
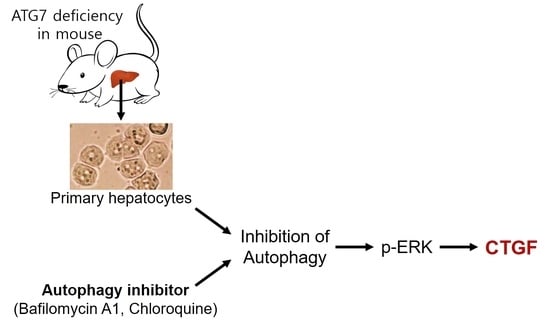
Increased Levels of Phosphorylated ERK Induce CTGF Expression in Autophagy-Deficient Mouse Hepatocytes

 , and
, and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Generation of ATG7flox/flox-Albumin-Cre Mice
2.2. Chemicals and Antibodies
2.3. Immunohistochemical Analysis
2.4. Cell Culture
2.5. Isolation of the Primary Hepatocytes
2.6. Depletion of SQSTM1/p62 Using siRNA
2.7. Western Blotting
2.8. Quantitative Real-Time RT-PCR
2.9. Immunofluorescence (IF) Analysis
2.10. Statistical Analysis
3. Results
3.1. Loss of ATG7 in the Liver Increases the Expression Levels of Fibrosis-Related Genes
3.2. CTGF Expression Is Increased in ATG7-Deficient Primary Hepatocytes
3.3. Inhibition of Autophagy Increases CTGF Expression in Hepatic Cell Lines
3.4. ATG7 Deficiency-Induced Expression of CTGF Does Not Rely on the TGF-β Pathway
3.5. Inhibition of Autophagy Increases ERK Expression
3.6. CTGF Increase in Autophagy Inhibition Is Associated with the Upregulation of Phospho-ERK
3.7. Inhibition of Autophagy Also Increases CTGF Expression in Kidney Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lipson, K.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair 2012, 5 (Suppl. 1), S24. [Google Scholar] [CrossRef] [PubMed]
- Gressner, O.A.; Gressner, A.M. Connective tissue growth factor: A fibrogenic master switch in fibrotic liver diseases. Liver Int. Off. J. Int. Assoc. Study Liver 2008, 28, 1065–1079. [Google Scholar] [CrossRef] [PubMed]
- Frazier, K.; Williams, S.; Kothapalli, D.; Klapper, H.; Grotendorst, G.R. Stimulation of fibroblast cell growth, matrix production, and granulation tissue formation by connective tissue growth factor. J. Investig. Dermatol. 1996, 107, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Parapuram, S.K.; Shi-Wen, X.; Abraham, D.J. Connective tissue growth factor (CTGF, CCN2) gene regulation: A potent clinical bio-marker of fibroproliferative disease? J. Cell Commun. Signal. 2009, 3, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef]
- Grotendorst, G.R.; Okochi, H.; Hayashi, N. A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ. 1996, 7, 469–480. [Google Scholar]
- Phanish, M.K.; Winn, S.K.; Dockrell, M.E.C. Connective Tissue Growth Factor-(CTGF, CCN2)—A Marker, Mediator and Therapeutic Target for Renal Fibrosis. Nephron Exp. Nephrol. 2010, 114, e83–e92. [Google Scholar] [CrossRef]
- Huang, G.; Brigstock, D.R. Regulation of hepatic stellate cells by connective tissue growth factor. Front. Biosci. 2012, 17, 2495–2507. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef]
- Madrigal-Matute, J.; Cuervo, A.M. Regulation of Liver Metabolism by Autophagy. Gastroenterology 2016, 150, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Allaire, M.; Rautou, P.-E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef]
- Gual, P.; Gilgenkrantz, H.; Lotersztajn, S. Autophagy in chronic liver diseases: The two faces of Janus. Am. J. Physiol. Cell Physiol. 2017, 312, C263–C273. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Woolbright, B.L.; Williams, J.; Copple, B.; Cui, W.; Luyendyk, J.P.; Jaeschke, H.; Ding, W.X. Nrf2 promotes the development of fibrosis and tumorigenesis in mice with defective hepatic autophagy. J. Hepatol. 2014, 61, 617–625. [Google Scholar] [CrossRef]
- Kim, K.H.; Jeong, Y.T.; Oh, H.; Kim, S.H.; Cho, J.M.; Kim, Y.N.; Kim, S.S.; Kim, D.H.; Hur, K.Y.; Kim, H.K.; et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013, 19, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.A.; Noon, L.A.; Akat, K.M.; Ybanez, M.D.; Lee, T.-F.; Berres, M.-L.; Fujiwara, N.; Goossens, N.; Chou, H.-I.; Parvin-Nejad, F.P.; et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nat. Commun. 2018, 9, 4962. [Google Scholar] [CrossRef]
