
All Quiet on the TE Front? The Role of Chromatin in Transposable Element Silencing


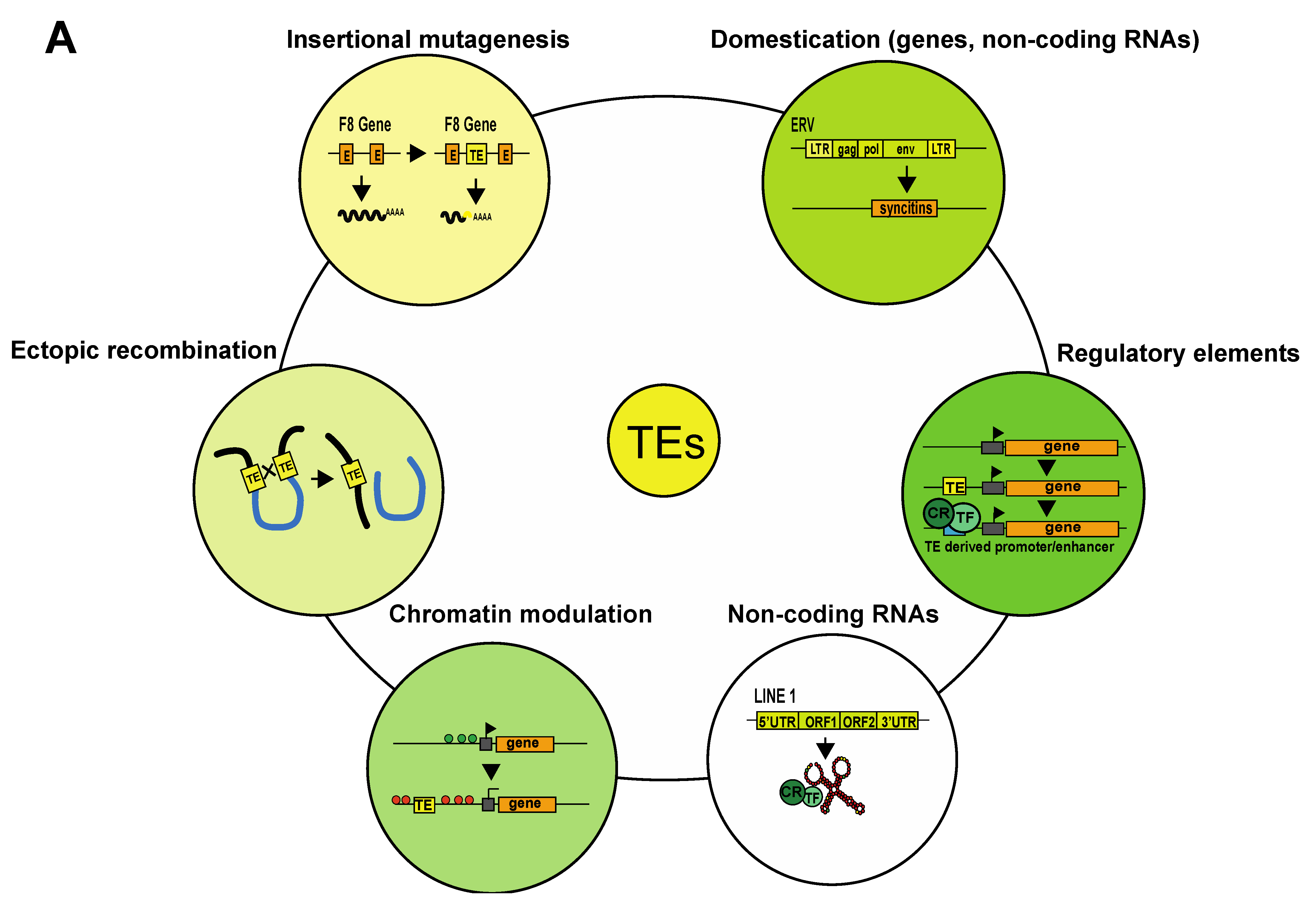{kind=link}
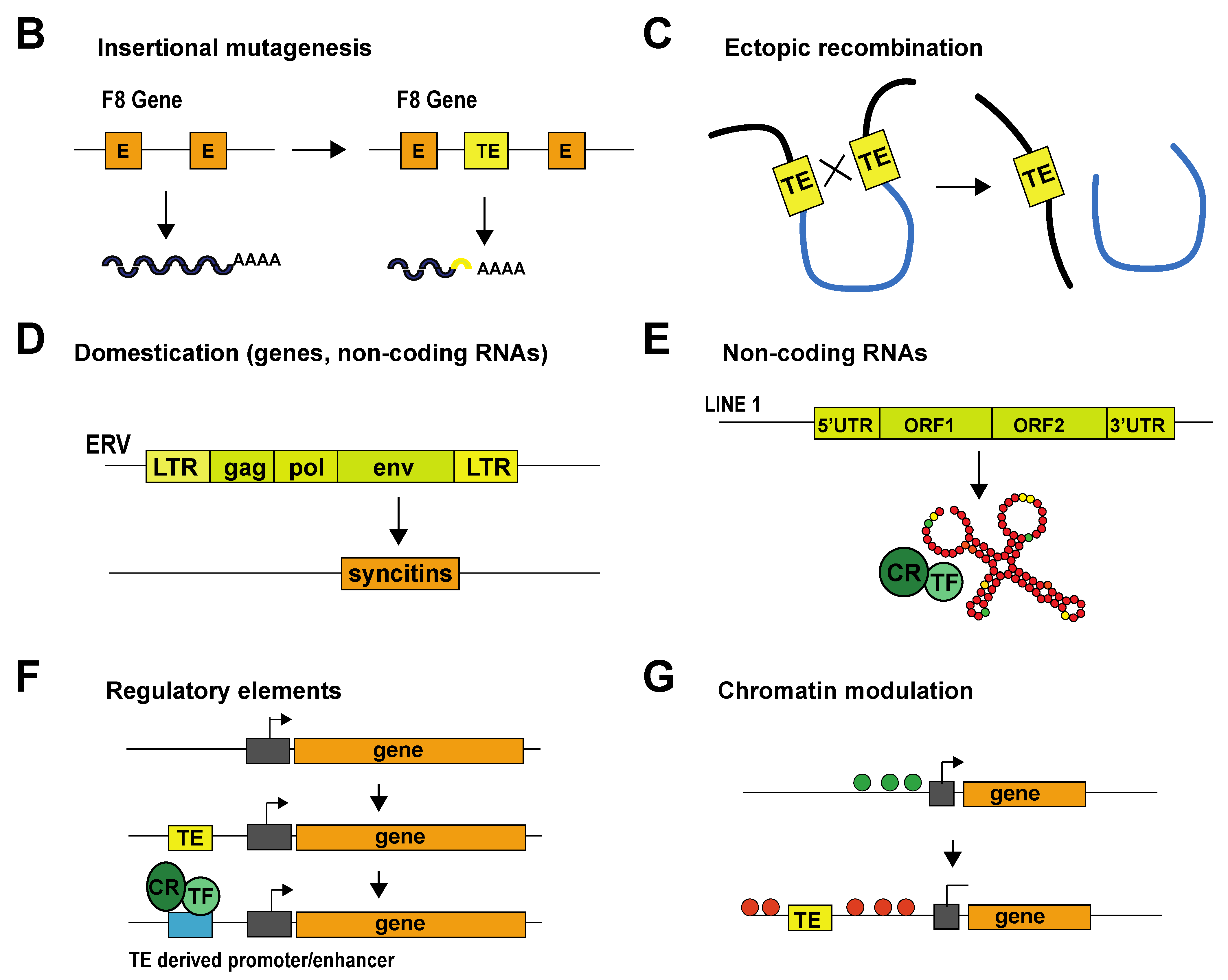{kind=link}
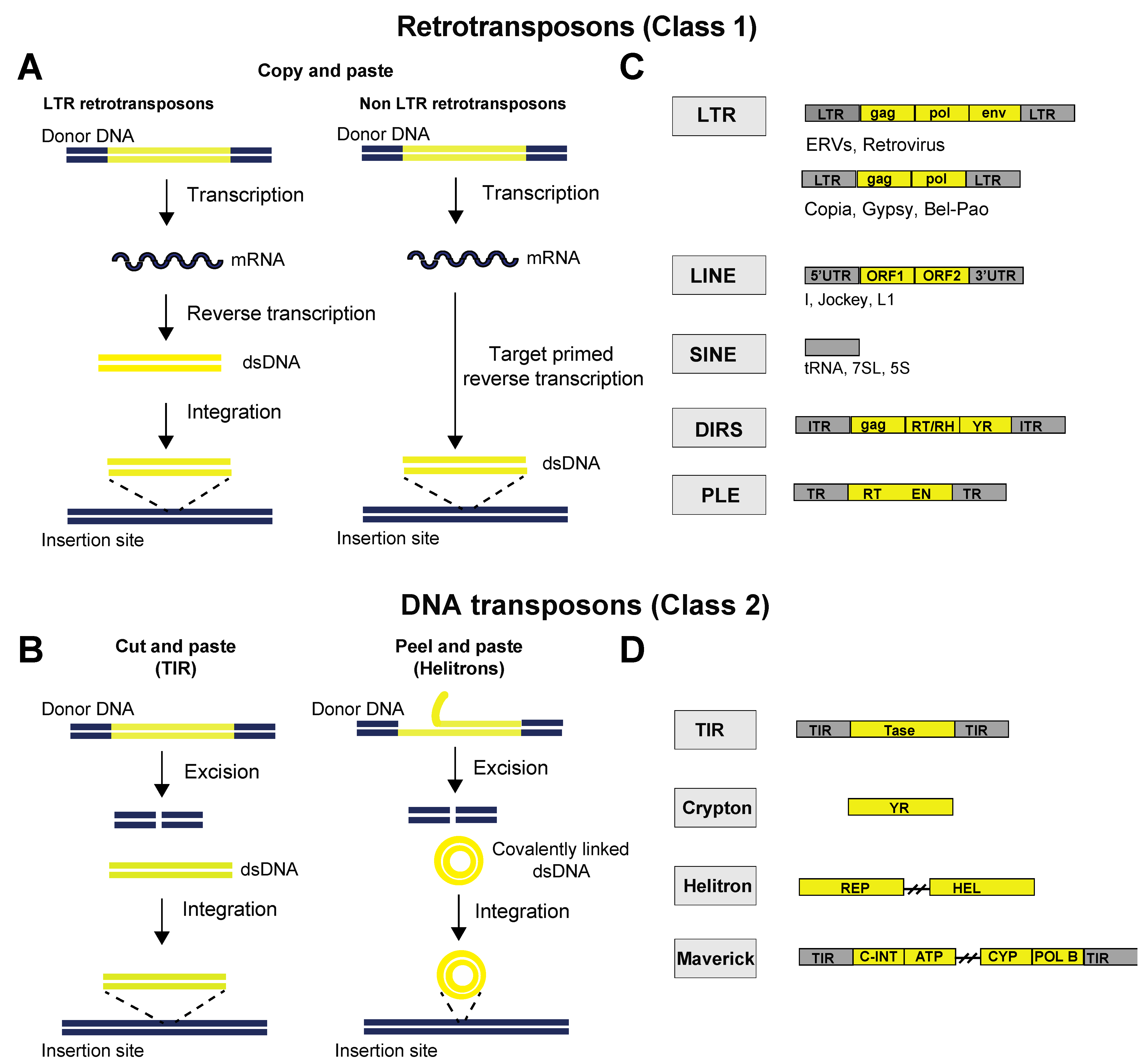{kind=link}
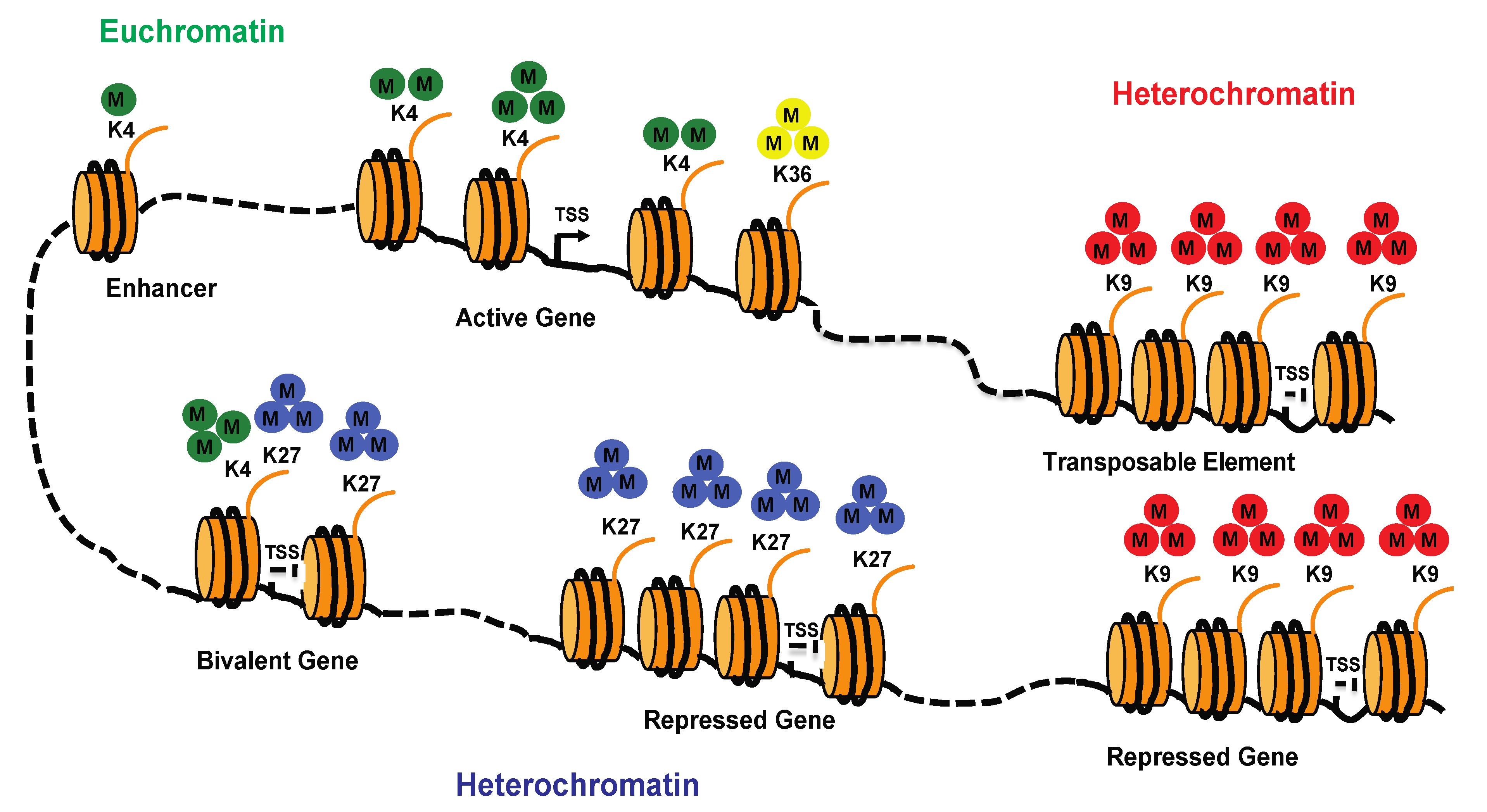{kind=link}
{kind=link}
Abstract
:1. Introduction
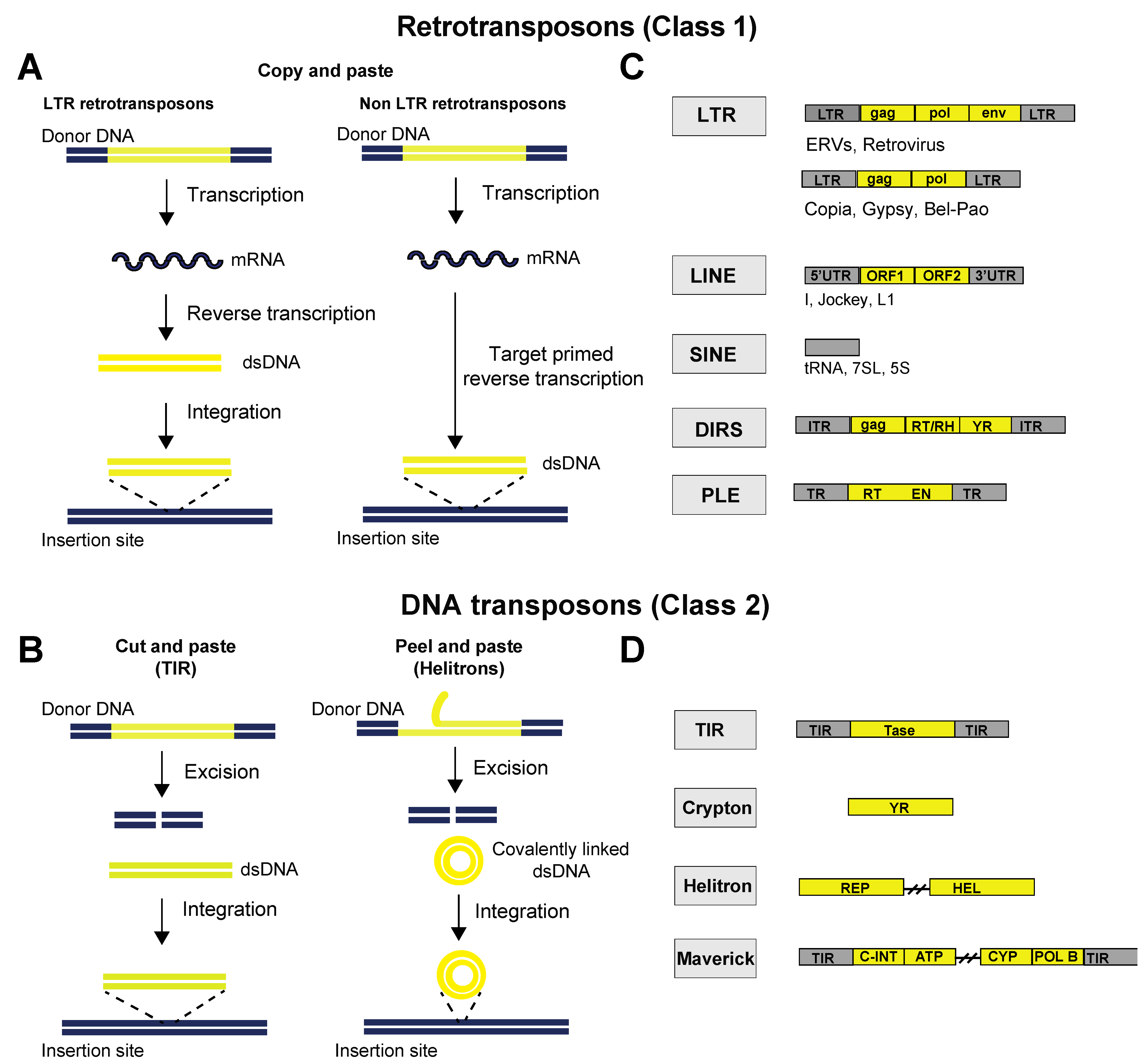
2. Classes of TEs
3. TE Silencing
4. Chromatin
4.1. Brief Overview of the Role of DNA Methylation in TE Silencing
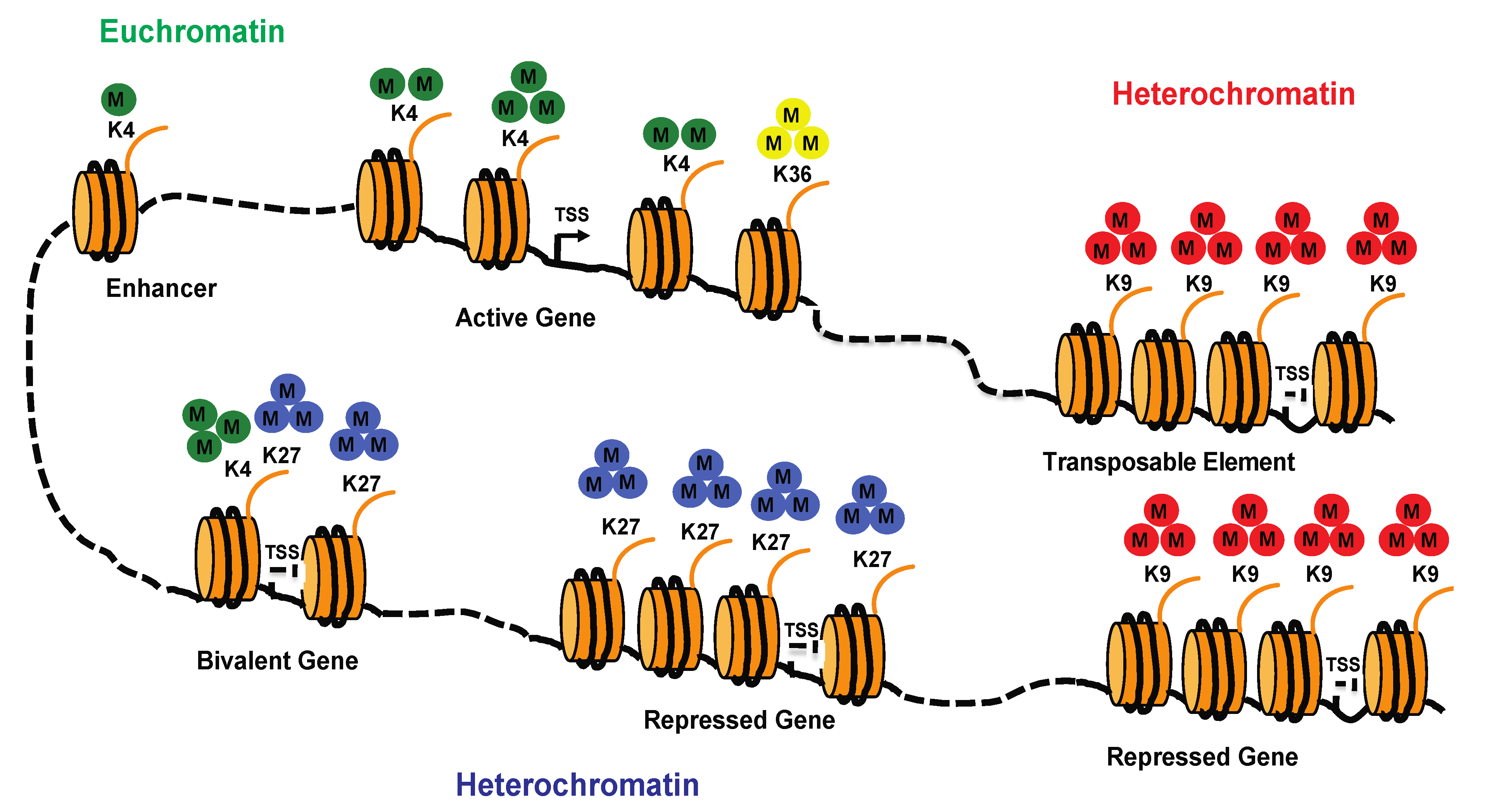
4.2. Histone Marks
4.3. Roles of Histone Marks
4.4. Histone Methylation
4.5. H3K9 Methylation, a Defining Feature of TEs?
4.6. Role of the KRAB-Znf Family of Transcription Factors in the Recruitment of H3K9 Methyltransferases
4.7. piRNA-Dependent Recruitment of H3K9 Methyltransferases
4.8. H3K27 Methylation, an Ancestral form of TE Silencing?
4.9. Role of Histone 4 in TE Silencing
4.10. Sumoylation
4.11. “Active” Histone Marks at TE Loci
4.12. H3K4 Methylation
4.13. H3K36 Methylation
4.14. H3.3
4.15. Histone Acetylation
4.16. The Emerging Role of Nuclear Architecture
4.17. Interplay between m6A RNA and Chromatin at TE loci
5. Summary, Significance and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kazazian, H.H., Jr.; Wong, C.; Youssoufian, H.; Scott, A.F.; Phillips, D.G.; Antonarakis, S.E. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature 1988, 332, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Robberecht, C.; Voet, T.; Zamani Esteki, M.; Nowakowska, B.A.; Vermeesch, J.R. Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res. 2013, 23, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Naville, M.; Henriet, S.; Warren, I.; Sumic, S.; Reeve, M.; Volff, J.N.; Chourrout, D. Massive Changes of Genome Size Driven by Expansions of Non-autonomous Transposable Elements. Curr. Biol. 2019, 29, 1161–1168.e6. [Google Scholar] [CrossRef]
- Shao, F.; Han, M.; Peng, Z. Evolution and diversity of transposable elements in fish genomes. Sci. Rep. 2019, 9, 15399. [Google Scholar] [CrossRef]
- Serrato-Capuchina, A.; Matute, D.R. The Role of Transposable Elements in Speciation. Genes 2018, 9, 254. [Google Scholar] [CrossRef]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvak, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Percharde, M.; Lin, C.J.; Yin, Y.; Guan, J.; Peixoto, G.A.; Bulut-Karslioglu, A.; Biechele, S.; Huang, B.; Shen, X.; Ramalho-Santos, M. A LINE1-Nucleolin Partnership Regulates Early Development and ESC Identity. Cell 2018, 174, 391–405.e19. [Google Scholar] [CrossRef]
- Goerner-Potvin, P.; Bourque, G. Computational tools to unmask transposable elements. Nat. Rev. Genet. 2018, 19, 688–704. [Google Scholar] [CrossRef]
- Morgan, H.D.; Sutherland, H.G.; Martin, D.I.; Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999, 23, 314–318. [Google Scholar] [CrossRef]
- Gualdrini, F.; Polletti, S.; Simonatto, M.; Prosperini, E.; Pileri, F.; Natoli, G. H3K9 trimethylation in active chromatin restricts the usage of functional CTCF sites in SINE B2 repeats. Genes Dev. 2022, 36, 414–432. [Google Scholar] [CrossRef]
- Kaaij, L.J.T.; Mohn, F.; van der Weide, R.H.; de Wit, E.; Buhler, M. The ChAHP Complex Counteracts Chromatin Looping at CTCF Sites that Emerged from SINE Expansions in Mouse. Cell 2019, 178, 1437–1451.e14. [Google Scholar] [CrossRef]
- Sentmanat, M.F.; Elgin, S.C. Ectopic assembly of heterochromatin in Drosophila melanogaster triggered by transposable elements. Proc. Natl. Acad. Sci. USA 2012, 109, 14104–14109. [Google Scholar] [CrossRef]
- Etchegaray, E.; Naville, M.; Volff, J.N.; Haftek-Terreau, Z. Transposable element-derived sequences in vertebrate development. Mob. DNA 2021, 12, 1. [Google Scholar] [CrossRef]
- Ninova, M.; Chen, Y.A.; Godneeva, B.; Rogers, A.K.; Luo, Y.; Fejes Toth, K.; Aravin, A.A. Su(var)2-10 and the SUMO Pathway Link piRNA-Guided Target Recognition to Chromatin Silencing. Mol. Cell 2020, 77, 556–570.e556. [Google Scholar] [CrossRef]
- Lee, Y.C.G.; Karpen, G.H. Pervasive epigenetic effects of Drosophila euchromatic transposable elements impact their evolution. Elife 2017, 6, e25762. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Koonin, E.V. Evolution of the RAG1-RAG2 locus: Both proteins came from the same transposon. Biol. Direct 2015, 10, 20. [Google Scholar] [CrossRef]
- Lanciano, S.; Cristofari, G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020, 21, 721–736. [Google Scholar] [CrossRef]
- Wells, J.N.; Feschotte, C. A Field Guide to Eukaryotic Transposable Elements. Annu. Rev. Genet. 2020, 54, 539–561. [Google Scholar] [CrossRef]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A unified classification system for eukaryotic transposable elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Makalowski, W.; Gotea, V.; Pande, A.; Makalowska, I. Transposable Elements: Classification, Identification, and Their Use As a Tool For Comparative Genomics. Methods Mol. Biol. 2019, 1910, 177–207. [Google Scholar] [CrossRef] [PubMed]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015, 162, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Sheng, W.; LaFleur, M.W.; Nguyen, T.H.; Chen, S.; Chakravarthy, A.; Conway, J.R.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.-H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef]
- Aravin, A.A.; Naumova, N.M.; Tulin, A.V.; Vagin, V.V.; Rozovsky, Y.M.; Gvozdev, V.A. Double-stranded RNA-mediated silencing of genomic tandem repeats and transposable elements in the D. melanogaster germline. Curr. Biol. 2001, 11, 1017–1027. [Google Scholar] [CrossRef]
- Aravin, A.A.; Sachidanandam, R.; Bourc’His, D.; Schaefer, C.; Pezic, D.; Toth, K.F.; Bestor, T.; Hannon, G.J. A piRNA Pathway Primed by Individual Transposons Is Linked to De Novo DNA Methylation in Mice. Mol. Cell 2008, 31, 785–799. [Google Scholar] [CrossRef]
