
Human Allogeneic Liver-Derived Progenitor Cells Significantly Improve NAFLD Activity Score and Fibrosis in Late-Stage NASH Animal Model

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Culture of HALPCs
2.2. Inflammation Priming and Generation of Conditioned Medium
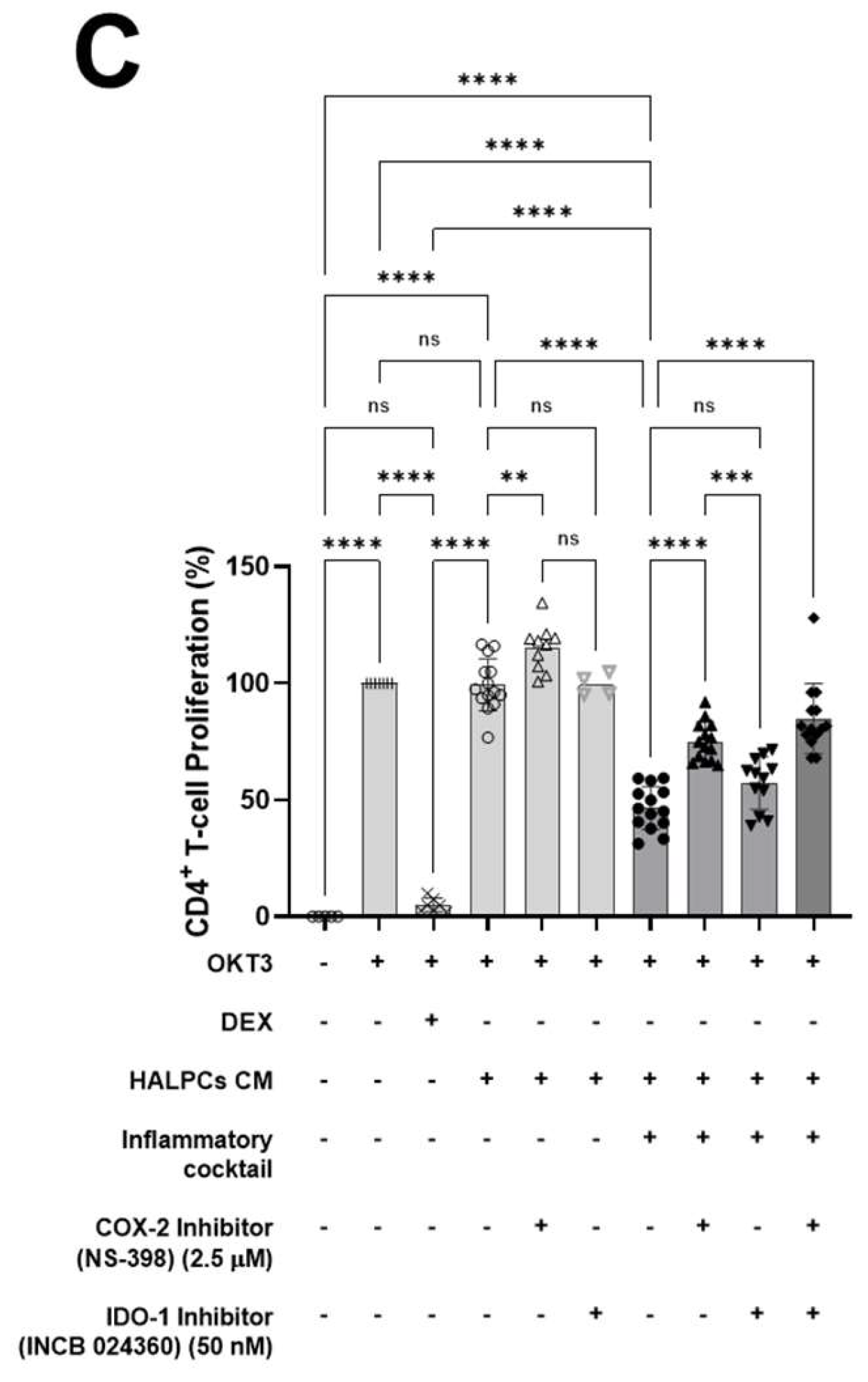
2.3. CD4+ T-Cell Activation Assay
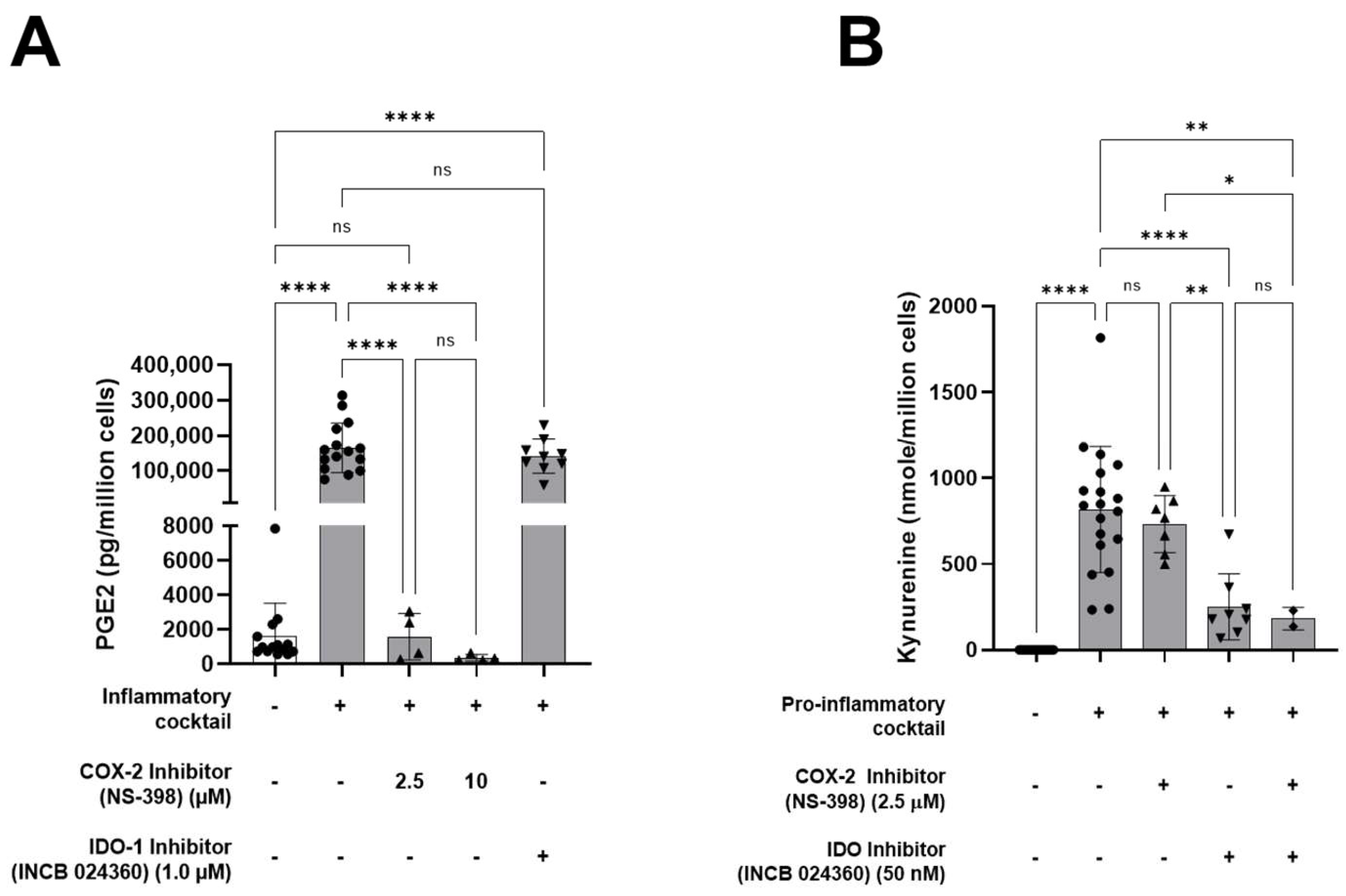
2.4. Evaluation of Secreted Levels of PGE2, Kynurenine, IFN-γ, and TNF-α in the Supernatants of HALPCs
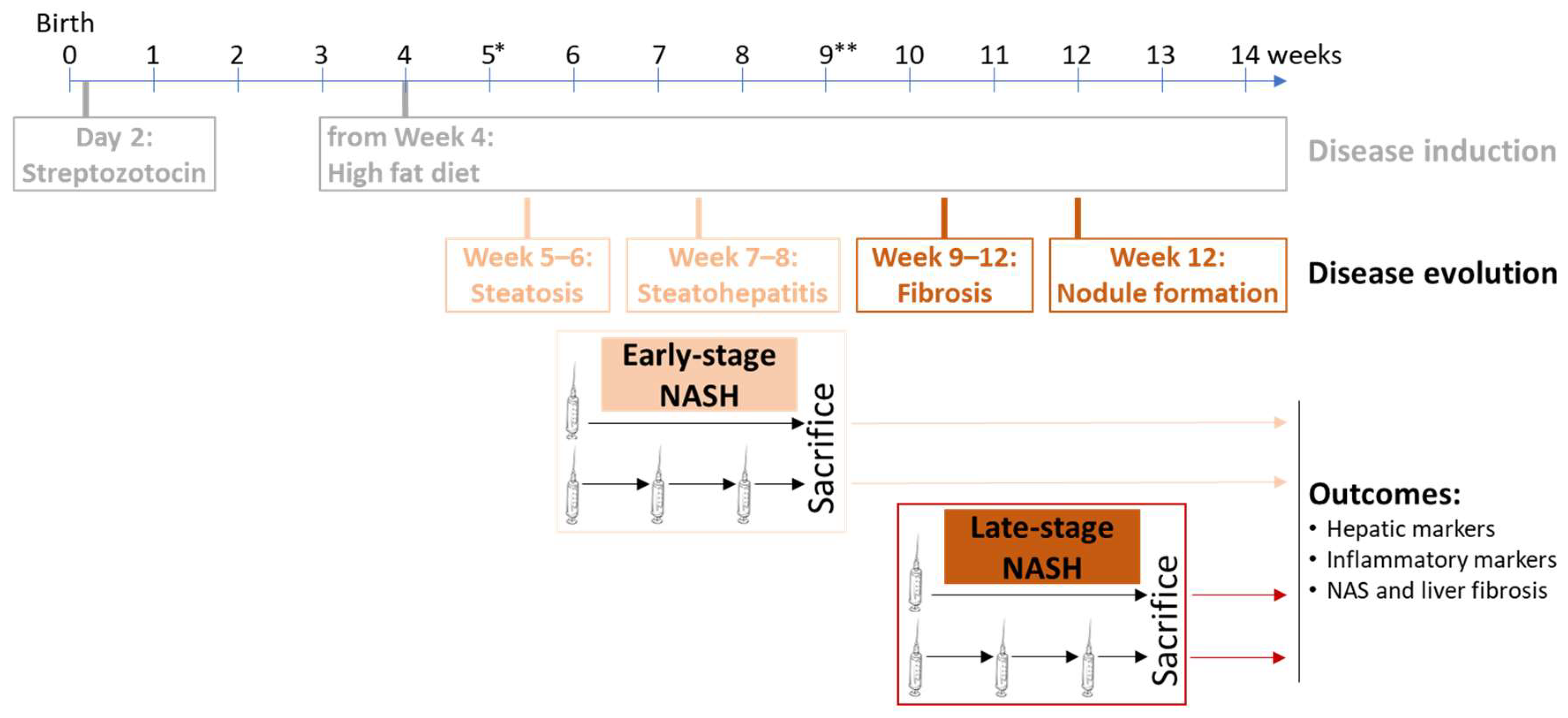
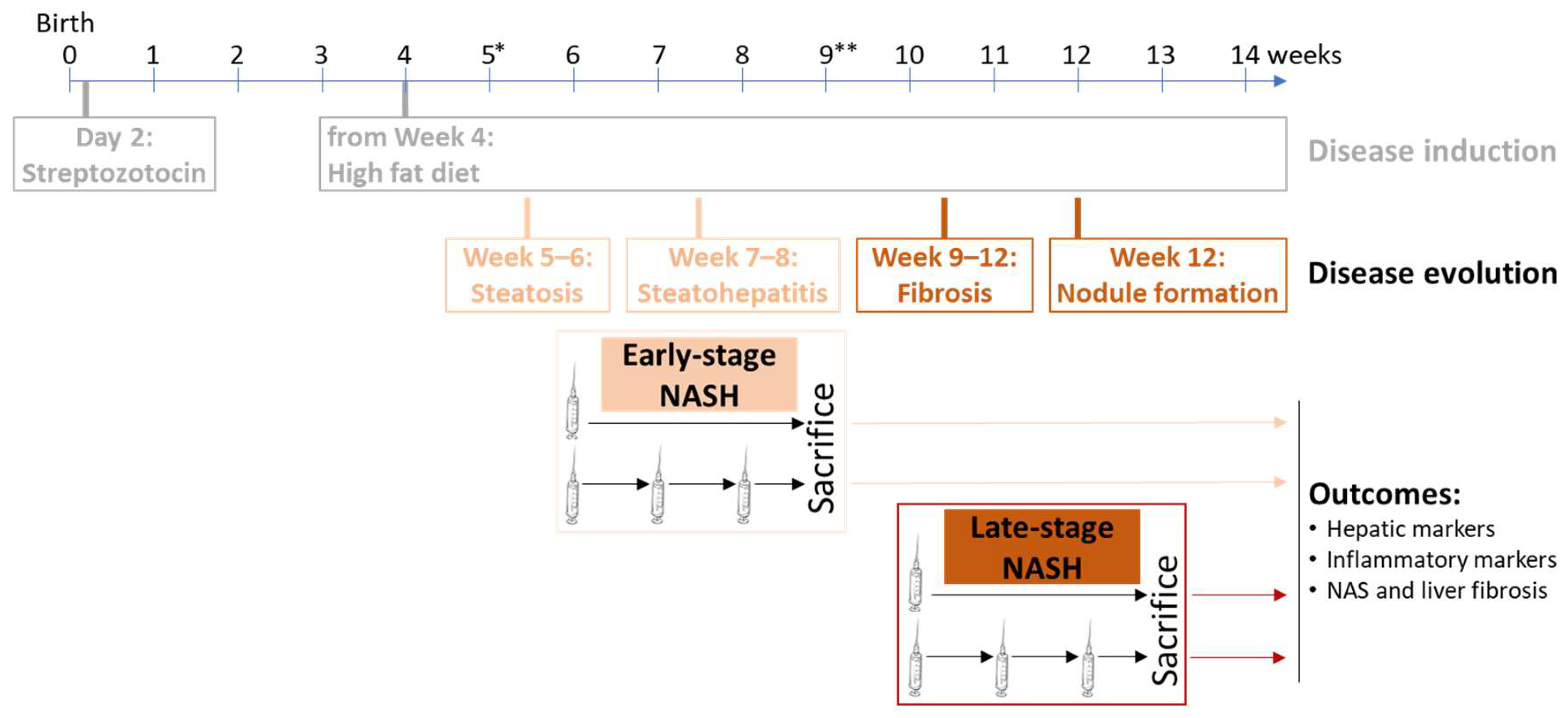
2.5. Transplantation of STAM Mice
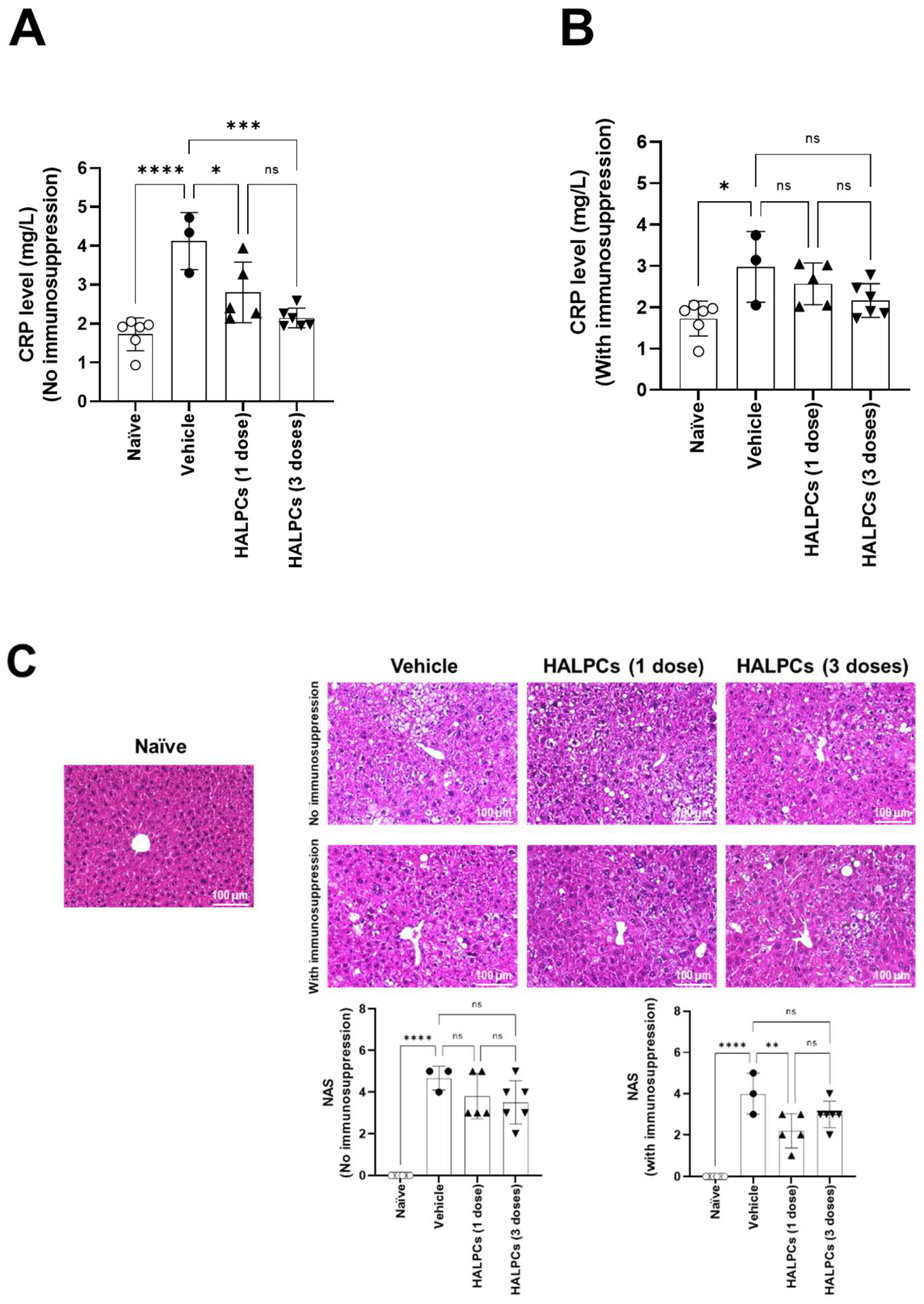
2.6. Blood and Serum Analyses
2.7. Liver Tissue Collection and Processing
2.8. Statistical Analyses
3. Results
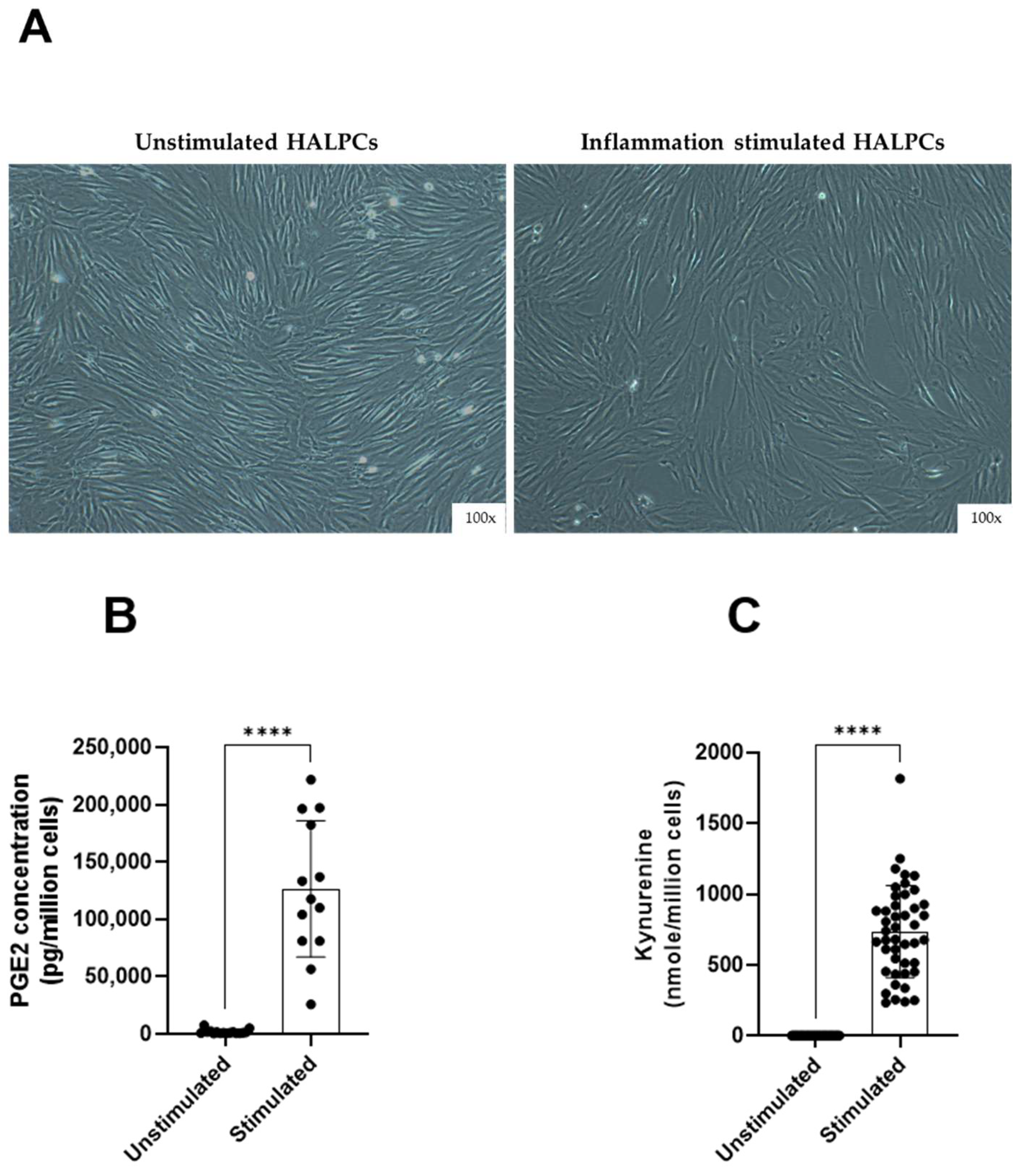
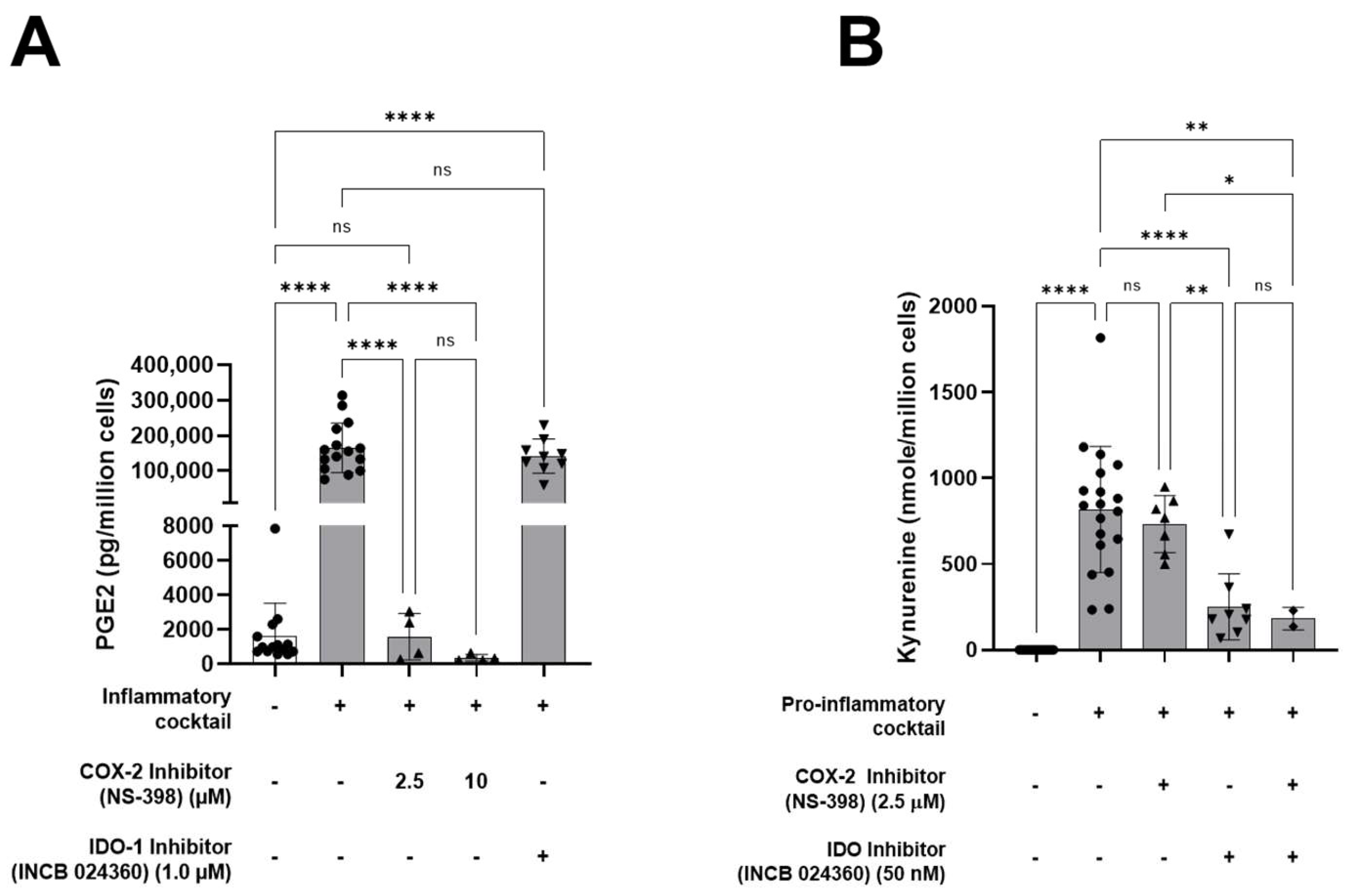
3.1. HALPCs Adapt to Inflammatory Environment by Secreting Potent Immunomodulatory Bioactive Molecules
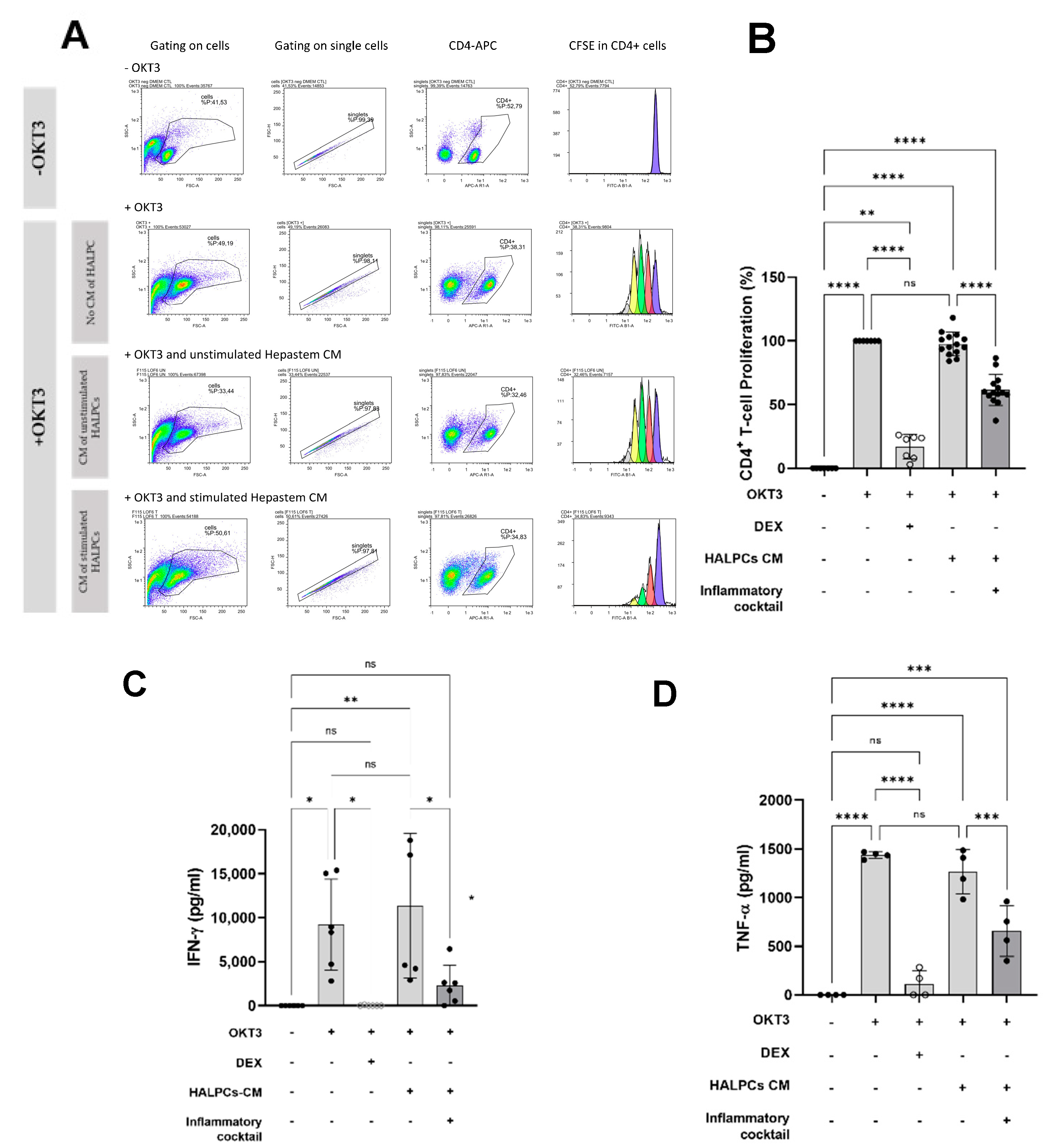
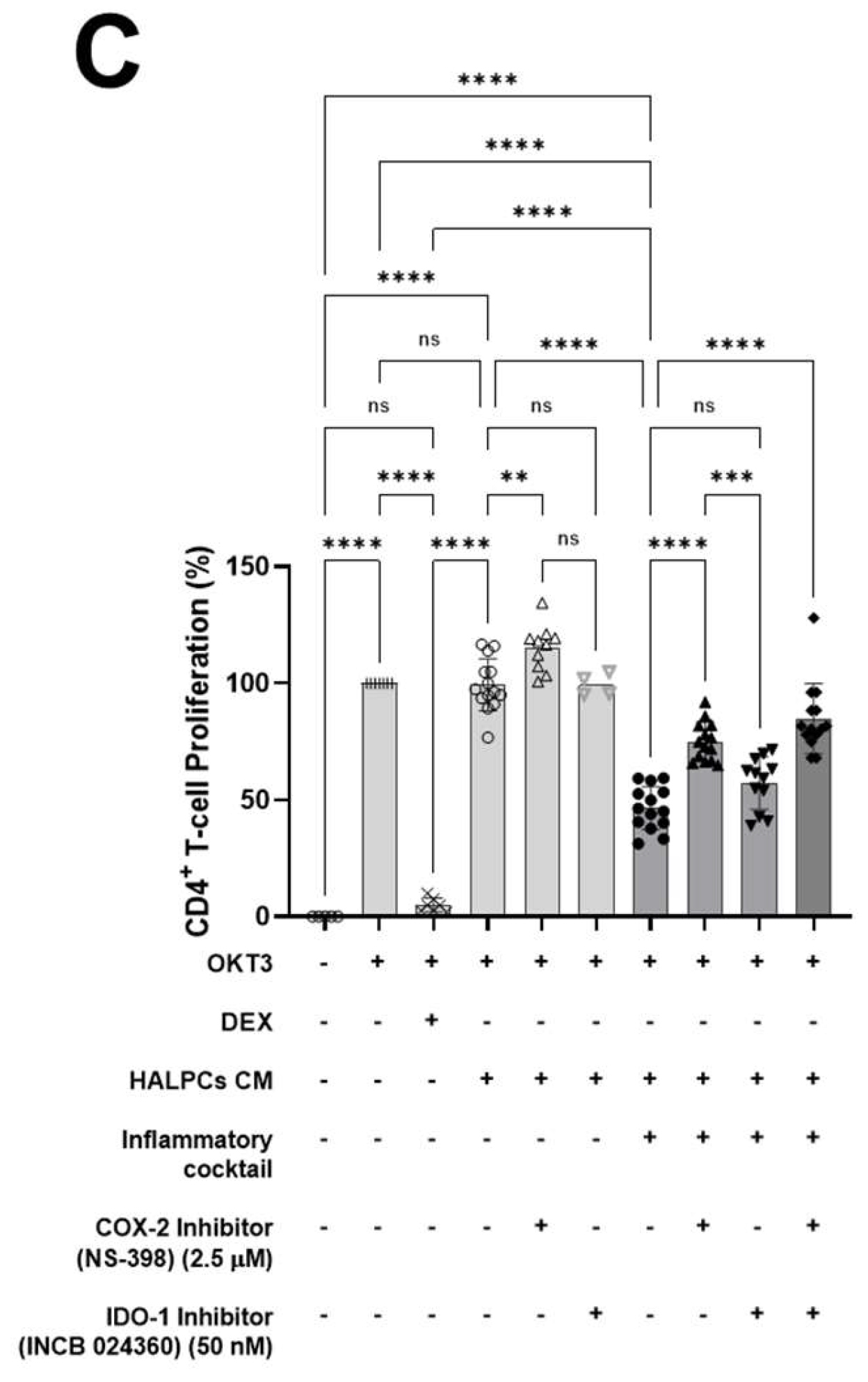
3.2. Effect of HALPC cm on Antigen-Presenting Cell-Induced T-Lymphocyte Proliferation
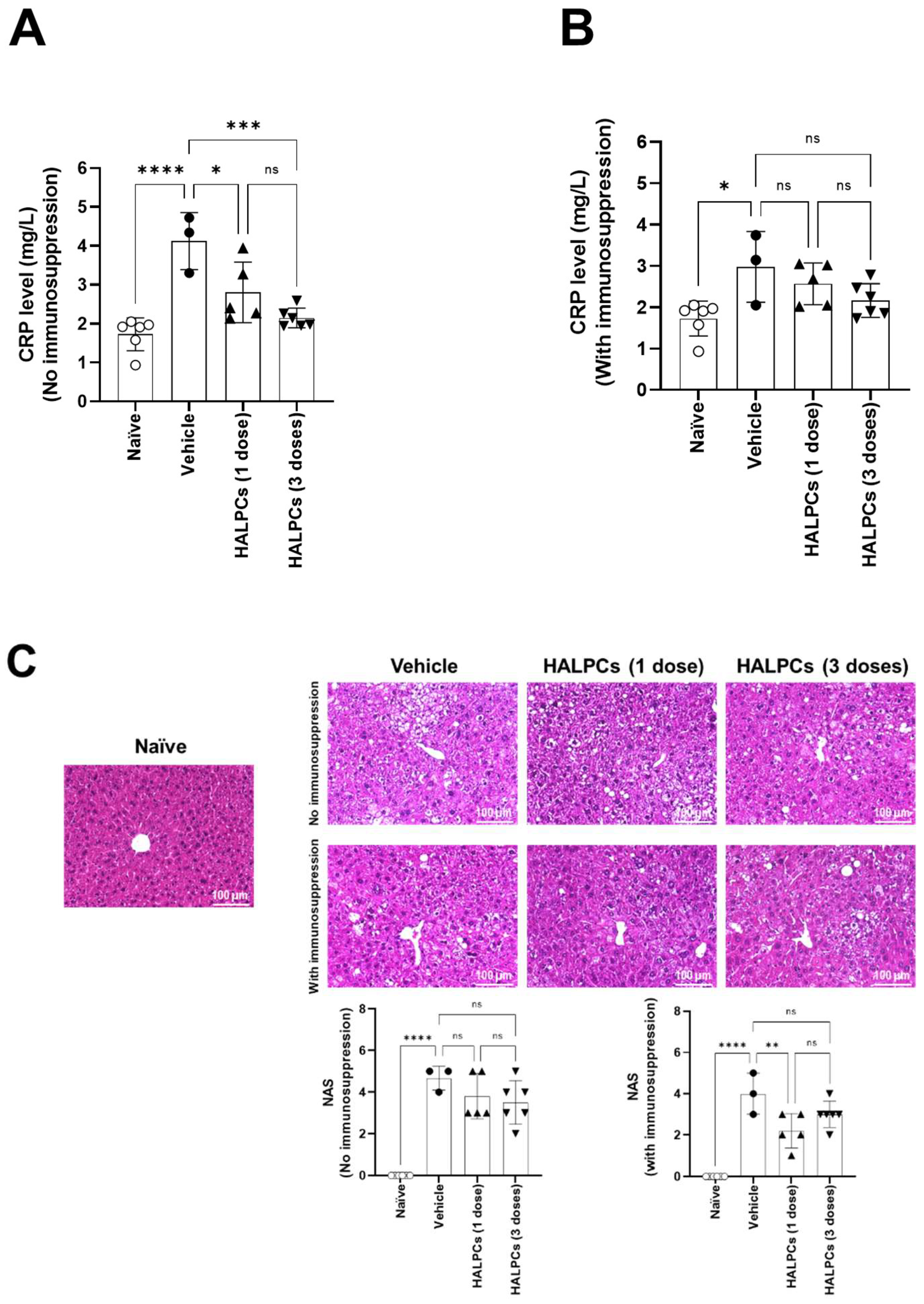
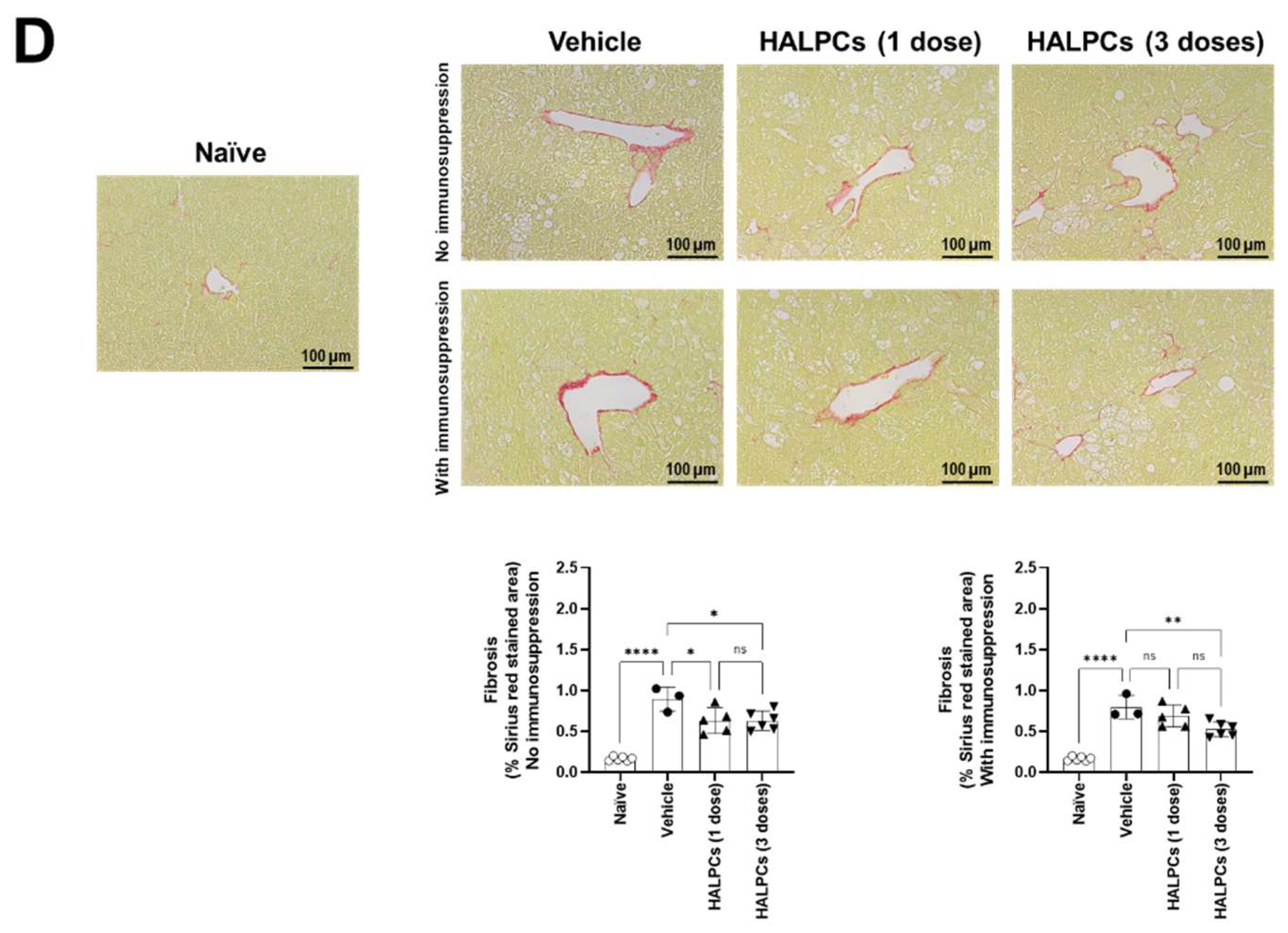
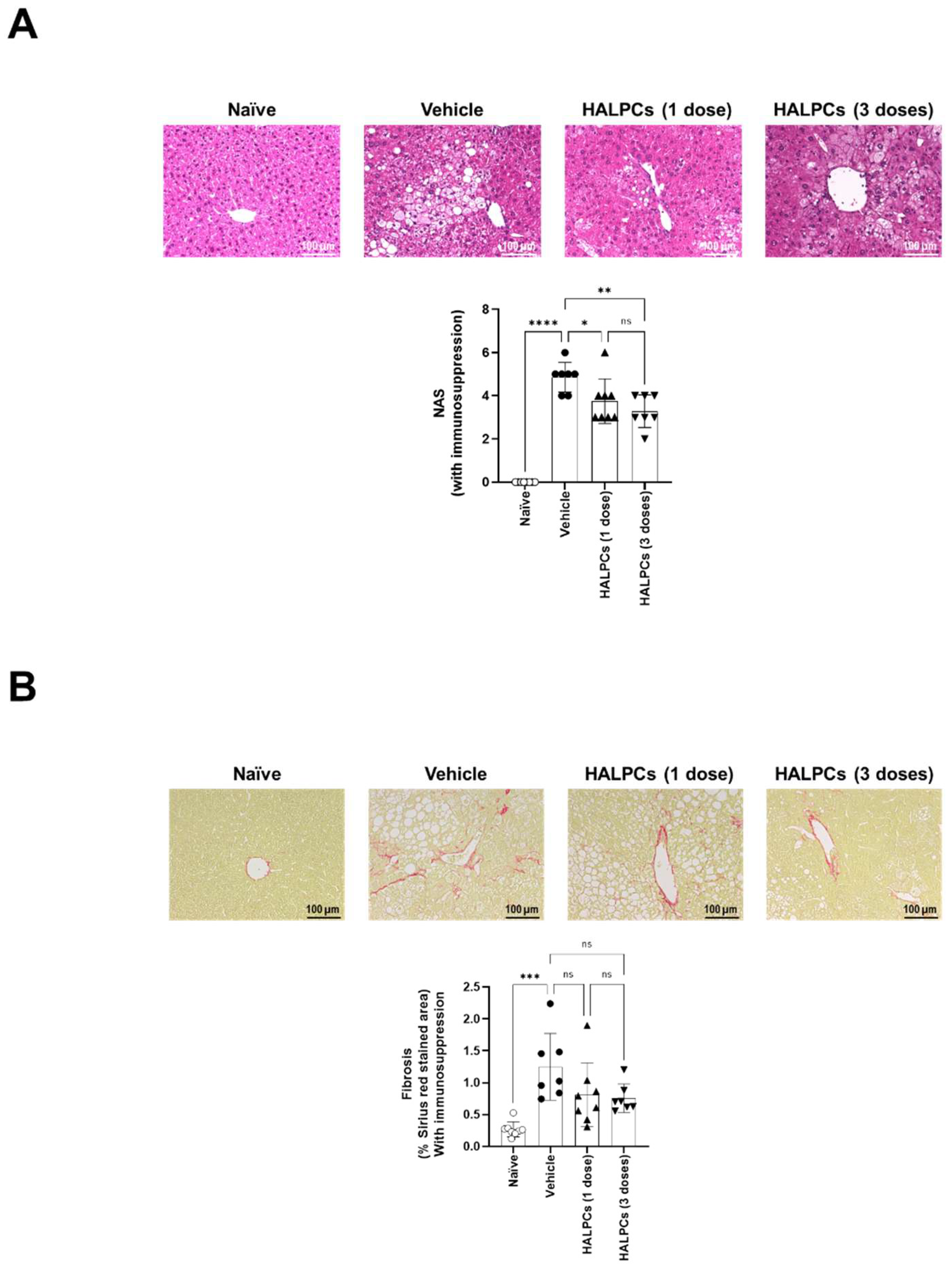
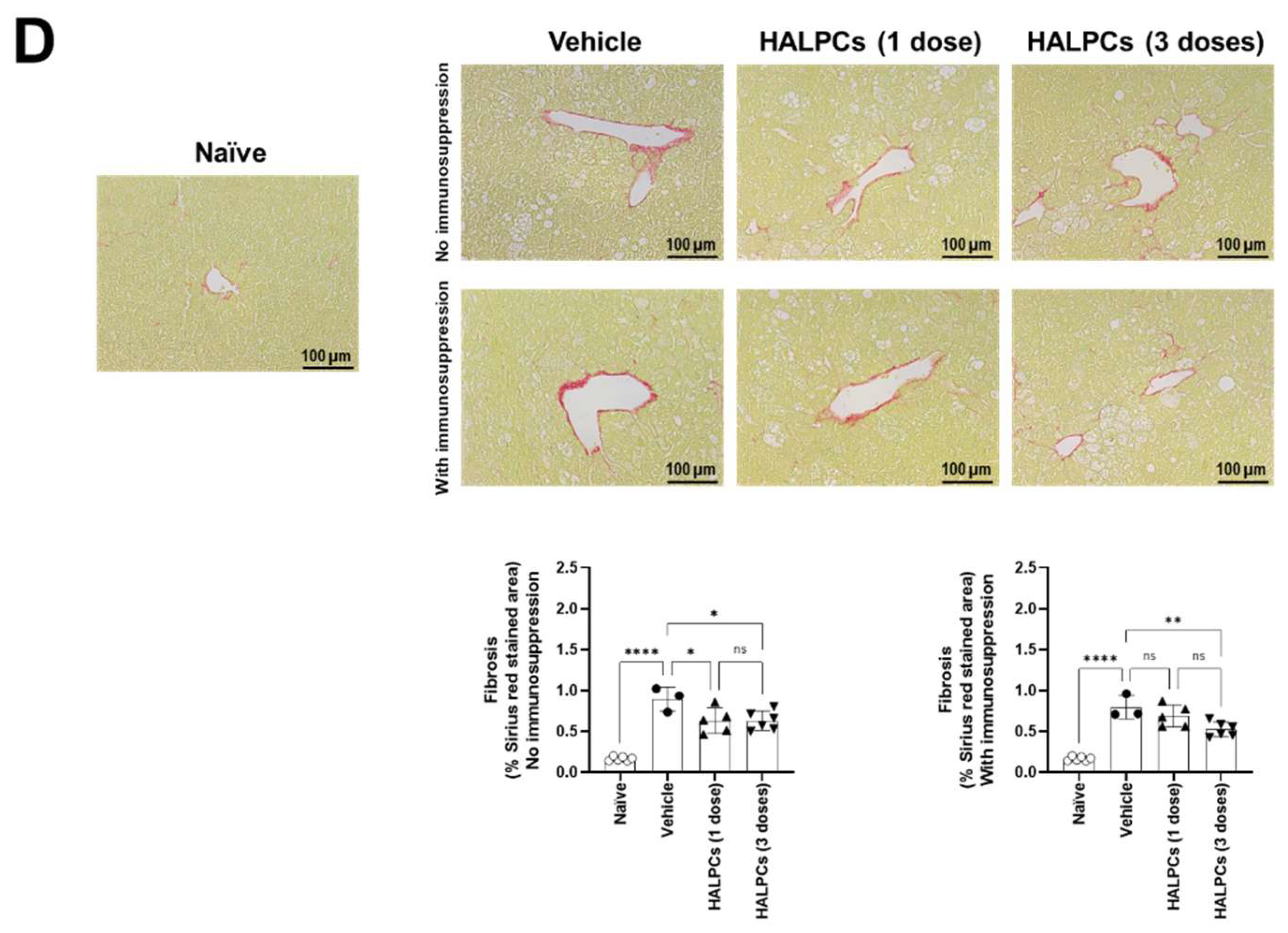
3.3. Effect of HALPCs on In Vivo Inflammation and Fibrosis in the NASH-STAM Mouse Model
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brunt, E.M.; Tiniakos, D.G. Histopathology of nonalcoholic fatty liver disease. World J. Gastroenterol. 2010, 16, 5286–5296. [Google Scholar] [CrossRef]
- Bhatt, H.B.; Smith, R.J. Fatty liver disease in diabetes mellitus. Hepatobiliary Surg. Nutr. 2015, 4, 101–108. [Google Scholar]
