

Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance

Abstract

1. Introduction
2. Therapeutic Options for PDAC Patients
3. Hallmarks of the Pancreatic Tumor Microenvironment
4. Pancreatic Stellate Cells and Therapy Resistance
5. Cancer-Associated Fibroblasts and Therapy Resistance
6. Tumor-Associated Macrophages and Therapy Resistance
7. Tumor-Associated Neutrophils and Therapy Resistance
8. Perspectives, Challenges, and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bengtsson, A.; Andersson, R.; Ansari, D. The actual 5-year survivors of pancreatic ductal adenocarcinoma based on real-world data. Sci. Rep. 2020, 10, 16425. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef]
- Hou, P.; Wang, Y.A. Conquering oncogenic KRAS and its bypass mechanisms. Theranostics 2022, 12, 5691–5709. [Google Scholar] [CrossRef]
- Ying, H.; Dey, P.; Yao, W.; Kimmelman, A.C.; Draetta, G.F.; Maitra, A.; DePinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2016, 30, 355–385. [Google Scholar] [CrossRef]
- Torres, C.; Grippo, P.J. Pancreatic cancer subtypes: A roadmap for precision medicine. Ann. Med. 2018, 50, 277–287. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]
- Truong, L.H.; Pauklin, S. Pancreatic Cancer Microenvironment and Cellular Composition: Current Understandings and Therapeutic Approaches. Cancers 2021, 13, 5028. [Google Scholar] [CrossRef]
- Opitz, F.V.; Haeberle, L.; Daum, A.; Esposito, I. Tumor Microenvironment in Pancreatic Intraepithelial Neoplasia. Cancers 2021, 13, 6188. [Google Scholar] [CrossRef]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Meads, M.B.; Gatenby, R.A.; Dalton, W.S. Environment-mediated drug resistance: A major contributor to minimal residual disease. Nat. Rev. Cancer 2009, 9, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, G.; Zhou, W.; Qiu, J.; Chen, G.; Luo, W.; Zhao, F.; You, L.; Zheng, L.; Zhang, T.; et al. Targeting hypoxic tumor microenvironment in pancreatic cancer. J. Hematol. Oncol. 2021, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.V.; Nyberg, K.D.; Scott, M.B.; Welsh, A.M.; Nguyen, A.H.; Wu, N.; Hohlbauch, S.V.; Geisse, N.A.; Gibb, E.A.; Robertson, A.G.; et al. Stiffness of pancreatic cancer cells is associated with increased invasive potential. Integr. Biol. 2016, 8, 1232–1245. [Google Scholar] [CrossRef]
- Ozdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.W.; et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.Y.; Doppler, H.; Necela, B.; Krishna, M.; Crawford, H.C.; Raimondo, M.; Storz, P. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J. Cell Biol. 2013, 202, 563–577. [Google Scholar] [CrossRef]
- Zhao, F.; Obermann, S.; von Wasielewski, R.; Haile, L.; Manns, M.P.; Korangy, F.; Greten, T.F. Increase in frequency of myeloid-derived suppressor cells in mice with spontaneous pancreatic carcinoma. Immunology 2009, 128, 141–149. [Google Scholar] [CrossRef]
- Tjomsland, V.; Niklasson, L.; Sandstrom, P.; Borch, K.; Druid, H.; Bratthall, C.; Messmer, D.; Larsson, M.; Spangeus, A. The desmoplastic stroma plays an essential role in the accumulation and modulation of infiltrated immune cells in pancreatic adenocarcinoma. Clin. Dev. Immunol. 2011, 2011, 212810. [Google Scholar] [CrossRef]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Das, S.; Handler, J.S.; Hajdu, C.H.; Coffre, M.; Koralov, S.B.; Bar-Sagi, D. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016, 6, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Hajdu, C.H.; Liot, C.; Miller, G.; Dustin, M.L.; Bar-Sagi, D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 2017, 20, 558–571. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lazarus, J.; Steele, N.G.; Yan, W.; Lee, H.J.; Nwosu, Z.C.; Halbrook, C.J.; Menjivar, R.E.; Kemp, S.B.; Sirihorachai, V.R.; et al. Regulatory T-cell Depletion Alters the Tumor Microenvironment and Accelerates Pancreatic Carcinogenesis. Cancer Discov. 2020, 10, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Zambirinis, C.P.; Seifert, L.; Akkad, N.; Mohan, N.; Werba, G.; Barilla, R.; Torres-Hernandez, A.; Hundeyin, M.; Mani, V.R.K.; et al. gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell 2016, 166, 1485–1499.e15. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef]
- Mathew, E.; Brannon, A.L.; Del Vecchio, A.; Garcia, P.E.; Penny, M.K.; Kane, K.T.; Vinta, A.; Buckanovich, R.J.; di Magliano, M.P. Mesenchymal Stem Cells Promote Pancreatic Tumor Growth by Inducing Alternative Polarization of Macrophages. Neoplasia 2016, 18, 142–151. [Google Scholar] [CrossRef]
- Li, J.; Yuan, S.; Norgard, R.J.; Yan, F.; Sun, Y.H.; Kim, I.K.; Merrell, A.J.; Sela, Y.; Jiang, Y.; Bhanu, N.V.; et al. Epigenetic and Transcriptional Control of the Epidermal Growth Factor Receptor Regulates the Tumor Immune Microenvironment in Pancreatic Cancer. Cancer Discov. 2021, 11, 736–753. [Google Scholar] [CrossRef]
