
Inhibition of Gap Junctional Intercellular Communication Upregulates Pluripotency Gene Expression in Endogenous Pluripotent Muse Cells

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Isolation of Muse Cells
2.2. Reagents Preparation
2.3. Quantitative PCR (qPCR)
2.4. Western Blot
2.5. Immunofluorescent Imaging
2.6. Apoptosis Assessment Assay
2.7. Cell Cycle Analysis
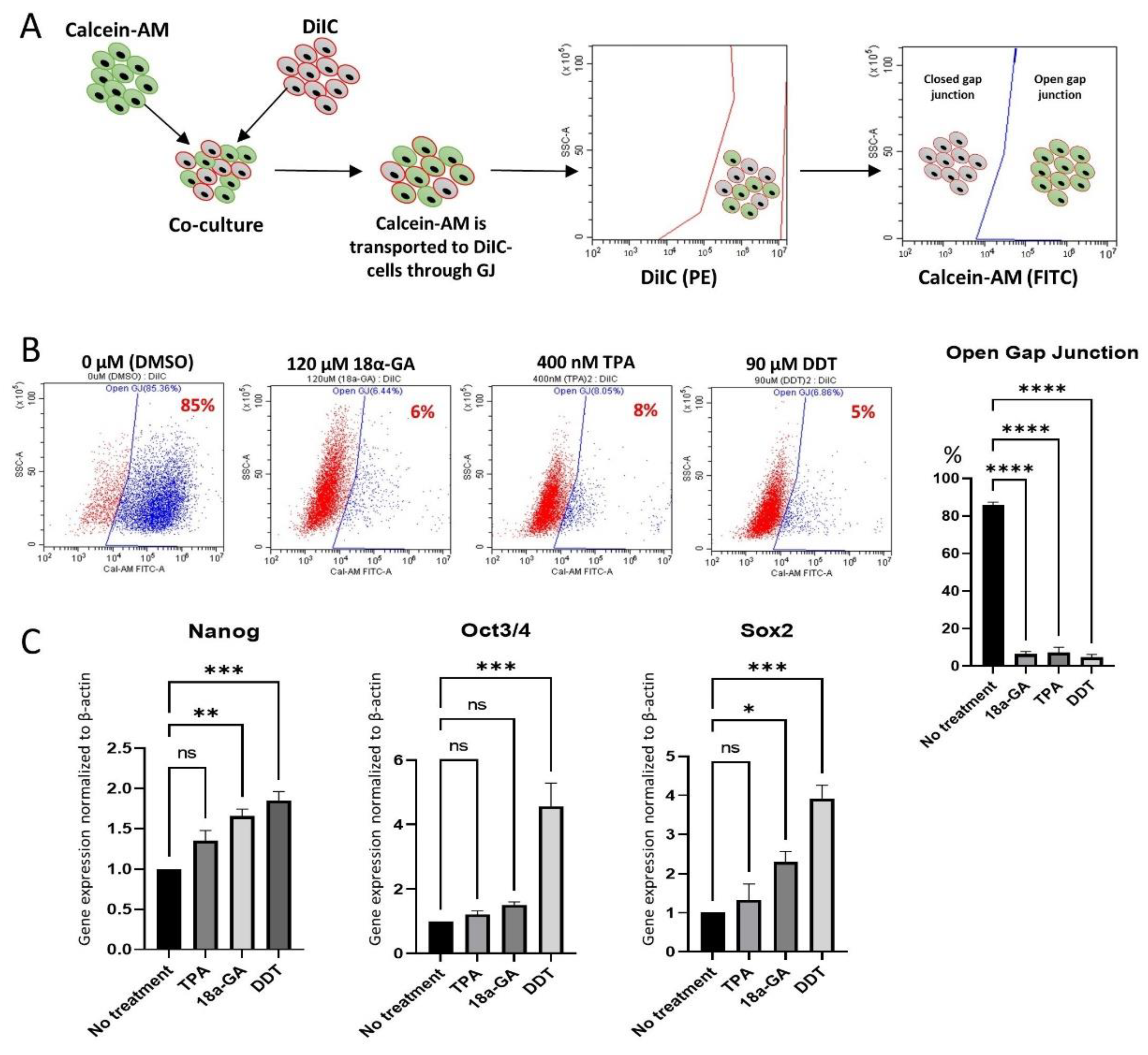
2.8. Dye Transfer Assay
2.9. RNA-Sequencing
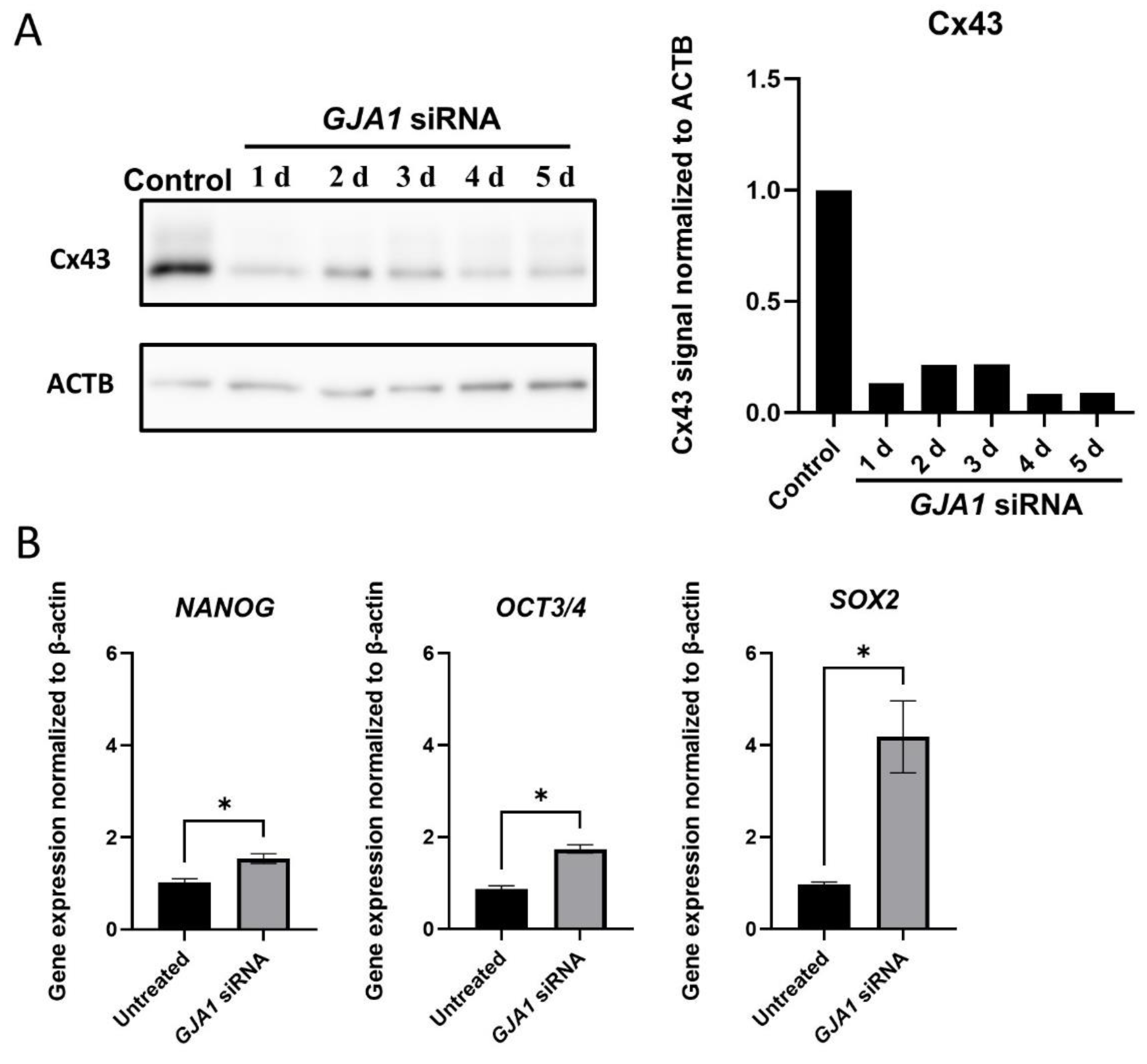
2.10. Cx43 Knockdown
2.11. Statistical Analysis
3. Results
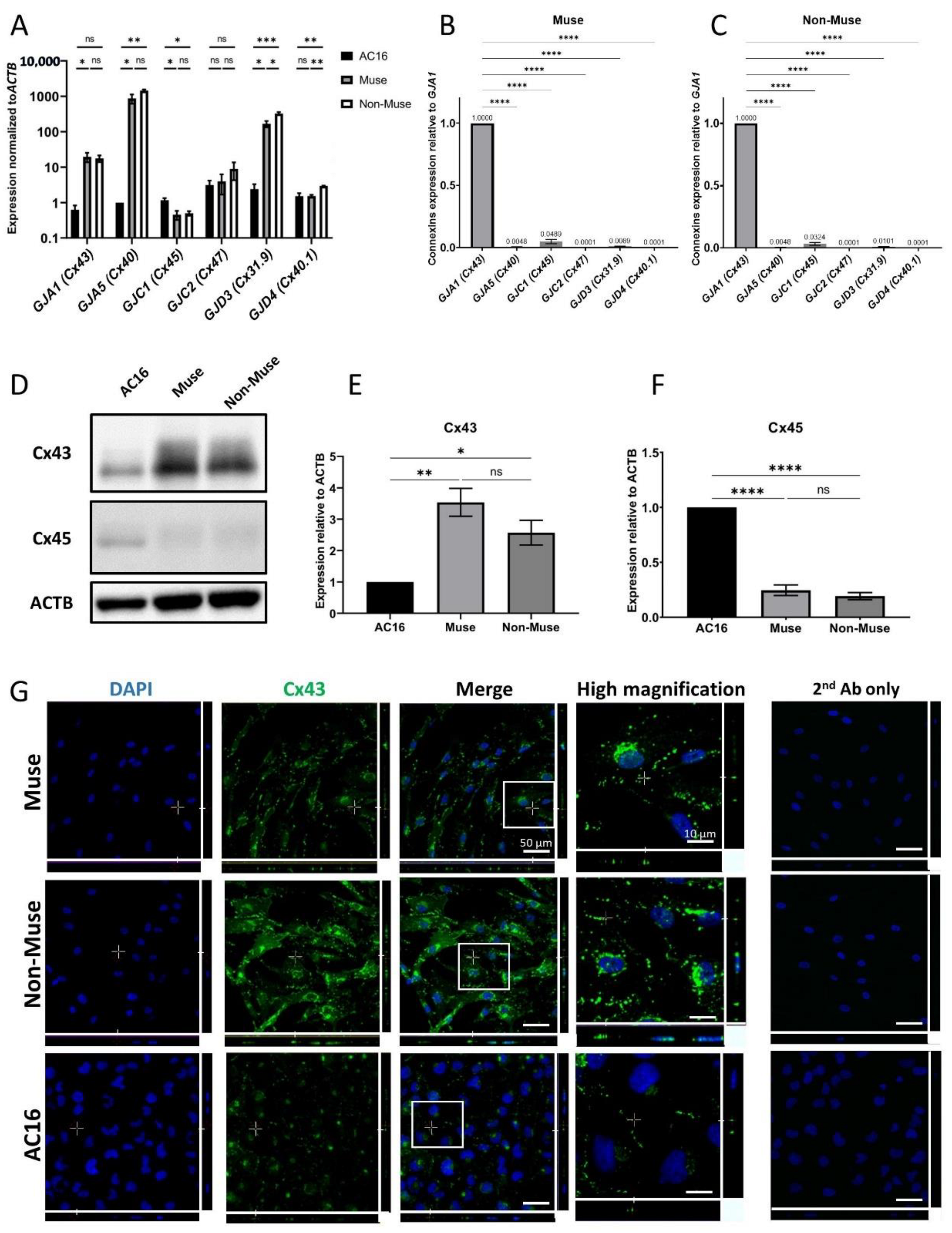
3.1. Connexin Gene Expression in Muse and Non-Muse Cells
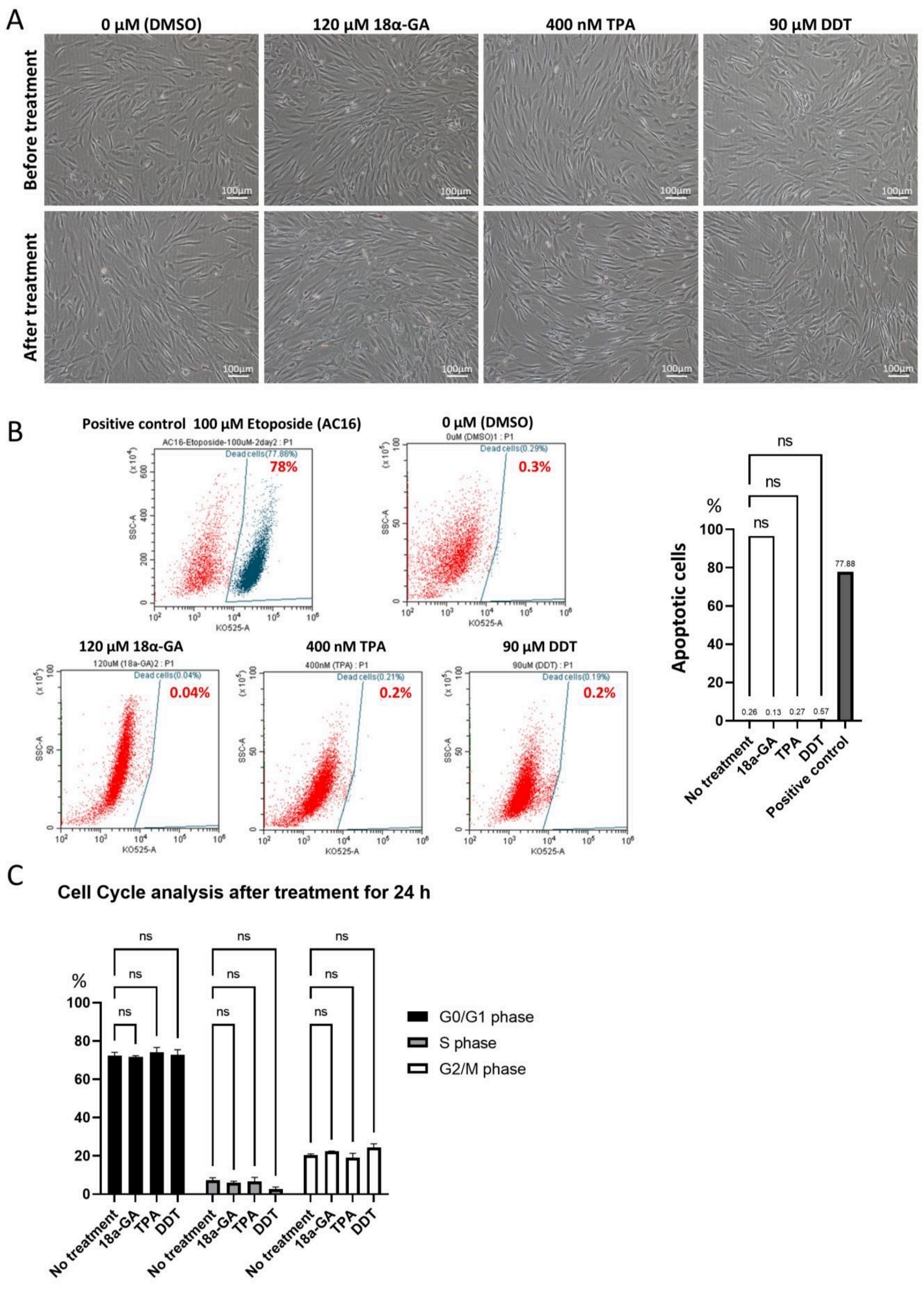
3.2. Chemical Inhibition of GJIC in Muse Cells
3.3. Effect of GJ Inhibition on Pluripotency Gene Expression
3.4. Pluripotency Gene Expression after GJA1 Knockdown in Muse Cells
3.5. RNA-Sequencing
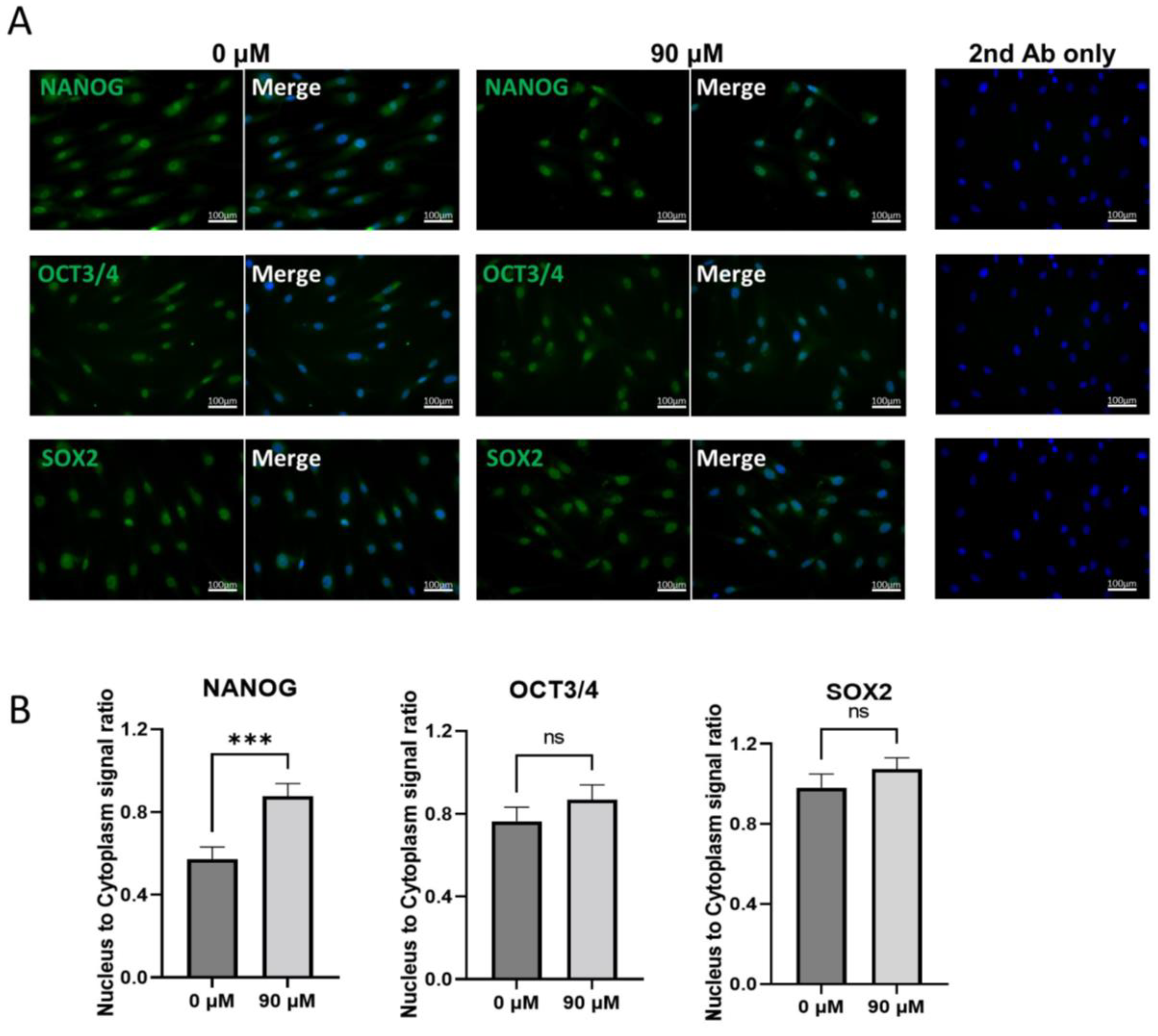
3.6. Localization of NANOG, OCT3/4, and SOX2 before and after DDT Treatment in Muse Cells
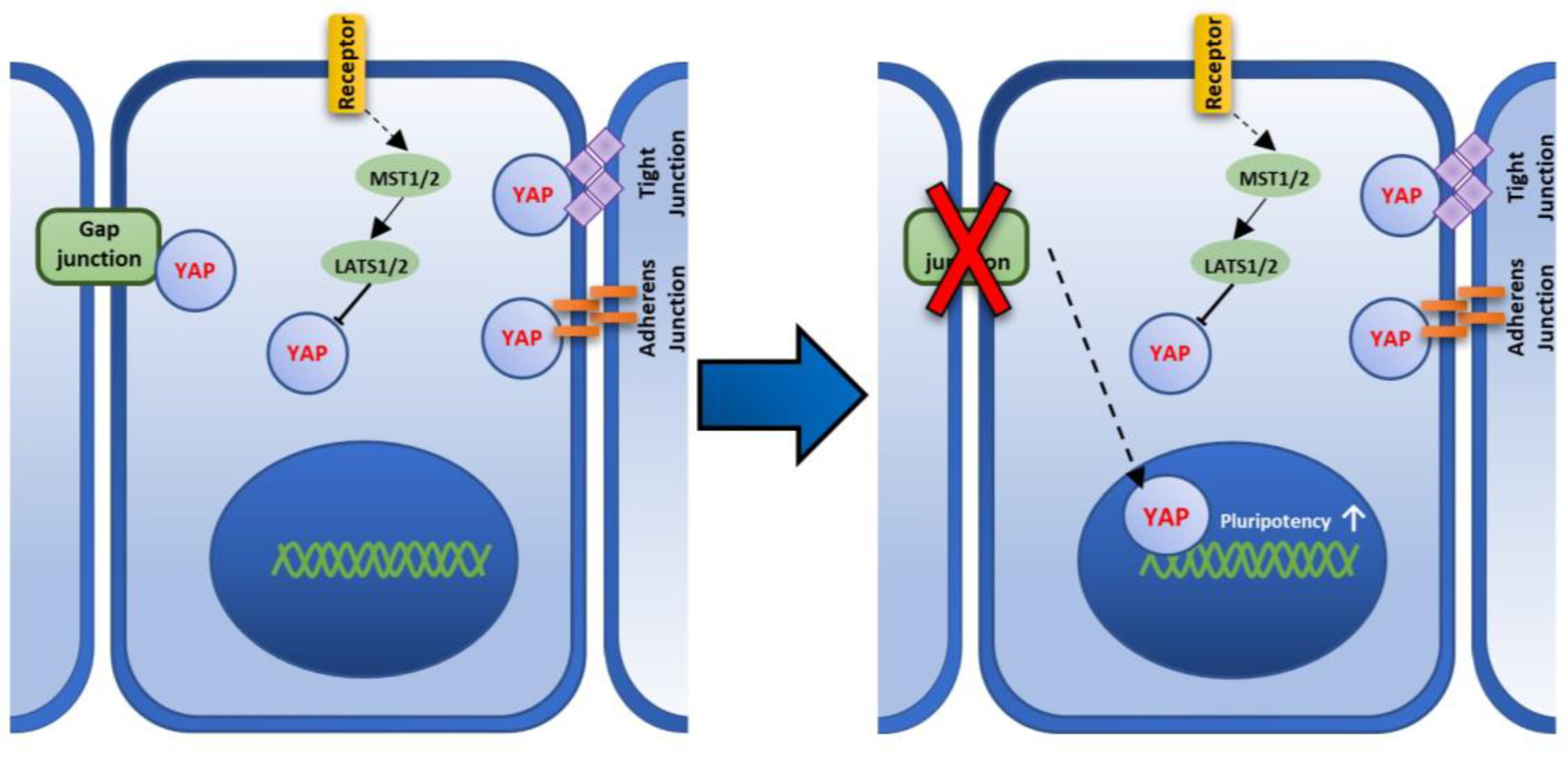
3.7. Localization of Cx43 and YAP in Muse Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Laird, D.W. Life cycle of connexins in health and disease. Biochem. J. 2006, 394, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Goodenough, D.A.; Paul, D.L. Gap Junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, 19. [Google Scholar] [CrossRef]
- Worsdorfer, P.; Bosen, F.; Gebhardt, M.; Russ, N.; Zimmermann, K.; Kessie, D.K.; Sekaran, T.; Egert, A.; Ergun, S.; Schorle, H.; et al. Abrogation of Gap Junctional Communication in ES Cells Results in a Disruption of Primitive Endoderm Formation in Embryoid Bodies. Stem Cells 2017, 35, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Sharovskaya, Y.Y.; Philonenko, E.S.; Kiselev, S.L.; Lagarkova, M.A. De Novo Reestablishment of Gap Junctional Intercellular Communications During Reprogramming to Pluripotency and Differentiation. Stem Cells Dev. 2012, 21, 2623–2629. [Google Scholar] [CrossRef]