- Rinella, M.E.; Sanyal, A.J. Management of NAFLD: A stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, J.M.; Woods, C.; Marjot, T.; Cobbold, J.F.; Tomlinson, J.W. Non-alcoholic fatty liver disease and diabetes. Metabolism 2016, 65, 1096–1108. [Google Scholar] [CrossRef]
- Tapper, E.B.; Lai, M. Weight loss results in significant improvements in quality of life for patients with nonalcoholic fatty liver disease: A prospective cohort study. Hepatology 2016, 63, 1184–1189. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef]
- Sumida, Y.; Yoneda, M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. [Google Scholar] [CrossRef]
- Baraniak, P.R.; McDevitt, T.C. Stem cell paracrine actions and tissue regeneration. Regen Med. 2010, 5, 121–143. [Google Scholar] [CrossRef]
- Fontaine, M.J.; Shih, H.; Schafer, R.; Pittenger, M.F. Unraveling the Mesenchymal Stromal Cells’ Paracrine Immunomodulatory Effects. Transfus. Med. Rev. 2016, 30, 37–43. [Google Scholar] [CrossRef]
- Han, Y.; Yang, J.; Fang, J.; Zhou, Y.; Candi, E.; Wang, J.; Hua, D.; Shao, C.; Shi, Y. The secretion profile of mesenchymal stem cells and potential applications in treating human diseases. Signal Transduct. Target Ther. 2022, 7, 92. [Google Scholar] [CrossRef]
- Yang, Y.; Zhao, Y.; Zhang, L.; Zhang, F.; Li, L. The Application of Mesenchymal Stem Cells in the Treatment of Liver Diseases: Mechanism, Efficacy, and Safety Issues. Front. Med. 2021, 8, 655268. [Google Scholar] [CrossRef] [PubMed]
- Najimi, M.; Khuu, D.N.; Lysy, P.A.; Jazouli, N.; Abarca, J.; Sempoux, C.; Sokal, E.M. Adult-derived human liver mesenchymal-like cells as a potential progenitor reservoir of hepatocytes? Cell Transplant. 2007, 16, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Khuu, D.N.; Scheers, I.; Ehnert, S.; Jazouli, N.; Nyabi, O.; Buc-Calderon, P.; Meulemans, A.; Nussler, A.; Sokal, E.; Najimi, M. In vitro differentiated adult human liver progenitor cells display mature hepatic metabolic functions: A potential tool for in vitro pharmacotoxicological testing. Cell Transplant. 2011, 20, 287–302. [Google Scholar] [CrossRef] [PubMed]