- Ceyhan, G.O.; Bergmann, F.; Kadihasanoglu, M.; Altintas, B.; Demir, I.E.; Hinz, U.; Muller, M.W.; Giese, T.; Buchler, M.W.; Giese, N.A.; et al. Pancreatic neuropathy and neuropathic pain--a comprehensive pathomorphological study of 546 cases. Gastroenterology 2009, 136, 177–186.e1. [Google Scholar] [CrossRef]
- Liebl, F.; Demir, I.E.; Mayer, K.; Schuster, T.; D’Haese, J.G.; Becker, K.; Langer, R.; Bergmann, F.; Wang, K.; Rosenberg, R.; et al. The impact of neural invasion severity in gastrointestinal malignancies: A clinicopathological study. Ann. Surg. 2014, 260, 900–907; discussion 907–908. [Google Scholar] [CrossRef]
- Bapat, A.A.; Munoz, R.M.; Von Hoff, D.D.; Han, H. Blocking Nerve Growth Factor Signaling Reduces the Neural Invasion Potential of Pancreatic Cancer Cells. PLoS ONE 2016, 11, e0165586. [Google Scholar] [CrossRef] [PubMed]
- Saloman, J.L.; Albers, K.M.; Li, D.; Hartman, D.J.; Crawford, H.C.; Muha, E.A.; Rhim, A.D.; Davis, B.M. Ablation of sensory neurons in a genetic model of pancreatic ductal adenocarcinoma slows initiation and progression of cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 3078–3083. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Fu, Y.Y.; Grimont, A.; Ketcham, M.; Lafaro, K.; Saglimbeni, J.A.; Askan, G.; Bailey, J.M.; Melchor, J.P.; Zhong, Y.; et al. PanIN Neuroendocrine Cells Promote Tumorigenesis via Neuronal Cross-talk. Cancer Res. 2017, 77, 1868–1879. [Google Scholar] [CrossRef] [PubMed]
- Kim-Fuchs, C.; Le, C.P.; Pimentel, M.A.; Shackleford, D.; Ferrari, D.; Angst, E.; Hollande, F.; Sloan, E.K. Chronic stress accelerates pancreatic cancer growth and invasion: A critical role for beta-adrenergic signaling in the pancreatic microenvironment. Brain Behav. Immun. 2014, 40, 40–47. [Google Scholar] [CrossRef]
- Renz, B.W.; Takahashi, R.; Tanaka, T.; Macchini, M.; Hayakawa, Y.; Dantes, Z.; Maurer, H.C.; Chen, X.; Jiang, Z.; Westphalen, C.B.; et al. β2 Adrenergic-Neurotrophin Feedforward Loop Promotes Pancreatic Cancer. Cancer Cell 2018, 33, 75–90.e7. [Google Scholar] [CrossRef]
- Renz, B.W.; Tanaka, T.; Sunagawa, M.; Takahashi, R.; Jiang, Z.; Macchini, M.; Dantes, Z.; Valenti, G.; White, R.A.; Middelhoff, M.A.; et al. Cholinergic Signaling via Muscarinic Receptors Directly and Indirectly Suppresses Pancreatic Tumorigenesis and Cancer Stemness. Cancer Discov. 2018, 8, 1458–1473. [Google Scholar] [CrossRef]
- Banh, R.S.; Biancur, D.E.; Yamamoto, K.; Sohn, A.S.W.; Walters, B.; Kuljanin, M.; Gikandi, A.; Wang, H.; Mancias, J.D.; Schneider, R.J.; et al. Neurons Release Serine to Support mRNA Translation in Pancreatic Cancer. Cell 2020, 183, 1202–1218.e25. [Google Scholar] [CrossRef] [PubMed]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norrie, I.C.; Miller, C.J.; Poulogiannis, G.; Lauffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 910–920. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Oon, C.; Kothari, A.; Horton, W.; Link, J.; Sears, R.C.; Sherman, M.H. Acidic fibroblast growth factor underlies microenvironmental regulation of MYC in pancreatic cancer. J. Exp. Med. 2020, 217, e20191805. [Google Scholar] [CrossRef]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef]
- De Monte, L.; Reni, M.; Tassi, E.; Clavenna, D.; Papa, I.; Recalde, H.; Braga, M.; Di Carlo, V.; Doglioni, C.; Protti, M.P. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J. Exp. Med. 2011, 208, 469–478. [Google Scholar] [CrossRef] [PubMed]
- De Monte, L.; Wormann, S.; Brunetto, E.; Heltai, S.; Magliacane, G.; Reni, M.; Paganoni, A.M.; Recalde, H.; Mondino, A.; Falconi, M.; et al. Basophil Recruitment into Tumor-Draining Lymph Nodes Correlates with Th2 Inflammation and Reduced Survival in Pancreatic Cancer Patients. Cancer Res. 2016, 76, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Simon, M.C. Cancer Cells Don’t Live Alone: Metabolic Communication within Tumor Microenvironments. Dev. Cell 2020, 54, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef]
- Hui, S.; Ghergurovich, J.M.; Morscher, R.J.; Jang, C.; Teng, X.; Lu, W.; Esparza, L.A.; Reya, T.; Le, Z.; Yanxiang Guo, J.; et al. Glucose feeds the TCA cycle via circulating lactate. Nature 2017, 551, 115–118. [Google Scholar] [CrossRef]
- Kamphorst, J.J.; Nofal, M.; Commisso, C.; Hackett, S.R.; Lu, W.; Grabocka, E.; Vander Heiden, M.G.; Miller, G.; Drebin, J.A.; Bar-Sagi, D.; et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015, 75, 544–553. [Google Scholar] [CrossRef]
- Commisso, C.; Davidson, S.M.; Soydaner-Azeloglu, R.G.; Parker, S.J.; Kamphorst, J.J.; Hackett, S.; Grabocka, E.; Nofal, M.; Drebin, J.A.; Thompson, C.B.; et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 2013, 497, 633–637. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef]