- Wakao, S.; Kushida, Y.; Dezawa, M. Basic Characteristics of Muse Cells. In Muse Cells: Endogenous Reparative Pluripotent Stem Cells; Dezawa, M., Ed.; Springer International Publishing Ag: Cham, Switzerland, 2018; Volume 1103, pp. 13–41. [Google Scholar] [CrossRef]
- Kuroda, Y.; Kitada, M.; Wakao, S.; Nishikawa, K.; Tanimura, Y.; Makinoshima, H.; Goda, M.; Akashi, H.; Inutsuka, A.; Niwa, A.; et al. Unique multipotent cells in adult human mesenchymal cell populations. Proc. Natl. Acad. Sci. USA 2010, 107, 8639–8643. [Google Scholar] [CrossRef]
- Sato, T.; Wakao, S.; Kushida, Y.; Tatsumi, K.; Kitada, M.; Abe, T.; Niizuma, K.; Tominaga, T.; Kushimoto, S.; Dezawa, M. A Novel Type of Stem Cells Double-Positive for SSEA-3 and CD45 in Human Peripheral Blood. Cell Transplant. 2020, 29, 963689720923574. [Google Scholar] [CrossRef]
- Alessio, N.; Ozcan, S.; Tatsumi, K.; Murat, A.; Peluso, G.; Dezawa, M.; Galderisi, U. The secretome of MUSE cells contains factors that may play a role in regulation of stemness, apoptosis and immunomodulation. Cell Cycle 2017, 16, 33–44. [Google Scholar] [CrossRef]
- Acar, M.B.; Aprile, D.; Ayaz-Guner, S.; Guner, H.; Tez, C.; Di Bernardo, G.; Peluso, G.; Ozcan, S.; Galderisi, U. Why Do Muse Stem Cells Present an Enduring Stress Capacity? Hints from a Comparative Proteome Analysis. Int. J. Mol. Sci. 2021, 22, 2064. [Google Scholar] [CrossRef]
- Aprile, D.; Alessio, N.; Demirsoy, I.H.; Squillaro, T.; Peluso, G.; Di Bernardo, G.; Galderisi, U. MUSE Stem Cells Can Be Isolated from Stromal Compartment of Mouse Bone Marrow, Adipose Tissue, and Ear Connective Tissue: A Comparative Study of Their In Vitro Properties. Cells 2021, 10, 761. [Google Scholar] [CrossRef]
- Ogura, F.; Wakao, S.; Kuroda, Y.; Tsuchiyama, K.; Bagheri, M.; Heneidi, S.; Chazenbalk, G.; Aiba, S.; Dezawa, M. Human adipose tissue possesses a unique population of pluripotent stem cells with nontumorigenic and low telomerase activities: Potential implications in regenerative medicine. Stem Cells Dev. 2014, 23, 717–728. [Google Scholar] [CrossRef]
