
Roads to Stat3 Paved with Cadherins


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Cell-to-Cell Adhesion Triggers a Dramatic Increase in the Levels of Rac and IL6, Leading to Stat3 Activation
2.1. The Signal Transducer and Activator of Transcription-3 (Stat3)
2.2. The Cadherin Family of Cell-to-Cell Adhesion Receptors
2.3. Cadherin Engagement, Rac, IL6 and Stat3 Activation in Nonneoplastic Cells
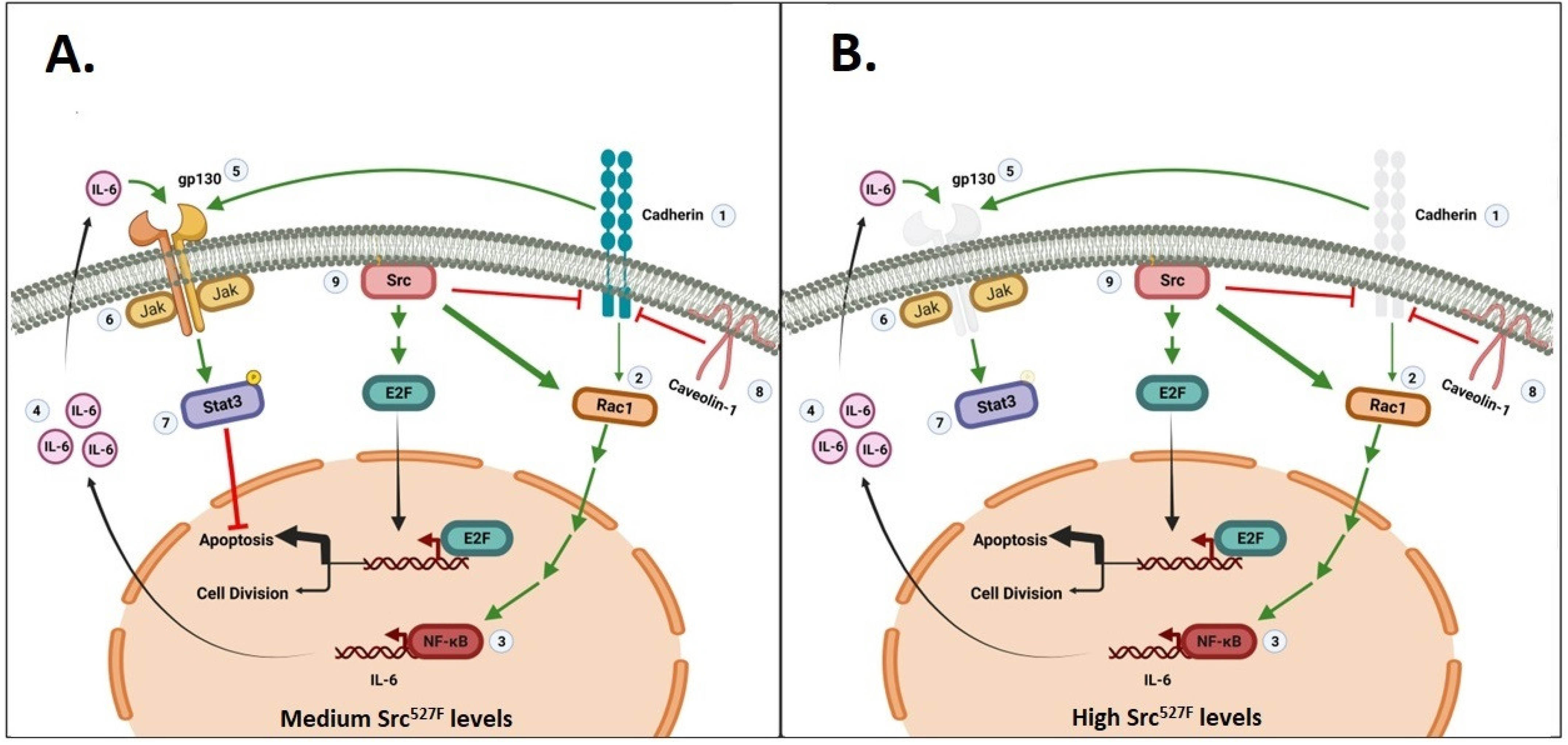
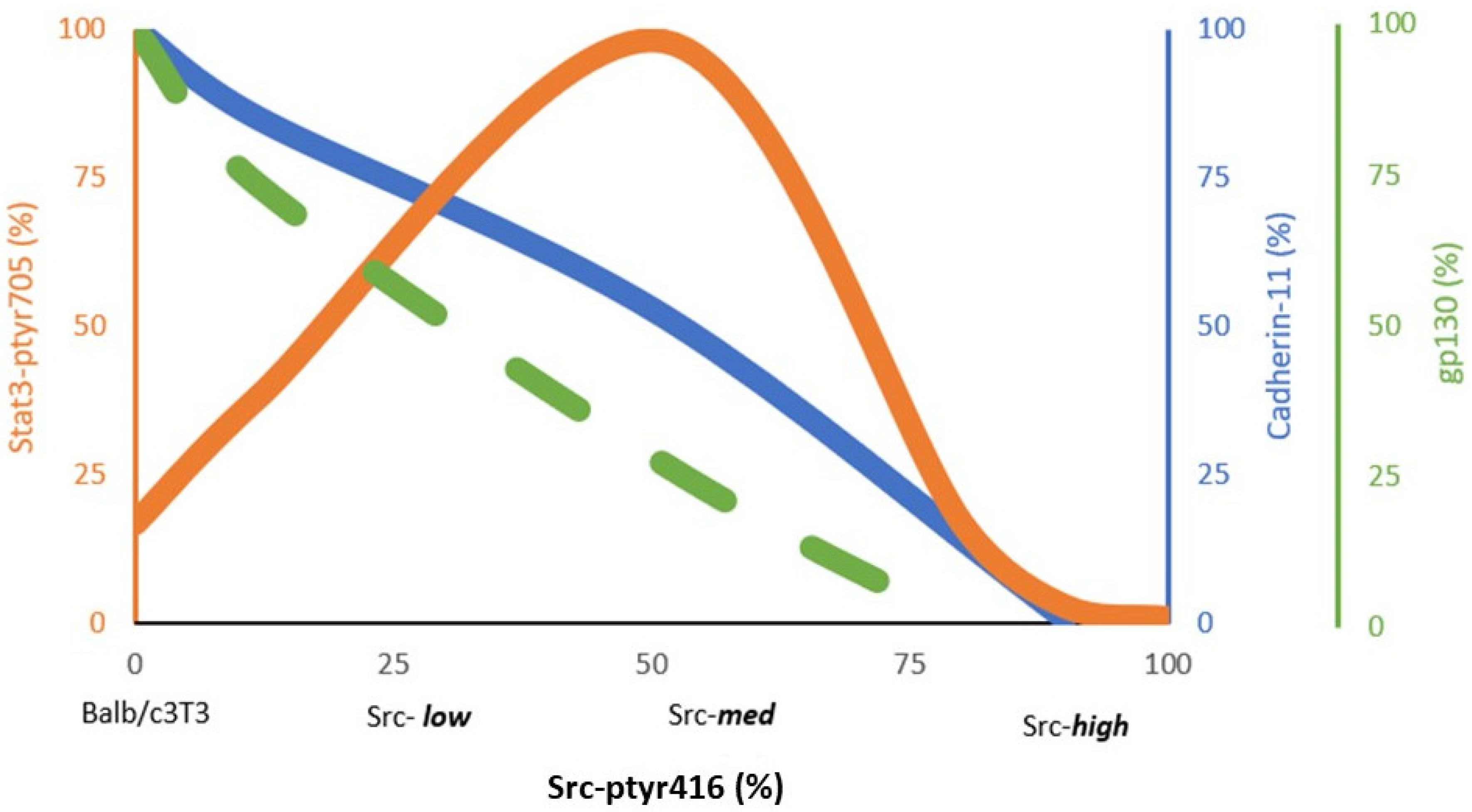
3. Activated Src527F Requires Rac, Cadherins and gp130 to Activate Stat3
3.1. The Src Oncogene and the Polyoma Virus Middle Tumor Antigen
3.2. Stat3 Activation by Src527F Requires Cadherin-11
3.3. Cadherins Are Required for gp130 Receptor Function and Stat3-ptyr705 Phosphorylation
4. Sparsely Growing, Nontransformed Cells: No Cadherin Engagement, No gp130, No Stat3-ptyr705
5. Caveolin-1 Reduces Stat3-ptyr705 through Cadherin Downregulation
5.1. Caveolae and Caveolins
5.2. Cav1 Negatively Regulates Stat3 through Cadherin-11 Downregulation in Mouse Fibroblasts and Lung Cancer Lines
6. Discussion
- What is the role of cadherins in the activation of Stat3?
- What are the implications of the cadherin-mediated activation of Stat3 for drug development?
6.1. Role of Cadherins in Stat3 Activation
6.2. Stat3 as a Cancer Therapy Target—Design and Testing of Inhibitors
