
Treatment of VLCAD-Deficient Patient Fibroblasts with Peroxisome Proliferator-Activated Receptor δ Agonist Improves Cellular Bioenergetics

Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Cell Culture and Treatments
2.3. Real-Time Quantitative Polymerase Chain Reaction (qPCR)
2.4. PPARδ Binding Site Analysis
2.5. Whole Cell Lysate, Protein Concentration, SDS-PAGE Gel, and Western Blot
2.6. Immunofluorescence Microscopy
2.7. Electron Transfer Flavoprotein (ETF) Fluorometric Reduction Assay
2.8. Fatty Acid Oxidation (FAO) Flux Analysis
2.9. Measurement of Mitochondrial Respiration
2.10. Measurement of ATP Production
2.11. Acylcarnitine Profile Analysis
2.12. Statistical Analysis
3. Results
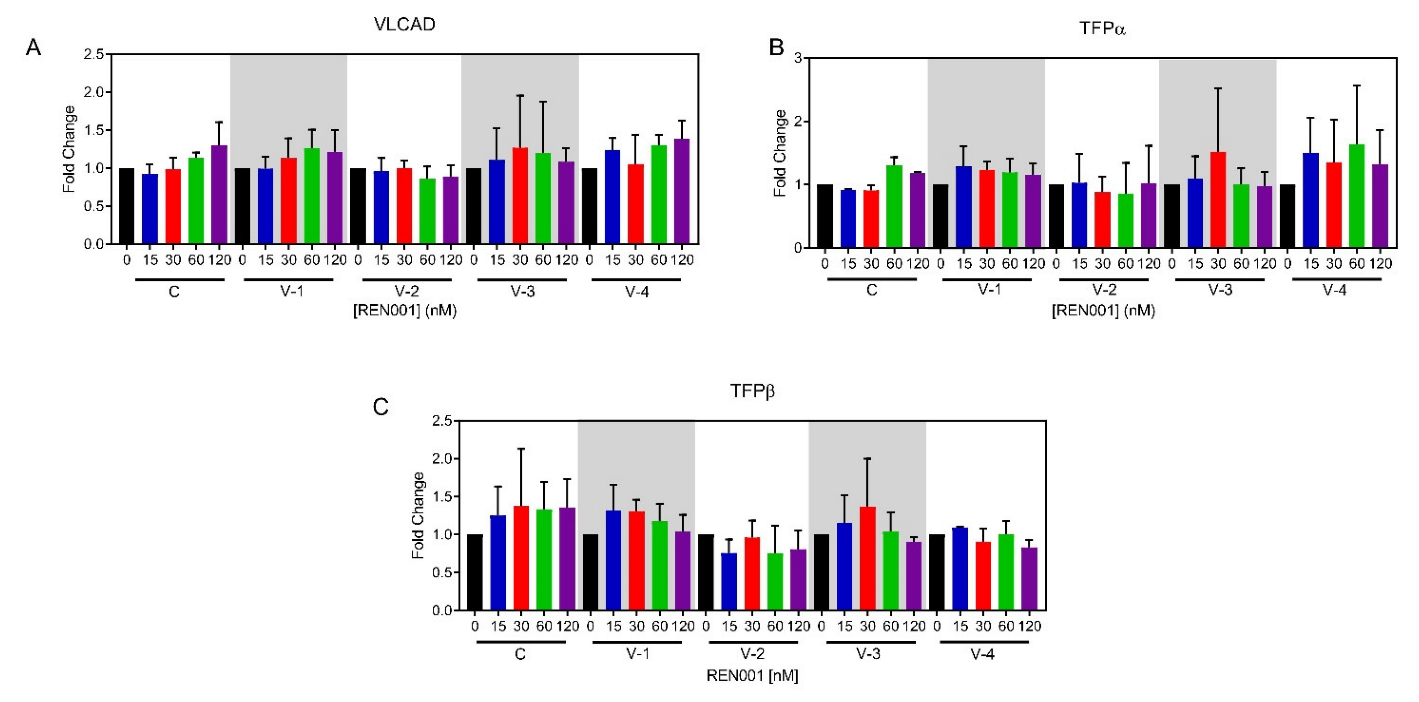
3.1. PPARδ Agonist Upregulates Genes Associated with Fatty Acid Oxidation and Mitochondrial ETC Complexes
3.2. Induction of Fatty Acid Oxidation Proteins
3.3. VLCAD Enzyme Activity
3.4. FAO Flux Assay
3.5. Whole-Cell Oximetry
3.6. ATP Production
3.7. Acylcarnitine Profile Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fillmore, N.; Alrob, O.A.; Lopaschuk, G.D. Fatty Acid beta-Oxidation; American Oil Chemistry Society—AOCS Lipid Library: Urbana, IL, USA, 2014. [Google Scholar]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Talley, J.T.; Mohiuddin, S.S. Biochemstry, Fatty Acid Oxidation; StatPearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.; Croix, C.S.; et al. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. Biol. Chem. 2019, 294, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.D.; Valencia, C.A.; Strauss, A.W.; Zhang, K. Very Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency; Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Eds.; GeneReviews®: Seattle, WA, USA, 1993. [Google Scholar]
- McHugh, D.M.S.; Cameron, C.A.; Abdenur, J.E.; Abdulrahman, M.; Adair, O.; Al Nuaimi, S.A.; Åhlman, H.; Allen, J.J.; Antonozzi, I.; Archer, S.; et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: A worldwide collaborative project. Genet. Med. 2011, 13, 230–254. [Google Scholar] [CrossRef] [PubMed]
- Watson, M.S.; Mann, M.Y.; Lloyd-Puryear, M.A.; Rinaldo, P.; Howell, R.R.; American College of Medical Genetics Newborn Screening Expert Group. Newborn screening: Toward a uniform screening panel and system—Executive summary. Pediatrics 2006, 117 Pt 2, S296–S307. [Google Scholar] [CrossRef] [PubMed]
