

A Novel Muscle Atrophy Mechanism: Myocyte Degeneration Due to Intracellular Iron Deprivation

 and
and
Abstract
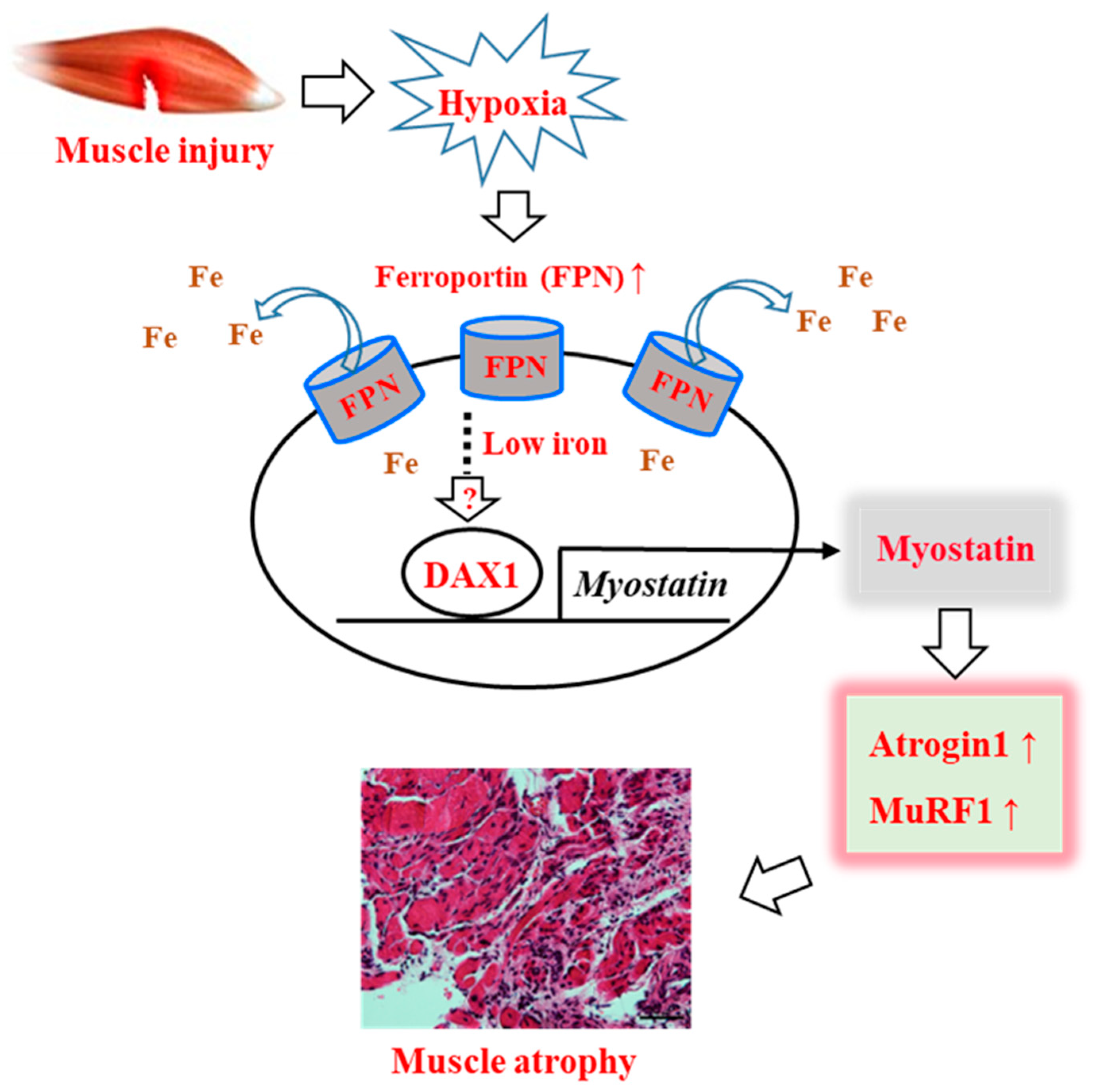
:1. Introduction
2. Materials and Methods
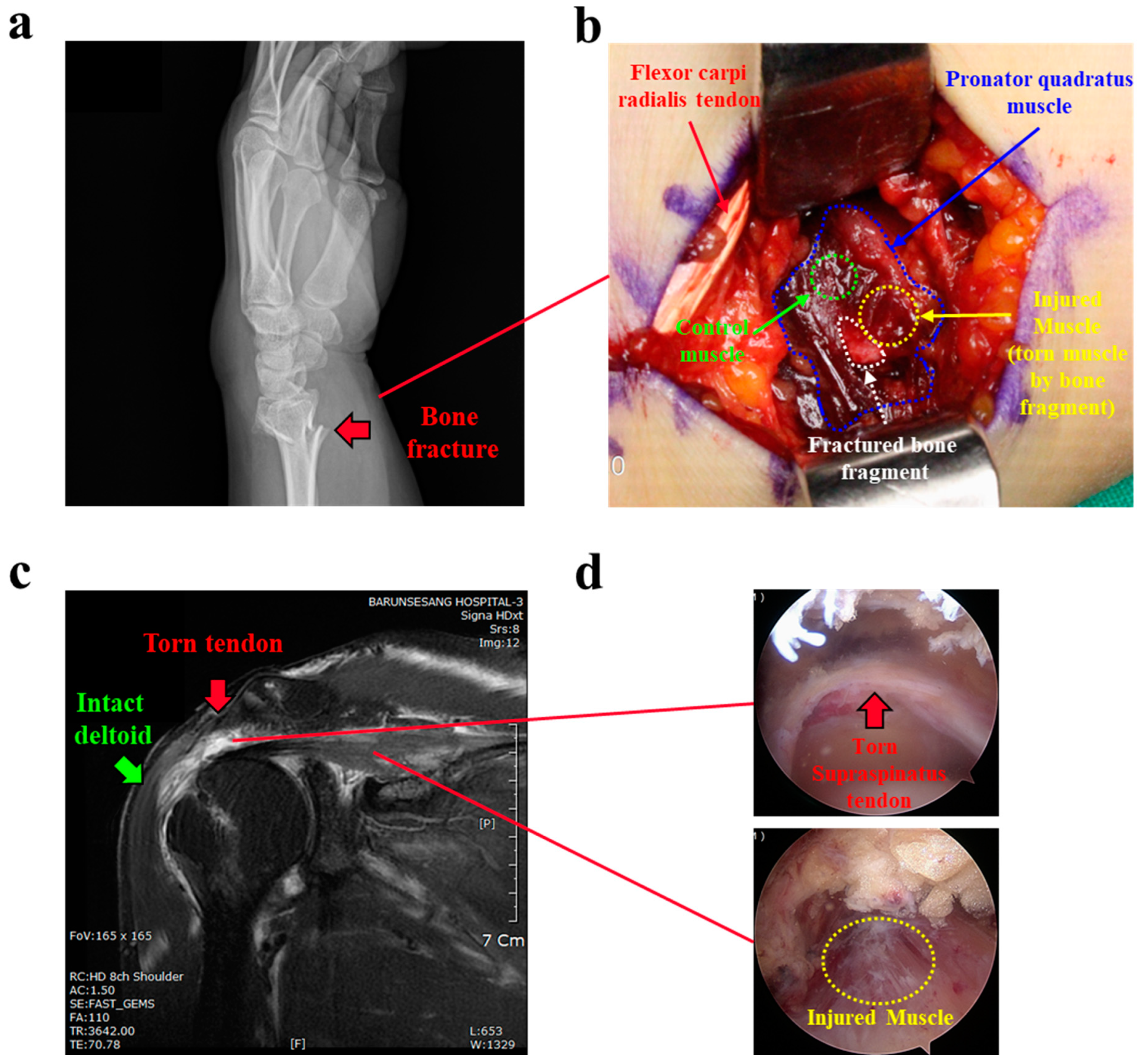
2.1. Subjects and Human Tissue Acquisition
2.2. Mouse Muscle Injury Model
2.3. Measurement of Iron Levels
2.4. Reverse Transcription Quantitative Real-Time PCR (RT-qPCR) Analysis
2.5. Western Blot Analysis
2.6. Tissue Histology
2.7. Cell Culture, Reagents, Hypoxic Incubation, Transient Transfection and Adenoviral Infection
2.8. Cell Viability Assay and Flow Cytometric Analysis
2.9. Statistical Analyses
3. Results
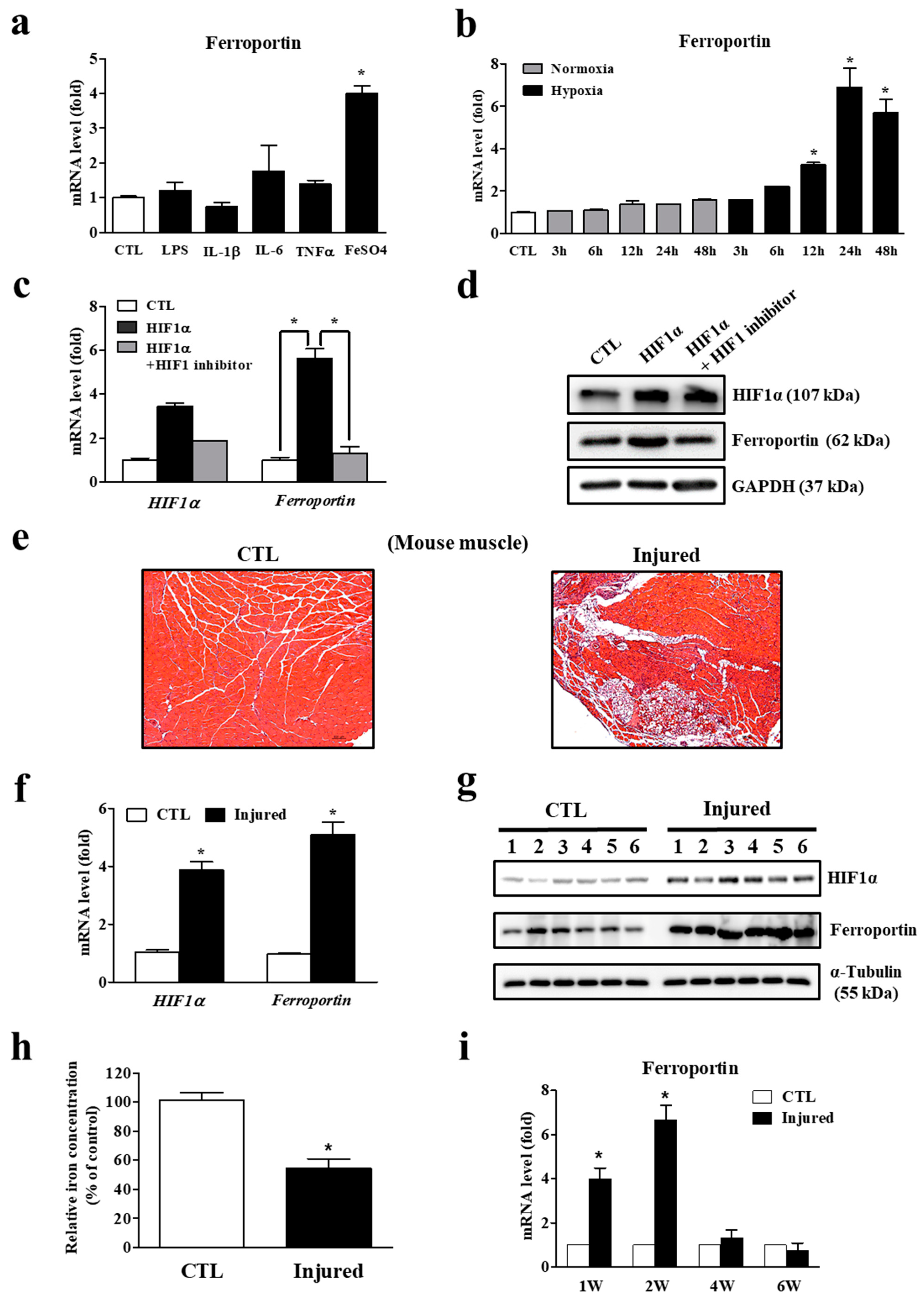
3.1. Decreasing Iron Levels Correlate with Increasing Ferroportin Expression in the Injured Muscles
3.2. Ferroportin Expression Is Regulated by Hypoxia, Leading to Lower Iron Concentration
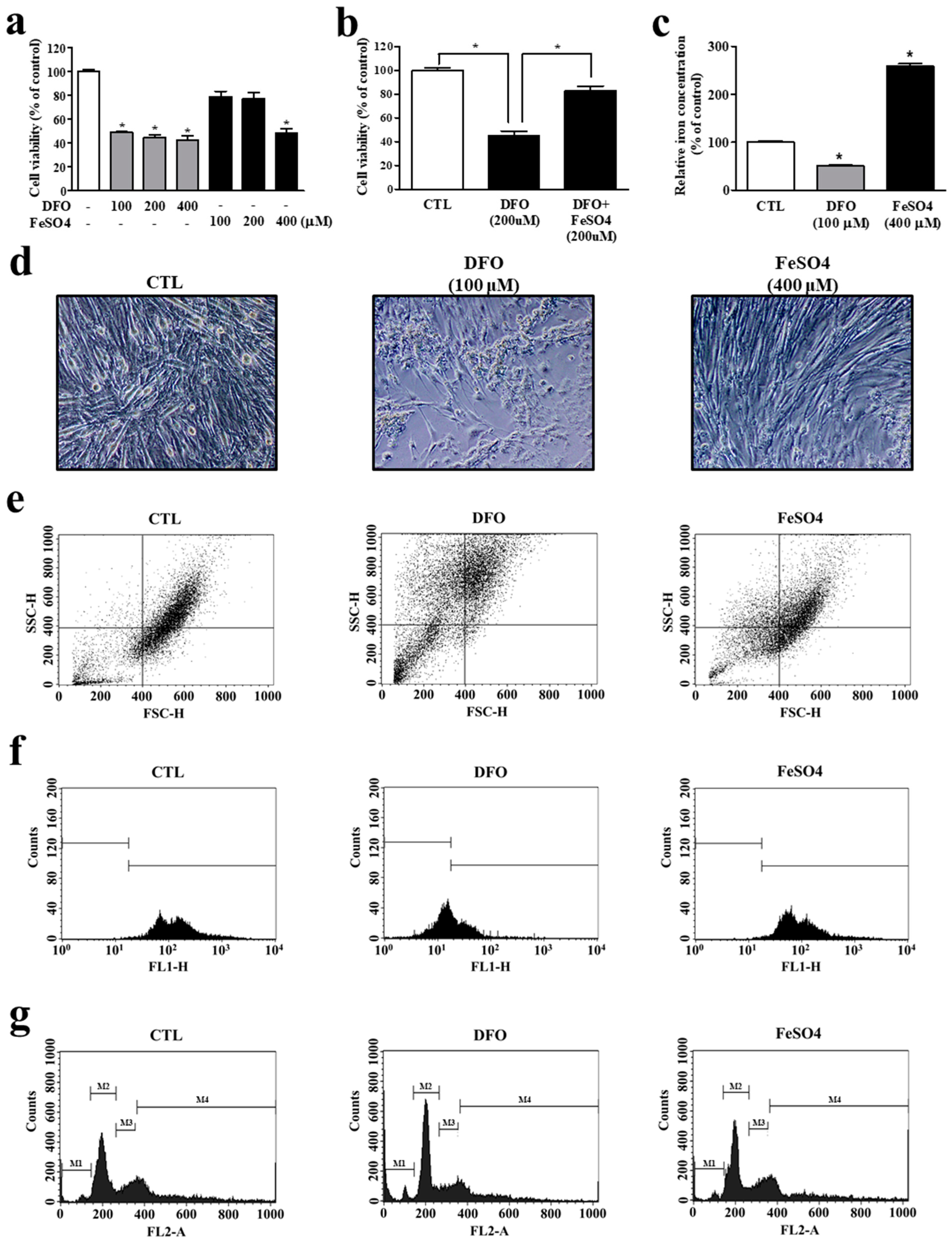
3.3. Iron Deprivation Leads to Smaller Cell Population and Myocyte Mitochondrial Dysfunction
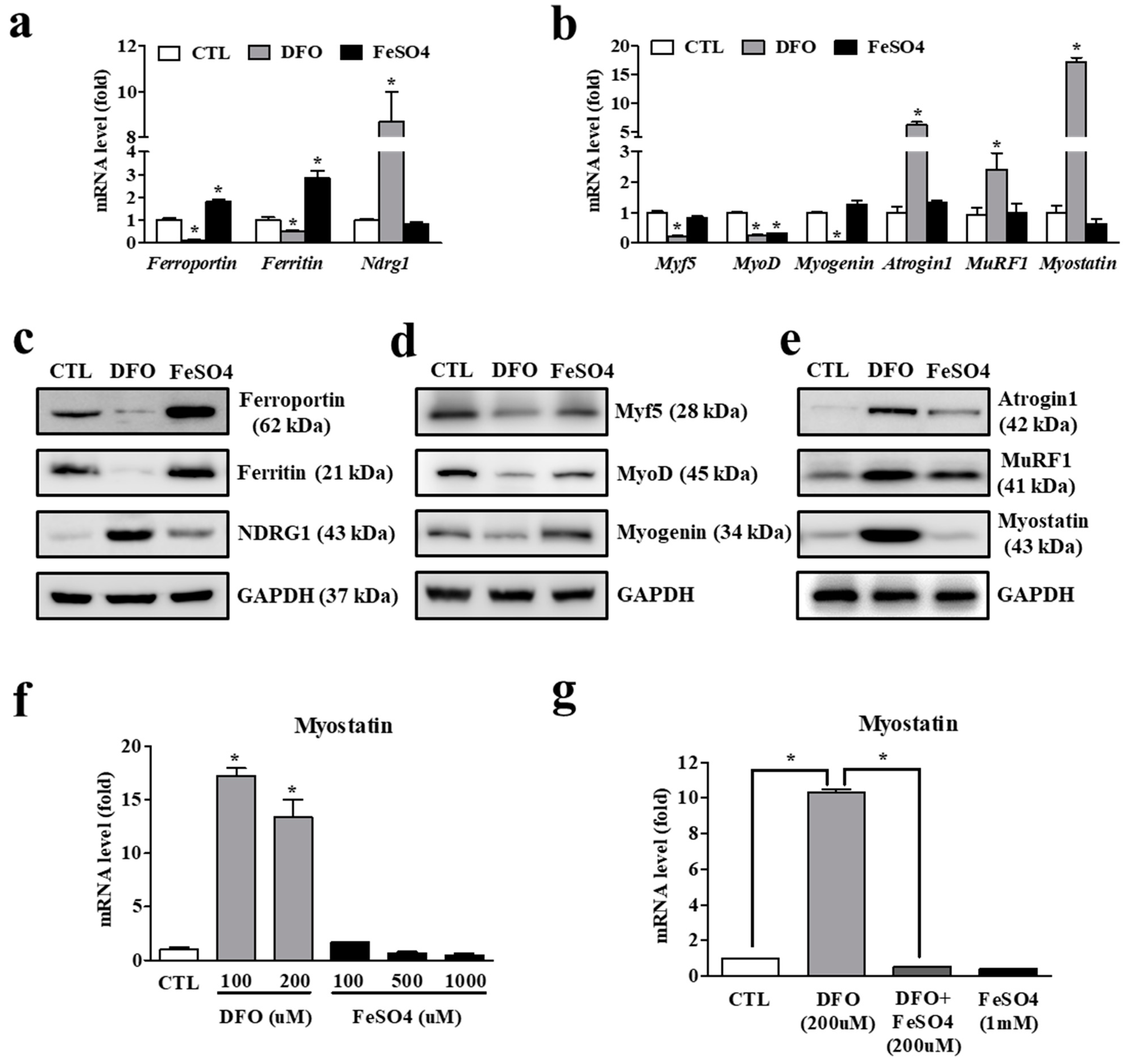
3.4. Iron Deficiency in Myocytes Is Accompanied by Increased Expression of Atrophy-Associated Genes
3.5. Iron Deficiency-Induced Myostatin Expression Is Mediated by an Orphan Nuclear Receptor, DAX1