- Uchida, H.; Niizuma, K.; Kushida, Y.; Wakao, S.; Tominaga, T.; Borlongan, C.V.; Dezawa, M. Human Muse Cells Reconstruct Neuronal Circuitry in Subacute Lacunar Stroke Model. Stroke 2017, 48, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.; Kushida, Y.; Wakao, S.; Kitada, M.; Tatsumi, K.; Dezawa, M. Cardiotrophic Growth Factor-Driven Induction of Human Muse Cells into Cardiomyocyte-Like Phenotype. Cell Transplant. 2018, 27, 285–298. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiyama, K.; Wakao, S.; Kuroda, Y.; Ogura, F.; Nojima, M.; Sawaya, N.; Yamasaki, K.; Aiba, S.; Dezawa, M. Functional melanocytes are readily reprogrammable from multilineage-differentiating stress-enduring (muse) cells, distinct stem cells in human fibroblasts. J. Investig. Dermatol. 2013, 133, 2425–2435. [Google Scholar] [CrossRef] [PubMed]
- Wakao, S.; Kitada, M.; Kuroda, Y.; Shigemoto, T.; Matsuse, D.; Akashi, H.; Tanimura, Y.; Tsuchiyama, K.; Kikuchi, T.; Goda, M.; et al. Multilineage-differentiating stress-enduring (Muse) cells are a primary source of induced pluripotent stem cells in human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 9875–9880. [Google Scholar] [CrossRef]
- Yamauchi, T.; Yamasaki, K.; Tsuchiyama, K.; Koike, S.; Aiba, S. The Potential of Muse Cells for Regenerative Medicine of Skin: Procedures to Reconstitute Skin with Muse Cell-Derived Keratinocytes, Fibroblasts, and Melanocytes. J. Investig. Dermatol. 2017, 137, 2639–2642. [Google Scholar] [CrossRef]
- Uchida, H.; Morita, T.; Niizuma, K.; Kushida, Y.; Kuroda, Y.; Wakao, S.; Sakata, H.; Matsuzaka, Y.; Mushiake, H.; Tominaga, T.; et al. Transplantation of Unique Subpopulation of Fibroblasts, Muse Cells, Ameliorates Experimental Stroke Possibly via Robust Neuronal Differentiation. Stem Cells 2016, 34, 160–173. [Google Scholar] [CrossRef]
