
Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer



Abstract
1. General Introduction
2. Mechanism of CMA
3. Targeting of Proteins for Degradation by CMA
4. Hsc70 and Its Interaction with Target Proteins
5. Assembly of the LAMP-2A/Hsc70/Protein Complexes on the Lysosomal Surface and Translocation into the Lysosomal Lumen
6. LAMP-2A—The Lysosomal Receptor for the Hsc70/Protein Complex and a Central Player in CMA
7. Visualising CMA
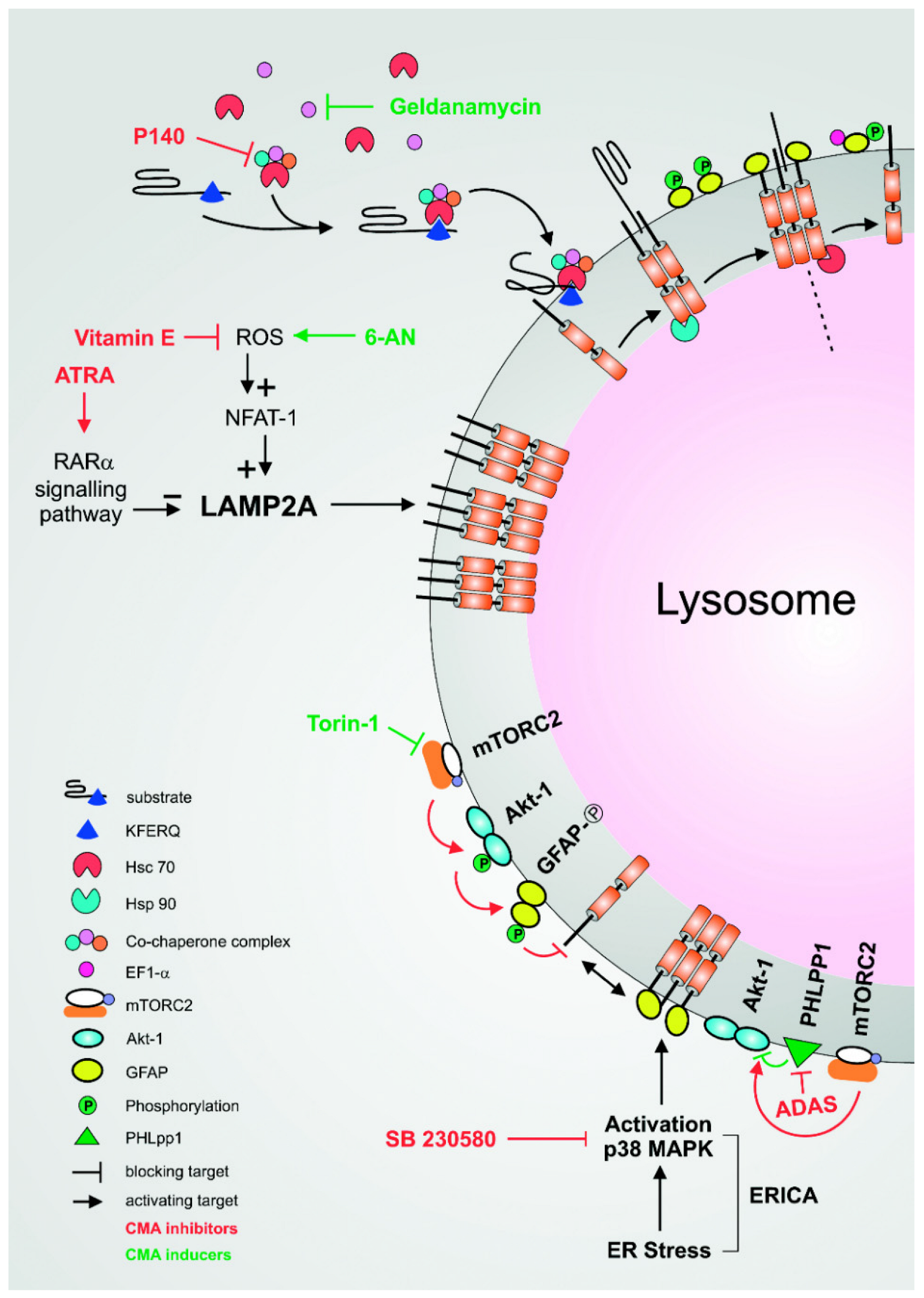
8. CMA Pathways and Their Pharmacological Modulation
8.1. The mTORC2/PHLPP1/AKT Pathway
8.2. The RARα Signalling Pathway and ATRA
8.3. The ERICA Pathway
8.4. Hsp90
8.5. Hsc70
8.6. LAMP-2A
8.7. Oxidative Stress
8.8. Cross-Talk between Macroautophagy and CMA
9. CMA and Cancer
9.1. Anti-Oncogenic Roles of CMA in Healthy Cells
9.2. The Roles of CMA in Malignant Neoplasias
9.2.1. Pro-Oncogenic Role of CMA in Cancer Cells
9.2.2. Glycolytic Capability
9.2.3. Cell Cycle and Proliferation
9.2.4. DNA Damage Response
10. CMA Modulation in Cancer
11. CMA, a New Target in Cancer Therapy?
12. Conclusions and Perspectives
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Klionsky, D.J. Autophagy Revisited: A Conversation with Christian de Duve. Autophagy 2008, 4, 740–743. [Google Scholar] [CrossRef]
- Clark, S.L. Cellular Differentiation in the Kidneys of Newborn Mice Studies with the Electron Microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349–362. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in Yeast Demonstrated with Proteinase-Deficient Mutants and Conditions for Its Induction. J. Cell Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in Mammalian Cells: Revisiting a 40-Year-Old Conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef]
- Lescat, L.; Herpin, A.; Mourot, B.; Véron, V.; Guiguen, Y.; Bobe, J.; Seiliez, I. CMA Restricted to Mammals and Birds: Myth or Reality? Autophagy 2018, 14, 1267–1270. [Google Scholar] [CrossRef]
- Lescat, L.; Véeron, V.; Mourot, B.; Péron, S.; Chenais, N.; Dias, K.; Riera-Heredia, N.; Beaumatin, F.; Pinel, K.; Priault, M.; et al. Chaperone-Mediated Autophagy in the Light of Evolution: Insight from Fish. Mol. Biol. Evol. 2020, 37, 2887–2899. [Google Scholar] [CrossRef]
- Reddy Bonam, S.; Wang, F.; Muller, S. Autophagy: A New Concept in Autoimmunity Regulation and a Novel Therpeutic Option. J. Autoimmun. 2018, 94, 16–32. [Google Scholar] [CrossRef]
- Rothaug, M.; Stroobants, S.; Schweizer, M.; Peters, J.; Zunke, F.; Allerding, M.; D’Hooge, R.; Saftig, P.; Blanz, J. LAMP-2 Deficiency Leads to Hippocampal Dysfunction but Normal Clearance of Neuronal Substrates of Chaperone-Mediated Autophagy in a Mouse Model for Danon Disease. Acta Neuropathol. Commun. 2015, 3, 6. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, B.; Wang, J.; Wu, H.; Xu, S.; Zhang, J.; Wang, L. Discovery of LAMP-2A as Potential Biomarkers for Glioblastoma Development by Modulating Apoptosis through N-CoR Degradation. Cell Commun. Signal. 2021, 19, 40. [Google Scholar] [CrossRef]