- Sokal, E.M.; Stephenne, X.; Ottolenghi, C.; Jazouli, N.; Clapuyt, P.; Lacaille, F.; Najimi, M.; de Lonlay, P.; Smets, F. Liver engraftment and repopulation by in vitro expanded adult derived human liver stem cells in a child with ornithine carbamoyltransferase deficiency. JIMD Rep. 2014, 13, 65–72. [Google Scholar] [PubMed]
- Defresne, F.; Tondreau, T.; Stephenne, X.; Smets, F.; Bourgois, A.; Najimi, M.; Jamar, F.; Sokal, E.M. Biodistribution of adult derived human liver stem cells following intraportal infusion in a 17-year-old patient with glycogenosis type 1A. Nucl. Med. Biol. 2014, 41, 371–375. [Google Scholar] [CrossRef]
- Smets, F.; Dobbelaere, D.; McKiernan, P.; Dionisi-Vici, C.; Broue, P.; Jacquemin, E.; Lopes, A.I.; Goncalves, I.; Mandel, H.; Pawlowska, J.; et al. Phase I/II Trial of Liver-derived Mesenchymal Stem Cells in Pediatric Liver-based Metabolic Disorders: A Prospective, Open Label, Multicenter, Partially Randomized, Safety Study of One Cycle of Heterologous Human Adult Liver-derived Progenitor Cells (HepaStem) in Urea Cycle Disorders and Crigler-Najjar Syndrome Patients. Transplantation 2019, 103, 1903–1915. [Google Scholar]
- Sokal, E.M.; Lombard, C.A.; Roelants, V.; Najimi, M.; Varma, S.; Sargiacomo, C.; Ravau, J.; Mazza, G.; Jamar, F.; Versavau, J.; et al. Biodistribution of Liver-Derived Mesenchymal Stem Cells After Peripheral Injection in a Hemophilia A Patient. Transplantation 2017, 101, 1845–1851. [Google Scholar] [CrossRef]