- Katagiri, H.; Kushida, Y.; Nojima, M.; Kuroda, Y.; Wakao, S.; Ishida, K.; Endo, F.; Kume, K.; Takahara, T.; Nitta, H.; et al. A Distinct Subpopulation of Bone Marrow Mesenchymal Stem Cells, Muse Cells, Directly Commit to the Replacement of Liver Components. Am. J. Transplant. 2016, 16, 468–483. [Google Scholar] [CrossRef]
- Uchida, N.; Kushida, Y.; Kitada, M.; Wakao, S.; Kumagai, N.; Kuroda, Y.; Kondo, Y.; Hirohara, Y.; Kure, S.; Chazenbalk, G.; et al. Beneficial Effects of Systemically Administered Human Muse Cells in Adriamycin Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 2946–2960. [Google Scholar] [CrossRef]
- Yamada, Y.; Wakao, S.; Kushida, Y.; Minatoguchi, S.; Mikami, A.; Higashi, K.; Baba, S.; Shigemoto, T.; Kuroda, Y.; Kanamori, H.; et al. S1P-S1PR2 Axis Mediates Homing of Muse Cells into Damaged Heart for Long-Lasting Tissue Repair and Functional Recovery After Acute Myocardial Infarction. Circ. Res. 2018, 122, 1069–1083. [Google Scholar] [CrossRef]
- Noda, T.; Nishigaki, K.; Minatoguchi, S. Safety and Efficacy of Human Muse Cell-Based Product for Acute Myocardial Infarction in a First-in-Human Trial. Circ. J. 2020, 84, 1189–1192. [Google Scholar] [CrossRef]
- Fujita, Y.; Nohara, T.; Takashima, S.; Natsuga, K.; Adachi, M.; Yoshida, K.; Shinkuma, S.; Takeichi, T.; Nakamura, H.; Wada, O.; et al. Intravenous allogeneic multilineage-differentiating stress-enduring cells in adults with dystrophic epidermolysis bullosa: A phase 1/2 open-label study. J. Eur. Acad. Dermatol. Venereol. 2021, 35, E528–E531. [Google Scholar] [CrossRef] [PubMed]
- Esseltine, J.L.; Brooks, C.R.; Edwards, N.A.; Subasri, M.; Sampson, J.; Seguin, C.; Betts, D.H.; Laird, D.W. Dynamic regulation of connexins in stem cell pluripotency. Stem Cells 2020, 38, 52–66. [Google Scholar] [CrossRef]