- Factors that promote cadherin disengagement, such as calcium chelators [34] or HAV peptides [45], would also rapidly reduce Jak and Stat3 activity (within 15–60 min [34]), with no effect upon Stat3 per se. The same would hold true with inhibitors of Rac, or IL6 family cytokines, or NFκB. In our hands, even changing the medium of HC11 mouse breast epithelial or Balb/c3T3 cells with calcium-free DMEM eliminated cadherin engagement and dramatically reduced Stat3-ptyr705 within 30–60 min, depending upon cell density (Raptis et al., unpublished).
- If an inhibitor causes growth retardation by any (unrelated) mechanism, then following treatment for 2–3 days the cells would be less confluent compared to their untreated controls, hence would have lower Stat3-ptyr705 levels due to the lower confluence, not due to a specific inhibition of Stat3 by the compound under study. The dramatic increase in Stat3 activity with cellular confluence dictates that experiments testing Stat3 inhibition must be conducted at a number of densities spanning the peak values of Stat3-ptyr705 (at 1–5 days post 100% confluence, depending upon the cell’s growth rate), and the peaks of treated and untreated cultures compared. The same holds true with experiments dealing with Stat3-activating oncogenes, e.g., the simian virus 40 large tumor antigen [33], adenovirus E1A [100,101], Src [48], or growth factors and cytokines.