- Andersen, R.M. Revisiting the behavioral model and access to medical care: Does it matter? J. Health Soc. Behav. 1995, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Solis, J.O.; Singh, R.H. Management of fatty acid oxidation disorders: A survey of current treatment strategies. J. Am. Diet Assoc. 2002, 102, 1800–1803. [Google Scholar] [CrossRef]
- Behrend, A.M.; Harding, C.O.; Shoemaker, J.D.; Matern, D.; Sahn, D.J.; Elliot, D.L.; Gillingham, M. Substrate oxidation and cardiac performance during exercise in disorders of long chain fatty acid oxidation. Mol. Genet. Metab. 2012, 105, 110–115. [Google Scholar] [CrossRef]
- Gillingham, M.B.; Heitner, S.B.; Martin, J.; Rose, S.; Goldstein, A.; El-Gharbawy, A.H.; DeWard, S.; Lasarev, M.R.; Pollaro, J.; DeLany, J.; et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: A double blinded, randomized controlled trial. J. Inherit. Metab. Dis. 2017, 40, 831–843. [Google Scholar] [CrossRef]
- Vockley, J.; Charrow, J.; Ganesh, J.; Eswara, M.; Diaz, G.; McCracken, E.; Conway, R.; Enns, G.; Starr, J.; Wang, R.; et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol. Genet. Metab. 2016, 119, 223–231. [Google Scholar] [CrossRef]
- Vockley, J. Long-chain fatty acid oxidation disorders and current management strategies. Am. J. Manag. Care 2020, 26 (Suppl. 7), S147–S154. [Google Scholar]
- Marsden, D.; Bedrosian, C.L.; Vockley, J. Impact of newborn screening on the reported incidence and clinical outcomes associated with medium- and long-chain fatty acid oxidation disorders. Genet. Med. 2021, 23, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barrish, G.D.; Wang, Y.-X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Michalik, L.; Auwerx, J.; Berger, J.P.; Chatterjee, V.K.; Glass, C.K.; Gonzalez, F.J.; Wahli, W. International Union of Pharmacology. LXI. Peroxisome proliferator-activated receptors. Pharmacol. Rev. 2006, 58, 726–741. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.L.C.; Siersbæk, M.; Mandrup, S. PPARs: Fatty acid sensors controlling metabolism. Semin. Cell Dev. Biol. 2012, 23, 631–639. [Google Scholar] [CrossRef]
- Burdick, A.D.; Kim, D.J.; Peraza, M.A.; Gonzalez, F.J.; Peters, J.M. The role of peroxisome proliferator-activated receptor-beta/delta in epithelial cell growth and differentiation. Cell Signal 2006, 18, 9–20. [Google Scholar] [CrossRef]
- Wang, Y.X.; Lee, C.-H.; Tiep, S.; Yu, R.T.; Ham, J.; Kang, H.; Evans, R.M. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003, 113, 159–170. [Google Scholar] [CrossRef]