- Sousa, C.M.; Biancur, D.E.; Wang, X.; Halbrook, C.J.; Sherman, M.H.; Zhang, L.; Kremer, D.; Hwang, R.F.; Witkiewicz, A.K.; Ying, H.; et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 2016, 536, 479–483. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Xu, J.; Liu, S. The Metabolism Symbiosis Between Pancreatic Cancer and Tumor Microenvironment. Front. Oncol. 2021, 11, 759376. [Google Scholar] [CrossRef]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 2016, 167, 829–842.e13. [Google Scholar] [CrossRef] [PubMed]
- Shah, V.M.; Sheppard, B.C.; Sears, R.C.; Alani, A.W. Hypoxia: Friend or Foe for drug delivery in Pancreatic Cancer. Cancer Lett. 2020, 492, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Daniel, S.K.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G. Hypoxia as a barrier to immunotherapy in pancreatic adenocarcinoma. Clin. Transl. Med. 2019, 8, 10. [Google Scholar] [CrossRef] [PubMed]
- Erkan, M.; Adler, G.; Apte, M.V.; Bachem, M.G.; Buchholz, M.; Detlefsen, S.; Esposito, I.; Friess, H.; Gress, T.M.; Habisch, H.J.; et al. StellaTUM: Current consensus and discussion on pancreatic stellate cell research. Gut 2012, 61, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhang, C.; Jiang, K.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. The Role of Stellate Cells in Pancreatic Ductal Adenocarcinoma: Targeting Perspectives. Front. Oncol. 2020, 10, 621937. [Google Scholar] [CrossRef]
- Kuninty, P.R.; Bansal, R.; De Geus, S.W.L.; Mardhian, D.F.; Schnittert, J.; van Baarlen, J.; Storm, G.; Bijlsma, M.F.; van Laarhoven, H.W.; Metselaar, J.M.; et al. ITGA5 inhibition in pancreatic stellate cells attenuates desmoplasia and potentiates efficacy of chemotherapy in pancreatic cancer. Sci. Adv. 2019, 5, eaax2770. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2013, 62, 112–120. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; Von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Buchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464. [Google Scholar] [CrossRef]
- Liu, Y.; Li, F.; Gao, F.; Xing, L.; Qin, P.; Liang, X.; Zhang, J.; Qiao, X.; Lin, L.; Zhao, Q.; et al. Periostin promotes the chemotherapy resistance to gemcitabine in pancreatic cancer. Tumour. Biol. 2016, 37, 15283–15291. [Google Scholar] [CrossRef] [PubMed]
- Dangi-Garimella, S.; Krantz, S.B.; Barron, M.R.; Shields, M.A.; Heiferman, M.J.; Grippo, P.J.; Bentrem, D.J.; Munshi, H.G. Three-dimensional collagen I promotes gemcitabine resistance in pancreatic cancer through MT1-MMP-mediated expression of HMGA2. Cancer Res. 2011, 71, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, T.; Packham, G.; Murphy, L.B.; Bateman, A.C.; Conti, J.A.; Fine, D.R.; Johnson, C.D.; Benyon, R.C.; Iredale, J.P. Type I collagen promotes the malignant phenotype of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 7427–7437. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, M.; Aasrum, M.; Verbeke, C.S.; Gladhaug, I.P. Secretion of fibronectin by human pancreatic stellate cells promotes chemoresistance to gemcitabine in pancreatic cancer cells. BMC Cancer 2019, 19, 596. [Google Scholar] [CrossRef]
- Ireland, L.; Santos, A.; Ahmed, M.S.; Rainer, C.; Nielsen, S.R.; Quaranta, V.; Weyer-Czernilofsky, U.; Engle, D.D.; Perez-Mancera, P.A.; Coupland, S.E.; et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016, 76, 6851–6863. [Google Scholar] [CrossRef]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef]
- Xu, J.; Liu, S.; Yang, X.; Cao, S.; Zhou, Y. Paracrine HGF promotes EMT and mediates the effects of PSC on chemoresistance by activating c-Met/PI3K/Akt signaling in pancreatic cancer in vitro. Life Sci. 2020, 263, 118523. [Google Scholar] [CrossRef]
- Hesler, R.A.; Huang, J.J.; Starr, M.D.; Treboschi, V.M.; Bernanke, A.G.; Nixon, A.B.; McCall, S.J.; White, R.R.; Blobe, G.C. TGF-beta-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis 2016, 37, 1041–1051. [Google Scholar] [CrossRef]
- Dalin, S.; Sullivan, M.R.; Lau, A.N.; Grauman-Boss, B.; Mueller, H.S.; Kreidl, E.; Fenoglio, S.; Luengo, A.; Lees, J.A.; Vander Heiden, M.G.; et al. Deoxycytidine Release from Pancreatic Stellate Cells Promotes Gemcitabine Resistance. Cancer Res. 2019, 79, 5723–5733. [Google Scholar] [CrossRef]
- Ene-Obong, A.; Clear, A.J.; Watt, J.; Wang, J.; Fatah, R.; Riches, J.C.; Marshall, J.F.; Chin-Aleong, J.; Chelala, C.; Gribben, J.G.; et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology 2013, 145, 1121–1132. [Google Scholar] [CrossRef]
- Tang, D.; Yuan, Z.; Xue, X.; Lu, Z.; Zhang, Y.; Wang, H.; Chen, M.; An, Y.; Wei, J.; Zhu, Y.; et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int. J. Cancer 2012, 130, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Mace, T.A.; Ameen, Z.; Collins, A.; Wojcik, S.; Mair, M.; Young, G.S.; Fuchs, J.R.; Eubank, T.D.; Frankel, W.L.; Bekaii-Saab, T.; et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013, 73, 3007–3018. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; Garcia, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [PubMed]