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fanzani, A.; Conraads, V.M.; Penna, F.; Martinet, W. Molecular and cellular mechanisms of skeletal muscle atrophy: An update. J. Cachexia Sarcopenia Muscle 2012, 3, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Signaling pathways perturbing muscle mass. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, R.R.; Corriere, M.; Ferrucci, L. Age-related and disease-related muscle loss: The effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2014, 2, 819–829. [Google Scholar] [CrossRef]
- Hornberger, T.A. Mechanotransduction and the regulation of mtorc1 signaling in skeletal muscle. Int. J. Biochem. Cell Biol. 2011, 43, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Wing, S.S.; Goldberg, A.L. Glucocorticoids activate the atp-ubiquitin-dependent proteolytic system in skeletal muscle during fasting. Am. J. Physiol. 1993, 264, E668–E676. [Google Scholar] [CrossRef] [PubMed]
- Bonaldo, P.; Sandri, M. Cellular and molecular mechanisms of muscle atrophy. Dis. Model Mech. 2013, 6, 25–39. [Google Scholar] [CrossRef]
- Kitajima, Y.; Yoshioka, K.; Suzuki, N. The ubiquitin-proteasome system in regulation of the skeletal muscle homeostasis and atrophy: From basic science to disorders. J. Physiol. Sci. 2020, 70, 40. [Google Scholar] [CrossRef]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Bodine, S.C.; Baehr, L.M. Skeletal muscle atrophy and the e3 ubiquitin ligases murf1 and mafbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E469–E484. [Google