6.3. Conclusions, Future Directions, and Clinical Relevance
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Halder, G.; Dupont, S.; Piccolo, S. Transduction of mechanical and cytoskeletal cues by yap and taz. Nat. Rev. Mol. Cell Biol. 2012, 13, 591–600. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Vignais, M.L.; Gilman, M. Distinct mechanisms of activation of stat1 and stat3 by platelet-derived growth factor receptor in a cell-free system. Mol. Cell. Biol. 1999, 19, 3727–3735. [Google Scholar] [CrossRef]
- Turkson, J.; Bowman, T.; Garcia, R.; Caldenhoven, E.; de Groot, R.P.; Jove, R. Stat3 activation by src induces specific gene regulation and is required for cell transformation. Mol. Cell. Biol. 1998, 18, 2545–2552. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Horvath, C.M.; Besser, D.; Lathem, W.W.; Darnell, J.E., Jr. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 1998, 18, 2553–2558. [Google Scholar] [CrossRef]
- Wang, Y.Z.; Wharton, W.; Garcia, R.; Kraker, A.; Jove, R.; Pledger, W.J. Activation of stat3 preassembled with platelet-derived growth factor beta receptors requires src kinase activity. Oncogene 2000, 19, 2075–2085. [Google Scholar] [CrossRef]
- Ma, J.H.; Qin, L.; Li, X. Role of stat3 signaling pathway in breast cancer. Cell Commun. Signal. 2020, 18, 33. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the il-6/jak/stat3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Decker, T.; Kovarik, P.; Meinke, A. Gas elements: A few nucleotides with a major impact on cytokine-induced gene expression. J. Interferon Cytokine Res. 1997, 17, 121–134. [Google Scholar] [CrossRef]
- Hung, W.; Elliott, B. Co-operative effect of c-src tyrosine kinase and stat3 in activation of hepatocyte growth factor expression in mammary carcinoma cells. J. Biol. Chem. 2001, 276, 12395–12403. [Google Scholar] [CrossRef]
- Sinibaldi, D.; Wharton, W.; Turkson, J.; Bowman, T.; Pledger, W.J.; Jove, R. Induction of p21waf1/cip1 and cyclin d1 expression by the src oncoprotein in mouse fibroblasts: Role of activated stat3 signaling. Oncogene 2000, 19, 5419–5427. [Google Scholar] [CrossRef]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Sullivan, D.; et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. Stats in cancer inflammation and immunity: A leading role for stat3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Demaria, M.; Poli, V. From the nucleus to the mitochondria and back: The odyssey of a multitask stat3. Cell Cycle 2011, 10, 3221–3222. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Ma, Y.; Wright, G.M.; Huang, M.; Irby, R.; Briggs, J.; Karras, J.; Cress, W.D.; Pardoll, D.; et al. Role of stat3 in regulating p53 expression and function. Mol. Cell. Biol. 2005, 25, 7432–7440. [Google Scholar] [CrossRef]
- Gao, X.; Wang, H.; Yang, J.J.; Liu, X.; Liu, Z.R. Pyruvate kinase m2 regulates gene transcription by acting as a protein kinase. Mol. Cell 2012, 45, 598–609. [Google Scholar] [CrossRef]
- Turkson, J.; Bowman, T.; Adnane, J.; Zhang, Y.; Djeu, J.Y.; Sekharam, M.; Frank, D.A.; Holzman, L.B.; Wu, J.; Sebti, S.; et al. Requirement for ras/rac1-mediated p38 and c-jun n-terminal kinase signaling in stat3 transcriptional activity induced by the src oncoprotein. Mol. Cell. Biol. 1999, 19, 7519–7528. [Google Scholar] [CrossRef]
- Geletu, M.; Guy, S.; Arulanandam, R.; Feracci, H.; Raptis, L. Engaged for survival: From cadherin ligation to stat3 activation. Jaks-Stat 2013, 2, e27363. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial stat3 supports ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Wegrzyn, J.; Potla, R.; Chwae, Y.J.; Sepuri, N.B.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of mitochondrial stat3 in cellular respiration. Science 2009, 323, 793–797. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E., Jr. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Yu, W.; Yang, L.; Li, T.; Zhang, Y. Cadherin signaling in cancer: Its functions and role as a therapeutic target. Front. Oncol. 2019, 9, 989. [Google Scholar] [CrossRef]
- Takeichi, M. Morphogenetic roles of classic cadherins. Curr. Opin. Cell Biol. 1995, 7, 619–627. [Google Scholar] [CrossRef]