- Froeling, F.E.; Feig, C.; Chelala, C.; Dobson, R.; Mein, C.E.; Tuveson, D.A.; Clevers, H.; Hart, I.R.; Kocher, H.M. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology 2011, 141, 1486–1497.e14. [Google Scholar] [CrossRef]
- Hessmann, E.; Patzak, M.S.; Klein, L.; Chen, N.; Kari, V.; Ramu, I.; Bapiro, T.E.; Frese, K.K.; Gopinathan, A.; Richards, F.M.; et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut 2018, 67, 497–507. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef]
- Hu, C.; Xia, R.; Zhang, X.; Li, T.; Ye, Y.; Li, G.; He, R.; Li, Z.; Lin, Q.; Zheng, S.; et al. circFARP1 enables cancer-associated fibroblasts to promote gemcitabine resistance in pancreatic cancer via the LIF/STAT3 axis. Mol. Cancer 2022, 21, 24. [Google Scholar] [CrossRef]
- Wei, L.; Lin, Q.; Lu, Y.; Li, G.; Huang, L.; Fu, Z.; Chen, R.; Zhou, Q. Cancer-associated fibroblasts-mediated ATF4 expression promotes malignancy and gemcitabine resistance in pancreatic cancer via the TGF-beta1/SMAD2/3 pathway and ABCC1 transactivation. Cell Death Dis. 2021, 12, 334. [Google Scholar] [CrossRef]
- Wei, L.; Ye, H.; Li, G.; Lu, Y.; Zhou, Q.; Zheng, S.; Lin, Q.; Liu, Y.; Li, Z.; Chen, R. Cancer-associated fibroblasts promote progression and gemcitabine resistance via the SDF-1/SATB-1 pathway in pancreatic cancer. Cell Death Dis. 2018, 9, 1065. [Google Scholar] [CrossRef]
- Long, K.B.; Tooker, G.; Tooker, E.; Luque, S.L.; Lee, J.W.; Pan, X.; Beatty, G.L. IL6 Receptor Blockade Enhances Chemotherapy Efficacy in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Ther. 2017, 16, 1898–1908. [Google Scholar] [CrossRef]
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zheng, S.; Hu, C.; Li, G.; Lin, H.; Xia, R.; Ye, Y.; He, R.; Li, Z.; Lin, Q.; et al. Cancer-associated fibroblast-induced lncRNA UPK1A-AS1 confers platinum resistance in pancreatic cancer via efficient double-strand break repair. Oncogene 2022, 41, 2372–2389. [Google Scholar] [CrossRef] [PubMed]
- Muerkoster, S.; Wegehenkel, K.; Arlt, A.; Witt, M.; Sipos, B.; Kruse, M.L.; Sebens, T.; Kloppel, G.; Kalthoff, H.; Folsch, U.R.; et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1beta. Cancer Res. 2004, 64, 1331–1337. [Google Scholar] [CrossRef] [PubMed]
- Liles, J.S.; Arnoletti, J.P.; Kossenkov, A.V.; Mikhaylina, A.; Frost, A.R.; Kulesza, P.; Heslin, M.J.; Frolov, A. Targeting ErbB3-mediated stromal-epithelial interactions in pancreatic ductal adenocarcinoma. Br. J. Cancer 2011, 105, 523–533. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ogier, C.; Colombo, P.E.; Bousquet, C.; Canterel-Thouennon, L.; Sicard, P.; Garambois, V.; Thomas, G.; Gaborit, N.; Jarlier, M.; Pirot, N.; et al. Targeting the NRG1/HER3 pathway in tumor cells and cancer-associated fibroblasts with an anti-neuregulin 1 antibody inhibits tumor growth in pre-clinical models of pancreatic cancer. Cancer Lett. 2018, 432, 227–236. [Google Scholar] [CrossRef]
- Hartmann, N.; Giese, N.A.; Giese, T.; Poschke, I.; Offringa, R.; Werner, J.; Ryschich, E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin. Cancer Res. 2014, 20, 3422–3433. [Google Scholar] [CrossRef]
- Zhang, A.; Qian, Y.; Ye, Z.; Chen, H.; Xie, H.; Zhou, L.; Shen, Y.; Zheng, S. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017, 6, 463–470. [Google Scholar] [CrossRef]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O.; Gopinathan, A.; Tuveson, D.A.; Fearon, D.T. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Gorchs, L.; Fernandez Moro, C.; Bankhead, P.; Kern, K.P.; Sadeak, I.; Meng, Q.; Rangelova, E.; Kaipe, H. Human Pancreatic Carcinoma-Associated Fibroblasts Promote Expression of Co-inhibitory Markers on CD4(+) and CD8(+) T-Cells. Front. Immunol. 2019, 10, 847. [Google Scholar] [CrossRef]
- Kerk, S.A.; Lin, L.; Myers, A.L.; Sutton, D.J.; Andren, A.; Sajjakulnukit, P.; Zhang, L.; Zhang, Y.; Jimenez, J.A.; Nelson, B.S.; et al. Metabolic requirement for GOT2 in pancreatic cancer depends on environmental context. Elife 2022, 11, e73245. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Sivanand, S.; Lau, A.N.; Florek, L.V.; Barbeau, A.M.; Wyckoff, J.; Skala, M.C.; Vander Heiden, M.G. Interactions with stromal cells promote a more oxidized cancer cell redox state in pancreatic tumors. Sci. Adv. 2022, 8, eabg6383. [Google Scholar] [CrossRef] [PubMed]
- Spek, C.A.; Aberson, H.L.; Duitman, J. Macrophage C/EBPdelta Drives Gemcitabine, but Not 5-FU or Paclitaxel, Resistance of Pancreatic Cancer Cells in a Deoxycytidine-Dependent Manner. Biomedicines 2022, 10, 219. [Google Scholar] [CrossRef] [PubMed]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; Ackroyd, J.J.; et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef]
- Quaranta, V.; Rainer, C.; Nielsen, S.R.; Raymant, M.L.; Ahmed, M.S.; Engle, D.D.; Taylor, A.; Murray, T.; Campbell, F.; Palmer, D.H.; et al. Macrophage-Derived Granulin Drives Resistance to Immune Checkpoint Inhibition in Metastatic Pancreatic Cancer. Cancer Res. 2018, 78, 4253–4269. [Google Scholar] [CrossRef]
