
PARA: A New Platform for the Rapid Assembly of gRNA Arrays for Multiplexed CRISPR Technologies

 ,
,  , and
, and 
Abstract
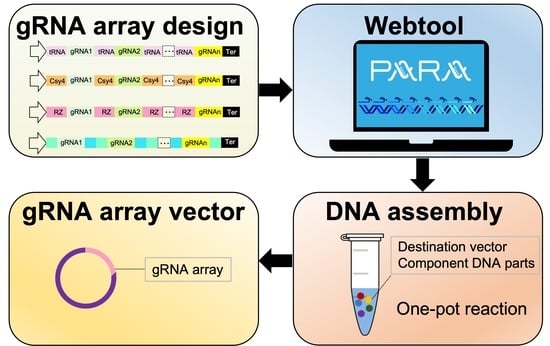
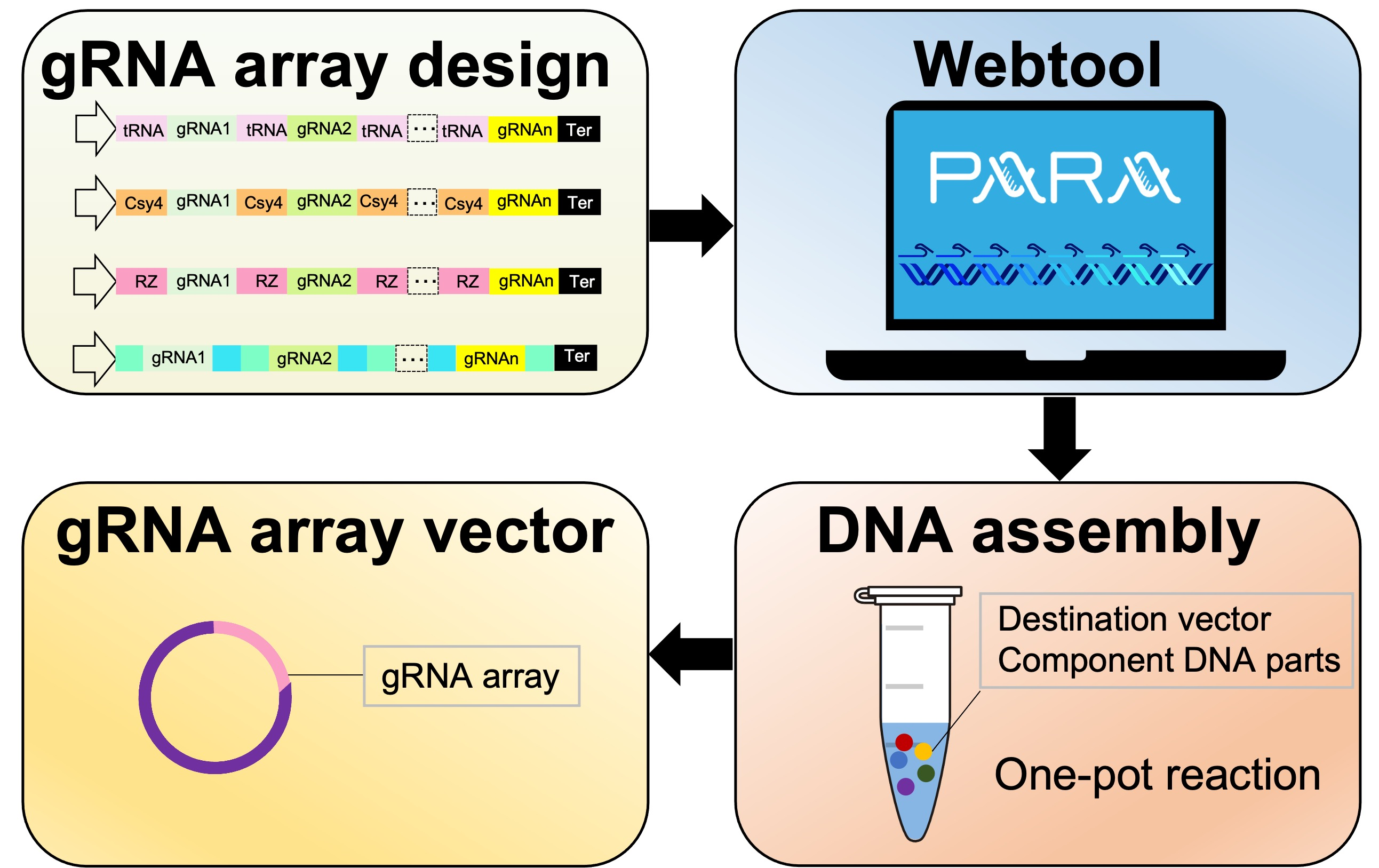
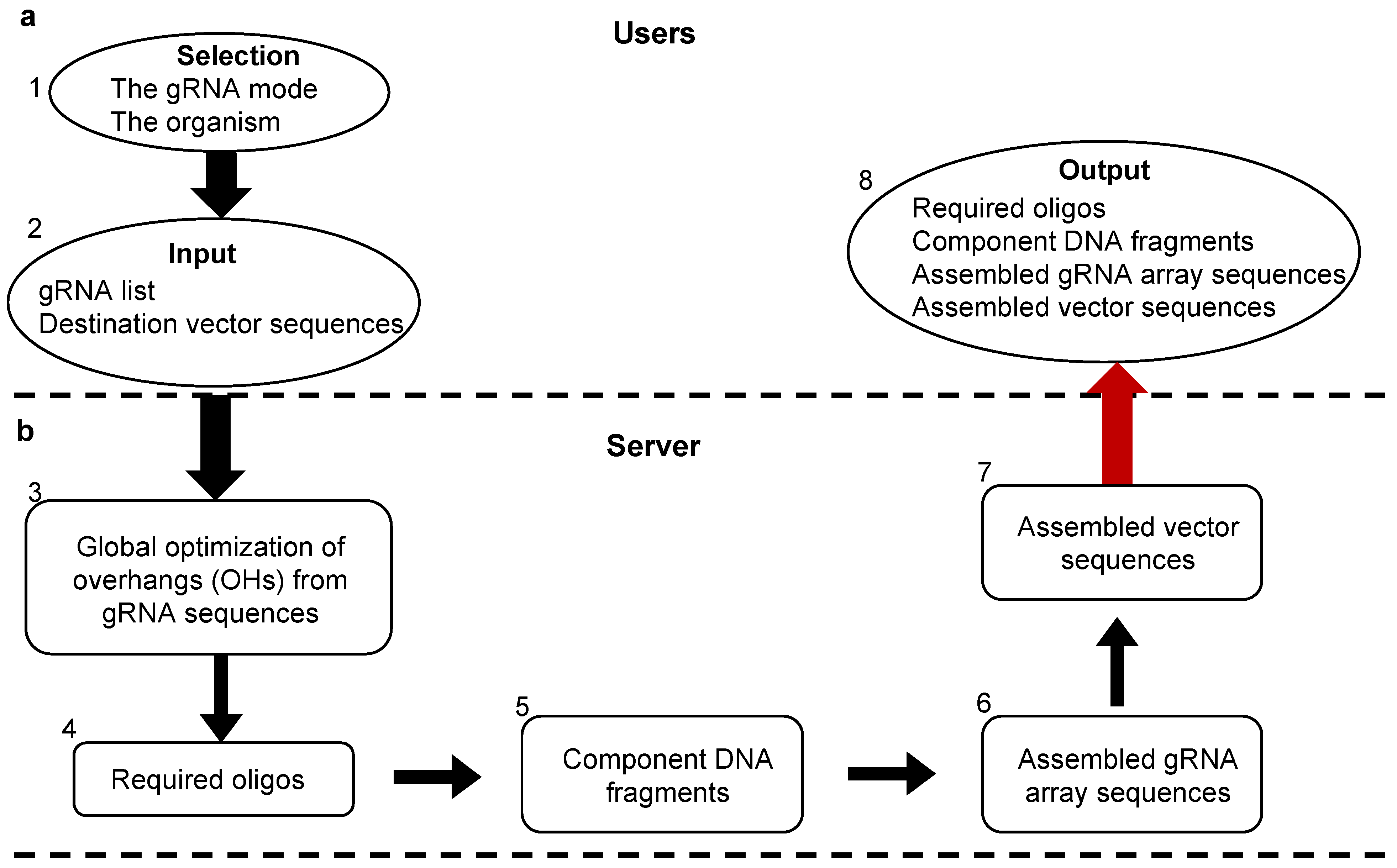
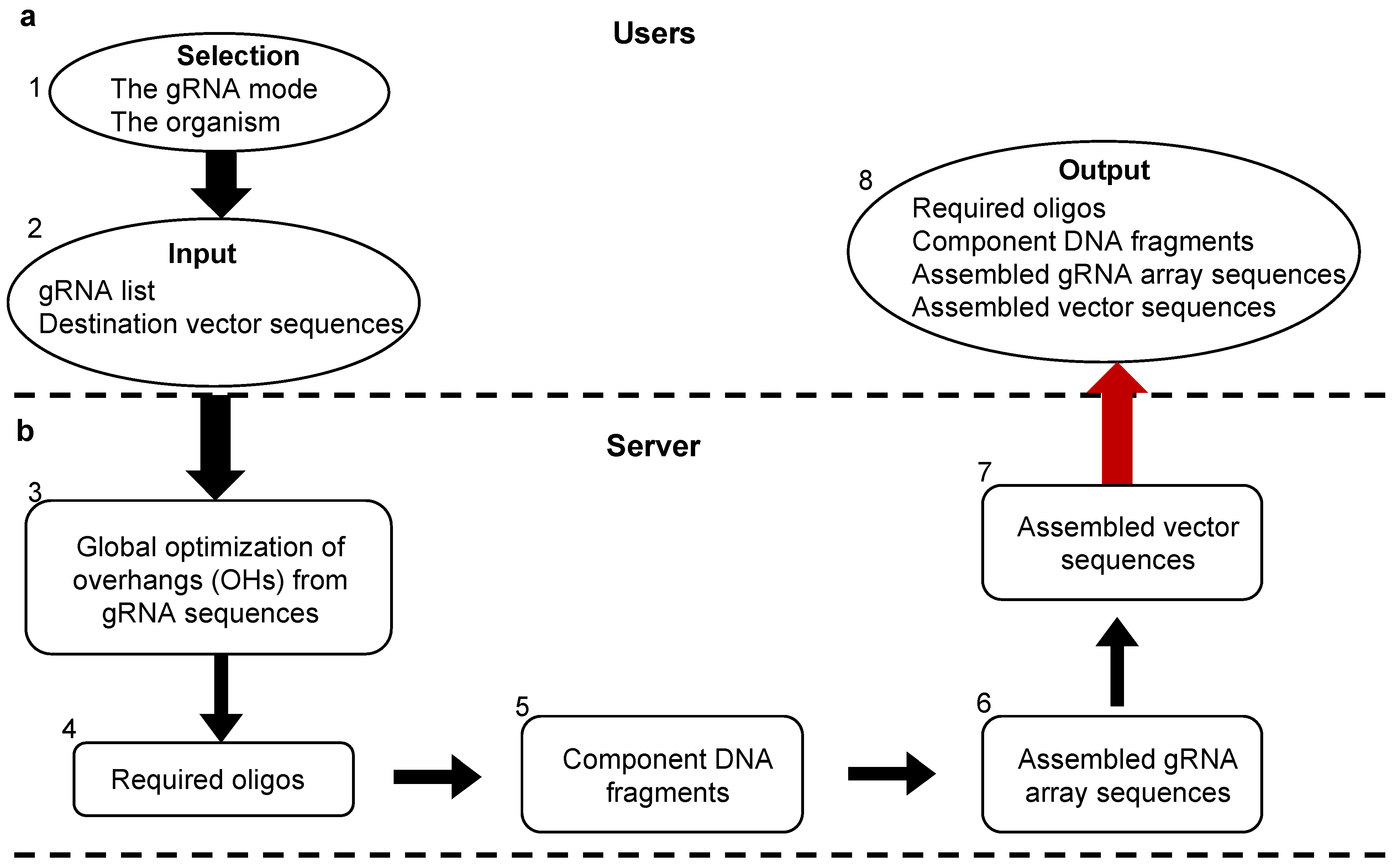
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
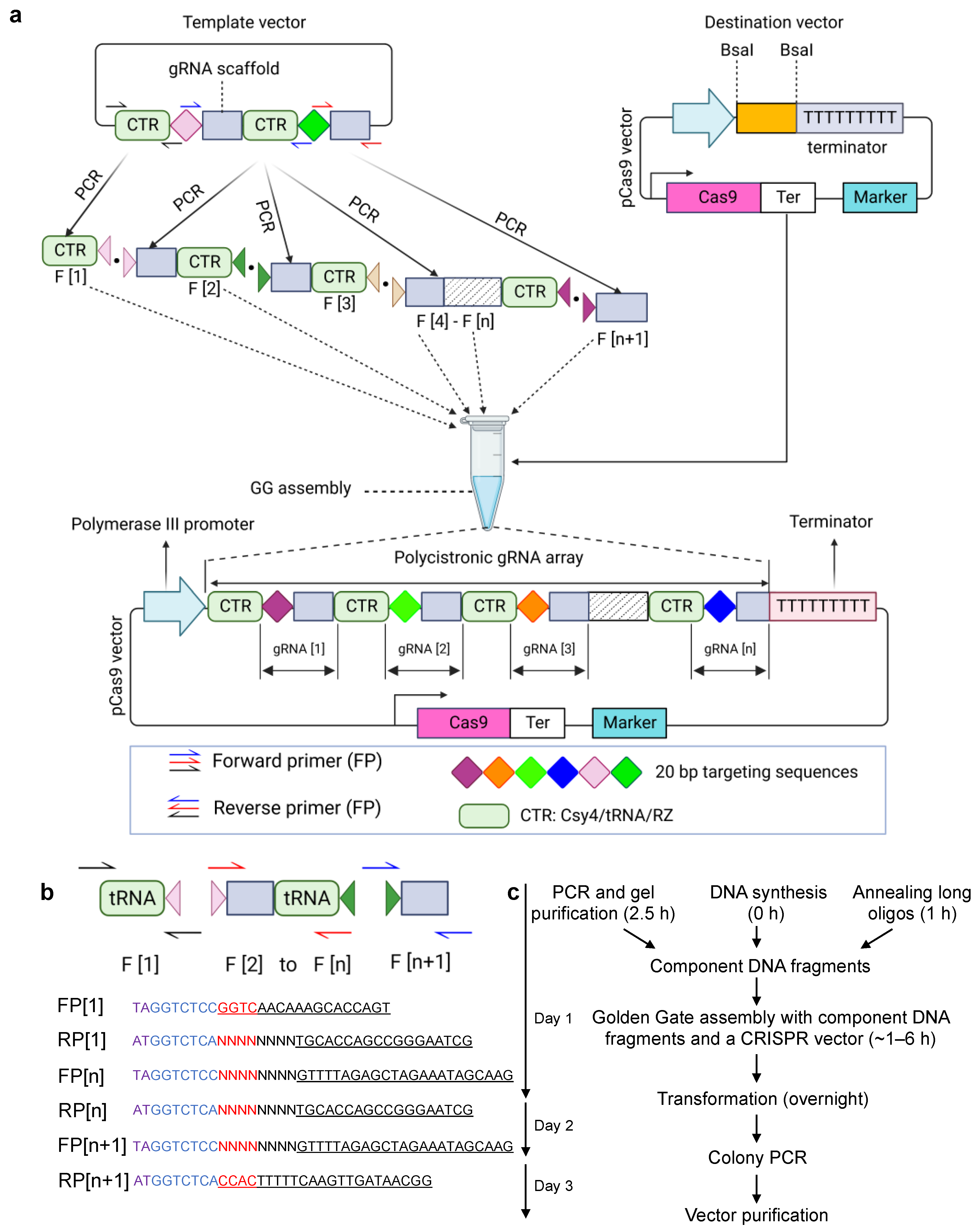
2. Materials and Methods
2.1. PCR-Based Cloning
2.2. Colony PCR
2.3. Restriction Digest of Plasmid DNA
2.4. Gel Purification
2.5. Golden Gate Assembly
2.6. Plasmid Sequencing
2.7. E. coli Transformation
2.8. Plasmid Isolation
2.9. Oligos Annealing
2.10. Vector Cloning
2.11. Web Tool Design
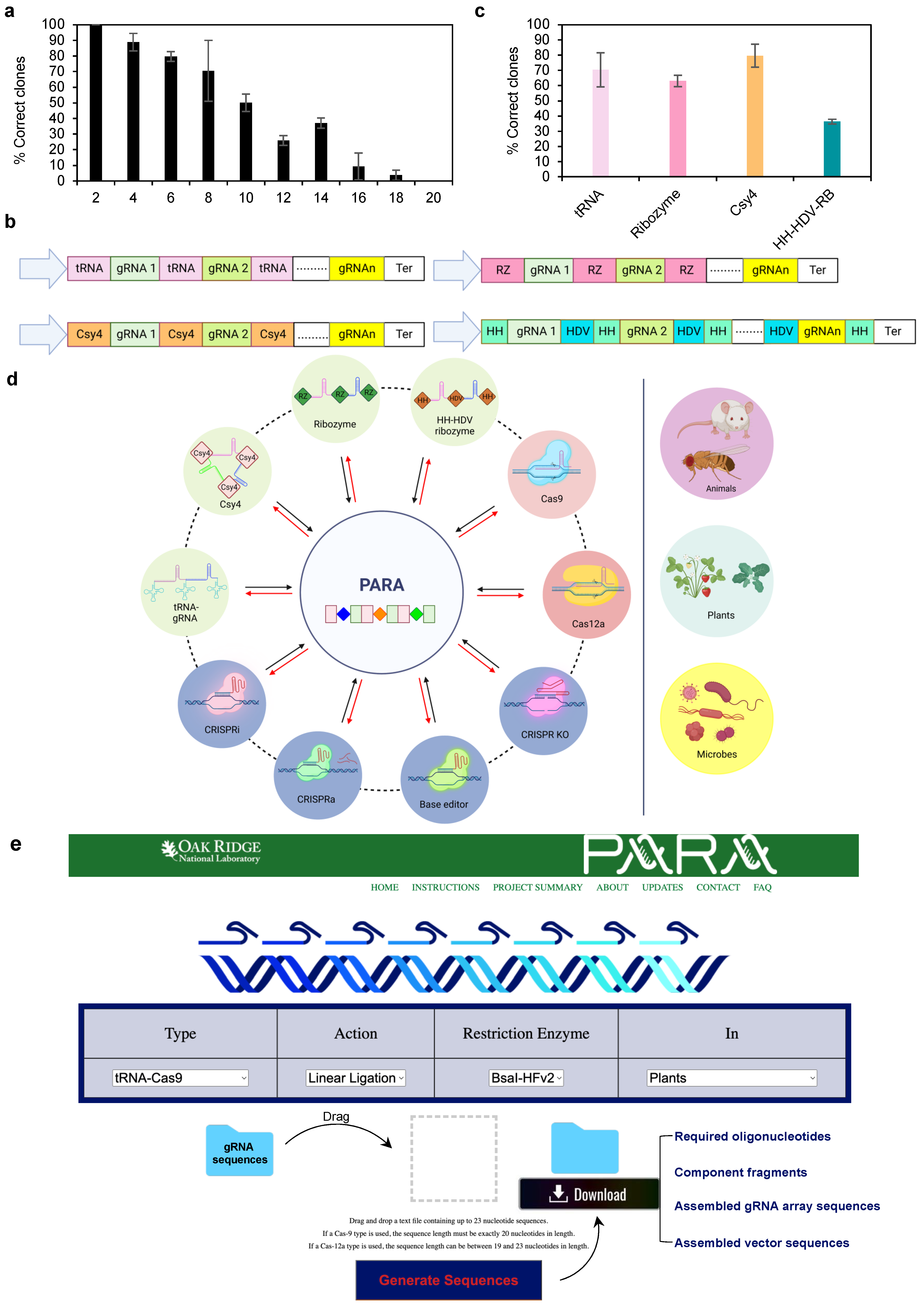
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCarty, N.S.; Graham, A.E.; Studena, L.; Ledesma-Amaro, R. Multiplexed CRISPR technologies for gene editing and transcriptional regulation. Nat. Commun. 