- Notomi, S.; Ishihara, K.; Efstathiou, N.E.; Lee, J.J.; Hisatomi, T.; Tachibana, T.; Konstantinou, E.K.; Ueta, T.; Murakami, Y.; Maidana, D.E.; et al. Genetic LAMP2 Deficiency Accelerates the Age-Associated Formation of Basal Laminar Deposits in the Retina. Proc. Natl. Acad. Sci. USA 2019, 116, 23724–23734. [Google Scholar] [CrossRef]
- Dong, S.; Wang, Q.; Kao, Y.R.; Diaz, A.; Tasset, I.; Kaushik, S.; Thiruthuvanathan, V.; Zintiridou, A.; Nieves, E.; Dzieciatkowska, M.; et al. Chaperone-Mediated Autophagy Sustains Haematopoietic Stem-Cell Function. Nature 2021, 591, 117–123. [Google Scholar] [CrossRef]
- Dice, J.F. Altered Degradation of Proteins Microinjected into Senescent Human Fibroblasts. J. Biol. Chem. 1982, 257, 14624–14627. [Google Scholar] [CrossRef]
- Dice, J.F.; Chiang, H.L.; Spencer, E.P.; Backer, J.M. Regulation of Catabolism of Microinjected Ribonuclease A. Identification of Residues 7-11 as the Essential Pentapeptide. J. Biol. Chem. 1986, 261, 6853–6859. [Google Scholar] [CrossRef]
- Chiang, H.-L.; Terlecky, S.; Plant, C.; Dice, J.F. A Role for a 70-Kilodaton Heat Shock Protein in Lysosomal Degradation of Intracellular Proteins. Science 1989, 246, 382–384. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. A Receptor for the Selective Uptake and Degradation of Proteins by Lysosomes. Science 1996, 273, 501–503. [Google Scholar] [CrossRef]
- Dice, J.F. Peptide Sequences That Target Cytosolic Proteins for Lysosomal Proteolysis. Trends Biochem. Sci. 1990, 8, 305–309. [Google Scholar] [CrossRef]
- Kirchner, P.; Bourdenx, M.; Madrigal-Matute, J.; Tiano, S.; Diaz, A.; Bartholdy, B.A.; Will, B.; Cuervo, A.M. Proteome-Wide Analysis of Chaperone-Mediated Autophagy Targeting Motifs. PLoS Biol. 2019, 17, e3000301. [Google Scholar] [CrossRef]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef]
- Thompson, L.M.; Aiken, C.T.; Kaltenbach, L.S.; Agrawal, N.; Illes, K.; Khoshnan, A.; Martinez-Vincente, M.; Arrasate, M.; O’Rourke, J.G.; Khashwji, H.; et al. IKK Phosphorylates Huntingtin and Targets It for Degradation by the Proteasome and Lysosome. J. Cell Biol. 2009, 187, 1083–1099. [Google Scholar] [CrossRef]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of Chaperone-Mediated Autophagy during Oxidative Stress. Mol. Biol. Cell 2004, 15, 5318–5328. [Google Scholar] [CrossRef]
- Agarraberes, F.A.; Dice, J.F. A Molecular Chaperone Complex at the Lysosomal Membrane Is Required for Protein Translocation. J. Cell Sci. 2001, 114, 2491–2499. [Google Scholar] [CrossRef]
- Shen, S.; Zhang, P.; Lovchik, M.A.; Li, Y.; Tang, L.; Chen, Z.; Zeng, R.; Ma, D.; Yuan, J.; Yu, Q. Cyclodepsipeptide Toxin Promotes the Degradation of Hsp90 Client Proteins through Chaperone-Mediated Autophagy. J. Cell Biol. 2009, 185, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Cuervo, A.M. The Coming of Age of Chaperone-Mediated Autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The Chaperone-Mediated Autophagy Receptor Organizes in Dynamic Protein Complexes at the Lysosomal Membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef]
- Gong, Z.; Tasset, I.; Diaz, A.; Anguiano, J.; Tas, E.; Cui, L.; Kuliawat, R.; Liu, H.; Kühn, B.; Cuervo, A.M.; et al. Humanin Is an Endogenous Activator of Chaperone-mediated Autophagy. J. Cell Biol. 2018, 217, 635–647. [Google Scholar] [CrossRef]