- Larue, L.; Antos, C.; Butz, S.; Huber, O.; Delmas, V.; Dominis, M.; Kemler, R. A role for cadherins in tissue formation. Development 1996, 122, 3185–3194. [Google Scholar] [CrossRef]
- Hulpiau, P.; van Roy, F. Molecular evolution of the cadherin superfamily. Int. J. Biochem. Cell Biol. 2009, 41, 349–369. [Google Scholar] [CrossRef]
- Gumbiner, B.M. Regulation of cadherin adhesive activity. J. Cell Biol. 2000, 148, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, J.M.; Nelson, W.J. Bench to bedside and back again: Molecular mechanisms of alpha-catenin function and roles in tumorigenesis. Semin. Cancer Biol. 2008, 18, 53–64. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Carmona, F.J.; Villanueva, A.; Vidal, A.; Munoz, C.; Puertas, S.; Penin, R.M.; Goma, M.; Lujambio, A.; Piulats, J.M.; Mesia, R.; et al. Epigenetic disruption of cadherin-11 in human cancer metastasis. J. Pathol. 2012, 228, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xiang, H.; Yu, S.; Lu, Y.; Wu, T. Research progress in the role and mechanism of cadherin-11 in different diseases. J. Cancer 2021, 12, 1190–1199. [Google Scholar] [CrossRef]
- Bruner, H.C.; Derksen, P.W.B. Loss of e-cadherin-dependent cell-cell adhesion and the development and progression of cancer. Cold Spring Harb Perspect. Biol. 2018, 10, a029330. [Google Scholar] [CrossRef] [PubMed]
- Raptis, L.; Brownell, H.L.; Wood, K.; Corbley, M.; Wang, D.; Haliotis, T. Cellular ras gene activity is required for full neoplastic transformation by simian virus 40. Cell Growth Differ. 1997, 8, 891–901. [Google Scholar] [PubMed]
- Vultur, A.; Arulanandam, R.; Turkson, J.; Niu, G.; Jove, R.; Raptis, L. Stat3 is required for full neoplastic transformation by the simian virus 40 large tumor antigen. Mol. Biol. Cell 2005, 16, 3832–3846. [Google Scholar] [CrossRef] [PubMed]
- Vultur, A.; Cao, J.; Arulanandam, R.; Turkson, J.; Jove, R.; Greer, P.; Craig, A.; Elliott, B.; Raptis, L. Cell-to-cell adhesion modulates stat3 activity in normal and breast carcinoma cells. Oncogene 2004, 23, 2600–2616. [Google Scholar] [CrossRef]
- Geletu, M.; Arulanandam, R.; Greer, S.; Trotman-Grant, A.; Tomai, E.; Raptis, L. Stat3 is a positive regulator of gap junctional intercellular communication in cultured, human lung carcinoma cells. BMC Cancer 2012, 12, 605. [Google Scholar] [CrossRef] [PubMed]
- Geletu, M.; Trotman-Grant, A.; Raptis, L. Mind the gap; regulation of gap junctional, intercellular communication by the src oncogene product and its effectors. Anticancer Res. 2012, 32, 4245–4250. [Google Scholar]
- Geletu, M.; Guy, S.; Raptis, L. Effects of src and stat3 upon gap junctional, intercellular communication in lung cancer lines. Anticancer Res. 2013, 33, 4401–4410. [Google Scholar]
- Niit, M.; Arulanandam, R.; Cass, J.; Geletu, M.; Hoskin, V.; Cote, G.; Gunning, P.; Elliott, B.; Raptis, L. Regulation of hc11 mouse breast epithelial cell differentiation by the e-cadherin/rac axis. Exp. Cell Res. 2017, 361, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Onishi, A.; Chen, Q.; Humtsoe, J.O.; Kramer, R.H. Stat3 signaling is induced by intercellular adhesion in squamous cell carcinoma cells. Exp. Cell Res. 2008, 314, 377–386. [Google Scholar] [CrossRef]
- Kreis, S.; Munz, G.A.; Haan, S.; Heinrich, P.C.; Behrmann, I. Cell density dependent increase of constitutive signal transducers and activators of transcription 3 activity in melanoma cells is mediated by janus kinases. Mol. Cancer Res. 2007, 5, 1331–1341. [Google Scholar] [CrossRef]
- Su, H.W.; Yeh, H.H.; Wang, S.W.; Shen, M.R.; Chen, T.L.; Kiela, P.R.; Ghishan, F.K.; Tang, M.J. Cell confluence-induced activation of signal transducer and activator of transcription-3 (stat3) triggers epithelial dome formation via augmentation of sodium hydrogen exchanger-3 (nhe3) expression. J. Biol. Chem. 2007, 282, 9883–9894. [Google Scholar] [CrossRef]
- Steinman, R.A.; Wentzel, A.; Lu, Y.; Stehle, C.; Grandis, J.R. Activation of stat3 by cell confluence reveals negative regulation of stat3 by cdk2. Oncogene 2003, 22, 3608–3615. [Google Scholar] [CrossRef] [PubMed]