Scholar] [CrossRef]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Li, J.; Zhang, Y.; Chang, Y.Z. Cellular iron metabolism and regulation. Adv. Exp. Med. Biol. 2019, 1173, 21–32. [Google Scholar]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Ordway, G.A.; Garry, D.J. Myoglobin: An essential hemoprotein in striated muscle. J. Exp. Biol. 2004, 207, 3441–3446. [Google Scholar] [CrossRef]
- Dziegala, M.; Josiak, K.; Kasztura, M.; Kobak, K.; von Haehling, S.; Banasiak, W.; Anker, S.D.; Ponikowski, P.; Jankowska, E. Iron deficiency as energetic insult to skeletal muscle in chronic diseases. J. Cachexia Sarcopenia Muscle 2018, 9, 802–815. [Google Scholar] [CrossRef]
- van der Meer, P.; van der Wal, H.H.; Melenovsky, V. Mitochondrial function, skeletal muscle metabolism, and iron deficiency in heart failure. Circulation 2019, 139, 2399–2402. [Google Scholar] [CrossRef]
- Leermakers, P.A.; Remels, A.H.V.; Zonneveld, M.I.; Rouschop, K.M.A.; Schols, A.; Gosker, H.R. Iron deficiency-induced loss of skeletal muscle mitochondrial proteins and respiratory capacity; the role of mitophagy and secretion of mitochondria-containing vesicles. FASEB J. 2020, 34, 6703–6717. [Google Scholar] [CrossRef]
- Hofer, T.; Marzetti, E.; Xu, J.; Seo, A.Y.; Gulec, S.; Knutson, M.D.; Leeuwenburgh, C.; Dupont-Versteegden, E.E. Increased iron content and rna oxidative damage in skeletal muscle with aging and disuse atrophy. Exp. Gerontol. 2008, 43, 563–570. [Google Scholar] [CrossRef]