- Seifert, L.; Werba, G.; Tiwari, S.; Giao Ly, N.N.; Alothman, S.; Alqunaibit, D.; Avanzi, A.; Barilla, R.; Daley, D.; Greco, S.H.; et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature 2016, 532, 245–249. [Google Scholar] [CrossRef]
- Wang, W.; Marinis, J.M.; Beal, A.M.; Savadkar, S.; Wu, Y.; Khan, M.; Taunk, P.S.; Wu, N.; Su, W.; Wu, J.; et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 2018, 34, 757–774.e7. [Google Scholar] [CrossRef]
- Kalbasi, A.; Komar, C.; Tooker, G.M.; Liu, M.; Lee, J.W.; Gladney, W.L.; Ben-Josef, E.; Beatty, G.L. Tumor-Derived CCL2 Mediates Resistance to Radiotherapy in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 137–148. [Google Scholar] [CrossRef]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef]
- Zhang, Y.; Chandra, V.; Riquelme Sanchez, E.; Dutta, P.; Quesada, P.R.; Rakoski, A.; Zoltan, M.; Arora, N.; Baydogan, S.; Horne, W.; et al. Interleukin-17-induced neutrophil extracellular traps mediate resistance to checkpoint blockade in pancreatic cancer. J. Exp. Med. 2020, 217, e20190354. [Google Scholar] [CrossRef] [PubMed]
- Nywening, T.M.; Belt, B.A.; Cullinan, D.R.; Panni, R.Z.; Han, B.J.; Sanford, D.E.; Jacobs, R.C.; Ye, J.; Patel, A.A.; Gillanders, W.E.; et al. Targeting both tumour-associated CXCR2(+) neutrophils and CCR2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut 2018, 67, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Merz, S.F.; Jansen, P.; Wang, B.; Bruderek, K.; Altenhoff, P.; Mattheis, S.; Lang, S.; Gunzer, M.; Klode, J.; et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci. Immunol. 2019, 4, eaaw9159. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, A.J.; Hong, C.S.; Lin, A.J.; Xiang, Y.; Cooper, D.E.; Zhang, J.; Xu, E.S.; Kuo, H.C.; Mowery, Y.M.; Carpenter, D.J.; et al. Neutrophils promote tumor resistance to radiation therapy. Proc. Natl. Acad. Sci. USA 2019, 116, 18584–18589. [Google Scholar] [CrossRef] [PubMed]
- Siolas, D.; Vucic, E.; Kurz, E.; Hajdu, C.; Bar-Sagi, D. Gain-of-function p53(R172H) mutation drives accumulation of neutrophils in pancreatic tumors, promoting resistance to immunotherapy. Cell Rep. 2021, 36, 109578. [Google Scholar] [CrossRef]
- Li, J.; Byrne, K.T.; Yan, F.; Yamazoe, T.; Chen, Z.; Baslan, T.; Richman, L.P.; Lin, J.H.; Sun, Y.H.; Rech, A.J.; et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity 2018, 49, 178–193.e7. [Google Scholar] [CrossRef]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef]
- Vaish, U.; Jain, T.; Are, A.C.; Dudeja, V. Cancer-Associated Fibroblasts in Pancreatic Ductal Adenocarcinoma: An Update on Heterogeneity and Therapeutic Targeting. Int. J. Mol. Sci. 2021, 22, 13408. [Google Scholar] [CrossRef]
- Ohlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Elyada, E.; Bolisetty, M.; Laise, P.; Flynn, W.F.; Courtois, E.T.; Burkhart, R.A.; Teinor, J.A.; Belleau, P.; Biffi, G.; Lucito, M.S.; et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019, 9, 1102–1123. [Google Scholar] [CrossRef]
- Hutton, C.; Heider, F.; Blanco-Gomez, A.; Banyard, A.; Kononov, A.; Zhang, X.; Karim, S.; Paulus-Hock, V.; Watt, D.; Steele, N.; et al. Single-cell analysis defines a pancreatic fibroblast lineage that supports anti-tumor immunity. Cancer Cell 2021, 39, 1227–1244.e20. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Wang, Z.; Zhang, Y.; Pradhan, R.N.; Ganguly, D.; Chandra, R.; Murimwa, G.; Wright, S.; Gu, X.; Maddipati, R.; et al. Mesothelial cell-derived antigen-presenting cancer-associated fibroblasts induce expansion of regulatory T cells in pancreatic cancer. Cancer Cell 2022, 40, 656–673.e7. [Google Scholar] [CrossRef]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Han, H.; Rong, Y.; Zhu, K.; Zhu, Z.; Tang, Z.; Xiong, C.; Tao, J. Hypoxia potentiates gemcitabine-induced stemness in pancreatic cancer cells through AKT/Notch1 signaling. J. Exp. Clin. Cancer Res. 2018, 37, 291. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, J.; Wei, W.; Shi, M.; Xin, B.; Zhang, T.; Shen, X. Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1alpha and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. Ther. 2016, 17, 188–198. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e12. [Google Scholar] [CrossRef]
- Garcia Garcia, C.J.; Huang, Y.; Fuentes, N.R.; Turner, M.C.; Monberg, M.E.; Lin, D.; Nguyen, N.D.; Fujimoto, T.N.; Zhao, J.; Lee, J.J.; et al. Stromal HIF2 Regulates Immune Suppression in the Pancreatic Cancer Microenvironment. Gastroenterology 2022, 162, 2018–2031. [Google Scholar] [CrossRef]
- Domen, A.; Quatannens, D.; Zanivan, S.; Deben, C.; Van Audenaerde, J.; Smits, E.; Wouters, A.; Lardon, F.; Roeyen, G.; Verhoeven, Y.; et al. Cancer-Associated Fibroblasts as a Common Orchestrator of Therapy Resistance in Lung and Pancreatic Cancer. Cancers 2021, 13, 987. [Google Scholar] [CrossRef]