- Gough, N.R.; Fambrough, D.M. Different Steady State Subcellular Distributions of the Three Splice Variants of Lysosome-Associated Membrane Protein LAMP-2 Are Determined Largely by the COOH-Terminal Amino Acid Residue. J. Cell Biol. 1997, 137, 1161–1169. [Google Scholar] [CrossRef]
- Bandyopad, U.; Cuervo, A.M. Entering the Lysosome. Autophagy 2010, 4, 1101–1103. [Google Scholar]
- Rout, A.K.; Strub, M.P.; Piszczek, G.; Tjandra, N. Structure of Transmembrane Domain of Lysosome-Associated Membrane Protein Type 2a (LAMP-2A) Reveals Key Features for Substrate Specificity in Chaperone-Mediated Autophagy. J. Biol. Chem. 2014, 289, 35111–35123. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Unique Properties of Lamp2a Compared to Other Lamp2 Isoforms. J. Cell Sci. 2000, 113, 4441–4450. [Google Scholar] [CrossRef]
- Arias, E.; Koga, H.; Diaz, A.; Mocholi, E.; Patel, B.; Cuervo, A.M. Lysosomal MTORC2/PHLPP1/Akt Regulate Chaperone-Mediated Autophagy. Mol. Cell 2015, 59, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Hall, M.N. Regulation of MTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Methods Mol. Biol. 2018, 1691, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, J.; Dou, J.; She, H.; Tao, K.; Xu, H.; Yang, Q.; Mao, Z. Phosphorylation of LAMP2A by P38 MAPK Couples ER Stress to Chaperone-Mediated Autophagy. Nat. Commun. 2017, 8, 1763. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Massey, A.C.; Cuervo, A.M. Lysosome Membrane Lipid Microdomains: Novel Regulators of Chaperone-Mediated Autophagy. EMBO J. 2006, 25, 3921–3933. [Google Scholar] [CrossRef]
- Kiffin, R.; Kaushik, S.; Zeng, M.; Bandyopadhyay, U.; Zhang, C.; Massey, A.C.; Martinez-Vicente, M.; Cuervo, A.M. Altered Dynamics of the Lysosomal Receptor for Chaperone-Mediated Autophagy with Age. J. Cell Sci. 2007, 120, 782–791. [Google Scholar] [CrossRef]
- Valdor, R.; Mocholi, E.; Botbol, Y.; Guerrero-Ros, I.; Chandra, D.; Koga, H.; Gravekamp, C.; Cuervo, A.M.; Macian, F.; Valdor, R.; et al. Chaperone-Mediated Autophagy Regulates T Cell Responses through Targeted Degradation of Negative Regulators of T Cell Activation. Nat. Immunol. 2014, 15, 1046–1054. [Google Scholar] [CrossRef]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical Modulation of Chaperone-Mediated Autophagy by Retinoic Acid Derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef]
- Gomez-Sintes, R.; Xin, Q.; Jimenez-Loygorri, J.I.; McCabe, M.; Diaz, A.; Garner, T.P.; Cotto-Rios, X.M.; Wu, Y.; Dong, S.; Reynolds, C.A.; et al. Targeting Retinoic Acid Receptor Alpha-Corepressor Interaction Activates Chaperone-Mediated Autophagy and Protects against Retinal Degeneration. Nat. Commun. 2022, 13, 4220. [Google Scholar] [CrossRef]
- Tang, F.L.; Erion, J.R.; Tian, Y.; Liu, W.; Yin, D.M.; Ye, J.; Tang, B.; Mei, L.; Xiong, W.C. VPS35 in Dopamine Neurons Is Required for Endosome-to-Golgi Retrieval of Lamp2a, a Receptor of Chaperone-Mediated Autophagy That Is Critical for α-Synuclein Degradation and Prevention of Pathogenesis of Parkinson’s Disease. J. Neurosci. 2015, 35, 10613–10628. [Google Scholar] [CrossRef]