- Vagin, V.V.; Sigova, A.; Li, C.; Seitz, H.; Gvozdev, V.; Zamore, P.D. A Distinct Small RNA Pathway Silences Selfish Genetic Elements in the Germline. Science 2006, 313, 320–324. [Google Scholar] [CrossRef]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef]
- Lau, N.C.; Seto, A.G.; Kim, J.; Kuramochi-Miyagawa, S.; Nakano, T.; Bartel, D.P.; Kingston, R.E. Characterization of the piRNA Complex from Rat Testes. Science 2006, 313, 363–367. [Google Scholar] [CrossRef]
- Aravin, A.; Gaidatzis, D.; Pfeffer, S.; Lagos-Quintana, M.; Landgraf, P.; Iovino, N.; Morris, P.; Brownstein, M.J.; Kuramochi-Miyagawa, S.; Nakano, T.; et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 2006, 442, 203–207. [Google Scholar] [CrossRef]
- Grivna, S.T.; Beyret, E.; Wang, Z.; Lin, H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev. 2006, 20, 1709–1714. [Google Scholar] [CrossRef]
- Saito, K.; Nishida, K.M.; Mori, T.; Kawamura, Y.; Miyoshi, K.; Nagami, T.; Siomi, H.; Siomi, M.C. Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev. 2006, 20, 2214–2222. [Google Scholar] [CrossRef]
- Houwing, S.; Kamminga, L.M.; Berezikov, E.; Cronembold, D.; Girard, A.; van den Elst, H.; Filippov, D.V.; Blaser, H.; Raz, E.; Moens, C.B.; et al. A Role for Piwi and piRNAs in Germ Cell Maintenance and Transposon Silencing in Zebrafish. Cell 2007, 129, 69–82. [Google Scholar] [CrossRef]
- Watanabe, T.; Takeda, A.; Tsukiyama, T.; Mise, K.; Okuno, T.; Sasaki, H.; Minami, N.; Imai, H. Identification and characterization of two novel classes of small RNAs in the mouse germline: Retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev. 2006, 20, 1732–1743. [Google Scholar] [CrossRef]
- Sienski, G.; Dönertas, D.; Brennecke, J. Transcriptional Silencing of Transposons by Piwi and Maelstrom and Its Impact on Chromatin State and Gene Expression. Cell 2012, 151, 964–980. [Google Scholar] [CrossRef]
- Le Thomas, A.; Rogers, A.K.; Webster, A.; Marinov, G.K.; Liao, S.E.; Perkins, E.M.; Hur, J.K.; Aravin, A.A.; Tóth, K.F. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev. 2013, 27, 390–399. [Google Scholar] [CrossRef]
- Rozhkov, N.V.; Hammell, M.; Hannon, G.J. Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev. 2013, 27, 400–412. [Google Scholar] [CrossRef]
- Yamashiro, H.; Siomi, M.C. PIWI-Interacting RNA in Drosophila: Biogenesis, Transposon Regulation, and Beyond. Chem Rev. 2018, 118, 4404–4421. [Google Scholar] [CrossRef]
- Kuramochi-Miyagawa, S.; Watanabe, T.; Gotoh, K.; Totoki, Y.; Toyoda, A.; Ikawa, M.; Asada, N.; Kojima, K.; Yamaguchi, Y.; Ijiri, T.W.; et al. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 2008, 22, 908–917. [Google Scholar] [CrossRef]
- Pezic, D.; Manakov, S.A.; Sachidanandam, R.; Aravin, A.A. piRNA pathway targets active LINE1 elements to establish the repressive H3K9me3 mark in germ cells. Genes Dev. 2014, 28, 1410–1428. [Google Scholar] [CrossRef]
- Molaro, A.; Malik, H.S. Hide and seek: How chromatin-based pathways silence retroelements in the mammalian germline. Curr. Opin. Genet. Dev. 2016, 37, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Onishi, R.; Yamanaka, S.; Siomi, M.C. piRNA- and siRNA-mediated transcriptional repression in Drosophila, mice, and yeast: New insights and biodiversity. EMBO Rep. 2021, 22, e53062. [Google Scholar] [CrossRef] [PubMed]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2018, 20, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Czech, B.; Munafo, M.; Ciabrelli, F.; Eastwood, E.L.; Fabry, M.H.; Kneuss, E.; Hannon, G.J. piRNA-Guided Genome Defense: From Biogenesis to Silencing. Annu. Rev. Genet. 2018, 52, 131–157. [Google Scholar] [CrossRef]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’His, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Jeltsch, A. Phylogeny of Methylomes. Science 2010, 328, 837–838. [Google Scholar] [CrossRef]
- Dor, Y.; Cedar, H. Principles of DNA methylation and their implications for biology and medicine. Lancet 2018, 392, 777–786. [Google Scholar] [CrossRef]
- Tsukahara, S.; Kobayashi, A.; Kawabe, A.; Mathieu, O.; Miura, A.; Kakutani, T. Bursts of retrotransposition reproduced in Arabidopsis. Nature 2009, 461, 423–426. [Google Scholar] [CrossRef]
- Lippman, Z.; Gendrel, A.-V.; Black, M.; Vaughn, M.; Dedhia, N.; McCombie, W.R.; Lavine, K.; Mittal, V.; May, B.; Kasschau, K.D.; et al. Role of transposable elements in heterochromatin and epigenetic control. Nature 2004, 430, 471–476. [Google Scholar] [CrossRef]
- Zhou, Y.; Cambareri, E.B.; Kinsey, J.A. DNA methylation inhibits expression and transposition of the Neurospora Tad retrotransposon. Mol. Genet. Genom. 2001, 265, 748–754. [Google Scholar] [CrossRef]
- Chernyavskaya, Y.; Mudbhary, R.; Tokarz, D.; Jacob, V.; Gopinath, S.; Zhang, C.; Sun, X.; Wang, S.; Magnani, E.; Madakashira, B.P.; et al. Loss of DNA methylation in zebrafish embryos activates retrotransposons to trigger antiviral signaling. Development 2017, 144, 2925–2939. [Google Scholar] [CrossRef]
- Walsh, C.; Chaillet, J.R.; Bestor, T.H. Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 1998, 20, 116–117. [Google Scholar] [CrossRef]
- Barau, J.; Teissandier, A.; Zamudio, N.; Roy, S.; Nalesso, V.; Hérault, Y.; Guillou, F.; Bourc’His, D. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 2016, 354, 909–912. [Google Scholar] [CrossRef]
- Haubold, B.; Wiehe, T. How repetitive are genomes? BMC Bioinformat. 2006, 7, 541. [Google Scholar] [CrossRef]
- Cuellar, T.L.; Herzner, A.-M.; Zhang, X.; Goyal, Y.; Watanabe, C.; Friedman, B.A.; Janakiraman, V.; Durinck, S.; Stinson, J.; Arnott, D.; et al. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J. Cell Biol. 2017, 216, 3535–3549. [Google Scholar] [CrossRef]
- Sanchez-Luque, F.J.; Kempen, M.-J.H.; Gerdes, P.; Vargas-Landin, D.B.; Richardson, S.R.; Troskie, R.-L.; Jesuadian, J.S.; Cheetham, S.W.; Carreira, P.E.; Salvador-Palomeque, C.; et al. LINE-1 Evasion of Epigenetic Repression in Humans. Mol. Cell 2019, 75, 590–604.e12. [Google Scholar] [CrossRef]