- El-Kehdy, H.; Sargiacomo, C.; Fayyad-Kazan, M.; Fayyad-Kazan, H.; Lombard, C.; Lagneaux, L.; Sokal, E.; Najar, M.; Najimi, M. Immunoprofiling of Adult-Derived Human Liver Stem/Progenitor Cells: Impact of Hepatogenic Differentiation and Inflammation. Stem Cells Int. 2017, 2017, 2679518. [Google Scholar] [CrossRef]
- Najimi, M.; Berardis, S.; El-Kehdy, H.; Rosseels, V.; Evraerts, J.; Lombard, C.; El Taghdouini, A.; Henriet, P.; van Grunsven, L.; Sokal, E.M. Human liver mesenchymal stem/progenitor cells inhibit hepatic stellate cell activation: In vitro and in vivo evaluation. Stem Cell Res. Ther. 2017, 8, 131. [Google Scholar] [CrossRef]
- Najar, M.; Crompot, E.; Raicevic, G.; Sokal, E.M.; Najimi, M.; Lagneaux, L. Cytokinome of adult-derived human liver stem/progenitor cells: Immunological and inflammatory features. Hepatobiliary Surg. Nutr. 2018, 7, 331–344. [Google Scholar] [CrossRef]
- Lombard, C.A.; Sana, G.; LeMaoult, J.; Najar, M.; Ravau, J.; Andre, F.; Bouhtit, F.; Daouya, M.; Loustau, M.; Najimi, M.; et al. Human Hepatocytes and Differentiated Adult-Derived Human Liver Stem/Progenitor Cells Display In Vitro Immunosuppressive Properties Mediated, at Least in Part, through the Nonclassical HLA Class I Molecule HLA-G. J. Immunol. Res. 2019, 2019, 8250584. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Gustot, T.; Laterre, P.F.; Lasser, L.L.; Haralampiev, L.E.; Vargas, V.; Lyubomirova, D.; Albillos, A.; Najimi, M.; Michel, S.; et al. A phase II study of human allogeneic liver-derived progenitor cell therapy for acute-on-chronic liver failure and acute decompensation. JHEP Rep. 2021, 3, 100291. [Google Scholar] [CrossRef] [PubMed]
- El-Kehdy, H.; Najar, M.; De Kock, J.; Agha, D.M.; Rogiers, V.; Merimi, M.; Lagneaux, L.; Sokal, E.M.; Najimi, M. Inflammation Differentially Modulates the Biological Features of Adult Derived Human Liver Stem/Progenitor Cells. Cells 2020, 9, 1640. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Shibazaki, Y.; Wakamatsu, K.; Honda, Y.; Kawauchi, Y.; Suzuki, K.; Arumugam, S.; Watanabe, K.; Ichida, T.; Asakura, H.; et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med. Mol. Morphol. 2013, 46, 141–152. [Google Scholar] [CrossRef]