- Nath, S.C.; Day, B.; Harper, L.; Yee, J.; Hsu, C.Y.M.; Larijani, L.; Rohani, L.; Duan, N.; Kallos, M.S.; Rancourt, D.E. Fluid shear stress promotes embryonic stem cell pluripotency via interplay between beta-catenin and vinculin in bioreactor culture. Stem Cells 2021, 39, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- Yoda, K.; Ohnuki, Y.; Masui, S.; Kurosawa, H. Optimized conditions for the supplementation of human-induced pluripotent stem cell cultures with a GSK-3 inhibitor during embryoid body formation with the aim of inducing differentiation into mesodermal and cardiac lineage. J. Biosci. Bioeng. 2020, 129, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, P.C.; Nihei, O.K.; Savino, W.; Spray, D.C.; Alves, L.A. Flow cytometry analysis of gap junction-mediated cell-cell communication: Advantages and pitfalls. Cytom. Part A 2006, 69A, 487–493. [Google Scholar] [CrossRef]
- Goldberg, G.S.; Moreno, A.P.; Bechberger, J.F.; Hearn, S.S.; Shivers, R.R.; MacPhee, D.J.; Zhang, Y.C.; Naus, C.C.G. Evidence that disruption of connexon particle arrangements in gap junction plaques is associated with inhibition of gap junctional communication by a glycyrrhetinic acid derivative. Exp. Cell Res. 1996, 222, 48–53. [Google Scholar] [CrossRef]
- Sovadinova, I.; Babica, P.; Boke, H.; Kumar, E.; Wilke, A.; Park, J.S.; Trosko, J.E.; Upham, B.L. Phosphatidylcholine Specific PLC-Induced Dysregulation of Gap Junctions, a Robust Cellular Response to Environmental Toxicants, and Prevention by Resveratrol in a Rat Liver Cell Model. PLoS ONE 2015, 10, 16. [Google Scholar] [CrossRef]
- Babica, P.; Ctverackova, L.; Lencesova, Z.; Trosko, J.E.; Upham, B.L. Chemopreventive Agents Attenuate Rapid Inhibition of Gap Junctional Intercellular Communication Induced by Environmental Toxicants. Nutr. Cancer Int. J. 2016, 68, 827–837. [Google Scholar] [CrossRef]
- Ramos, A.; Camargo, F.D. The Hippo signaling pathway and stem cell biology. Trends Cell Biol. 2012, 22, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ren, J.; Sun, Y.H.; Xue, Y.; Zhang, Z.J.; Gong, A.H.; Wang, B.F.; Zhong, Z.H.; Cui, Z.W.; Xi, Z.Y.; et al. A connexin43/YAP axis regulates astroglial-mesenchymal transition in hemoglobin induced astrocyte activation. Cell Death Differ. 2018, 25, 1870–1884. [Google Scholar] [CrossRef]