- De Cecco, M.; Criscione, S.W.; Peterson, A.L.; Neretti, N.; Sedivy, J.M.; Kreiling, J.A. Transposable elements become active and mobile in the genomes of aging mammalian somatic tissues. Aging 2013, 5, 867–883. [Google Scholar] [CrossRef]
- Burns, K.H. Transposable elements in cancer. Nat. Cancer 2017, 17, 415–424. [Google Scholar] [CrossRef]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921, Correction in Nature 2001, 412, 565–566; Erratum in Nature 2001, 7, 720. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef]
- Wu, T.P.; Wang, T.; Seetin, M.G.; Lai, Y.; Zhu, S.; Lin, K.; Liu, Y.; Byrum, S.D.; Mackintosh, S.G.; Zhong, M.; et al. DNA methylation on N6-adenine in mammalian embryonic stem cells. Nature 2016, 532, 329–333. [Google Scholar] [CrossRef]
- Yao, B.; Cheng, Y.; Wang, Z.; Li, Y.; Chen, L.; Huang, L.; Zhang, W.; Chen, D.; Wu, H.; Tang, B.; et al. DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef]
- Zhu, S.; Beaulaurier, J.; Deikus, G.; Wu, T.P.; Strahl, M.; Hao, Z.; Luo, G.; Gregory, J.; Chess, A.; He, C.; et al. Mapping and characterizing N6-methyladenine in eukaryotic genomes using single-molecule real-time sequencing. Genome Res. 2018, 28, 1067–1078. [Google Scholar] [CrossRef]
- Deniz, Ö.; Frost, J.M.; Branco, M.R. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 2019, 20, 417–431. [Google Scholar] [CrossRef]
- Jansz, N. DNA methylation dynamics at transposable elements in mammals. Essays Biochem. 2019, 63, 677–689. [Google Scholar] [CrossRef]
- Allshire, R.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2017, 19, 229–244. [Google Scholar] [CrossRef]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation & Co: An expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019, 63, 97–107. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. The Yin and Yang of Histone Marks in Transcription. Annu. Rev. Genom. Hum. Genet. 2021, 22, 147–170. [Google Scholar] [CrossRef]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mESCs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Shilatifard, A. Epigenetic modifications of histones in cancer. Genome Biol. 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- modENCODE Consortium; Roy, S.; Ernst, J.; Kharchenko, P.V.; Kheradpour, P.; Negre, N.; Eaton, M.L.; Landolin, J.M.; Bristow, C.A.; Ma, L.; et al. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 2010, 330, 1787–1797. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Fu, K.; Bonora, G.; Pellegrini, M. Interactions between core histone marks and DNA methyltransferases predict DNA methylation patterns observed in human cells and tissues. Epigenetics 2019, 15, 272–282. [Google Scholar] [CrossRef]
- Li, Y.; Chen, X.; Lu, C. The interplay between DNA and histone methylation: Molecular mechanisms and disease implications. EMBO Rep. 2021, 22, e51803. [Google Scholar] [CrossRef]
- Walter, M.; Teissandier, A.; Pérez-Palacios, R.; Bourc’His, D. An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. eLife 2016, 5, e11418. [Google Scholar] [CrossRef]
- Berrens, R.V.; Andrews, S.; Spensberger, D.; Santos, F.; Dean, W.; Gould, P.; Sharif, J.; Olova, N.; Chandra, T.; Koseki, H.; et al. An endosiRNA-Based Repression Mechanism Counteracts Transposon Activation during Global DNA Demethylation in Embryonic Stem Cells. Cell Stem Cell 2017, 21, 694–703.e7. [Google Scholar] [CrossRef]
- Molaro, A.; Falciatori, I.; Hodges, E.; Aravin, A.A.; Marran, K.; Rafii, S.; McCombie, W.R.; Smith, A.D.; Hannon, G.J. Two waves of de novo methylation during mouse germ cell development. Genes Dev. 2014, 28, 1544–1549. [Google Scholar] [CrossRef]
- Fadloun, A.; Eid, A.; Torres-Padilla, M.E. Mechanisms and dynamics of heterochromatin formation during mammalian development: Closed paths and open questions. Curr. Top. Dev. Biol. 2013, 104, 1–45. [Google Scholar] [CrossRef]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of Histone H3K4 Methylation Patterns. Mol. Cell 2017, 68, 773–785.e6. [Google Scholar] [CrossRef]
- Rada-Iglesias, A. Is H3K4me1 at enhancers correlative or causative? Nat. Genet. 2018, 50, 4–5. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A Bivalent Chromatin Structure Marks Key Developmental Genes in Embryonic Stem Cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef]
- Nicetto, D.; Donahue, G.; Jain, T.; Peng, T.; Sidoli, S.; Sheng, L.; Montavon, T.; Becker, J.S.; Grindheim, J.M.; Blahnik, K.; et al. H3K9me3-heterochromatin loss at protein-coding genes enables developmental lineage specification. Science 2019, 363, 294–297. [Google Scholar] [CrossRef]
- Methot, S.P.; Padeken, J.; Brancati, G.; Zeller, P.; Delaney, C.E.; Gaidatzis, D.; Kohler, H.; van Oudenaarden, A.; Großhans, H.; Gasser, S.M. H3K9me selectively blocks transcription factor activity and ensures differentiated tissue integrity. Nature 2021, 23, 1163–1175. [Google Scholar] [CrossRef]
- Bannister, A.J.; Zegerman, P.; Partridge, J.F.; Miska, E.A.; Thomas, J.O.; Allshire, R.C.; Kouzarides, T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 2001, 410, 120–124. [Google Scholar] [CrossRef]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef]
- Nakayama, J.-I.; Rice, J.C.; Strahl, B.D.; Allis, C.D.; Grewal, S.I.S. Role of Histone H3 Lysine 9 Methylation in Epigenetic Control of Heterochromatin Assembly. Science 2001, 292, 110–113. [Google Scholar] [CrossRef]
- Grewal, S.I.S.; Jia, S. Heterochromatin revisited. Nat. Rev. Genet. 2007, 8, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Elgin, S.C.; Reuter, G. Position-Effect Variegation, Heterochromatin Formation, and Gene Silencing in Drosophila. Cold Spring Harb. Perspect. Biol. 2013, 5, a017780. [Google Scholar] [CrossRef] [PubMed]