2020, 11, 1281. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Zhang, Y.; Yuan, G.; De, K.; Chen, J.-G.; Muchero, W.; Tuskan, G.A.; Qi, Y.; Yang, X. Construct design for CRISPR/Cas-based genome editing in plants. Trends Plant Sci. 2021, 26, 1133–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Wang, J.; Wang, Z.B.; Zhang, Y.M.; Shi, S.B.; Nielsen, J.; Liu, Z.H. A gRNA-tRNA array for CRISPR-Cas9 based rapid multiplexed genome editing in Saccharomyces cerevisiae. Nat. Commun. 2019, 10, 1053. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Bullock, S.L. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat. Methods 2016, 13, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Curtin, S.J.; Gil-Humanes, J.; Cegan, R.; Kono, T.J.Y.; Konecna, E.; Belanto, J.J.; Starker, C.G.; Mathre, J.W.; Greenstein, R.L.; et al. A Multipurpose toolkit to enable advanced genome engineering in plants. Plant Cell 2017, 29, 1196–1217. [Google Scholar] [CrossRef] [PubMed]
- Potapov, V.; Ong, J.L.; Kucera, R.B.; Langhorst, B.W.; Bilotti, K.; Pryor, J.M.; Cantor, E.J.; Canton, B.; Knight, T.F.; Evans, T.C.; et al. Comprehensive profiling of four base overhang ligation fidelity by T4 DNA ligase and application to DNA assembly. ACS Synth. Biol. 2018, 7, 2665–2674. [Google Scholar] [CrossRef] [PubMed]
- Pryor, J.M.; Potapov, V.; Kucera, R.B.; Bilotti, K.; Cantor, E.J.; Lohman, G.J.S. Enabling one-pot Golden Gate assemblies of unprecedented complexity using data-optimized assembly design. PLoS ONE 2020, 15, e0238592. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.B.; Minkenberg, B.; Yang, Y.N. Boosting CRISPR/Cas9 multiplex editing capability with the endogenous tRNA-processing system. Proc. Natl. Acad. Sci. USA 2015, 112, 3570–3575. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Ren, Q.R.; Tang, X.; Liu, S.S.; Malzahn, A.A.; Zhou, J.P.; Wang, J.H.; Yin, D.S.; Pan, C.T.; Yuan, M.Z.; et al. Expanding the scope of plant genome engineering with Cas12a orthologs and highly multiplexable editing systems. Nat. Commun. 2021, 12, 1994. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.; Danzer, C.; Rechsteiner, M.; Lehmann, H.; Brandt, L.P.; Hejhal, T.; Catalano, A.; Busenhart, P.; Gonçalves, A.F.; Brandt, S.; et al. A versatile modular vector system for rapid combinatorial mammalian genetics. J. Clin. Investig. 2015, 125, 1603–1619. [Google Scholar] [CrossRef] [PubMed]
- Kabadi, A.M.; Ousterout, D.G.; Hilton, I.B.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014, 42, e147. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Nishikawa, A.; Kume, S.; Chayama, K.; Yamamoto, T. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci. Rep. 2014, 4, 5400. [Google Scholar] [CrossRef] [PubMed]
- Goodstein, D.M.; Shu, S.; Howson, R.; Neupane, R.; Hayes, R.D.; Fazo, J.; Mitros, T.; Dirks, W.; Hellsten, U.; Putnam, N.; et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 2011, 40, D1178–D1186. [Google Scholar] [CrossRef] [PubMed]
- Labun, K.; Montague, T.G.; Krause, M.; Torres Cleuren, Y.N.; Tjeldnes, H.; Valen, E. CHOPCHOP v3: Expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019, 47, W171–W174. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.-L.; Dong, L.; Wang, Z.-P.; Zhang, H.-Y.; Han, C.-Y.; Liu, B.; Wang, X.-C.; Chen, Q.-J. A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol. 2014, 14, 327. [Google Scholar] [CrossRef] [PubMed]