- Gervois, P.; Fruchart, J.-C.; Staels, B. Drug Insight: Mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors. Nat. Clin. Pr. Endocrinol. Metab. 2007, 3, 145–156. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Tanaka, T.; Yamamoto, J.; Iwasaki, S.; Asaba, H.; Hamura, H.; Ikeda, Y.; Sakai, J. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc. Natl. Acad. Sci. USA 2003, 100, 15924–15929. [Google Scholar] [CrossRef]
- Fredenrich, A.; Grimaldi, P. PPAR delta: An uncompletely known nuclear receptor. Diabetes Metab. 2005, 31, 23–27. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Endo, N.; Rutledge, S.J.; Vogel, R.; Shinar, D.; Rodan, G.A. Identification of a new member of the steroid hormone receptor superfamily that is activated by a peroxisome proliferator and fatty acids. Mol. Endocrinol. 1992, 6, 1634–1641. [Google Scholar] [PubMed]
- Riserus, U.; Sprecher, D.; Johnson, T.; Olson, E.; Hirschberg, S.; Liu, A.; Karpe, F. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008, 57, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Oliver, W.R., Jr.; Shenk, J.L.; Snaith, M.R.; Russell, C.S.; Plunket, K.D.; Bodkin, N.L.; Willson, T.M. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc. Natl. Acad. Sci. USA 2001, 98, 5306–5311. [Google Scholar] [CrossRef]
- Fedorova, L.V.; Sodhi, K.; Gatto-Weis, C.; Puri, N.; Hinds, T.D., Jr.; Shapiro, J.I.; Malhotra, D. Peroxisome proliferator-activated receptor delta agonist, HPP593, prevents renal necrosis under chronic ischemia. PLoS ONE 2013, 8, e64436. [Google Scholar] [CrossRef]
- Misawa, K.; Hashizume, K.; Yamamoto, M.; Minegishi, Y.; Hase, T.; Shimotoyodome, A. Ginger extract prevents high-fat diet-induced obesity in mice via activation of the peroxisome proliferator-activated receptor delta pathway. J. Nutr. Biochem. 2015, 26, 1058–1067. [Google Scholar] [CrossRef]
- Feng, L.; Luo, H.; Xu, Z.; Yang, Z.; Du, G.; Zhang, Y.; Li, Y. Bavachinin, as a novel natural pan-PPAR agonist, exhibits unique synergistic effects with synthetic PPAR-gamma and PPAR-alpha agonists on carbohydrate and lipid metabolism in db/db and diet-induced obese mice. Diabetologia 2016, 59, 1276–1286. [Google Scholar] [CrossRef]
- Information, N.C.f.B. Bezafibrate, CID=39042. PubChem Database. 2019. Available online: https://pubchem.ncbi.nlm.nih.gov (accessed on 6 June 2022).