- Montavon, T.; Shukeir, N.; Erikson, G.; Engist, B.; Onishi-Seebacher, M.; Ryan, D.; Musa, Y.; Mittler, G.; Meyer, A.G.; Genoud, C.; et al. Complete loss of H3K9 methylation dissolves mouse heterochromatin organization. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Padeken, J.; Zeller, P.; Towbin, B.; Katic, I.; Kalck, V.; Methot, S.P.; Gasser, S.M. Synergistic lethality between BRCA1 and H3K9me2 loss reflects satellite derepression. Genes Dev. 2019, 33, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Penke, T.J.; McKay, D.J.; Strahl, B.D.; Matera, A.G.; Duronio, R.J. Direct interrogation of the role of H3K9 in metazoan heterochromatin function. Genes Dev. 2016, 30, 1866–1880. [Google Scholar] [CrossRef] [PubMed]
- Tachibana, M.; Sugimoto, K.; Nozaki, M.; Ueda, J.; Ohta, T.; Ohki, M.; Fukuda, M.; Takeda, N.; Niida, H.; Kato, H.; et al. G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis. Genes Dev. 2002, 16, 1779–1791. [Google Scholar] [CrossRef] [PubMed]
- Sienski, G.; Batki, J.; Senti, K.A.; Donertas, D.; Tirian, L.; Meixner, K.; Brennecke, J. Silencio/CG9754 connects the Piwi-piRNA complex to the cellular heterochromatin machinery. Genes Dev. 2015, 29, 2258–2271. [Google Scholar] [CrossRef]
- Agger, K.; Christensen, J.; Cloos, P.A.; Helin, K. The emerging functions of histone demethylases. Curr. Opin. Genet. Dev. 2008, 18, 159–168. [Google Scholar] [CrossRef]
- Cloos, P.A.; Christensen, J.; Agger, K.; Helin, K. Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 2008, 22, 1115–1140. [Google Scholar] [CrossRef]
- Nottke, A.; Colaiácovo, M.P.; Shi, Y. Developmental roles of the histone lysine demethylases. Development 2009, 136, 879–889. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.F.M.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Zeller, P.; Padeken, J.; van Schendel, R.; Kalck, V.; Tijsterman, M.; Gasser, S.M. Histone H3K9 methylation is dispensable for Caenorhabditis elegans development but suppresses RNA:DNA hybrid-associated repeat instability. Nat. Genet. 2016, 48, 1385–1395. [Google Scholar] [CrossRef]
- Jacobs, F.M.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 2014, 516, 242–245. [Google Scholar] [CrossRef]
- Ecco, G.; Cassano, M.; Kauzlaric, A.; Duc, J.; Coluccio, A.; Offner, S.; Imbeault, M.; Rowe, H.M.; Turelli, P.; Trono, D. Transposable Elements and Their KRAB-ZFP Controllers Regulate Gene Expression in Adult Tissues. Dev. Cell 2016, 36, 611–623. [Google Scholar] [CrossRef]
- Tan, X.; Xu, X.; Elkenani, M.; Smorag, L.; Zechner, U.; Nolte, J.; Engel, W.; Pantakani, D.K. Zfp819, a novel KRAB-zinc finger protein, interacts with KAP1 and functions in genomic integrity maintenance of mouse embryonic stem cells. Stem Cell Res. 2013, 11, 1045–1059. [Google Scholar] [CrossRef]
- Najafabadi, H.; Mnaimneh, S.; Schmitges, F.W.; Garton, M.; Lam, K.; Yang, A.; Albu, M.; Weirauch, M.T.; Radovani, E.; Kim, P.M.; et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat. Biotechnol. 2015, 33, 555–562. [Google Scholar] [CrossRef]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef]
- Peng, H.; Begg, G.E.; Harper, S.L.; Friedman, J.R.; Speicher, D.W.; Rauscher, F.J. Biochemical Analysis of the Kruppel-associated Box (KRAB) Transcriptional Repression Domain. J. Biol. Chem. 2000, 275, 18000–18010. [Google Scholar] [CrossRef]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., III. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef]
- Ecco, G.; Rowe, H.M.; Trono, D. A Large-Scale Functional Screen to Identify Epigenetic Repressors of Retrotransposon Expression. Transposons Retrotransposons 2016, 1400, 403–417. [Google Scholar] [CrossRef]
- Imbeault, M.; Helleboid, P.-Y.; Trono, M.I.P.-Y.H.D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017, 543, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD Domain-Mediated E3 Ligase Activity Directs Intramolecular Sumoylation of an Adjacent Bromodomain Required for Gene Silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Rowe, H.M.; Friedli, M.; Offner, S.; Verp, S.; Mesnard, D.; Marquis, J.; Aktas, T.; Trono, D. De novo DNA methylation of endogenous retroviruses is shaped by KRAB-ZFPs/KAP1 and ESET. Development 2013, 140, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Tchasovnikarova, I.A.; Timms, R.T.; Douse, C.H.; Roberts, R.C.; Dougan, G.; Kingston, R.E.; Modis, Y.; Lehner, P.J. Hyperactivation of HUSH complex function by Charcot-Marie-Tooth disease mutation in MORC2. Nat. Genet. 2017, 49, 1035–1044. [Google Scholar] [CrossRef]
- Tchasovnikarova, I.A.; Timms, R.T.; Matheson, N.J.; Wals, K.; Antrobus, R.; Göttgens, B.; Dougan, G.; Dawson, M.A.; Lehner, P.J. Epigenetic silencing by the HUSH complex mediates position-effect variegation in human cells. Science 2015, 348, 1481–1485. [Google Scholar] [CrossRef]
- Robbez-Masson, L.; Tie, C.H.C.; Conde, L.; Tunbak, H.; Husovsky, C.; Tchasovnikarova, I.A.; Timms, R.T.; Herrero, J.; Lehner, P.J.; Rowe, H.M. The HUSH complex cooperates with TRIM28 to repress young retrotransposons and new genes. Genome Res. 2018, 28, 836–845. [Google Scholar] [CrossRef]
- Seczynska, M.; Bloor, S.; Cuesta, S.M.; Lehner, P.J. Genome surveillance by HUSH-mediated silencing of intronless mobile elements. Nature 2021, 601, 440–445. [Google Scholar] [CrossRef]
- Akkouche, A.; Mugat, B.; Barckmann, B.; Varela-Chavez, C.; Li, B.; Raffel, R.; Pélisson, A.; Chambeyron, S. Piwi Is Required during Drosophila Embryogenesis to License Dual-Strand piRNA Clusters for Transposon Repression in Adult Ovaries. Mol. Cell 2017, 66, 411–419.e4. [Google Scholar] [CrossRef]
- Gu, T.; Elgin, S.C.R. Maternal Depletion of Piwi, a Component of the RNAi System, Impacts Heterochromatin Formation in Drosophila. PLoS Genet. 2013, 9, e1003780. [Google Scholar] [CrossRef]
- Yu, Y.; Gu, J.; Jin, Y.; Luo, Y.; Preall, J.B.; Ma, J.; Czech, B.; Hannon, G.J. Panoramix enforces piRNA-dependent cotranscriptional silencing. Science 2015, 350, 339–342. [Google Scholar] [CrossRef]
- Dönertas, D.; Sienski, G.; Brennecke, J. Drosophila Gtsf1 is an essential component of the Piwi-mediated transcriptional silencing complex. Genes Dev. 2013, 27, 1693–1705. [Google Scholar] [CrossRef]