- Wyvekens, N.; Topkar, V.V.; Khayter, C.; Joung, J.K.; Tsai, S.Q. Dimeric CRISPR RNA-guided FokI-dCas9 nucleases directed by truncated gRNAs for highly specific genome editing. Hum. Gene Ther. 2015, 26, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.B.; Zhao, Y.D. Self- processing of ribozyme- flanked RNAs into guide RNAs in vitro and in vivo for CRISPR- mediated genome editing. J. Integr. Plant Biol. 2014, 56, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Skrekas, C.; Nielsen, J.; David, F. Multiplexed CRISPR/Cas9 genome editing and gene regulation using Csy4 in saccharomyces cerevisiae. ACS Synth. Biol. 2018, 7, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Kurata, M.; Wolf, N.K.; Lahr, W.S.; Weg, M.T.; Kluesner, M.G.; Lee, S.; Hui, K.; Shiraiwa, M.; Webber, B.R.; Moriarity, B.S. Highly multiplexed genome engineering using CRISPR/Cas9 gRNA arrays. PLoS ONE 2018, 13, e019871. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, G.; Martin, S.; Hassan, M.M.; Tuskan, G.A.; Yang, X. PARA: A New Platform for the Rapid Assembly of gRNA Arrays for Multiplexed CRISPR Technologies. Cells 2022, 11, 2467. https://doi.org/10.3390/cells11162467
Yuan G, Martin S, Hassan MM, Tuskan GA, Yang X. PARA: A New Platform for the Rapid Assembly of gRNA Arrays for Multiplexed CRISPR Technologies. Cells. 2022; 11(16):2467. https://doi.org/10.3390/cells11162467
Chicago/Turabian StyleYuan, Guoliang, Stanton Martin, Md Mahmudul Hassan, Gerald A. Tuskan, and Xiaohan Yang. 2022. "PARA: A New Platform for the Rapid Assembly of gRNA Arrays for Multiplexed CRISPR Technologies" Cells 11, no. 16: 2467. https://doi.org/10.3390/cells11162467
APA StyleYuan, G., Martin, S., Hassan, M. M., Tuskan, G. A., & Yang, X. (2022). PARA: A New Platform for the Rapid Assembly of gRNA Arrays for Multiplexed CRISPR Technologies. Cells, 11(16), 2467. https://doi.org/10.3390/cells11162467