- Ikeda, Y.; Imao, M.; Satoh, A.; Watanabe, H.; Hamano, H.; Horinouchi, Y.; Izawa-Ishizawa, Y.; Kihira, Y.; Miyamoto, L.; Ishizawa, K.; et al. Iron-induced skeletal muscle atrophy involves an akt-forkhead box o3-e3 ubiquitin ligase-dependent pathway. J. Trace Elem. Med. Biol. 2016, 35, 66–76. [Google Scholar] [CrossRef]
- Halon-Golabek, M.; Borkowska, A.; Herman-Antosiewicz, A.; Antosiewicz, J. Iron metabolism of the skeletal muscle and neurodegeneration. Front Neurosci. 2019, 13, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Kim, J.Y.; Oh, K.S.; Chung, S.W. Fatty acid-binding protein 4 regulates fatty infiltration after rotator cuff tear by hypoxia-inducible factor 1 in mice. J. Cachexia Sarcopenia Muscle 2017, 8, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Scheerer, N.; Dehne, N.; Stockmann, C.; Swoboda, S.; Baba, H.A.; Neugebauer, A.; Johnson, R.S.; Fandrey, J. Myeloid hypoxia-inducible factor-1alpha is essential for skeletal muscle regeneration in mice. J. Immunol. 2013, 191, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Reversing cachexia. Cell 2010, 142, 511–512. [Google Scholar] [CrossRef]
- Peyssonnaux, C.; Zinkernagel, A.S.; Schuepbach, R.A.; Rankin, E.; Vaulont, S.; Haase, V.H.; Nizet, V.; Johnson, R.S. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (hifs). J. Clin. Investig. 2007, 117, 1926–1932. [Google Scholar] [CrossRef]