- Djouadi, F.; Aubey, F.; Schlemmer, D.; Ruiter, J.P.N.; Wanders, R.J.A.; Strauss, A.W.; Bastin, J. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum. Mol. Genet. 2005, 14, 2695–2703. [Google Scholar] [CrossRef]
- Djouadi, F.; Aubey, F.; Schlemmer, D.; Bastin, J. Peroxisome proliferator activated receptor delta (PPARdelta) agonist but not PPARalpha corrects carnitine palmitoyl transferase 2 deficiency in human muscle cells. J. Clin. Endocrinol. Metab. 2005, 90, 1791–1797. [Google Scholar] [CrossRef]
- Djouadi, F.; Habarou, F.; Le Bachelier, C.; Ferdinandusse, S.; Schlemmer, D.; Benoist, J.F.; Boutron, A.; Andresen, B.S.; Visser, G.; De Lonlay, P.; et al. Mitochondrial trifunctional protein deficiency in human cultured fibroblasts: Effects of bezafibrate. J. Inherit. Metab. Dis. 2015, 39, 47–58. [Google Scholar] [CrossRef]
- Yamada, K.; Shiraishi, H.; Oki, E.; Ishige, M.; Fukao, T.; Hamada, Y.; Sakai, N.; Ochi, F.; Watanabe, A.; Kawakami, S.; et al. Open-label clinical trial of bezafibrate treatment in patients with fatty acid oxidation disorders in Japan. Mol. Genet. Metab. Rep. 2018, 15, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, H.; Yamada, K.; Oki, E.; Ishige, M.; Fukao, T.; Hamada, Y.; Sakai, N.; Ochi, F.; Watanabe, A.; Kawakami, S.; et al. Open-label clinical trial of bezafibrate treatment in patients with fatty acid oxidation disorders in Japan; 2nd report QOL survey. Mol. Genet. Metab. Rep. 2019, 20, 100496. [Google Scholar] [CrossRef] [PubMed]
- Ørngreen, M.C.; Madsen, K.L.; Preisler, N.; Andersen, G.; Vissing, J.; Laforêt, P. Bezafibrate in skeletal muscle fatty acid oxidation disorders: A randomized clinical trial. Neurology 2014, 82, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Seed, B. A PCR primer bank for quantitative gene expression analysis. Nucleic. Acids Res. 2003, 31, e154. [Google Scholar] [CrossRef] [PubMed]
- Spandidos, A.; Wang, X.; Wang, H.; Dragnev, S.; Thurber, T.; Seed, B. A comprehensive collection of experimentally validated primers for Polymerase Chain Reaction quantitation of murine transcript abundance. BMC Genom. 2008, 9, 633. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Spandidos, A.; Wang, H.; Seed, B. PrimerBank: A PCR primer database for quantitative gene expression analysis, 2012 update. Nucleic Acids Res. 2011, 40, D1144–D1149. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Schiff, M.; Mohsen, A.W.; Karunanidhi, A.; McCracken, E.; Yeasted, R.; Vockley, J. Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2013, 109, 21–27. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Frerman, F.E.; Goodman, S.I. Fluorometric assay of acyl-CoA dehydrogenases in normal and mutant human fibroblasts. Biochem. Med. 1985, 33, 38–44. [Google Scholar] [CrossRef]
- Leipnitz, G.; Mohsen, A.W.; Karunanidhi, A.; Seminotti, B.; Roginskaya, V.Y.; Markantone, D.M.; Grings, M.; Mihalik, S.J.; Wipf, P.; Van Houten, B.; et al. Evaluation of mitochondrial bioenergetics, dynamics, endoplasmic reticulum-mitochondria crosstalk, and reactive oxygen species in fibroblasts from patients with complex I deficiency. Sci Rep. 2018, 8, 1165. [Google Scholar] [CrossRef] [PubMed]
- Watkins, P.A.; Ferrell, E.V., Jr.; Pedersen, J.I.; Hoefler, G. Peroxisomal fatty acid beta-oxidation in HepG2 cells. Arch. Biochem. Biophys. 1991, 289, 329–336. [Google Scholar] [CrossRef]
- Okun, J.G.; Kölker, S.; Schulze, A.; Kohlmüller, D.; Olgemöller, K.; Lindner, M.; Hoffmann, G.F.; Wanders, R.J.; Mayatepek, E. A method for quantitative acylcarnitine profiling in human skin fibroblasts using unlabelled palmitic acid: Diagnosis of fatty acid oxidation disorders and differentiation between biochemical phenotypes of MCAD deficiency. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2002, 1584, 91–98. [Google Scholar] [CrossRef]