- Zhang, J.; Johnson, J.L.; He, J.; Napolitano, G.; Ramadass, M.; Rocca, C.; Kiosses, W.B.; Bucci, C.; Xin, Q.; Gavathiotis, E.; et al. Cystinosin, the Small GTPase Rab11, and the Rab7 Effector RILP Regulate Intracellular Trafficking of the Chaperone-Mediated Autophagy Receptor LAMP2A. J. Biol. Chem. 2017, 292, 10328–10346. [Google Scholar] [CrossRef]
- Arias, E. Methods to Study Chaperone-Mediated Autophagy. Methods Enzymol. 2017, 588, 283–305. [Google Scholar] [CrossRef]
- Wen, X.; Yang, Y.; Klionsky, D.J. Moments in Autophagy and Disease: Past and Present. Mol. Asp. Med. 2021, 82, 100966. [Google Scholar] [CrossRef]
- Koga, H.; Martinez-Vicente, M.; Macian, F.; Verkhusha, V.V.; Cuervo, A.M. A Photoconvertible Fluorescent Reporter to Track Chaperone-Mediated Autophagy. Nat. Commun. 2011, 2, 386. [Google Scholar] [CrossRef]
- Sato, M.; Seki, T.; Konno, A.; Hirai, H.; Kurauchi, Y.; Hisatsune, A.; Katsuki, H. Fluorescent-Based Evaluation of Chaperone-Mediated Autophagy and Microautophagy Activities in Cultured Cells. Genes Cells 2016, 21, 861–873. [Google Scholar] [CrossRef]
- Seki, T.; Yoshino, K.-i.; Tanaka, S.; Dohi, E.; Onji, T.; Yamamoto, K.; Hide, I.; Paulson, H.L.; Saito, N.; Sakai, N. Establishment of a Novel Fluorescence-Based Method to Evaluate Chaperone-Mediated Autophagy in a Single Neuron. PLoS ONE 2012, 7, e31232. [Google Scholar] [CrossRef]
- Dong, S.; Aguirre-Hernandez, C.; Scrivo, A.; Eliscovich, C.; Arias, E.; Bravo-Cordero, J.J.; Cuervo, A.M. Monitoring Spatiotemporal Changes in Chaperone-Mediated Autophagy In Vivo. Nat. Commun. 2020, 11, 645. [Google Scholar] [CrossRef]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-Propionylpiperazin-1-Yl)-3-(trifluoromethyl)phenyl)-9-(quinolin-3-Yl)benzo[h][1,6]naphthyridin-2(1 H)-one as a Highly Potent, Selective Mammalian Target of Rapamycin (MTOR) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef]
- Bradley, E.W.; Taylor, E.L.; Becerra, C.C.; Lorang, I.M.; Burhow, S.A.; Reid, J.M.; Westendorf, J.J. Inhibitors of Phlpp Phosphatases Are Highly Stable and Suppress Pain Following Joint Injury. Osteoarthr. Cartil. 2018, 26, S356–S357. [Google Scholar] [CrossRef]
- Hu, Y.; Chan, E.; Wang, S.X.; Li, B. Activation of P38 Mitogen-Activated Protein Kinase Is Required for Osteoblast Differentiation. Endocrinology 2003, 144, 2068–2074. [Google Scholar] [CrossRef]
- Brown, K.K.; Heitmeyer, S.A.; Hookfin, E.B.; Hsieh, L.; Buchalova, M.; Taiwo, Y.O.; Janusz, M.J. P38 MAP Kinase Inhibitors as Potential Therapeutics for the Treatment of Joint Degeneration and Pain Associated with Osteoarthritis. J. Inflamm. 2008, 5, 22. [Google Scholar] [CrossRef]
- Kankaanranta, H.; De Souza, P.M.; Barnes, P.J.; Salmon, M.; Giembycz, M.A.; Lindsay, M.A. SB 203580, an Inhibitor of P38 Mitogen-Activated Protein Kinase, Enhances Constitutive Apoptosis of Cytokine-Deprived Human Eosinophils. J. Pharmacol. Exp. Ther. 1999, 290, 621–628. [Google Scholar]
- Finn, P.F.; Mesires, N.T.; Vine, M.; Dice, J.F. Effects of Small Molecules on Chaperone-Mediated Autophagy. Autophagy 2005, 1, 141–145. [Google Scholar] [CrossRef]