- Saito, K.; Uebanso, T.; Maekawa, K.; Ishikawa, M.; Taguchi, R.; Nammo, T.; Nishimaki-Mogami, T.; Udagawa, H.; Fujii, M.; Shibazaki, Y.; et al. Characterization of hepatic lipid profiles in a mouse model with nonalcoholic steatohepatitis and subsequent fibrosis. Sci. Rep. 2015, 5, 12466. [Google Scholar] [CrossRef]
- Berardis, S.; Dwisthi Sattwika, P.; Najimi, M.; Sokal, E.M. Use of mesenchymal stem cells to treat liver fibrosis: Current situation and future prospects. World J. Gastroenterol. 2015, 21, 742–758. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, H.; Shi, M.; Xu, R.; Fu, J.; Lv, J.; Chen, L.; Lv, S.; Li, Y.; Yu, S.; et al. Human umbilical cord mesenchymal stem cells improve liver function and ascites in decompensated liver cirrhosis patients. J. Gastroenterol. Hepatol. 2012, 27, 112–120. [Google Scholar] [CrossRef]
- Wang, L.; Li, J.; Liu, H.; Li, Y.; Fu, J.; Sun, Y.; Xu, R.; Lin, H.; Wang, S.; Lv, S.; et al. Pilot study of umbilical cord-derived mesenchymal stem cell transfusion in patients with primary biliary cirrhosis. J. Gastroenterol. Hepatol. 2013, 28, 85–92. [Google Scholar] [CrossRef]
- Zimmermann, E.; Anty, R.; Tordjman, J.; Verrijken, A.; Gual, P.; Tran, A.; Iannelli, A.; Gugenheim, J.; Bedossa, P.; Francque, S.; et al. C-reactive protein levels in relation to various features of non-alcoholic fatty liver disease among obese patients. J. Hepatol. 2011, 55, 660–665. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Nonalcoholic Steatohepatitis Clinical Research, N., Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Segal-Salto, M.; Barashi, N.; Katav, A.; Edelshtein, V.; Aharon, A.; Hashmueli, S.; George, J.; Maor, Y.; Pinzani, M.; Haberman, D.; et al. A blocking monoclonal antibody to CCL24 alleviates liver fibrosis and inflammation in experimental models of liver damage. JHEP Rep. 2020, 2, 100064. [Google Scholar] [CrossRef] [PubMed]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Animal Models of Fibrosis in Nonalcoholic Steatohepatitis: Do They Reflect Human Disease? Adv. Nutr. 2020, 11, 1696–1711. [Google Scholar] [CrossRef]
- Merimi, M.; Lagneaux, L.; Lombard, C.A.; Agha, D.M.; Bron, D.; Lewalle, P.; Meuleman, N.; Najimi, M.; Sokal, E.M.; Najar, M. Immuno-comparative screening of adult-derived human liver stem/progenitor cells for immune-inflammatory-associated molecules. Inflamm. Res. 2021, 70, 229–239. [Google Scholar] [CrossRef]
- Chan, M.M.; Moore, A.R. Resolution of inflammation in murine autoimmune arthritis is disrupted by cyclooxygenase-2 inhibition and restored by prostaglandin E2-mediated lipoxin A4 production. J. Immunol. 2010, 184, 6418–6426. [Google Scholar] [CrossRef] [PubMed]
- Fabricius, D.; Neubauer, M.; Mandel, B.; Schutz, C.; Viardot, A.; Vollmer, A.; Jahrsdorfer, B.; Debatin, K.M. Prostaglandin E2 inhibits IFN-alpha secretion and Th1 costimulation by human plasmacytoid dendritic cells via E-prostanoid 2 and E-prostanoid 4 receptor engagement. J. Immunol. 2010, 184, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Brea, R.; Motino, O.; Frances, D.; Garcia-Monzon, C.; Vargas, J.; Fernandez-Velasco, M.; Bosca, L.; Casado, M.; Martin-Sanz, P.; Agra, N. PGE2 induces apoptosis of hepatic stellate cells and attenuates liver fibrosis in mice by downregulating miR-23a-5p and miR-28a-5p. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 325–337. [Google Scholar] [CrossRef]