- Taylor, M.; Qu, A.; Anderson, E.R.; Matsubara, T.; Martin, A.; Gonzalez, F.J.; Shah, Y.M. Hypoxia-inducible factor-2alpha mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology 2011, 140, 2044–2055. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Polonifi, A.; Politou, M.; Kalotychou, V.; Xiromeritis, K.; Tsironi, M.; Berdoukas, V.; Vaiopoulos, G.; Aessopos, A. Iron metabolism gene expression in human skeletal muscle. Blood Cells Mol. Dis. 2010, 45, 233–237. [Google Scholar] [CrossRef]
- Chen, W.; Huang, F.W.; de Renshaw, T.B.; Andrews, N.C. Skeletal muscle hemojuvelin is dispensable for systemic iron homeostasis. Blood 2011, 117, 6319–6325. [Google Scholar] [CrossRef]
- Corna, G.; Caserta, I.; Monno, A.; Apostoli, P.; Manfredi, A.A.; Camaschella, C.; Rovere-Querini, P. The repair of skeletal muscle requires iron recycling through macrophage ferroportin. J. Immunol. 2016, 197, 1914–1925. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Elkina, Y.; von Haehling, S.; Anker, S.D.; Springer, J. The role of myostatin in muscle wasting: An overview. J. Cachexia Sarcopenia Muscle 2011, 2, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; McFarlane, C.; Kambadur, R.; Kukreti, H.; Bonala, S.; Srinivasan, S. Myostatin: Expanding horizons. IUBMB Life 2015, 67, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Niakan, K.K.; McCabe, E.R. Dax1 origin, function, and novel role. Mol. Genet. Metab. 2005, 86, 70–83. [Google Scholar] [CrossRef] [PubMed]