- Xu, Y.; Wallace, M.A.G.; Fitzgerald, M.C. Thermodynamic Analysis of the Geldanamycin–Hsp90 Interaction in a Whole Cell Lysate Using a Mass Spectrometry-Based Proteomics Approach. J. Am. Soc. Mass Spectrom. 2016, 27, 1670–1676. [Google Scholar] [CrossRef]
- Sõti, C.; Nagy, E.; Giricz, Z.; Vígh, L.; Csermely, P.; Ferdinandy, P. Heat Shock Proteins as Emerging Therapeutic Targets. Br. J. Pharmacol. 2005, 146, 769–780. [Google Scholar] [CrossRef]
- Wang, F.; Bonam, S.R.; Schall, N.; Kuhn, L.; Hammann, P.; Chaloin, O.; Madinier, J.B.; Briand, J.P.; Page, N.; Muller, S. Blocking Nuclear Export of HSPA8 after Heat Shock Stress Severely Alters Cell Survival. Sci. Rep. 2018, 8, 16820. [Google Scholar] [CrossRef]
- Muller, S.; Monneaux, F.; Schal, N.; Rashkov, R.K.; Oparanov, B.A.; Wiesel, P.; Geiger, J.M.; Zimmer, R. Spliceosomal Peptide P140 for Immunotherapy of Systemic Lupus Erythematosus: Results of an Early Phase II Clinical Trial. Arthritis Rheum. 2008, 58, 3873–3883. [Google Scholar] [CrossRef]
- Page, N.; Gros, F.; Schall, N.; Décossas, M.; Bagnard, D.; Briand, J.-P.P.; Muller, S. HSC70 Blockade by the Therapeutic Peptide P140 Affects Autophagic Processes and Endogenous MHCII Presentation in Murine Lupus. Ann. Rheum. Dis. 2011, 70, 837–843. [Google Scholar] [CrossRef]
- Macri, C.; Wang, F.; Tasset, I.; Schall, N.; Page, N.; Briand, J.P.; Cuervo, A.M.; Muller, S.; Briand, P.; Cuervo, A.M.; et al. Modulation of Deregulated Chaperone-Mediated Autophagy by a Phosphopeptide. Autophagy 2015, 11, 472–486. [Google Scholar] [CrossRef]
- Zhou, D.; Li, P.; Lin, Y.; Lott, J.M.; Hislop, A.D.; Canaday, D.H.; Brutkiewicz, R.R.; Blum, J.S. Lamp-2a Facilitates MHC Class II Presentation of Cytoplasmic Antigens. Immunity 2005, 22, 571–581. [Google Scholar] [CrossRef]
- Wang, F.; Tasset, I.; Cuervo, A.M.; Muller, S. In Vivo Remodeling of Altered Autophagy-Lysosomal Pathway by a Phosphopeptide in Lupus. Cells 2020, 9, 2328. [Google Scholar] [CrossRef]
- Zhang, C.; Cuervo, A.M. Restoration of Chaperone-Mediated Autophagy in Aging Liver Improves Cellular Maintenance and Hepatic Function. Nat. Med. 2008, 14, 959–965. [Google Scholar] [CrossRef]
- Hubert, V.; Peschel, A.; Langer, B.; Groeger, M.; Rees, A.J.; Kain, R.; Gröger, M.; Rees, A.J.; Kain, R.; Gro, M.; et al. LAMP-2 Is Required for Incorporating Syntaxin-17 into Autophagosomes and for Their Fusion with Lysosomes. Biol. Open 2016, 5, 1516–1529. [Google Scholar] [CrossRef]
- Tanak, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lullmann-Rauch, R.; Janssen, P.M.; Blanz, J.; von Figura, K.; Saftig, P. Accumulation of Autophagic Vacuoles Anc Cardiomyopathy in LAMP-2 Deficient Mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Nishino, I.; Fu, J.; Tanji, K.; Yamada, T.; Shimojo, S.; Koori, T.; Mora, M.; Riggs, J.E.J.E.; Oh, S.J.; Koga, Y.; et al. Primary LAMP-2 Deficiency Causes X-Linked Vacuolar Cardiomyopathy and Myopathy (Danon Disease). Nature 2000, 406, 906–910. [Google Scholar] [CrossRef]