- Muerdter, F.; Guzzardo, P.M.; Gillis, J.; Luo, Y.; Yu, Y.; Chen, C.; Fekete, R.; Hannon, G.J. A Genome-wide RNAi Screen Draws a Genetic Framework for Transposon Control and Primary piRNA Biogenesis in Drosophila. Mol. Cell 2013, 50, 736–748. [Google Scholar] [CrossRef]
- Ohtani, H.; Iwasaki, Y.W.; Shibuya, A.; Siomi, H.; Siomi, M.C.; Saito, K. DmGTSF1 is necessary for Piwi-piRISC-mediated transcriptional transposon silencing in the Drosophila ovary. Genes Dev. 2013, 27, 1656–1661. [Google Scholar] [CrossRef]
- Rangan, P.; Malone, C.D.; Navarro, C.; Newbold, S.P.; Hayes, P.S.; Sachidanandam, R.; Hannon, G.J.; Lehmann, R. piRNA Production Requires Heterochromatin Formation in Drosophila. Curr. Biol. 2011, 21, 1373–1379. [Google Scholar] [CrossRef]
- Osumi, K.; Sato, K.; Murano, K.; Siomi, H.; Siomi, M.C. Essential roles of Windei and nuclear monoubiquitination of Eggless/SETDB1 in transposon silencing. EMBO Rep. 2019, 20, e48296. [Google Scholar] [CrossRef]
- Andreev, V.I.; Yu, C.; Wang, J.; Schnabl, J.; Tirian, L.; Gehre, M.; Handler, D.; Duchek, P.; Novatchkova, M.; Baumgartner, L.; et al. Panoramix SUMOylation on chromatin connects the piRNA pathway to the cellular heterochromatin machinery. Nat. Struct. Mol. Biol. 2022, 29, 130–142. [Google Scholar] [CrossRef]
- Klenov, M.S.; Lavrov, S.A.; Korbut, A.P.; Stolyarenko, A.D.; Yakushev, E.Y.; Reuter, M.; Pillai, R.S.; Gvozdev, V.A. Impact of nuclear Piwi elimination on chromatin state in Drosophila melanogaster ovaries. Nucleic Acids Res. 2014, 42, 6208–6218. [Google Scholar] [CrossRef]
- Lepesant, J.M.J.; Iampietro, C.; Galeota, E.; Augé, B.; Aguirrenbengoa, M.; Mercé, C.; Chaubet, C.; Rocher, V.; Haenlin, M.; Waltzer, L.; et al. A dual role of dLsd1 in oogenesis: Regulating developmental genes and repressing transposons. Nucleic Acids Res. 2019, 48, 1206–1224. [Google Scholar] [CrossRef]
- Yang, F.; Quan, Z.; Huang, H.; He, M.; Liu, X.; Cai, T.; Xi, R. Ovaries absent links dLsd1 to HP1a for local H3K4 demethylation required for heterochromatic gene silencing. eLife 2019, 8, e40806. [Google Scholar] [CrossRef]
- Mugat, B.; Nicot, S.; Varela-Chavez, C.; Jourdan, C.; Sato, K.; Basyuk, E.; Juge, F.; Siomi, M.C.; Pelisson, A.; Chambeyron, S. The Mi-2 nucleosome remodeler and the Rpd3 histone deacetylase are involved in piRNA-guided heterochromatin formation. Nat. Commun. 2020, 11, 2818. [Google Scholar] [CrossRef]
- Zhao, K.; Cheng, S.; Miao, N.; Xu, P.; Lu, X.; Zhang, Y.; Wang, M.; Ouyang, X.; Yuan, X.; Liu, W.; et al. A Pandas complex adapted for piRNA-guided transcriptional silencing and heterochromatin formation. Nat. Cell Biol. 2019, 21, 1261–1272. [Google Scholar] [CrossRef]
- Fabry, M.H.; Ciabrelli, F.; Munafo, M.; Eastwood, E.L.; Kneuss, E.; Falciatori, I.; Falconio, F.A.; Hannon, G.J.; Czech, B. piRNA-guided co-transcriptional silencing coopts nuclear export factors. Elife 2019, 8, e47999. [Google Scholar] [CrossRef]
- Murano, K.; Iwasaki, Y.W.; Ishizu, H.; Mashiko, A.; Shibuya, A.; Kondo, S.; Adachi, S.; Suzuki, S.; Saito, K.; Natsume, T.; et al. Nuclear RNA export factor variant initiates piRNA-guided co-transcriptional silencing. EMBO J. 2019, 38, e102870. [Google Scholar] [CrossRef]
- Batki, J.; Schnabl, J.; Wang, J.; Handler, D.; Andreev, V.I.; Stieger, C.E.; Novatchkova, M.; Lampersberger, L.; Kauneckaite, K.; Xie, W.; et al. The nascent RNA binding complex SFiNX licenses piRNA-guided heterochromatin formation. Nat. Struct. Mol. Biol. 2019, 26, 720–731. [Google Scholar] [CrossRef]
- Eastwood, E.L.; Jara, K.A.; Bornelov, S.; Munafo, M.; Frantzis, V.; Kneuss, E.; Barbar, E.J.; Czech, B.; Hannon, G.J. Dimerisation of the PICTS complex via LC8/Cut-up drives co-transcriptional transposon silencing in Drosophila. Elife 2021, 10, e65557. [Google Scholar] [CrossRef]
- Schopp, T.; Zoch, A.; Berrens, R.V.; Auchynnikava, T.; Kabayama, Y.; Vasiliauskaite, L.; Rappsilber, J.; Allshire, R.C.; O’Carroll, D. TEX15 is an essential executor of MIWI2-directed transposon DNA methylation and silencing. Nat. Commun. 2020, 11, 3739. [Google Scholar] [CrossRef]
- Zoch, A.; Auchynnikava, T.; Berrens, R.V.; Kabayama, Y.; Schopp, T.; Heep, M.; Vasiliauskaite, L.; Perez-Rico, Y.A.; Cook, A.G.; Shkumatava, A.; et al. SPOCD1 is an essential executor of piRNA-directed de novo DNA methylation. Nature 2020, 584, 635–639. [Google Scholar] [CrossRef]
- Klattenhoff, C.; Xi, H.; Li, C.; Lee, S.; Xu, J.; Khurana, J.S.; Zhang, F.; Schultz, N.; Koppetsch, B.S.; Nowosielska, A.; et al. The Drosophila HP1 Homolog Rhino Is Required for Transposon Silencing and piRNA Production by Dual-Strand Clusters. Cell 2009, 138, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Mohn, F.; Sienski, G.; Handler, D.; Brennecke, J. The Rhino-Deadlock-Cutoff Complex Licenses Noncanonical Transcription of Dual-Strand piRNA Clusters in Drosophila. Cell 2014, 157, 1364–1379. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Schultz, N.; Zhang, F.; Parhad, S.S.; Tu, S.; Vreven, T.; Zamore, P.D.; Weng, Z.; Theurkauf, W.E. The HP1 Homolog Rhino Anchors a Nuclear Complex that Suppresses piRNA Precursor Splicing. Cell 2014, 157, 1353–1363. [Google Scholar] [CrossRef]
- ElMaghraby, M.F.; Andersen, P.R.; Pühringer, F.; Hohmann, U.; Meixner, K.; Lendl, T.; Tirian, L.; Brennecke, J. A Heterochromatin-Specific RNA Export Pathway Facilitates piRNA Production. Cell 2019, 178, 964–979.e20. [Google Scholar] [CrossRef] [PubMed]