- Raptis, L.; Arulanandam, R.; Vultur, A.; Geletu, M.; Chevalier, S.; Feracci, H. Beyond structure, to survival: Stat3 activation by cadherin engagement. Biochem. Cell Biol. 2009, 87, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Makagiansar, I.T.; Avery, M.; Hu, Y.; Audus, K.L.; Siahaan, T.J. Improving the selectivity of hav-peptides in modulating e-cadherin-e-cadherin interactions in the intercellular junction of mdck cell monolayers. Pharm. Res. 2001, 18, 446–453. [Google Scholar] [CrossRef]
- Arulanandam, R.; Vultur, A.; Cao, J.; Carefoot, E.; Elliott, B.E.; Truesdell, P.F.; Larue, L.; Feracci, H.; Raptis, L. Cadherin-cadherin engagement promotes cell survival via rac1/cdc42 and signal transducer and activator of transcription-3. Mol. Cancer Res. 2009, 7, 1310–1327. [Google Scholar] [CrossRef] [PubMed]
- Geletu, M.; Arulanandam, R.; Chevalier, S.; Saez, B.; Larue, L.; Feracci, H.; Raptis, L. Classical cadherins control survival through the gp130/stat3 axis. BBA-Mol. Cell Res. 2013, 1833, 1947–1959. [Google Scholar] [CrossRef] [PubMed]
- Arulanandam, R.; Geletu, M.; Feracci, H.; Raptis, L. Racv12 requires gp130 for stat3 activation, cell proliferation and migration. Exp. Cell Res. 2010, 316, 875–886. [Google Scholar] [CrossRef]
- Adan, H.; Guy, S.; Arulanandam, R.; Geletu, M.; Daniel, J.; Raptis, L. Activated src requires cadherin-11, rac, and gp130 for stat3 activation and survival of mouse balb/c3t3 fibroblasts. Cancer Gene Ther. 2022; online ahead of print. [Google Scholar] [CrossRef]
- Anagnostopoulou, A.; Vultur, A.; Arulanandam, R.; Cao, J.; Turkson, J.; Jove, R.; Kim, J.S.; Glenn, M.; Hamilton, A.D.; Raptis, L. Differential effects of stat3 inhibition in sparse vs confluent normal and breast cancer cells. Cancer Lett. 2006, 242, 120–132. [Google Scholar] [CrossRef]
- Geletu, M.; Mohan, R.; Arulanandam, R.; Berger-Becvar, A.; Nabi, I.R.; Gunning, P.T.; Raptis, L. Reciprocal regulation of the cadherin-11/stat3 axis by caveolin-1 in mouse fibroblasts and lung carcinoma cells. Biochim. Biophys. Acta 2018, 1865, 794–802. [Google Scholar] [CrossRef]
- Espada, J.; Martin-Perez, J. An update on src family of nonreceptor tyrosine kinases biology. Int. Rev. Cell Mol. Biol. 2017, 331, 83–122. [Google Scholar] [PubMed]
- Elsberger, B.; Tan, B.A.; Mitchell, T.J.; Brown, S.B.; Mallon, E.A.; Tovey, S.M.; Cooke, T.G.; Brunton, V.G.; Edwards, J. Is expression or activation of src kinase associated with cancer-specific survival in er-, pr- and her2-negative breast cancer patients? Am. J. Pathol. 2009, 175, 1389–1397. [Google Scholar] [CrossRef] [PubMed]
- Irby, R.B.; Yeatman, T.J. Role of src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [PubMed]
- Courtneidge, S.A.; Smith, A.E. The complex of polyoma virus middle t antigen and pp60c-src. EMBO J. 1984, 3, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Courtneidge, S.A. Transformation by polyoma virus middle t antigen. Cancer Surv. 1986, 5, 173–182. [Google Scholar]
- Campbell, K.S.; Ogris, E.; Burke, B.; Su, W.; Auger, K.R.; Druker, B.J.; Schaffhausen, B.S.; Roberts, T.M.; Pallas, D.C. Polyoma middle tumor antigen interacts with shc protein via the npty (asn-pro-thr-tyr) motif in middle tumor antigen. Proc. Nat. Acad. Sci. USA 1994, 91, 6344–6348. [Google Scholar] [CrossRef]
- Dilworth, S.M.; Brewster, C.E.; Jones, M.D.; Lanfrancone, L.; Pelicci, G.; Pelicci, P.G. Transformation by polyoma virus middle t-antigen involves the binding and tyrosine phosphorylation of shc. Nature 1994, 367, 87–90. [Google Scholar] [CrossRef]
- Courtneidge, S.A.; Heber, A. An 81 kd protein complexed with middle t antigen and pp60c-src: A possible phosphatidylinositol kinase. Cell 1987, 50, 1031–1037. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Whitman, M.; Schaffhausen, B.S.; Pallas, D.C.; White, M.; Cantley, L.; Roberts, T.M. Common elements in growth factor stimulation and oncogenic transformation: 85 kd phosphoprotein and phosphatidylinositol kinase activity. Cell 1987, 50, 1021–1029. [Google Scholar] [CrossRef]
- Whitman, M.; Kaplan, D.R.; Schaffhausen, B.S.; Cantley, L.; Roberts, T.M. Association of phosphatidylinositol kinase activity with polyoma mt competent for transformation. Nature 1985, 315, 239–242. [Google Scholar] [CrossRef]
- Talmage, D.A.; Freund, R.; Young, A.T.; Dahl, J.; Dawe, C.J.; Benjamin, T.L. Phosphorylation of middle t by pp60c-src: A switch for binding of phosphatidylinositol 3-kinase and optimal tumorigenesis. Cell 1989, 59, 55–65. [Google Scholar] [CrossRef]