- Yu, H.L.; Cao, X.; Li, W.; Liu, P.Y.; Zhao, Y.Y.; Song, L.L.; Chen, J.; Chen, B.L.; Yu, W.K.; Xu, Y. Targeting connexin 43 provides anti-inflammatory effects after intracerebral hemorrhage injury by regulating YAP signaling. J. Neuroinflamm. 2020, 17, 19. [Google Scholar] [CrossRef] [PubMed]
- Iseki, M.; Kushida, Y.; Wakao, S.; Akimoto, T.; Mizuma, M.; Motoi, F.; Asada, R.; Shimizu, S.; Unno, M.; Chazenbalk, G.; et al. Human Muse Cells, Nontumorigenic Pluripotent-Like Stem Cells, Have Liver Regeneration Capacity Through Specific Homing and Cell Replacement in a Mouse Model of Liver Fibrosis. Cell Transplant. 2017, 26, 821–840. [Google Scholar] [CrossRef] [PubMed]
- Loewenstein, W. Junctional intercellular communication and the control of growth. Biochim. Biophys. Acta BBA Rev. Cancer 1979, 560, 1–65. [Google Scholar] [CrossRef]
- Yamasaki, H.; Naus, C.C.G. Role of connexin genes in growth control. Carcinogenesis 1996, 17, 1199–1213. [Google Scholar] [CrossRef] [PubMed]
- Mesnil, M.; Crespin, S.; Avanzo, J.L.; Zaidan-Dagli, M.L. Defective gap junctional intercellular communication in the carcinogenic process. Biochim. Biophys. Acta Biomembr. 2005, 1719, 125–145. [Google Scholar] [CrossRef]
- Trosko, J.E. The role of stem cells and gap junctional intercellular communication in carcinogenesis. J. Biochem. Mol. Biol. 2003, 36, 43–48. [Google Scholar] [CrossRef]
- Rivedal, E.; Opsahl, H. Role of PKC and MAP kinase in EGF- and TPA-induced connexin43 phosphorylation and inhibition of gap junction intercellular communication in rat liver epithelial cells. Carcinogenesis 2001, 22, 1543–1550. [Google Scholar] [CrossRef]
- Talukdar, S.; Emdad, L.; Das, S.K.; Fisher, P.B. GAP junctions: Multifaceted regulators of neuronal differentiation. Tissue Barriers 2022, 10, 22. [Google Scholar] [CrossRef]