- Han, X.; Zhang, W.H.; Wang, W.Q.; Yu, X.J.; Liu, L. Cancer-associated fibroblasts in therapeutic resistance of pancreatic cancer: Present situation, predicaments, and perspectives. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188444. [Google Scholar] [CrossRef]
- Beatty, G.L.; Werba, G.; Lyssiotis, C.A.; Simeone, D.M. The biological underpinnings of therapeutic resistance in pancreatic cancer. Genes Dev. 2021, 35, 940–962. [Google Scholar] [CrossRef]
- Geng, X.; Chen, H.; Zhao, L.; Hu, J.; Yang, W.; Li, G.; Cheng, C.; Zhao, Z.; Zhang, T.; Li, L.; et al. Cancer-Associated Fibroblast (CAF) Heterogeneity and Targeting Therapy of CAFs in Pancreatic Cancer. Front. Cell Dev. Biol. 2021, 9, 655152. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.; Ferrer, C.; Dominguez-Hormaetxe, S.; Bockorny, B.; Murias, L.; Seifert, O.; Eisler, S.A.; Kontermann, R.E.; Pfizenmaier, K.; Lee, S.Y.; et al. OMTX705, a Novel FAP-Targeting ADC Demonstrates Activity in Chemotherapy and Pembrolizumab-Resistant Solid Tumor Models. Clin. Cancer Res. 2020, 26, 3420–3430. [Google Scholar] [CrossRef]
- Wang, L.C.; Lo, A.; Scholler, J.; Sun, J.; Majumdar, R.S.; Kapoor, V.; Antzis, M.; Cotner, C.E.; Johnson, L.A.; Durham, A.C.; et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014, 2, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Cox, N.; Pokrovskii, M.; Vicario, R.; Geissmann, F. Origins, Biology, and Diseases of Tissue Macrophages. Annu. Rev. Immunol. 2021, 39, 313–344. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Guan, R.; Hong, W.; Zhou, Y.; Lin, Y.; Jin, H.; Hou, B.; Jian, Z. Prognostic value of tumor-associated macrophages in pancreatic cancer: A meta-analysis. Cancer Manag. Res. 2019, 11, 4041–4058. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2020, 8, 607209. [Google Scholar] [CrossRef]
- Atanasov, G.; Potner, C.; Aust, G.; Schierle, K.; Dietel, C.; Benzing, C.; Krenzien, F.; Bartels, M.; Eichfeld, U.; Schmelzle, M.; et al. TIE2-expressing monocytes and M2-polarized macrophages impact survival and correlate with angiogenesis in adenocarcinoma of the pancreas. Oncotarget 2018, 9, 29715–29726. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Ruffell, B.; Coussens, L.M. Macrophages and therapeutic resistance in cancer. Cancer Cell 2015, 27, 462–472. [Google Scholar] [CrossRef]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef]
- Larionova, I.; Cherdyntseva, N.; Liu, T.; Patysheva, M.; Rakina, M.; Kzhyshkowska, J. Interaction of tumor-associated macrophages and cancer chemotherapy. Oncoimmunology 2019, 8, 1596004. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef]
- Adachi, Y.; Ito, K.; Hayashi, Y.; Kimura, R.; Tan, T.Z.; Yamaguchi, R.; Ebi, H. Epithelial-to-Mesenchymal Transition is a Cause of Both Intrinsic and Acquired Resistance to KRAS G12C Inhibitor in KRAS G12C-Mutant Non-Small Cell Lung Cancer. Clin. Cancer Res. 2020, 26, 5962–5973. [Google Scholar] [CrossRef] [PubMed]
- Tekin, C.; Aberson, H.L.; Waasdorp, C.; Hooijer, G.K.J.; de Boer, O.J.; Dijk, F.; Bijlsma, M.F.; Spek, C.A. Macrophage-secreted MMP9 induces mesenchymal transition in pancreatic cancer cells via PAR1 activation. Cell Oncol. 2020, 43, 1161–1174. [Google Scholar] [CrossRef]
- Zuo, C.; Baer, J.M.; Knolhoff, B.L.; Belle, J.I.; Liu, X.; Hogg, G.D.; Fu, C.; Kingston, N.L.; Brenden, M.A.; De La Lastra, A.A.; et al. Macrophage proliferation machinery leads to PDAC progression, but susceptibility to innate immunotherapy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Neutrophils in Cancer: Two Sides of the Same Coin. J. Immunol. Res. 2015, 2015, 983698. [Google Scholar] [CrossRef]
- McFarlane, A.J.; Fercoq, F.; Coffelt, S.B.; Carlin, L.M. Neutrophil dynamics in the tumor microenvironment. J. Clin. Investig. 2021, 131, e143759. [Google Scholar] [CrossRef]
- Jin, L.; Kim, H.S.; Shi, J. Neutrophil in the Pancreatic Tumor Microenvironment. Biomolecules 2021, 11, 1170. [Google Scholar] [CrossRef] [PubMed]
- Faget, J.; Peters, S.; Quantin, X.; Meylan, E.; Bonnefoy, N. Neutrophils in the era of immune checkpoint blockade. J. Immunother. Cancer 2021, 9, e002242. [Google Scholar] [CrossRef] [PubMed]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Andzinski, L.; Kasnitz, N.; Stahnke, S.; Wu, C.F.; Gereke, M.; von Kockritz-Blickwede, M.; Schilling, B.; Brandau, S.; Weiss, S.; Jablonska, J. Type I IFNs induce anti-tumor polarization of tumor associated neutrophils in mice and human. Int. J. Cancer 2016, 138, 1982–1993. [Google Scholar] [CrossRef]
- Singhal, S.; Bhojnagarwala, P.S.; O’Brien, S.; Moon, E.K.; Garfall, A.L.; Rao, A.S.; Quatromoni, J.G.; Stephen, T.L.; Litzky, L.; Deshpande, C.; et al. Origin and Role of a Subset of Tumor-Associated Neutrophils with Antigen-Presenting Cell Features in Early-Stage Human Lung Cancer. Cancer Cell 2016, 30, 120–135. [Google Scholar] [CrossRef]