- Kneuss, E.; Munafò, M.; Eastwood, E.L.; Deumer, U.-S.; Preall, J.B.; Hannon, G.J.; Czech, B. Specialization of the Drosophila nuclear export family protein Nxf3 for piRNA precursor export. Genes Dev. 2019, 33, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, T.-Y.; Lee, C.-H.; Chan, L.-W.; Shen, C.-K.J. Epigenetic regulation of the Drosophila chromosome 4 by the histone H3K9 methyltransferase dSETDB1. Proc. Natl. Acad. Sci. USA 2007, 104, 12691–12696. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Fu, X.; Zhang, M.; He, F.; Li, W.; Abdul, M.; Zhou, J.; Sun, L.; Chang, C.; Li, Y.; et al. Transposable elements are regulated by context-specific patterns of chromatin marks in mouse embryonic stem cells. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- van Mierlo, G.; Dirks, R.A.M.; De Clerck, L.; Brinkman, A.B.; Huth, M.; Kloet, S.L.; Saksouk, N.; Kroeze, L.I.; Willems, S.; Farlik, M.; et al. Integrative Proteomic Profiling Reveals PRC2-Dependent Epigenetic Crosstalk Maintains Ground-State Pluripotency. Cell Stem Cell 2019, 24, 123–137.e8. [Google Scholar] [CrossRef]
- Miller, S.A.; Damle, M.; Kim, J.; Kingston, R.E. Full methylation of H3K27 by PRC2 is dispensable for initial embryoid body formation but required to maintain differentiated cell identity. Development 2021, 148. [Google Scholar] [CrossRef]
- Jain, P.; Di Croce, L. Mutations and deletions of PRC2 in prostate cancer. BioEssays 2016, 38, 446–454. [Google Scholar] [CrossRef]
- Deleris, A.; Stroud, H.; Bernatavichute, Y.; Johnson, E.; Klein, G.; Schubert, D.; Jacobsen, S.E. Loss of the DNA Methyltransferase MET1 Induces H3K9 Hypermethylation at PcG Target Genes and Redistribution of H3K27 Trimethylation to Transposons in Arabidopsis thaliana. PLoS Genet. 2012, 8, e1003062. [Google Scholar] [CrossRef]
- Déléris, A.; Berger, F.; Duharcourt, S. Role of Polycomb in the control of transposable elements. Trends Genet. 2021, 37, 882–889. [Google Scholar] [CrossRef]
- Frapporti, A.; Pina, C.M.; Arnaiz, O.; Holoch, D.; Kawaguchi, T.; Humbert, A.; Eleftheriou, E.; Lombard, B.; Loew, D.; Sperling, L.; et al. The Polycomb protein Ezl1 mediates H3K9 and H3K27 methylation to repress transposable elements in Paramecium. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef]
- Miró-Pina, C.; Charmant, O.; Kawaguchi, T.; Holoch, D.; Michaud, A.; Cohen, I.; Humbert, A.; Jaszczyszyn, Y.; Chevreux, G.; Del Maestro, L.; et al. Paramecium Polycomb repressive complex 2 physically interacts with the small RNA-binding PIWI protein to repress transposable elements. Dev. Cell 2022, 57, 1037–1052.e8. [Google Scholar] [CrossRef]
- Leeb, M.; Pasini, D.; Novatchkova, M.; Jaritz, M.; Helin, K.; Wutz, A. Polycomb complexes act redundantly to repress genomic repeats and genes. Genes Dev. 2010, 24, 265–276. [Google Scholar] [CrossRef]
- Ozturk, N.; Dansranjavin, T.; Gies, S.; Calay, D.; Shiplu, S.; Creppe, C.; Hendrickx, J.; Schagdarsurengin, U. H4K20me3 marks distal intergenic and repetitive regions in human mature spermatozoa. Development 2021, 148. [Google Scholar] [CrossRef]
- Kurup, J.T.; Han, Z.; Jin, W.; Kidder, B.L. H4K20me3 methyltransferase SUV420H2 shapes the chromatin landscape of pluripotent embryonic stem cells. Development 2020. [Google Scholar] [CrossRef]
- Schotta, G.; Sengupta, R.; Kubicek, S.; Malin, S.; Kauer, M.; Callén, E.; Celeste, A.; Pagani, M.; Opravil, S.; De La Rosa-Velazquez, I.A.; et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008, 22, 2048–2061. [Google Scholar] [CrossRef]
- Ren, W.; Fan, H.; Grimm, S.A.; Kim, J.J.; Li, L.; Guo, Y.; Petell, C.J.; Tan, X.-F.; Zhang, Z.-M.; Coan, J.P.; et al. DNMT1 reads heterochromatic H4K20me3 to reinforce LINE-1 DNA methylation. Nat. Commun. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- Schotta, G.; Lachner, M.; Sarma, K.; Ebert, A.; Sengupta, R.; Reuter, G.; Reinberg, D.; Jenuwein, T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004, 18, 1251–1262. [Google Scholar] [CrossRef]
- Kourmouli, N.; Jeppesen, P.; Mahadevhaiah, S.; Burgoyne, P.; Wu, R.; Gilbert, D.M.; Bongiorni, S.; Prantera, G.; Fanti, L.; Pimpinelli, S.; et al. Heterochromatin and tri-methylated lysine 20 of histone H4 in animals. J. Cell Sci. 2004, 117 Pt 12, 2491–2501. [Google Scholar] [CrossRef]
- van Kruijsbergen, I.; Hontelez, S.; Elurbe, D.M.; van Heeringen, S.J.; Huynen, M.A.; Veenstra, G.J.C. Heterochromatic histone modifications at transposons in Xenopus tropicalis embryos. Dev. Biol. 2017, 426, 460–471. [Google Scholar] [CrossRef]
- Girardot, M.; Hirasawa, R.; Kacem, S.; Fritsch, L.; Pontis, J.; Kota, S.K.; Filipponi, D.; Fabbrizio, E.; Sardet, C.; Lohmann, F.; et al. PRMT5-mediated histone H4 arginine-3 symmetrical dimethylation marks chromatin at G + C-rich regions of the mouse genome. Nucleic Acids Res. 2014, 42, 235–248. [Google Scholar] [CrossRef]
- Kim, S.; Günesdogan, U.; Zylicz, J.J.; Hackett, J.A.; Cougot, D.; Bao, S.; Lee, C.; Dietmann, S.; Allen, G.E.; Sengupta, R.; et al. PRMT5 Protects Genomic Integrity during Global DNA Demethylation in Primordial Germ Cells and Preimplantation Embryos. Mol. Cell 2014, 56, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.-Y.; Hochstrasser, M. Histone sumoylation and chromatin dynamics. Nucleic Acids Res. 2021, 49, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Maison, C.; Bailly, D.; Quivy, J.-P.; Almouzni, G. The methyltransferase Suv39h1 links the SUMO pathway to HP1α marking at pericentric heterochromatin. Nat. Commun. 2016, 7, 12224. [Google Scholar] [CrossRef]
- Maison, C.; Bailly, D.; Roche, D.; de Oca, R.M.; Probst, A.V.; Vassias, I.; Dingli, F.; Lombard, B.; Loew, D.; Quivy, J.-P.; et al. SUMOylation promotes de novo targeting of HP1α to pericentric heterochromatin. Nat. Genet. 2011, 43, 220–227. [Google Scholar] [CrossRef]
- Shin, J.A.; Choi, E.S.; Kim, H.S.; Ho, J.C.; Watts, F.Z.; Park, S.D.; Jang, Y.K. SUMO Modification Is Involved in the Maintenance of Heterochromatin Stability in Fission Yeast. Mol. Cell 2005, 19, 817–828. [Google Scholar] [CrossRef]
- Stielow, B.; Sapetschnig, A.; Wink, C.; Krüger, I.; Suske, G. SUMO-modified Sp3 represses transcription by provoking local heterochromatic gene silencing. EMBO Rep. 2008, 9, 899–906. [Google Scholar] [CrossRef]
- Thompson, P.J.; Dulberg, V.; Moon, K.-M.; Foster, L.J.; Chen, C.; Karimi, M.M.; Lorincz, M.C. hnRNP K Coordinates Transcriptional Silencing by SETDB1 in Embryonic Stem Cells. PLoS Genet. 2015, 11, e1004933. [Google Scholar] [CrossRef]