- Yoakim, M.; Hou, W.; Liu, Y.; Carpenter, C.L.; Kapeller, R.; Schaffhausen, B.S. Interactions of polyomavirus middle t with the sh2 domains of the pp85 subunit of phosphatidylinositol-3-kinase. J. Virol. 1992, 66, 5485–5491. [Google Scholar] [CrossRef] [PubMed]
- Songyang, Z.; Shoelson, S.E.; Chaudhuri, M.; Gish, G.; Pawson, T.; Haser, W.G.; King, F.; Roberts, T.; Ratnofsky, S.; Lechleider, R.J.; et al. Sh2 domains recognize specific phosphopeptide sequences. Cell 1993, 72, 767–778. [Google Scholar] [PubMed]
- Smolar, N.; Griffin, B.E. DNA sequences of polyoma virus early deletion mutants. J. Virol. 1981, 38, 958–967. [Google Scholar] [CrossRef]
- Ichaso, N.; Dilworth, S.M. Cell transformation by the middle t-antigen of polyoma virus. Oncogene 2001, 20, 7908–7916. [Google Scholar] [CrossRef]
- Raptis, L.; Lamfrom, H.; Benjamin, T.L. Regulation of cellular phenotype and expression of polyomavirus middle t antigen in rat fibroblasts. Mol. Cell. Biol. 1985, 5, 2476–2486. [Google Scholar]
- Wadhawan, A.; Smith, C.; Nicholson, R.I.; Barrett-Lee, P.; Hiscox, S. Src-mediated regulation of homotypic cell adhesion: Implications for cancer progression and opportunities for therapeutic intervention. Cancer Treat. Rev. 2011, 37, 234–241. [Google Scholar] [CrossRef]
- Servitja, J.M.; Marinissen, M.J.; Sodhi, A.; Bustelo, X.R.; Gutkind, J.S. Rac1 function is required for src-induced transformation. Evidence of a role for tiam1 and vav2 in rac activation by src. J. Biol. Chem. 2003, 278, 34339–34346. [Google Scholar] [CrossRef]
- del Valle, I.; Rudloff, S.; Carles, A.; Li, Y.; Liszewska, E.; Vogt, R.; Kemler, R. E-cadherin is required for the proper activation of the lifr/gp130 signaling pathway in mouse embryonic stem cells. Development 2013, 140, 1684–1692. [Google Scholar] [CrossRef]
- Sears, R.C.; Nevins, J.R. Signaling networks that link cell proliferation and cell fate. J. Biol. Chem. 2002, 277, 11617–11620. [Google Scholar] [CrossRef]
- Hynes, R.O. Integrins: Versatility, modulation, and signaling in cell adhesion. Cell 1992, 69, 11–25. [Google Scholar] [CrossRef]
- Friedman, S.L.; Roll, F.J.; Boyles, J.; Arenson, D.M.; Bissell, D.M. Maintenance of differentiated phenotype of cultured rat hepatic lipocytes by basement membrane matrix. J. Biol. Chem. 1989, 264, 10756–10762. [Google Scholar] [CrossRef]
- Geletu, M.; Adan, H.; Niit, M.; Arulanandam, R.; Carefoot, E.; Hoskin, V.; Sina, D.; Elliott, B.; Gunning, P.; Raptis, L. Modulation of akt vs stat3 activity by the focal adhesion kinase in non-neoplastic mouse fibroblasts. Exp. Cell Res. 2021, 404, 112601. [Google Scholar] [CrossRef] [PubMed]
- Niit, M.; Hoskin, V.; Geletu, M.; Arulanandam, R.; Elliott, B.; Raptis, L. Cell-cell and cell-matrix adhesion in survival and metastasis: Stat3 vs akt. Biomol. Concepts 2015, 6, 383–399. [Google Scholar] [CrossRef]
- Patel, H.H.; Murray, F.; Insel, P.A. Caveolae as organizers of pharmacologically relevant signal transduction molecules. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 359–391. [Google Scholar] [CrossRef]
- Cohen, A.W.; Hnasko, R.; Schubert, W.; Lisanti, M.P. Role of caveolae and caveolins in health and disease. Physiol. Rev. 2004, 84, 1341–1379. [Google Scholar] [CrossRef]
- Goetz, J.G.; Lajoie, P.; Wiseman, S.M.; Nabi, I.R. Caveolin-1 in tumor progression: The good, the bad and the ugly. Cancer Metastasis Rev. 2008, 27, 715–735. [Google Scholar] [CrossRef]
- Boscher, C.; Nabi, I.R. Caveolin-1: Role in cell signaling. Adv. Exp. Med. Biol. 2012, 729, 29–50. [Google Scholar]
- Galbiati, F.; Volonte, D.; Engelman, J.A.; Watanabe, G.; Burk, R.; Pestell, R.G.; Lisanti, M.P. Targeted downregulation of caveolin-1 is sufficient to drive cell transformation and hyperactivate the p42/44 map kinase cascade. EMBO J. 1998, 17, 6633–6648. [Google Scholar] [CrossRef]
- Williams, T.M.; Lisanti, M.P. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am. J. Physiol. Cell Physiol. 2005, 288, C494–C506. [Google Scholar] [CrossRef]