- Mellor, A.L.; Lemos, H.; Huang, L. Indoleamine 2,3-Dioxygenase and Tolerance: Where Are We Now? Front. Immunol. 2017, 8, 1360. [Google Scholar] [CrossRef]
- Nagano, J.; Shimizu, M.; Hara, T.; Shirakami, Y.; Kochi, T.; Nakamura, N.; Ohtaki, H.; Ito, H.; Tanaka, T.; Tsurumi, H.; et al. Effects of indoleamine 2,3-dioxygenase deficiency on high-fat diet-induced hepatic inflammation. PLoS ONE 2013, 8, e73404. [Google Scholar] [CrossRef]
- Hoogduijn, M.J.; Popp, F.; Verbeek, R.; Masoodi, M.; Nicolaou, A.; Baan, C.; Dahlke, M.H. The immunomodulatory properties of mesenchymal stem cells and their use for immunotherapy. Int. Immunopharmacol. 2010, 10, 1496–1500. [Google Scholar] [CrossRef]
- Williams, E.L.; White, K.; Oreffo, R.O. Isolation and enrichment of Stro-1 immunoselected mesenchymal stem cells from adult human bone marrow. Methods Mol. Biol. 2013, 1035, 67–73. [Google Scholar]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Mias, C.; Lairez, O.; Trouche, E.; Roncalli, J.; Calise, D.; Seguelas, M.H.; Ordener, C.; Piercecchi-Marti, M.D.; Auge, N.; Salvayre, A.N.; et al. Mesenchymal stem cells promote matrix metalloproteinase secretion by cardiac fibroblasts and reduce cardiac ventricular fibrosis after myocardial infarction. Stem Cells 2009, 27, 2734–2743. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Brunt, E.M.; Kleiner, D.E.; Kowdley, K.V.; Chalasani, N.; Lavine, J.E.; Ratziu, V.; McCullough, A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology 2011, 54, 344–353. [Google Scholar] [CrossRef] [Green Version]
- Ekstedt, M.; Hagstrom, H.; Nasr, P.; Fredrikson, M.; Stal, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- DeWoskin, R.S.; Knudsen, T.B.; Shah, I. Virtual Models (vM). In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Elsevier Inc.; Academic Press: Cambridge, MA, USA, 2014; Volume 4, pp. 948–957. [Google Scholar]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef]