- Uchimura, Y.; Ichimura, T.; Uwada, J.; Tachibana, T.; Sugahara, S.; Nakao, M.; Saitoh, H. Involvement of SUMO Modification in MBD1- and MCAF1-mediated Heterochromatin Formation. J. Biol. Chem. 2006, 281, 23180–23190. [Google Scholar] [CrossRef]
- Psakhye, I.; Jentsch, S. Protein Group Modification and Synergy in the SUMO Pathway as Exemplified in DNA Repair. Cell 2012, 151, 807–820. [Google Scholar] [CrossRef]
- Efroni, S.; Duttagupta, R.; Cheng, J.; Dehghani, H.; Hoeppner, D.J.; Dash, C.; Bazett-Jones, D.P.; Le Grice, S.; McKay, R.D.; Buetow, K.H.; et al. Global Transcription in Pluripotent Embryonic Stem Cells. Cell Stem Cell 2008, 2, 437–447. [Google Scholar] [CrossRef]
- Jachowicz, J.; Bing, X.; Pontabry, J.; Bošković, A.; Rando, O.J.; Torres-Padilla, M.-E. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat. Genet. 2017, 49, 1502–1510. [Google Scholar] [CrossRef]
- Baillie, J.K.; Barnett, M.W.; Upton, K.R.; Gerhardt, D.J.; Richmond, T.A.; De Sapio, F.; Brennan, P.M.; Rizzu, P.; Smith, S.; Fell, M.; et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature 2011, 479, 534–537. [Google Scholar] [CrossRef]
- Perrat, P.N.; DasGupta, S.; Wang, J.; Theurkauf, W.; Weng, Z.; Rosbash, M.; Waddell, S. Transposition-Driven Genomic Heterogeneity in the Drosophila Brain. Science 2013, 340, 91–95. [Google Scholar] [CrossRef]
- Tam, O.H.; Ostrow, L.; Hammell, M.G. Diseases of the nERVous system: Retrotransposon activity in neurodegenerative disease. Mob. DNA 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Jang, H.S.; Shah, N.; Du, A.; Dailey, Z.Z.; Pehrsson, E.; Godoy, P.M.; Zhang, D.; Li, D.; Xing, X.; Kim, S.; et al. Transposable elements drive widespread expression of oncogenes in human cancers. Nat. Genet. 2019, 51, 611–617. [Google Scholar] [CrossRef]
- Chuong, E.; Elde, N.C.; Feschotte, E.B.C.N.C.E.C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2016, 18, 71–86. [Google Scholar] [CrossRef]
- Pehrsson, E.C.; Choudhary, M.N.K.; Sundaram, V.; Wang, T. The epigenomic landscape of transposable elements across normal human development and anatomy. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Fadloun, A.; Le Gras, S.; Jost, B.; Ziegler-Birling, C.; Takahashi, H.; Gorab, E.; Carninci, P.; Torres-Padilla, M.-E. Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat. Struct. Mol. Biol. 2013, 20, 332–338. [Google Scholar] [CrossRef]
- Sze, C.C.; Shilatifard, A. MLL3/MLL4/COMPASS Family on Epigenetic Regulation of Enhancer Function and Cancer. Cold Spring Harb. Perspect. Med. 2016, 6, a026427. [Google Scholar] [CrossRef]
- Macfarlan, T.S.; Gifford, W.D.; Agarwal, S.; Driscoll, S.; Lettieri, K.; Wang, J.; Andrews, S.E.; Franco, L.; Rosenfeld, M.G.; Ren, B.; et al. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev. 2011, 25, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Ancelin, K.; Syx, L.; Borensztein, M.; Ranisavljevic, N.; Vassilev, I.; Briseno-Roa, L.; Liu, T.; Metzger, E.; Servant, N.; Barillot, E.; et al. Maternal LSD1/KDM1A is an essential regulator of chromatin and transcription landscapes during zygotic genome activation. eLife 2016, 5, e08851. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Shen, Y.; Wang, Y.; Wang, X.; Francisco, J.C.; Luo, Z.; Lin, C. Striking a balance: Regulation of transposable elements by Zfp281 and Mll2 in mouse embryonic stem cells. Nucleic Acids Res. 2017, 45, 12301–12310. [Google Scholar] [CrossRef] [PubMed]
- Chaouch, A.; Berlandi, J.; Chen, C.C.; Frey, F.; Badini, S.; Harutyunyan, A.S.; Chen, X.; Krug, B.; Hébert, S.; Jeibmann, A.; et al. Histone H3.3 K27M and K36M mutations de-repress transposable elements through perturbation of antagonistic chromatin marks. Mol. Cell 2021, 81, 4876–4890.e7. [Google Scholar] [CrossRef]
- Ahmad, K.; Henikoff, S. Histone H3 variants specify modes of chromatin assembly. Proc. Natl. Acad. Sci. USA 2002, 99, 16477–16484. [Google Scholar] [CrossRef]
- Elsässer, S.J.; Noh, K.-M.; Diaz, N.; Allis, C.D.; Banaszynski, L.A. Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 2015, 522, 240–244. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, X.; He, K.; Charron, J.-B.F.; Elling, A.A.; Deng, X.W. Genome-wide profiling of histone H3 lysine 9 acetylation and dimethylation in Arabidopsis reveals correlation between multiple histone marks and gene expression. Plant Mol. Biol. 2010, 72, 585–595. [Google Scholar] [CrossRef]
- Yang, J.; Yuan, L.; Yen, M.R.; Zheng, F.; Ji, R.; Peng, T.; Gu, D.; Yang, S.; Cui, Y.; Chen, P.Y.; et al. SWI3B and HDA6 interact and are required for transposon silencing in Arabidopsis. Plant. J. 2020, 102, 809–822. [Google Scholar] [CrossRef]
- Liu, X.; Yu, C.-W.; Duan, J.; Luo, M.; Wang, K.; Tian, G.; Cui, Y.; Wu, K. HDA6 Directly Interacts with DNA Methyltransferase MET1 and Maintains Transposable Element Silencing in Arabidopsis. Plant Physiol. 2011, 158, 119–129. [Google Scholar] [CrossRef]
- Lennartsson, A.; Arner, E.; Fagiolini, M.; Saxena, A.; Andersson, R.; Takahashi, H.; Noro, Y.; Sng, J.; Sandelin, A.; Hensch, T.K.; et al. Remodeling of retrotransposon elements during epigenetic induction of adult visual cortical plasticity by HDAC inhibitors. Epigenet. Chromatin 2015, 8, 1–15. [Google Scholar] [CrossRef]
- Szabo, Q.; Bantignies, F.; Cavalli, G. Principles of genome folding into topologically associating domains. Sci. Adv. 2019, 5, eaaw1668. [Google Scholar] [CrossRef]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef]
- Iwasaki, Y.W.; Sriswasdi, S.; Kinugasa, Y.; Adachi, J.; Horikoshi, Y.; Shibuya, A.; Iwasaki, W.; Tashiro, S.; Tomonaga, T.; Siomi, H. Piwi-piRNA complexes induce stepwise changes in nuclear architecture at target loci. EMBO J. 2021, 40, e108345. [Google Scholar] [CrossRef]
- Choudhary, M.N.; Friedman, R.Z.; Wang, J.T.; Jang, H.S.; Zhuo, X.; Wang, T. Co-opted transposons help perpetuate conserved higher-order chromosomal structures. Genome Biol. 2020, 21, 16. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef]