- Chiu, W.T.; Lee, H.T.; Huang, F.J.; Aldape, K.D.; Yao, J.; Steeg, P.S.; Chou, C.Y.; Lu, Z.; Xie, K.; Huang, S. Caveolin-1 upregulation mediates suppression of primary breast tumor growth and brain metastases by stat3 inhibition. Cancer Res. 2011, 71, 4932–4943. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Toya, Y.; Schwencke, C.; Lisanti, M.P.; Myers, M.G., Jr.; Ishikawa, Y. Caveolin is an activator of insulin receptor signaling. J. Biol. Chem. 1998, 273, 26962–26968. [Google Scholar] [CrossRef] [PubMed]
- Jasmin, J.F.; Mercier, I.; Hnasko, R.; Cheung, M.W.; Tanowitz, H.B.; Dupuis, J.; Lisanti, M.P. Lung remodeling and pulmonary hypertension after myocardial infarction: Pathogenic role of reduced caveolin expression. Cardiovasc. Res. 2004, 63, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.M.; Lee, H.; Cheung, M.W.; Cohen, A.W.; Razani, B.; Iyengar, P.; Scherer, P.E.; Pestell, R.G.; Lisanti, M.P. Combined loss of ink4a and caveolin-1 synergistically enhances cell proliferation and oncogene-induced tumorigenesis: Role of ink4a/cav-1 in mammary epithelial cell hyperplasia. J. Biol. Chem. 2004, 279, 24745–24756. [Google Scholar] [CrossRef]
- Savage, K.; Lambros, M.B.; Robertson, D.; Jones, R.L.; Jones, C.; Mackay, A.; James, M.; Hornick, J.L.; Pereira, E.M.; Milanezi, F.; et al. Caveolin 1 is overexpressed and amplified in a subset of basal-like and metaplastic breast carcinomas: A morphologic, ultrastructural, immunohistochemical, and in situ hybridization analysis. Clin. Cancer Res. 2007, 13, 90–101. [Google Scholar] [CrossRef]
- Obrink, B. Epithelial cell adhesion molecules. Exp. Cell Res. 1986, 163, 1–21. [Google Scholar] [CrossRef]
- Zhang, Y.; Turkson, J.; Carter-Su, C.; Smithgall, T.; Levitzki, A.; Kraker, A.; Krolewski, J.J.; Medveczky, P.; Jove, R. Activation of stat3 in v-src transformed fibroblasts requires cooperation of jak1 kinase activity. J. Biol. Chem. 2000, 275, 24935–24944. [Google Scholar] [CrossRef]
- Madarampalli, B.; Watts, G.F.M.; Panipinto, P.M.; Nguygen, H.N.; Brenner, M.B.; Noss, E.H. Interactions between cadherin-11 and platelet-derived growth factor receptor-alpha signaling link cell adhesion and proliferation. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1516–1524. [Google Scholar] [CrossRef]
- Wong, J.V.; Dong, P.; Nevins, J.R.; Mathey-Prevot, B.; You, L. Network calisthenics: Control of e2f dynamics in cell cycle entry. Cell Cycle 2011, 10, 3086–3094. [Google Scholar] [CrossRef]
- Young, A.P.; Nagarajan, R.; Longmore, G.D. Mechanisms of transcriptional regulation by rb-e2f segregate by biological pathway. Oncogene 2003, 22, 7209–7217. [Google Scholar] [CrossRef]
- Moroni, M.C.; Hickman, E.S.; Lazzerini, D.E.; Caprara, G.; Colli, E.; Cecconi, F.; Muller, H.; Helin, K. Apaf-1 is a transcriptional target for e2f and p53. Nat. Cell Biol. 2001, 3, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.; Marin, M.C.; Phillips, A.C.; Seelan, R.S.; Smith, D.I.; Liu, W.; Flores, E.R.; Tsai, K.Y.; Jacks, T.; Vousden, K.H.; et al. Role for the p53 homologue p73 in e2f-1-induced apoptosis. Nature 2000, 407, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Chu, K.; Cheng, C.J.; Ye, X.; Lee, Y.C.; Zurita, A.J.; Chen, D.T.; Yu-Lee, L.Y.; Zhang, S.; Yeh, E.T.; Hu, M.C.; et al. Cadherin-11 promotes the metastasis of prostate cancer cells to bone. Mol. Cancer Res. 2008, 6, 1259–1267. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.F.; Lira, C.; Chu, K.; Bilen, M.A.; Lee, Y.C.; Ye, X.; Kim, S.M.; Ortiz, A.; Wu, F.L.; Logothetis, C.J.; et al. Cadherin-11 increases migration and invasion of prostate cancer cells and enhances their interaction with osteoblasts. Cancer Res. 2010, 70, 4580–4589. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Bilen, M.A.; Yu, G.; Lin, S.C.; Huang, C.F.; Ortiz, A.; Cho, H.; Song, J.H.; Satcher, R.L.; Kuang, J.; et al. Inhibition of cell adhesion by a cadherin-11 antibody thwarts bone metastasis. Mol. Cancer Res. 2013, 11, 1401–1411. [Google Scholar] [CrossRef]
- Tanaka, H.; Kono, E.; Tran, C.P.; Miyazaki, H.; Yamashiro, J.; Shimomura, T.; Fazli, L.; Wada, R.; Huang, J.; Vessella, R.L.; et al. Monoclonal antibody targeting of n-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 2010, 16, 1414–1420. [Google Scholar] [CrossRef]