- Sana, G.; Lombard, C.; Vosters, O.; Jazouli, N.; Andre, F.; Stephenne, X.; Smets, F.; Najimi, M.; Sokal, E.M. Adult human hepatocytes promote CD4(+) T-cell hyporesponsiveness via interleukin-10-producing allogeneic dendritic cells. Cell Transplant. 2014, 23, 1127–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najimi, M.; Michel, S.; Binda, M.M.; Gellynck, K.; Belmonte, N.; Mazza, G.; Gordillo, N.; Vainilovich, Y.; Sokal, E. Human Allogeneic Liver-Derived Progenitor Cells Significantly Improve NAFLD Activity Score and Fibrosis in Late-Stage NASH Animal Model. Cells 2022, 11, 2854. https://doi.org/10.3390/cells11182854
Najimi M, Michel S, Binda MM, Gellynck K, Belmonte N, Mazza G, Gordillo N, Vainilovich Y, Sokal E. Human Allogeneic Liver-Derived Progenitor Cells Significantly Improve NAFLD Activity Score and Fibrosis in Late-Stage NASH Animal Model. Cells. 2022; 11(18):2854. https://doi.org/10.3390/cells11182854
Chicago/Turabian StyleNajimi, Mustapha, Sébastien Michel, Maria M. Binda, Kris Gellynck, Nathalie Belmonte, Giuseppe Mazza, Noelia Gordillo, Yelena Vainilovich, and Etienne Sokal. 2022. "Human Allogeneic Liver-Derived Progenitor Cells Significantly Improve NAFLD Activity Score and Fibrosis in Late-Stage NASH Animal Model" Cells 11, no. 18: 2854. https://doi.org/10.3390/cells11182854
APA StyleNajimi, M., Michel, S., Binda, M. M., Gellynck, K., Belmonte, N., Mazza, G., Gordillo, N., Vainilovich, Y., & Sokal, E. (2022). Human Allogeneic Liver-Derived Progenitor Cells Significantly Improve NAFLD Activity Score and Fibrosis in Late-Stage NASH Animal Model. Cells, 11(18), 2854. https://doi.org/10.3390/cells11182854