- Evrard, M.; Kwok, I.W.H.; Chong, S.Z.; Teng, K.W.W.; Becht, E.; Chen, J.; Sieow, J.L.; Penny, H.L.; Ching, G.C.; Devi, S.; et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity 2018, 48, 364–379.e8. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Zilionis, R.; Da Silva Martins, J.; Bos, S.A.; Courties, G.; Rickelt, S.; Severe, N.; Baryawno, N.; Faget, J.; et al. Osteoblasts remotely supply lung tumors with cancer-promoting SiglecF(high) neutrophils. Science 2017, 358, eaal5081. [Google Scholar] [CrossRef]
- Wang, W.Q.; Liu, L.; Xu, H.X.; Wu, C.T.; Xiang, J.F.; Xu, J.; Liu, C.; Long, J.; Ni, Q.X.; Yu, X.J. Infiltrating immune cells and gene mutations in pancreatic ductal adenocarcinoma. Br. J. Surg. 2016, 103, 1189–1199. [Google Scholar] [CrossRef]
- Nielsen, S.R.; Strobech, J.E.; Horton, E.R.; Jackstadt, R.; Laitala, A.; Bravo, M.C.; Maltese, G.; Jensen, A.R.D.; Reuten, R.; Rafaeva, M.; et al. Suppression of tumor-associated neutrophils by lorlatinib attenuates pancreatic cancer growth and improves treatment with immune checkpoint blockade. Nat. Commun. 2021, 12, 3414. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e7. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Horner, J.W.; Paul, E.; Shang, X.; Troncoso, P.; Deng, P.; Jiang, S.; Chang, Q.; Spring, D.J.; Sharma, P.; et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature 2017, 543, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Eisenberg, P.D.; Sachdev, J.C.; Weise, A.M.; Tse, A.N.; Hutchinson, M.; Aromin, I.; West, B.; Tong, S.; Ribas, A.; et al. Phase 1/2a study of double immune suppression blockade by combining a CSF1R inhibitor (Pexidartinib/PLX3397) with an anti PD-1 antibody (Pembrolizumab) to treat advanced, solid tumors. J. Clin. Oncol. 2016, 34, TPS11618-TPS. [Google Scholar] [CrossRef]
- Wang-Gillam, A.; O’Reilly, E.M.; Bendell, J.C.; Wainberg, Z.A.; Borazanci, E.H.; Bahary, N.; O’Hara, M.H.; Beatty, G.L.; Pant, S.; Cohen, D.J.; et al. A randomized phase II study of cabiralizumab (cabira) + nivolumab (nivo) ± chemotherapy (chemo) in advanced pancreatic ductal adenocarcinoma (PDAC). J. Clin. Oncol. 2019, 37, TPS465-TPS. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
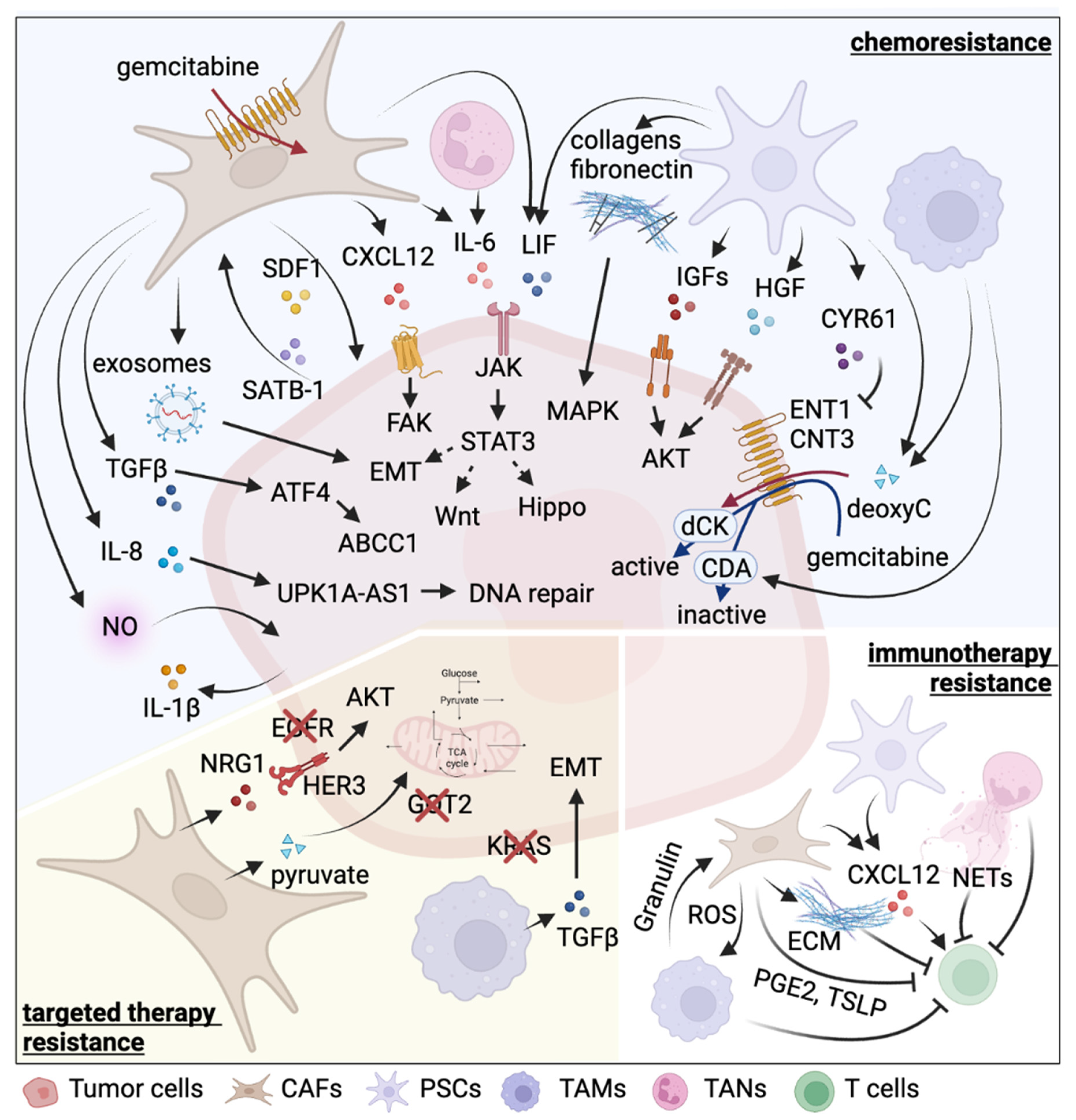{kind=link}
| Cell Type | Therapy | Resistance Inducer | Detailed Mechanism | Reference |
|---|---|---|---|---|
| PSCs | chemotherapy (gemcitabine) | Collagen I | Promote proliferation by MAPK pathway activation and chromatin remodeling | [62,63] |
| Periostin | Induce ECM molecules, including collagen I | [60,61] | ||
| fibronectin | Promote proliferation by MAPK pathway activation | [64] | ||
| IGF1, IGF2 | Activate IGFR-PI3K-AKT pathway | [65] | ||
| LIF | Activate Wnt and Hippo signaling pathways and induce EMT | [66] | ||
| HGF | Activate c-Met-PI3K-Akt pathway and induce EMT | [67] | ||
| CYR61 | Downregulate nucleoside transporters ENT1 and CNT3 | [68] | ||
| Deoxycytidine | Compete with gemcitabine for deoxycytidine kinase-mediated phosphorylation | [69] | ||
| immunotherapy | CXCL12 | Chemoattract CD8+ T cells via CXCL12-CXCR4 axis to sequester them in the panstromal compartment | [70] | |
| Galectin-1 | Induce T cell apoptosis and Th2 differentiation | [71] | ||
| IL-6 | Promote MDSC differentiation via STAT3 activation and suppress T cell proliferation | [72] | ||
| CAFs | chemotherapy (gemcitabine) | 5′-nucleotidases | Entrap active gemcitabine intracellularly via downregulation of Nt5c1A, Nt5c3 | [75] |
| Exosomes | Deliver SNAI1 and miR-146a to tumor cells via exosomes | [76] | ||
| circFARP1 | Enhance LIF expression and secretion | [77] | ||
| TGF-β | Upregulate ATF4 in tumor cells to activate ABCC1 expression | [78] | ||
| SDF-1 | Form a reciprocal feedback loop with tumor cells via SDF-1/SATB-1 axis | [79] | ||
| IL-6 | Activate JAK-STAT3 signaling pathway | [80] | ||
| CXCL12 | Bind to CXCR4 to activate FAK, AKT, and ERK pathways | [81] | ||
| chemotherapy (oxaliplatin) | IL-8 | Upregulate UPK1A-AS1 to facilitate DNA repair | [82] | |
| chemotherapy (etoposide) | NO | Elevate IL-1β production in tumor cells | [83] | |
| targeted therapy (EGFRi erlotinib) | NRG-1 | Activate ERBB3-AKT signaling pathway | [84,85] | |
| immunotherapy | ECM | Form a physical barrier to impede T cell-tumor cell contact | [86] | |
| ROS | Induce M2 TAM polarization | [87] | ||
| / | Suppress immunogenic activities | [88] | ||
| CXCL12 | Exclude T cells from tumor region by binding to CXCR4 | [89] | ||
| PGE2 | Induce expression of immune checkpoints on CD4+ and CD8+ T cells | [90] | ||
| TSLP | Induce Th2 cell polarization through dendritic cell conditioning | [41] | ||
| targeted therapy (GOT2i) | Pyruvate | Provide tumor cells with pyruvate to maintain redox balance | [91,92] | |
| TAMs | chemotherapy (gemcitabine) | Deoxycytidine | Interfere the uptake and metabolism of gemcitabine | [93] |
| Cytidine deaminase | Elevate cytidine deaminase expression in tumor cells to inactivate gemcitabine | [94] | ||