- Helguero, L.A.; Hedengran Faulds, M.; Forster, C.; Gustafsson, J.A.; Haldosen, L.A. Dax-1 expression is regulated during mammary epithelial cell differentiation. Endocrinology 2006, 147, 3249–3259. [Google Scholar] [CrossRef] [PubMed]
- Khalfallah, O.; Rouleau, M.; Barbry, P.; Bardoni, B.; Lalli, E. Dax-1 knockdown in mouse embryonic stem cells induces loss of pluripotency and multilineage differentiation. Stem Cells 2009, 27, 1529–1537. [Google Scholar] [CrossRef]
- Lalli, E.; Alonso, J. Targeting dax-1 in embryonic stem cells and cancer. Expert Opin. Ther. Targets 2010, 14, 169–177. [Google Scholar] [CrossRef]
- Stugiewicz, M.; Tkaczyszyn, M.; Kasztura, M.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. The influence of iron deficiency on the functioning of skeletal muscles: Experimental evidence and clinical implications. Eur. J. Heart Fail 2016, 18, 762–773. [Google Scholar] [CrossRef]
- Kobak, K.; Kasztura, M.; Dziegala, M.; Bania, J.; Kapusniak, V.; Banasiak, W.; Ponikowski, P.; Jankowska, E.A. Iron limitation promotes the atrophy of skeletal myocytes, whereas iron supplementation prevents this process in the hypoxic conditions. Int. J. Mol. Med. 2018, 41, 2678–2686. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Full Name | Gene Symbol | Sequences; Forward (F)/Reverse (R) |
|---|---|---|
| (human) Ferritin, heavy polypeptide 1 | FTH1 | (F) 5′-CCCCCATTTGTGTGACTTCAT-3′ (R) 5′-GCCCGAGGCTTAGCTTTCATT-3′ |