- Koizumi, J.I.; Kojima, T.; Kamekura, R.; Kurose, M.; Harimaya, A.; Murata, M.; Osanai, M.; Chiba, H.; Himi, T.; Sawada, N. Changes of gap and tight junctions during differentiation of human nasal epithelial cells using primary human nasal epithelial cells and primary human nasal fibroblast cells in a noncontact coculture system. J. Membr. Biol. 2007, 218, 1–7. [Google Scholar] [CrossRef]
- Gu, S.M.; Yu, X.S.; Yin, X.Y.; Jiang, J.X. Stimulation of lens cell differentiation by gap junction protein connexin 45.6. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2103–2111. [Google Scholar] [CrossRef]
- Hirschi, K.K.; Burt, J.M.; Hirschi, K.D.; Dai, C.P. Gap junction communication mediates transforming growth factor-beta activation and endothelial-induced mural cell differentiation. Circ. Res. 2003, 93, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The Biology of Yap/Taz: Hippo Signaling and Beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lu, C.Y.; Cheng, T.Y.; Pan, S.H.; Chen, H.F.; Chang, N.S. WW Domain-Containing Proteins YAP and TAZ in the Hippo Pathway as Key Regulators in Stemness Maintenance, Tissue Homeostasis, and Tumorigenesis. Front. Oncol. 2019, 9, 20. [Google Scholar] [CrossRef]
- Xu, Y.X.; Wang, X.Y.; Yu, M.; Ruan, Y.; Zhang, J.L.; Tian, Y.P.; Xiong, J.X.; Liu, L.L.; Cheng, Y.D.; Yang, Y.; et al. Identification, subcellular localization, and functional comparison of novel Yap splicing isoforms in mouse embryonic stem cells. Iubmb Life 2021, 73, 1432–1445. [Google Scholar] [CrossRef] [PubMed]
- Tamm, C.; Bower, N.; Anneren, C. Regulation of mouse embryonic stem cell self-renewal by a Yes-YAP-TEAD2 signaling pathway downstream of LIF. J. Cell Sci. 2011, 124, 1136–1144. [Google Scholar] [CrossRef]
- Wang, X.Y.; Ruan, Y.; Zhang, J.L.; Tian, Y.P.; Liu, L.L.; Wang, J.L.; Liu, G.K.; Cheng, Y.D.; Xu, Y.X.; Yang, Y.; et al. Expression levels and activation status of Yap splicing isoforms determine self-renewal and differentiation potential of embryonic stem cells. Stem Cells 2021, 39, 1178–1191. [Google Scholar] [CrossRef]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.G.; Zhao, B.; Yu, J.D.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.B.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef]