- Shen, J.J.; Matern, D.; Millington, D.S.; Hillman, S.; Feezor, M.D.; Bennett, M.J.; Van Hove, J.L.K. Acylcarnitines in fibroblasts of patients with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and other fatty acid oxidation disorders. J. Inherit. Metab. Dis. 2000, 23, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.H.; Matern, D. Acylcarnitine Analysis by Tandem Mass Spectrometry. Curr. Protoc. Hum. Genet. 2010, 64, 17.8.1–17.8.20. [Google Scholar] [CrossRef]
- Fang, L.; Zhang, M.; Li, Y.; Liu, Y.; Cui, Q.; Wang, N. PPARgene: A Database of Experimentally Verified and Computationally Predicted PPAR Target Genes. PPAR Res. 2016, 2016, 6042162. [Google Scholar] [CrossRef]
- Diekman, E.F.; Ferdinandusse, S.; van der Pol, L.; Waterham, H.R.; Ruiter, J.P.; Ijlst, L.; Wanders, R.J.; Houten, S.M.; Wijburg, F.A.; Blank, A.C.; et al. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet. Med. 2015, 17, 989–994. [Google Scholar] [CrossRef]
- Seminotti, B.; Leipnitz, G.; Karunanidhi, A.; Kochersperger, C.; Roginskaya, V.Y.; Basu, S.; Vockley, J. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum. Mol. Genet. 2019, 28, 928–941. [Google Scholar] [CrossRef]
- Gobin-Limballe, S.; Djouadi, F.; Aubey, F.; Olpin, S.; Andresen, B.; Yamaguchi, S.; Mandel, H.; Fukao, T.; Ruiter, J.; Wanders, R.; et al. Genetic Basis for Correction of Very-Long-Chain Acyl–Coenzyme A Dehydrogenase Deficiency by Bezafibrate in Patient Fibroblasts: Toward a Genotype-Based Therapy. Am. J. Hum. Genet. 2007, 81, 1133–1143. [Google Scholar] [CrossRef]
- Gobin-Limballe, S.; McAndrew, R.P.; Djouadi, F.; Kim, J.J.; Bastin, J. Compared effects of missense mutations in Very-Long-Chain Acyl-CoA Dehydrogenase deficiency: Combined analysis by structural, functional and pharmacological approaches. Biochim. Biophys. Acta 2010, 1802, 478–484. [Google Scholar] [CrossRef]
- Gillingham, M.B.; Scott, B.; Elliott, D.; Harding, C.O. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol. Genet. Metab. 2006, 89, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, M.A.; Kendal, F.; Hegde, M.; Singh, R.H. MCT oil-based diet reverses hypertrophic cardiomyopathy in a patient with very long chain acyl-coA dehydrogenase deficiency. Indian J. Hum. Genet. 2011, 17, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Keeler, A.M.; Conlon, T.; Walter, G.; Zeng, H.; Shaffer, S.A.; Dungtao, F.; Flotte, T.R. Long-term correction of very long-chain acyl-coA dehydrogenase deficiency in mice using AAV9 gene therapy. Mol. Ther. 2012, 20, 1131–1138. [Google Scholar] [CrossRef]
- Vockley, J.; Burton, B.; Berry, G.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Cataldo, J. Effects of triheptanoin (UX007) in patients with long-chain fatty acid oxidation disorders: Results from an open-label, long-term extension study. J. Inherit. Metab. Dis. 2021, 44, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Calleri, E.; Pochetti, G.; Dossou, K.S.S.; Laghezza, A.; Montanari, R.; Capelli, D.; Prada, E.; Loiodice, F.; Massolini, G.; Bernier, M.; et al. Resveratrol and Its Metabolites Bind to PPARs. ChemBioChem 2014, 15, 1154–1160. [Google Scholar] [CrossRef]
- Storgaard, J.H.; Løkken, N.; Madsen, K.L.; Voermans, N.C.; Laforêt, P.; Nadaj-Pakleza, A.; Tard, C.; van Hall, G.; Vissing, J.; Ørngreen, M.C. No effect of resveratrol on fatty acid oxidation or exercise capacity in patients with fatty acid oxidation disorders: A randomized clinical cross-over trial. J. Inherit. Metab. Dis. 2022, 45, 517–528. [Google Scholar] [CrossRef]
- Aoyama, T.; Uchida, Y.; Kelley, R.I.; Marble, M.; Hofman, K.; Tonsgard, J.H.; Hashimoto, T. A novel disease with deficiency of mitochondrial very-long-chain acyl-CoA dehydrogenase. Biochem. Biophys. Res. Commun. 1993, 191, 1369–1372. [Google Scholar] [CrossRef]