- Pérez, L.; McLetchie, S.; Gardiner, G.J.; Sarah, N.; Zhou, D.; Blum, J.S.; Deffit, S.N.; Zhou, D.; Blum, J.S. LAMP-2C Inhibits MHC Class II Presentation of Cytoplasmic Antigens by Disrupting Chaperone-Mediated Autophagy. J. Immunol. 2016, 196, 2457–2465. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Knecht, E.; Terlecky, S.R.; Dice, J.F. Activation of a Selective Pathway of Lysosomal Proteolysis in Rat Liver by Prolonged Starvation. Am. J. Physiol. Cell Physiol. 1995, 269, 1200–1208. [Google Scholar] [CrossRef]
- Dohi, E.; Tanaka, S.; Seki, T.; Miyagi, T.; Hide, I.; Takahashi, T.; Matsumoto, M.; Sakai, N. Hypoxic Stress Activates Chaperone-Mediated Autophagy and Modulates Neuronal Cell Survival. Neurochem. Int. 2012, 60, 431–442. [Google Scholar] [CrossRef]
- Tasset, I.; Cuervo, A.M. Role of Chaperone-Mediated Autophagy in Metabolism. FEBS J. 2016, 283, 2403–2413. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, J.A.; Kaushik, S.; Koga, H.; Dall’Armi, C.; Shui, G.; Wenk, M.R.; Di Paolo, G.; Cuervo, A.M. Inhibitory Effect of Dietary Lipids on Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, E705–E714. [Google Scholar] [CrossRef]
- Aw, T.Y.; Rhoads, C.A. Glucose Regulation of Hydroperoxide Metabolism in Rat Intestinal Cells. Stimulation of Reduced Nicotinamide Adenine Dinucleotide Phosphate Supply. J. Clin. Investig. 1994, 94, 2426–2434. [Google Scholar] [CrossRef]
- Yu, Y.; Hou, L.; Song, H.; Xu, P.; Sun, Y.; Wu, K. Akt/AMPK/MTOR Pathway Was Involved in the Autophagy Induced by Vitamin E Succinate in Human Gastric Cancer SGC-7901 Cells. Mol. Cell. Biochem. 2017, 424, 173–183. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Dice, J.F. Age-Related Decline in Chaperone-Mediated Autophagy. J. Biol. Chem. 2000, 275, 31505–31513. [Google Scholar] [CrossRef]
- Okada, A.A.; Dice, J.F. Altered Degradation of Intracellular Proteins in Aging Human Fibroblast. Mech. Aging Dev. 1984, 26, 341–356. [Google Scholar] [CrossRef]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the Selective Blockage of Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Talloczy, Z.; Wong, E.; Tang, G.; Koga, H.; Kaushik, S.; De Vries, R.; Arias, E.; Harris, S.; Sulzer, D.; et al. Cargo Recognition Failure Is Responsible for Inefficient Autophagy in Huntington’s Disease. Nat. Neurosci. 2010, 13, 567–576. [Google Scholar] [CrossRef]
- Wu, H.; Chen, S.; Ammar, A.B.; Xu, J.; Wu, Q.; Pan, K.; Zhang, J.; Hong, Y. Crosstalk Between Macroautophagy and Chaperone-Mediated Autophagy: Implications for the Treatment of Neurological Diseases. Mol. Neurobiol. 2015, 52, 1284–1296. [Google Scholar] [CrossRef]
- Gomes, L.R.; Menck, C.F.M.; Cuervo, A.M. Chaperone-Mediated Autophagy Prevents Cellular Transformation by Regulating MYC Proteasomal Degradation. Autophagy 2017, 13, 928–940. [Google Scholar] [CrossRef]
- Kon, M.; Kiffin, R.; Koga, H.; Chapochnick, J.; Macian, F.; Varticovski, L.; Cuervo, A.M. Chaperone-Mediated Autophagy Is Required for Tumor Growth. Sci. Transl. Med. 2011, 3, 109ra117. [Google Scholar] [CrossRef]
- Saha, T. LAMP2A Overexpression in Breast Tumors Promotes Cancer Cell Survival via Chaperone-Mediated Autophagy. Autophagy 2012, 8, 1643–1656. [Google Scholar] [CrossRef]
- Schneider, J.L.; Villarroya, J.; Diaz-Carretero, A.; Patel, B.; Urbanska, A.M.; Thi, M.M.; Villarroya, F.; Santambrogio, L.; Cuervo, A.M. Loss of Hepatic Chaperone-Mediated Autophagy Accelerates Proteostasis Failure in Aging. Aging Cell 2015, 14, 249–264. [Google Scholar] [CrossRef]
