilumab | IL-4Ra | Approved for some allergic disease |
| Rituximab | CD20 | Approved for some autoimmune diseases and B cell malignancies |
| Lirentelimab | Siglec-8 | Phase 3 trials for allergic disease |
| MTPS9579A | Tryptase | Phase 2 trials for allergic disease |
| CDX-0159 | c-Kit | Phase 1 and 2 trials for allergic disease |
| Drug | Target | FDA Status |
|---|---|---|
| Imatinib | BCR-ABL | Approved for Chronic Myeloid Leukemia (CML); Phase 2 clinical trial for Asthma |
| Nilotinib | BCR-ABL | Approved for CML |
| Dasatinib | BCR-ABL | Approved for CML and Philadelphia chromosome-positive acute lymphoblastic leukemia (ALL) |
| Bosutinib | BCR-ABL | Accelerated approval for CML and ALL |
| Ponatinib | BCR-ABL | Approved for CML and ALL |
| Ibrutinib | BTK | Multiple FDA approvals; Phase 2 clinical trial for Food Allergy |
| Acalabrutinib | BTK | Approved for Chronic lymphocytic leukemia (CLL) and Small lymphocytic lymphoma (SLL); Phase 2 clinical trials for Food Allergy |
| AZD7762 | Lyn/Fyn/Syk | Phase I clinical trials for Solid Tumors |
| WZ3146 | Lyn/Fyn/Syk | No US trials listed |
| Dasatinib | Lyn/Fyn/Syk | Approved for leukemia |
| Pictilisib | PI3K | Multiple clinical trials for cancer treatment |
| MK2206 | Akt | Multiple clinical trials |
| Rapamycin | mTOR | Multiple FDA approvals |
| MHY1485 | mTOR | No US trials listed |
| Compound | Target | FDA Status |
|---|---|---|
| DHMQ | NFkB | No US trials listed |
| Pyrrolidine dithiocarbamate | NFkB | No US trials listed |
| Tacrolimus (FK506) | Calcineurin | Multiple FDA approvals |
| Cyclosporine A | Calcineurin | Multiple FDA approvals |
| Pimozide | STAT5 (off-target) | Approved for Tourette’s syndrome |
| C188-9 (TTI-101) | STAT3 | Phase 1 for multiple cancers |
| AS1517499 | STAT6 | No US trials listed |
| Ruxolitinib | JAK1/2 | Multiple FDA approvals |
| Tofacitinib | JAK3 | Multiple FDA approvals |
| Oclacitinib | JAK1 | Approved for use in canines |
| Momelotinib | JAK1/2 | Phase 3 for myelofibrosis |
| Upadacitinib | JAK1 | Multiple FDA approvals |
| Abrocitinib | JAK1 | Approved for atopic dermatitis |
| Tamoxifen | Estrogen E2 receptor | Approved for breast cancer |
| Perphenazine | Dopamine D1 and D2 receptors | Approved for psychotic disorders and severe nausea |
| Berberine | Dopamine D1 and D2 receptors | Multiple stages of trials for metabolic disorders |
| Pioglitazone | PPAR-g (agonist) | Approved for Type 2 diabetes |
| Rosiglitazone | PPAR-g (agonist) | Approved for Type 2 diabetes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burchett, J.R.; Dailey, J.M.; Kee, S.A.; Pryor, D.T.; Kotha, A.; Kankaria, R.A.; Straus, D.B.; Ryan, J.J. Targeting Mast Cells in Allergic Disease: Current Therapies and Drug Repurposing. Cells 2022, 11, 3031. https://doi.org/10.3390/cells11193031
Burchett JR, Dailey JM, Kee SA, Pryor DT, Kotha A, Kankaria RA, Straus DB, Ryan JJ. Targeting Mast Cells in Allergic Disease: Current Therapies and Drug Repurposing. Cells. 2022; 11(19):3031. https://doi.org/10.3390/cells11193031
Chicago/Turabian StyleBurchett, Jason R., Jordan M. Dailey, Sydney A. Kee, Destiny T. Pryor, Aditya Kotha, Roma A. Kankaria, David B. Straus, and John J. Ryan. 2022. "Targeting Mast Cells in Allergic Disease: Current Therapies and Drug Repurposing" Cells 11, no. 19: 3031. https://doi.org/10.3390/cells11193031
APA StyleBurchett, J. R., Dailey, J. M., Kee, S. A., Pryor, D. T., Kotha, A., Kankaria, R. A., Straus, D. B., & Ryan, J. J. (2022). Targeting Mast Cells in Allergic Disease: Current Therapies and Drug Repurposing. Cells, 11(19), 3031. https://doi.org/10.3390/cells11193031