| (mouse) Ferritin, light polypeptide 1 | Ftl1 | (F) 5′-AATCAGGCCCTCTTGGATCT-3′ (R) 5′-GGCGCTCAAAGAGATACTCG-3′ |
| (human) Hepcidin antimicrobial peptide | HAMP | (F) 5′-ATGGCACTGAGCTCCCAGAT-3′ (R) 5′-CACATCCCACACTTTGATCG-3′ |
| (mouse) Hepcidin antimicrobial peptide | Hamp | (F) 5′-TGCCTGTCTCCTGCTTCTCCT-3′ (R) 5′-GATGGGGAAGTTGGTGTCTC-3′ |
| (human/mouse) Ferroportin | FTN | (F) 5′-CTGCAAGAAAATGTAATTGAATCTG-3′ (R) 5′-ATTGCCACAAAGGAGACTGAAATC-3′ |
| (mouse) Hypoxia inducible factor 1 alpha | Hif1a | (F) 5′-ACCTTCATCGGAAACTCCAAAG-3′ (R) 5′-CTGTTAGGCTGGGAAAAGTTAGG-3′ |
| (mouse) N-myc downstream regulated gene 1 | Ndrg1 | (F) 5′-ATGTCCCGAGAGCTACATGAC-3′ (R) 5′-CCTGCTCCTGAACATCGAACT-3′ |
| (mouse) Myogenic factor 5 | Myf5 | (F) 5′-CCTCATGTGGGCCTGCAAAG-3′ (R) 5′-CATTCCTGAGGATCTCCACC-3′ |
| (mouse) Myogenic differentiation 1 | Myod1 | (F) 5′-CAAGCGCAAGACCACCAACG-3′ (R) 5′-ATATAGCGGATGGCGTTGC-3′ |
| (mouse) Myogenin | Myog | (F) 5′-GAGACATCCCCCTATTTCTACCA-3′ (R) 5′-GCTCAGTCCGCTCATAGCC-3′ |
| (mouse) F-box protein 32 (Atrogin 1) | Fbxo32 | (F) 5′-ATGCACACTGGTGCAGAGAG-3′ (R) 5′-TGTAAGCACACAGGCAGGTC-3′ |
| (mouse) Tripartite motif-containing 63 (MuRF1) | Trim63 | (F) 5′-ACCTGCTGGTGGAAAACATC-3′ (R) 5′-AGGAGCAAGTAGGCACCTCA-3′ |
| (mouse) Myostatin | Mstn | (F) 5′-AGTGGATCTAAATGAGGGCAGT-3′ (R) 5′-GTTTCCAGGCGCAGCTTAC-3′ |