- Christopher, G.A.; Noort, R.J.; Esseltine, J.L. Connexin 43 Gene Ablation Does Not Alter Human Pluripotent Stem Cell Germ Lineage Specification. Biomolecules 2022, 12, 15. [Google Scholar] [CrossRef]
- Trosko, J.E. Commentary: “Re-programming or selecting adult stem cells?”. Stem Cell Rev. 2008, 4, 81–88. [Google Scholar] [CrossRef]
- Trosko, J.E. On the potential origin and characteristics of cancer stem cells. Carcinogenesis 2021, 42, 905–912. [Google Scholar] [CrossRef]
- Tai, M.H.; Chang, C.C.; Olson, L.K.; Trosko, J.E. Oct4 expression in adult human stem cells: Evidence in support of the stem cell theory of carcinogenesis. Carcinogenesis 2005, 26, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hatabi, K.; Hirohara, Y.; Kushida, Y.; Kuroda, Y.; Wakao, S.; Trosko, J.; Dezawa, M. Inhibition of Gap Junctional Intercellular Communication Upregulates Pluripotency Gene Expression in Endogenous Pluripotent Muse Cells. Cells 2022, 11, 2701. https://doi.org/10.3390/cells11172701
Hatabi K, Hirohara Y, Kushida Y, Kuroda Y, Wakao S, Trosko J, Dezawa M. Inhibition of Gap Junctional Intercellular Communication Upregulates Pluripotency Gene Expression in Endogenous Pluripotent Muse Cells. Cells. 2022; 11(17):2701. https://doi.org/10.3390/cells11172701
Chicago/Turabian StyleHatabi, Khaled, Yukari Hirohara, Yoshihiro Kushida, Yasumasa Kuroda, Shohei Wakao, James Trosko, and Mari Dezawa. 2022. "Inhibition of Gap Junctional Intercellular Communication Upregulates Pluripotency Gene Expression in Endogenous Pluripotent Muse Cells" Cells 11, no. 17: 2701. https://doi.org/10.3390/cells11172701
APA StyleHatabi, K., Hirohara, Y., Kushida, Y., Kuroda, Y., Wakao, S., Trosko, J., & Dezawa, M. (2022). Inhibition of Gap Junctional Intercellular Communication Upregulates Pluripotency Gene Expression in Endogenous Pluripotent Muse Cells. Cells, 11(17), 2701. https://doi.org/10.3390/cells11172701