| targeted therapy (KRASi) | TGFβ | Activate canonical SMAD3/4 pathway and promote EMT | [95] | |
| immunotherapy | Granulin | Induce fibrosis to prevent T cell infiltration | [96] | |
| Mincle | Ligate to SAP130 expressed by tumor cells to suppress cancer immunity | [97] | ||
| RIP1 | Regulate M2 TAM polarization | [98] | ||
| radiotherapy, immunotherapy | / | n/a | [99,100] | |
| TANs | chemotherapy (gemcitabine) | IL-6 | Activate JAK-STAT3 signaling pathway | [80] |
| immunotherapy | NETs | Cause tumor CD8+ T cell inactivation and spatial exclusion | [101] | |
| chemotherapy (FOLFIRINOX, gemcitabine, nab-paclitaxel), radiotherapy, immunotherapy | / | n/a | [102,103,104,105,106,107] |
| Target | Agent | Combined Agent | Selected Clinical Trials |
|---|---|---|---|
| ECM or membrane proteins | |||
| Hyaluronic acid | PEGPH20 | Avelumab, chemotherapy, pembrolizumab | NCT03481920, NCT01453153, NCT01839487, NCT04058964 |
| Plectin | ZB131 | NCT05074472 | |
| Galectin-9 | LYT-200 | NCT04666688 | |
| CTLA-4 | Zalifrelimab | NCT04827953 | |
| RARα/β | Am80 | NCT05064618 | |
| Receptors | |||
| IGF1R | MK-0646 | Chemotherapy + TKI | NCT00769483 |
| Cixutumumab | NCT00617708 | ||
| AMG 479 | Chemotherapy, radiotherapy, AMG 655 | NCT00630552, NCT01298401, NCT00819169, NCT01231347 | |
| Metformin | Everolimus, octreotide LAR | NCT01971034, NCT02431676 | |
| MM-141 | Chemotherapy | NCT02399137 | |
| HER3 | Seribantumab | NCT04790695, NCT04383210 | |
| HMBD-001 | NCT05057013 | ||
| HER2/3 | Zenocutuzumab (MCLA-128) | NCT02912949 | |
| IL6R | Tocilizumab | Chemotherapy | NCT02767557, NCT04258150 |
| CNTO 328 | NCT00841191 | ||
| CXCR4 | MB1707 | NCT05465590 | |
| Plerixafor | Cemiplimab | NCT03277209, NCT02179970 | |
| IL1RAP | CAN04 | FOLFIRINOX | NCT04990037 |
| TGFβR | PF-06952229 | NCT03685591 | |
| SHR-1701 | Chemotherapy | NCT04624217 | |
| CSF1R | Cabiralizumab | Nivolumab, chemotherapy | NCT02526017, NCT03697564 |
| Pexidartinib | Durvalumab | NCT02777710 | |
| IMC-CS4 | Pembrolizumab, GVAX | NCT03153410 | |
| CXCR2 | SX-682 | Nivolumab | NCT04477343 |
| Enzymes | |||
| COX | Etodolac | NCT03838029 | |
| Celecoxib | Chemotherapy, irinotecan, interferon α-2b, DC vaccine | NCT00198081, NCT00068432, NCT00177853, NCT01111591 | |
| RIPK1 | GSK3145095 | NCT03681951 | |
| Cytokines, chemokines, or growth factors | |||
| LIF | MSC-1 | NCT03490669 | |
| HGF | Ficlatuzumab | NCT03316599 | |
| CXCL12 | Olaptesed pegol (NOX-A12) | Pembrolizumab | NCT03168139, NCT04901741 |
| IL-6 | Siltuximab | Spartalizumab | NCT04191421 |
| IL-12 | VG161 | Nivolumab | NCT05162118 |
| IL-15 | ALT-803 | NCT02559674 | |
| IL-1β | Canakinumab | Spartalizumab, nab-paclitaxel, gemcitabine | NCT04581343, NCT04229004 |
| IL-2 | Aldesleukin | chemotherapy, anti-KRAS G12D mTCR PBL, anti-KRAS G12V mTCR PBL, pembrolizumab, anti-hCD70 CAR-transduced PBL, HER2Bi-armed T cells, sargramostim, ALVAC-CEA vaccine, neoantigen-specific TCR-T | NCT05194735, NCT02620865, NCT01583686, NCT01212887, NCT03745326, NCT01174121, NCT03190941, NCT02830724, NCT02662348, NCT00003125, NCT05194735, NCT04426669 |
| IL-8 | BMS-986253 | Nivolumab | NCT02451982 |
| VEGF | Bevacizumab | Chemotherapy, radiotherapy, TKI, cetuximab, ALT-803, cancer vaccine, immunotherapy, pembrolizumab, ZN-c3, PEGPH20, durvalumab, TGR-1202 | NCT00047710, NCT00417976, NCT00614653, NCT00460174, NCT00365144, NCT00602602, NCT00410774, NCT00126633 |
| Bevacizumab-800CW | NCT02743975 | ||
| Avastin | Chemotherapy, NANT-008, radiotherapy | NCT03127124, NCT00735306, NCT00609765 | |
| rhuMAB-VEGF | Chemotherapy | NCT00066677 | |
| TGF-β | HCW9218 | NCT05304936 | |
| BCA101 | NCT04429542 | ||
| NIS793 | PDR001, chemotherapy | NCT02947165, NCT05417386 | |
| AP 12009 | NCT00844064 | ||
| M-CSF | MCS110 | Spartalizumab | NCT02807844 |
| GM-CSF | Sargramostim | Carcinoembryonic antigen peptide 1-6D | NCT00669734, NCT00012246 |
| GM-CSF | iNeo-Vac-P01, TG-01 | NCT04810910, NCT03645148 | |
| OH2 injection | NCT04637698 | ||
| PANC 10.05 pcDNA-1/GM-Neo | NCT01088789 | ||
| PANVAC™-VF | NCT00088660 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, D.; Patel, R.; Chiang, C.-Y.; Hou, P. Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance. Cells 2022, 11, 2952. https://doi.org/10.3390/cells11192952
Deng D, Patel R, Chiang C-Y, Hou P. Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance. Cells. 2022; 11(19):2952. https://doi.org/10.3390/cells11192952
Chicago/Turabian StyleDeng, Daiyong, Riya Patel, Cheng-Yao Chiang, and Pingping Hou. 2022. "Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance" Cells 11, no. 19: 2952. https://doi.org/10.3390/cells11192952
APA StyleDeng, D., Patel, R., Chiang, C.-Y., & Hou, P. (2022). Role of the Tumor Microenvironment in Regulating Pancreatic Cancer Therapy Resistance. Cells, 11(19), 2952. https://doi.org/10.3390/cells11192952