- Lu, T.J.L.; Huang, G.J.; Wang, H.J.; Chen, J.L.; Hsu, H.P.; Lu, T.J.L. Hispolon Promotes MDM2 Downregulation through Chaperone-Mediated Autophagy. Biochem. Biophys. Res. Commun. 2010, 398, 26–31. [Google Scholar] [CrossRef]
- Kielbik, M.; Szulc-Kielbik, I.; Klink, M. Calreticulin—Multifunctional Chaperone in Immunogenic Cell Death: Potential Significance as a Prognostic Biomarker in Ovarian Cancer Patients. Cells 2021, 10, 130. [Google Scholar] [CrossRef]
- Serrano-del Valle, A.; Anel, A.; Naval, J.; Marzo, I. Immunogenic Cell Death and Immunotherapy of Multiple Myeloma. Front. Cell Dev. Biol. 2019, 7, 50. [Google Scholar] [CrossRef]
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Calreticulin Surface Exposure Is Abrogated in Cells Lacking, Chaperone-Mediated Autophagy-Essential Gene, LAMP2A. Cell Death Dis. 2013, 4, e826. [Google Scholar] [CrossRef]
- Franch, H.A.; Sooparb, S.; Du, J. A Mechanism Regulating Proteolysis of Specific Proteins during Renal Tubular Cell Growth. J. Biol. Chem. 2001, 276, 19126–19131. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Riquelme, D.; Martinez, C.M.; Moraleda, J.M.; Cuervo, A.M.; Macian, F.; Martinez, S. Glioblastoma Ablates Pericytes Antitumor Immune Function through Aberrant Up-Regulation of Chaperone-Mediated Autophagy. Proc. Natl. Acad. Sci. USA 2019, 116, 20655–20665. [Google Scholar] [CrossRef]
- Arias, E.; Cuervo, A.M. Pros and Cons of Chaperone-Mediated Autophagy in Cancer Biology. Trends Endocrinol. Metab. 2020, 31, 53–66. [Google Scholar] [CrossRef]
- Ding, Z.-B.; Fu, X.T.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Song, K.; et al. Lamp2a Is Required for Tumor Growth and Promotes Tumor Recurrence of Hepatocellular Carcinoma. Int. J. Oncol. 2016, 49, 2367–2376. [Google Scholar] [CrossRef]
- Zhou, J.; Yang, J.; Fan, X.; Hu, S.; Zhou, F.; Dong, J.; Zhang, S.; Shang, Y.; Jiang, X.; Guo, H.; et al. Chaperone-Mediated Autophagy Regulates Proliferation by Targeting RND3 in Gastric Cancer. Autophagy 2016, 12, 515–528. [Google Scholar] [CrossRef]
- Rios, J.; Sequeida, A.; Albornoz, A.; Budini, M. Chaperone Mediated Autophagy Substrates and Components in Cancer. Front. Oncol. 2021, 10, 614677. [Google Scholar] [CrossRef]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.-G.; Najafov, A.; Geng, J.; Galan-Acosta, L.; Han, X.; Guo, Y.; Shan, B.; Zhang, Y.; Norberg, E.; Zhang, T.; et al. Degradation of HK2 by Chaperone-Mediated Autophagy Promotes Metabolic Catastrophe and Cell Death. J. Cell Biol. 2015, 210, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Ren, C.; Qiao, P.; Han, X.; Wang, L.; Lv, S.; Sun, Y.; Liu, Z.; Du, Y.; Yu, Z. PIM2-Mediated Phosphorylation of Hexokinase 2 Is Critical for Tumor Growth and Paclitaxel Resistance in Breast Cancer. Oncogene 2018, 37, 5997–6009. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Hu, W.; Lim, B.; Dice, J.F. IκB Is a Substrate for a Selective Pathway of Lysosomal Proteolysis. Mol. Biol. Cell 1998, 9, 1995–2010. [Google Scholar] [CrossRef]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, Death, and Autophagy in Cancer: NF-ΚB Turns up Everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef]