- Pishvaian, M.J.; Feltes, C.M.; Thompson, P.; Bussemakers, M.J.; Schalken, J.A.; Byers, S.W. Cadherin-11 is expressed in invasive breast cancer cell lines. Cancer Res. 1999, 59, 947–952. [Google Scholar]
- Tamura, D.; Hiraga, T.; Myoui, A.; Yoshikawa, H.; Yoneda, T. Cadherin-11-mediated interactions with bone marrow stromal/osteoblastic cells support selective colonization of breast cancer cells in bone. Int. J. Oncol. 2008, 33, 17–24. [Google Scholar] [CrossRef]
- Furtek, S.L.; Backos, D.S.; Matheson, C.J.; Reigan, P. Strategies and approaches of targeting stat3 for cancer treatment. ACS Chem. Biol. 2016, 11, 308–318. [Google Scholar] [CrossRef]
- Cao, J.; Arulanandam, R.; Vultur, A.; Anagnostopoulou, A.; Raptis, L. Adenovirus e1a requires c-ras for full neoplastic transformation or suppression of differentiation of murine preadipocytes. Mol. Carcinog. 2007, 46, 284–302. [Google Scholar] [CrossRef]
- Cao, J.; Arulanandam, R.; Vultur, A.; Preston, T.; Jaronczyk, K.; Tomai, E.; Zandi, K.; Raptis, L. Adenovirus-5 e1a suppresses differentiation of 3t3 l1 preadipocytes at lower levels than required for induction of apoptosis. Mol. Carcinog. 2005, 43, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Arulanandam, R.; Vultur, A.; Raptis, L. Transfection techniques affecting stat3 activity levels. Anal. Biochem. 2005, 338, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Herold, C.I.; Chadaram, V.; Peterson, B.L.; Marcom, P.K.; Hopkins, J.; Kimmick, G.G.; Favaro, J.; Hamilton, E.; Welch, R.A.; Bacus, S.; et al. Phase ii trial of dasatinib in patients with metastatic breast cancer using real-time pharmacodynamic tissue biomarkers of src inhibition to escalate dosing. Clin. Cancer Res. 2011, 17, 6061–6070. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Bengala, C.; Ibrahim, N.; Roche, H.; Sparano, J.; Strauss, L.C.; Fairchild, J.; Sy, O.; Goldstein, L.J. Dasatinib as a single agent in triple-negative breast cancer: Results of an open-label phase 2 study. Clin. Cancer Res. 2011, 17, 6905–6913. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.L.; Baurain, J.F.; Sparano, J.; Strauss, L.; Campone, M.; Fumoleau, P.; Rugo, H.; Awada, A.; Sy, O.; Llombart-Cussac, A. A phase 2 trial of dasatinib in patients with advanced her2-positive and/or hormone receptor-positive breast cancer. Clin. Cancer Res. 2011, 17, 6897–6904. [Google Scholar] [CrossRef]
- Tian, J.; Raffa, F.A.; Dai, M.; Moamer, A.; Khadang, B.; Hachim, I.Y.; Bakdounes, K.; Ali, S.; Jean-Claude, B.; Lebrun, J.J. Dasatinib sensitises triple negative breast cancer cells to chemotherapy by targeting breast cancer stem cells. Br. J. Cancer 2018, 119, 1495–1507. [Google Scholar] [CrossRef]
- Chen, Q.; Watson, J.T.; Marengo, S.R.; Decker, K.S.; Coleman, I.; Nelson, P.S.; Sikes, R.A. Gene expression in the lncap human prostate cancer progression model: Progression associated expression in vitro corresponds to expression changes associated with prostate cancer progression in vivo. Cancer Lett. 2006, 244, 274–288. [Google Scholar] [CrossRef]
- Wei, C.J.; Francis, R.; Xu, X.; Lo, C.W. Connexin43 associated with an n-cadherin-containing multiprotein complex is required for gap junction formation in nih3t3 cells. J. Biol. Chem. 2005, 280, 19925–19936. [Google Scholar] [CrossRef]
- Frenzel, E.M.; Johnson, R.G. Gap junction formation between cultured embryonic lens cells is inhibited by antibody to n-cadherin. Dev. Biol. 1996, 179, 1–16. [Google Scholar] [CrossRef]
- Greer, S.; Honeywell, R.; Geletu, M.; Arulanandam, R.; Raptis, L. Housekeeping gene products; levels may change with confluence of cultured cells. J. Immunol. Methods 2010, 355, 76–79. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adan, H.; Daniel, J.; Raptis, L. Roads to Stat3 Paved with Cadherins. Cells 2022, 11, 2537. https://doi.org/10.3390/cells11162537
Adan H, Daniel J, Raptis L. Roads to Stat3 Paved with Cadherins. Cells. 2022; 11(16):2537. https://doi.org/10.3390/cells11162537
Chicago/Turabian StyleAdan, Hanad, Juliet Daniel, and Leda Raptis. 2022. "Roads to Stat3 Paved with Cadherins" Cells 11, no. 16: 2537. https://doi.org/10.3390/cells11162537
APA StyleAdan, H., Daniel, J., & Raptis, L. (2022). Roads to Stat3 Paved with Cadherins. Cells, 11(16), 2537. https://doi.org/10.3390/cells11162537