- Liu, J.; Dou, X.; Chen, C.; Chen, C.; Liu, C.; Xu, M.M.; Zhao, S.; Shen, B.; Gao, Y.; Han, D.; et al. N6 -methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 2020, 367, 580–586. [Google Scholar] [CrossRef]
- Shi, H.; Wei, J.; He, C. Where, When, and How: Context-Dependent Functions of RNA Methylation Writers, Readers, and Erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef]
- Bokar, J.A.; Shambaugh, M.E.; Polayes, D.; Matera, A.G.; Rottman, F.M. Purification and cDNA cloning of the AdoMet-binding subunit of the human mRNA (N6-adenosine)-methyltransferase. RNA 1997, 3, 1233–1247. [Google Scholar]
- Liu, J.; Yue, Y.; Han, D.; Wang, X.; Fu, Y.; Zhang, L.; Jia, G.; Yu, M.; Lu, Z.; Deng, X.; et al. A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2013, 10, 93–95. [Google Scholar] [CrossRef]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O.V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 snRNA m 6 A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824–835.e14. [Google Scholar] [CrossRef]
- Ma, H.; Wang, X.; Cai, J.; Dai, Q.; Natchiar, S.K.; Lv, R.; Chen, K.; Lu, Z.; Chen, H.; Shi, Y.G.; et al. N6-Methyladenosine methyltransferase ZCCHC4 mediates ribosomal RNA methylation. Nat. Chem. Biol. 2018, 15, 88–94. [Google Scholar] [CrossRef]
- Fu, Y.; Jia, G.; Pang, X.; Wang, R.N.; Wang, X.; Li, C.J.; Smemo, S.; Dai, Q.; Bailey, K.A.; Nobrega, M.A.; et al. FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat. Commun. 2013, 4, 1–8. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887, Erratum in Nat. Chem. Biol. 2012, 8, 1008. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Jang, K.H.; Heras, C.R.; Lee, G. m(6)A in the Signal Transduction Network. Mol. Cells 2022, 45, 435–443. [Google Scholar] [CrossRef]
- Xu, W.; Li, J.; He, C.; Wen, J.; Ma, H.; Rong, B.; Diao, J.; Wang, L.; Wang, J.; Wu, F.; et al. METTL3 regulates heterochromatin in mouse embryonic stem cells. Nature 2021, 591, 317–321. [Google Scholar] [CrossRef]
- Chelmicki, T.; Roger, E.; Teissandier, A.; Dura, M.; Bonneville, L.; Rucli, S.; Dossin, F.; Fouassier, C.; Lameiras, S.; Bourc’His, D. m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 2021, 591, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gao, M.; He, J.; Wu, K.; Lin, S.; Jin, L.; Chen, Y.; Liu, H.; Shi, J.; Wang, X.; et al. The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 2021, 591, 322–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, W.; Guo, J.; Liu, Y.; Liu, X.; Liu, J.; Dou, X.; Le, R.; Huang, Y.; Li, C.; et al. Correction to: Nuclear m6A reader YTHDC1 regulates the scaffold function of LINE1 RNA in mouse ESCs and early embryos. Protein Cell 2021, 13, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Lerat, E.; Casacuberta, J.; Chaparro, C.; Vieira, C. On the Importance to Acknowledge Transposable Elements in Epigenomic Analyses. Genes 2019, 10, 258. [Google Scholar] [CrossRef]
- He, J.; Babarinde, I.A.; Sun, L.; Xu, S.; Chen, R.; Shi, J.; Wei, Y.; Li, Y.; Ma, G.; Zhuang, Q.; et al. Identifying transposable element expression dynamics and heterogeneity during development at the single-cell level with a processing pipeline scTE. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Shao, W.; Wang, T. Transcript assembly improves expression quantification of transposable elements in single-cell RNA-seq data. Genome Res. 2020, 31, 88–100. [Google Scholar] [CrossRef]
- Anwar, S.L.; Wulaningsih, W.; Lehmann, U. Transposable Elements in Human Cancer: Causes and Consequences of Deregulation. Int. J. Mol. Sci. 2017, 18, 974. [Google Scholar] [CrossRef]
- Scott, E.C.; Gardner, E.; Masood, A.; Chuang, N.T.; Vertino, P.M.; Devine, S.E. A hot L1 retrotransposon evades somatic repression and initiates human colorectal cancer. Genome Res. 2016, 26, 745–755. [Google Scholar] [CrossRef]
- Miki, Y.; Nishisho, I.; Horii, A.; Miyoshi, Y.; Utsunomiya, J.; Kinzler, K.W.; Vogelstein, B.; Nakamura, Y. Disruption of the APC gene by a retrotransposal insertion of L1 sequence in a colon cancer. Cancer Res. 1992, 52. [Google Scholar]
- Cruickshanks, H.A.; Vafadar-Isfahani, N.; Dunican, D.S.; Lee, A.; Sproul, D.; Lund, J.; Meehan, R.R.; Tufarelli, C. Expression of a large LINE-1-driven antisense RNA is linked to epigenetic silencing of the metastasis suppressor gene TFPI-2 in cancer. Nucleic Acids Res. 2013, 41, 6857–6869. [Google Scholar] [CrossRef]
- Erwin, J.; Marchetto, M.C.; Gage, F.H. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat. Rev. Neurosci. 2014, 15, 497–506. [Google Scholar] [CrossRef]
- Ravel-Godreuil, C.; Znaidi, R.; Bonnifet, T.; Joshi, R.L.; Fuchs, J. Transposable elements as new players in neurodegenerative diseases. FEBS Lett. 2021, 595, 2733–2755. [Google Scholar] [CrossRef]
- Wood, J.G.; Jones, B.C.; Jiang, N.; Chang, C.; Hosier, S.; Wickremesinghe, P.; Garcia, M.; Hartnett, D.A.; Burhenn, L.; Neretti, N.; et al. Chromatin-modifying genetic interventions suppress age-associated transposable element activation and extend life span in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, 11277–11282. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Stefano, L. All Quiet on the TE Front? The Role of Chromatin in Transposable Element Silencing. Cells 2022, 11, 2501. https://doi.org/10.3390/cells11162501
Di Stefano L. All Quiet on the TE Front? The Role of Chromatin in Transposable Element Silencing. Cells. 2022; 11(16):2501. https://doi.org/10.3390/cells11162501
Chicago/Turabian StyleDi Stefano, Luisa. 2022. "All Quiet on the TE Front? The Role of Chromatin in Transposable Element Silencing" Cells 11, no. 16: 2501. https://doi.org/10.3390/cells11162501
APA StyleDi Stefano, L. (2022). All Quiet on the TE Front? The Role of Chromatin in Transposable Element Silencing. Cells, 11(16), 2501. https://doi.org/10.3390/cells11162501