| (mouse) Nuclear receptor subfamily 0, group B, member 1 | Dax1 | (F) 5′-AGGGCAGCATCCTCTACAAC-3′ (R) 5′-TGGTCTTCACCACAAAAGCA-3′ |
| (mouse) Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | (F) 5′-ATAATACCGATCCCCGAAGG-3′ (R) 5′-CTGGATGGTGTATGCACAGG-3′ |
| (human/mouse) Actin, beta | Actb | (F) 5′-TCTGGCACCACACCTTCTAC-3′ (R) 5′-TCGTAGATGGGCACAGTGTGG-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suh, D.K.; Lee, W.-Y.; Yeo, W.J.; Kyung, B.S.; Jung, K.W.; Seo, H.K.; Lee, Y.-S.; Suh, D.W. A Novel Muscle Atrophy Mechanism: Myocyte Degeneration Due to Intracellular Iron Deprivation. Cells 2022, 11, 2853. https://doi.org/10.3390/cells11182853
Suh DK, Lee W-Y, Yeo WJ, Kyung BS, Jung KW, Seo HK, Lee Y-S, Suh DW. A Novel Muscle Atrophy Mechanism: Myocyte Degeneration Due to Intracellular Iron Deprivation. Cells. 2022; 11(18):2853. https://doi.org/10.3390/cells11182853
Chicago/Turabian StyleSuh, Dae Keun, Won-Young Lee, Woo Jin Yeo, Bong Soo Kyung, Koo Whang Jung, Hye Kyung Seo, Yong-Soo Lee, and Dong Won Suh. 2022. "A Novel Muscle Atrophy Mechanism: Myocyte Degeneration Due to Intracellular Iron Deprivation" Cells 11, no. 18: 2853. https://doi.org/10.3390/cells11182853
APA StyleSuh, D. K., Lee, W.-Y., Yeo, W. J., Kyung, B. S., Jung, K. W., Seo, H. K., Lee, Y.-S., & Suh, D. W. (2022). A Novel Muscle Atrophy Mechanism: Myocyte Degeneration Due to Intracellular Iron Deprivation. Cells, 11(18), 2853. https://doi.org/10.3390/cells11182853