- Song, Z.M.; Liu, F.; Chen, Y.M.; Liu, Y.J.; Wang, X.D.; Du, S.Y. CTGF-mediated ERK signaling pathway influences the inflammatory factors and intestinal flora in ulcerative colitis. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 111, 1429–1437. [Google Scholar] [CrossRef]
- Kato, S.; Yokoyama, S.; Hayakawa, Y.; Li, L.; Iwakami, Y.; Sakurai, H.; Saiki, I. P38 pathway as a key downstream signal of connective tissue growth factor to regulate metastatic potential in non-small-cell lung cancer. Cancer Sci. 2016, 107, 1416–1421. [Google Scholar] [CrossRef]
- Shi, L.; Chang, Y.; Yang, Y.; Zhang, Y.; Yu, F.S.; Wu, X. Activation of JNK signaling mediates connective tissue growth factor expression and scar formation in corneal wound healing. PLoS ONE 2012, 7, e32128. [Google Scholar] [CrossRef]
- Cirit, M.; Grant, K.G.; Haugh, J.M. Systemic perturbation of the ERK signaling pathway by the proteasome inhibitor, MG132. PLoS ONE 2012, 7, e50975. [Google Scholar] [CrossRef]
- He, L.; Livingston, M.J.; Dong, Z. Autophagy in acute kidney injury and repair. Nephron Clin. Pract. 2014, 127, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Koike, M.; Sou, Y.S.; Ueno, T.; Hara, T.; Mizushima, N.; Iwata, J.; Ezaki, J.; Murata, S.; et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007, 131, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Inami, Y.; Waguri, S.; Sakamoto, A.; Kouno, T.; Nakada, K.; Hino, O.; Watanabe, S.; Ando, J.; Iwadate, M.; Yamamoto, M.; et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J. Cell Biol. 2011, 193, 275–284. [Google Scholar] [CrossRef]
- Bernard, M.; Dieudé, M.; Yang, B.; Hamelin, K.; Underwood, K.; Hébert, M.J. Autophagy fosters myofibroblast differentiation through MTORC2 activation and downstream upregulation of CTGF. Autophagy 2014, 10, 2193–2207. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.T.; Tan, H.L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.N.; Codogno, P.; Shen, H.M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef]
- Mahli, A.; Saugspier, M.; Koch, A.; Sommer, J.; Dietrich, P.; Lee, S.; Thasler, R.; Schulze-Luehrmann, J.; Luehrmann, A.; Thasler, W.E.; et al. ERK activation and autophagy impairment are central mediators of irinotecan-induced steatohepatitis. Gut 2018, 67, 746–756. [Google Scholar] [CrossRef]
- Martinez-Lopez, N.; Athonvarangkul, D.; Mishall, P.; Sahu, S.; Singh, R. Autophagy proteins regulate ERK phosphorylation. Nat. Commun. 2013, 4, 2799. [Google Scholar] [CrossRef]
- Montford, J.R.; Furgeson, S.B. A new CTGF target in renal fibrosis. Kidney Int. 2017, 92, 784–786. [Google Scholar] [CrossRef]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef]
- Sánchez-López, E.; Rayego, S.; Rodrigues-Díez, R.; Rodriguez, J.S.; Rodrigues-Díez, R.; Rodríguez-Vita, J.; Carvajal, G.; Aroeira, L.S.; Selgas, R.; Mezzano, S.A.; et al. CTGF promotes inflammatory cell infiltration of the renal interstitium by activating NF-kappaB. J. Am. Soc. Nephrol. JASN 2009, 20, 1513–1526. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seo, H.-Y.; Lee, S.-H.; Han, E.; Hwang, J.S.; Kim, M.K.; Jang, B.K. Increased Levels of Phosphorylated ERK Induce CTGF Expression in Autophagy-Deficient Mouse Hepatocytes. Cells 2022, 11, 2704. https://doi.org/10.3390/cells11172704
Seo H-Y, Lee S-H, Han E, Hwang JS, Kim MK, Jang BK. Increased Levels of Phosphorylated ERK Induce CTGF Expression in Autophagy-Deficient Mouse Hepatocytes. Cells. 2022; 11(17):2704. https://doi.org/10.3390/cells11172704
Chicago/Turabian StyleSeo, Hye-Young, So-Hee Lee, Eugene Han, Jae Seok Hwang, Mi Kyung Kim, and Byoung Kuk Jang. 2022. "Increased Levels of Phosphorylated ERK Induce CTGF Expression in Autophagy-Deficient Mouse Hepatocytes" Cells 11, no. 17: 2704. https://doi.org/10.3390/cells11172704
APA StyleSeo, H.-Y., Lee, S.-H., Han, E., Hwang, J. S., Kim, M. K., & Jang, B. K. (2022). Increased Levels of Phosphorylated ERK Induce CTGF Expression in Autophagy-Deficient Mouse Hepatocytes. Cells, 11(17), 2704. https://doi.org/10.3390/cells11172704