- Wood, J.C.; Magera, M.J.; Rinaldo, P.; Seashore, M.R.; Strauss, A.W.; Friedman, A. Diagnosis of very long chain acyl-dehydrogenase deficiency from an infant’s newborn screening card. Pediatrics 2001, 108, E19. [Google Scholar] [CrossRef]
- Miller, M.J.; Burrage, L.C.; Gibson, J.B.; Strenk, M.E.; Lose, E.J.; Bick, D.P.; Wong, L.J.C. Recurrent ACADVL molecular findings in individuals with a positive newborn screen for very long chain acyl-coA dehydrogenase (VLCAD) deficiency in the United States. Mol. Genet. Metab. 2015, 116, 139–145. [Google Scholar] [CrossRef]
- Wanders, R.J.; Vreken, P.; Den Boer, M.E.J.; Wijburg, F.A.; Van Gennip, A.H. Disorders of mitochondrial fatty acyl-CoA beta-oxidation. J. Inherit. Metab. Dis. 1999, 22, 442–487. [Google Scholar] [CrossRef]
- Cox, K.B.; Hamm, D.A.; Millington, D.S.; Matern, D.; Vockley, J.; Rinaldo, P.; Wood, P.A. Gestational, pathologic and biochemical differences between very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency in the mouse. Hum. Mol. Genet. 2001, 10, 2069–2077. [Google Scholar] [CrossRef] [PubMed]
- Cox, K.B.; Liu, J.; Tian, L.; Barnes, S.; Yang, Q.; Wood, P.A. Cardiac hypertrophy in mice with long-chain acyl-CoA dehydrogenase or very long-chain acyl-CoA dehydrogenase deficiency. Lab. Investig. 2009, 89, 1348–1354. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Kraft, K.; Geuer, S.; Will, A.J.; Chan, W.L.; Paliou, C.; Borschiwer, M.; Harabula, I.; Wittler, L.; Franke, M.; Ibrahim, D.M.; et al. Deletions, Inversions, Duplications: Engineering of Structural Variants using CRISPR/Cas in Mice. Cell Rep. 2015, 10, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, K.; Kunihiro, Y.; Kaneko, T.; Nagahora, H.; Voigt, B.; Mashimo, T. ssODN-mediated knock-in with CRISPR-Cas for large genomic regions in zygotes. Nat. Commun. 2016, 7, 10431. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Laboratory Designation | Mutations | Phenotypic Severity |
|---|---|---|---|
| Control-1 | FB826 | N/A | Control |
| Control-2 | FB549 | N/A | Control |
| Control-3 | FB902 | N/A | Control |
| VLCAD-1 | FB833 | c.520G > A (p.Val174Met)/c.1825G > A (p.Glu609Lys) | Mild |
| VLCAD-2 | FB671 | c.1619T > C (p.Leu540Pro)/c.1707–1715dup (p.Asp570_Ala572dup) | Severe |
| VLCAD-3 | FB863 | c.896_898del (p.Lys299del)/c.1147C > G (p.Leu383Val) | Mild |
| VLCAD-4 | FB782 | c.848T > C (p.Val283Ala)/c.1258A > C (p.Ile420Leu) | Mild |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Annibale, O.M.; Phua, Y.L.; Van’t Land, C.; Karunanidhi, A.; Dorenbaum, A.; Mohsen, A.-W.; Vockley, J. Treatment of VLCAD-Deficient Patient Fibroblasts with Peroxisome Proliferator-Activated Receptor δ Agonist Improves Cellular Bioenergetics. Cells 2022, 11, 2635. https://doi.org/10.3390/cells11172635
D’Annibale OM, Phua YL, Van’t Land C, Karunanidhi A, Dorenbaum A, Mohsen A-W, Vockley J. Treatment of VLCAD-Deficient Patient Fibroblasts with Peroxisome Proliferator-Activated Receptor δ Agonist Improves Cellular Bioenergetics. Cells. 2022; 11(17):2635. https://doi.org/10.3390/cells11172635
Chicago/Turabian StyleD’Annibale, Olivia M., Yu Leng Phua, Clinton Van’t Land, Anuradha Karunanidhi, Alejandro Dorenbaum, Al-Walid Mohsen, and Jerry Vockley. 2022. "Treatment of VLCAD-Deficient Patient Fibroblasts with Peroxisome Proliferator-Activated Receptor δ Agonist Improves Cellular Bioenergetics" Cells 11, no. 17: 2635. https://doi.org/10.3390/cells11172635
APA StyleD’Annibale, O. M., Phua, Y. L., Van’t Land, C., Karunanidhi, A., Dorenbaum, A., Mohsen, A.-W., & Vockley, J. (2022). Treatment of VLCAD-Deficient Patient Fibroblasts with Peroxisome Proliferator-Activated Receptor δ Agonist Improves Cellular Bioenergetics. Cells, 11(17), 2635. https://doi.org/10.3390/cells11172635