- Jiang, L.; Huang, S.; Wang, J.; Zhang, Y.; Xiong, Y.; Zeng, S.X.; Lu, H. Inactivating P53 Is Essential for Nerve Growth Factor Receptor to Promote Melanoma-Initiating Cell-Stemmed Tumorigenesis. Cell Death Dis. 2020, 11, 550. [Google Scholar] [CrossRef]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz, K.; Ahmed, I.; Semenza, G.L. Cyclin-Dependent Kinases Regulate Lysosomal Degradation of Hypoxia-Inducible Factor 1α to Promote Cell-Cycle Progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef]
- Park, C.; Suh, Y.; Cuervo, A.M. Regulated Degradation of Chk1 by Chaperone-Mediated Autophagy in Response to DNA Damage. Nat. Commun. 2015, 6, 6823. [Google Scholar] [CrossRef]
- Galan-Acosta, L.; Xia, H.; Yuan, J.; Vakifahmetoglu-Norberg, H. Activation of Chaperone-Mediated Autophagy as a Potential Anticancer Therapy. Autophagy 2015, 11, 2370–2371. [Google Scholar] [CrossRef][Green Version]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.G.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-Mediated Autophagy Degrades Mutant P53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Method | Pros | Cons |
|---|---|---|
| Immunoblot of LAMP-2A | - Technically simple | - Overall estimate of CMA changes |
| Quantification of CMA active lysosomes | - Technically simple | - Overall estimate of CMA changes - Methanol fixation |
| Monitoring the degradation of radiolabelled long-lived protein | - Measures CMA flux | - Involves use of radioactivity - Requires inhibition of other autophagic pathways - Measures CMA and microautophagy |
| Measurement of the uptake of CMA substrate by isolated lysosomes | - Reconstitution of CMA in vitro - Measures functional CMA definitively | - Requires lysosome isolation - Requires large volume of cells or tissues |
| Photoswitchable/photoactivable CMA reporter | - Measures CMA flux | - Overall estimate of CMA changes - Must be complemented by other methods - Difficult to distinguish surface bound from translocated substrate |
| GAPDH-Halo tag fluorescence-based method | - Measures CMA flux | - Overall estimate of CMA changes - Must be complemented by other methods - Measures CMA and microautophagy |
| KFERQ Dendra reporter | - Measures CMA flux in vitro and in vivo | - Must be complemented by other methods |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hubert, V.; Weiss, S.; Rees, A.J.; Kain, R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells 2022, 11, 2562. https://doi.org/10.3390/cells11162562
Hubert V, Weiss S, Rees AJ, Kain R. Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells. 2022; 11(16):2562. https://doi.org/10.3390/cells11162562
Chicago/Turabian StyleHubert, Virginie, Sebastian Weiss, Andrew Jackson Rees, and Renate Kain. 2022. "Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer" Cells 11, no. 16: 2562. https://doi.org/10.3390/cells11162562
APA StyleHubert, V., Weiss, S., Rees, A. J., & Kain, R. (2022). Modulating Chaperone-Mediated Autophagy and Its Clinical Applications in Cancer. Cells, 11(16), 2562. https://doi.org/10.3390/cells11162562