
Genome Editing Approaches with CRISPR/Cas9 for Cancer Treatment: Critical Appraisal of Preclinical and Clinical Utility, Challenges, and Future Research


 , , and
, , and 
Abstract
1. Introduction
2. From Bacteria Defense System to Mammalian Genome Engineering
3. Approaches for Therapeutic Genome Editing in Human Malignant Cells Using CRISPR/Cas9
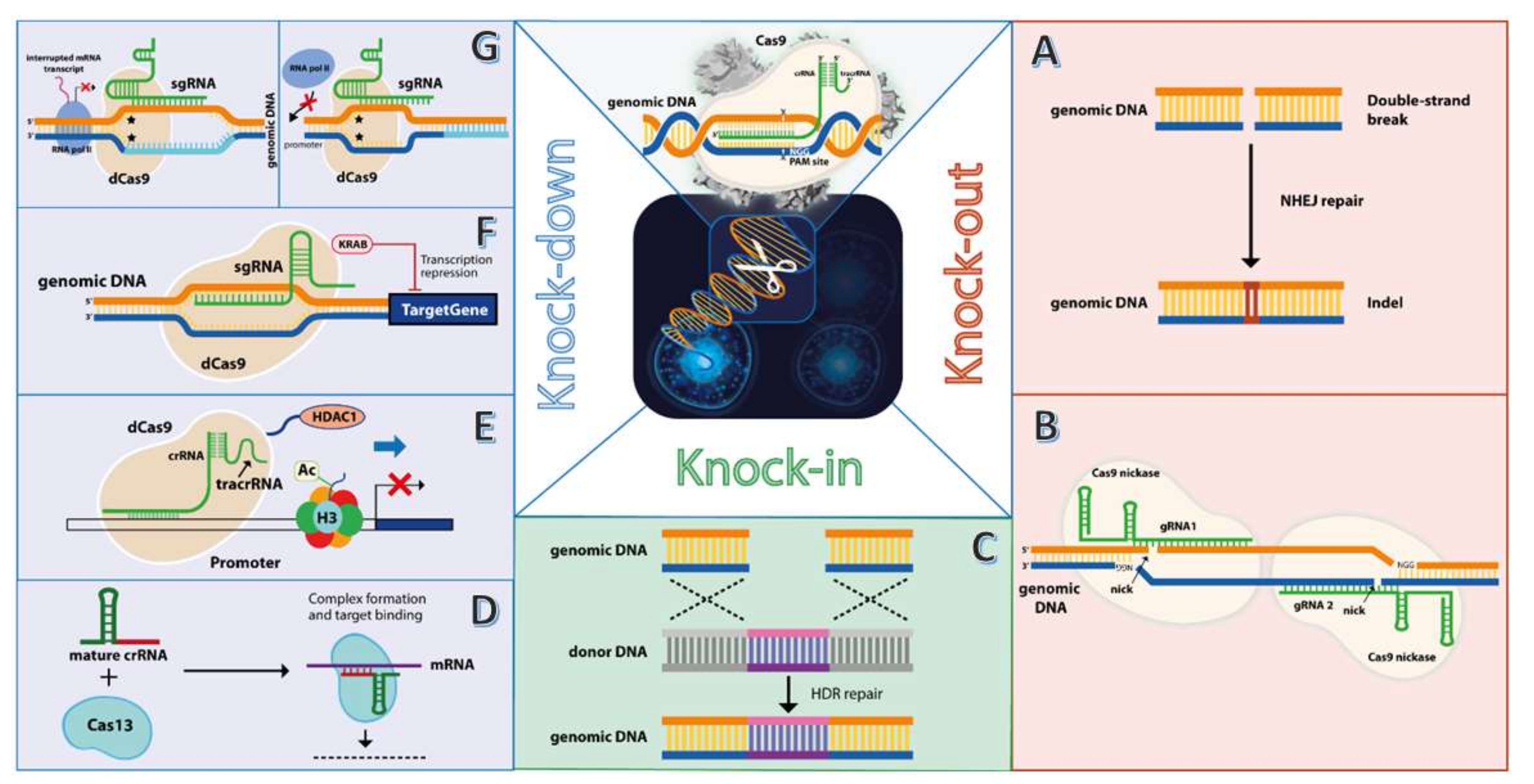
3.1. CRISPR/Cas9 Knockout of Oncogenes—Double-Strand Break (DBS) Approach
3.2. CRISPR/Cas9 Knockdown of Oncogenes—Transcription Interference Approach
3.3. CRISPR/Cas9 Knock-in of Exogenous DNA
4. CRISPR Clinical Trials for the Treatment of Malignant Disorders
5. Concluding Remarks and Future Perspective
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Hendijani, F. Human Mesenchymal Stromal Cell Therapy for Prevention and Recovery of Chemo/Radiotherapy Adverse Reactions. Cytotherapy 2015, 17, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From Chemotherapy to Biological Therapy: A Review of Novel Concepts to Reduce the Side Effects of Systemic Cancer Treatment (Review). Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.K.; Vapiwala, N. Secondary Malignancy after Radiotherapy: Not Always a Secondary Concern. Nat. Rev. Urol. 2021, 18, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Woodward, W.A.; Strom, E.A.; McNeese, M.D.; Perkins, G.H.; Outlaw, E.L.; Hortobagyi, G.N.; Buzdar, A.U.; Buchholz, T.A. Cardiovascular Death and Second Non-Breast Cancer Malignancy after Postmastectomy Radiation and Doxorubicin-Based Chemotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2003, 57, 327–335. [Google Scholar] [CrossRef]
- André, M.; Mounier, N.; Leleu, X.; Sonet, A.; Brice, P.; Henry-Amar, M.; Tilly, H.; Coiffier, B.; Bosly, A.; Morel, P.; et al. Second Cancers and Late Toxicities after Treatment of Aggressive Non-Hodgkin Lymphoma with the ACVBP Regimen: A GELA Cohort Study on 2837 Patients. Blood 2004, 103, 1222–1228. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.; Burmester, J.K. Gene Therapy for Cancer Treatment: Past, Present and Future. Clin. Med. Res. 2006, 4, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Libutti, S.K. Recording 25 Years of Progress in Cancer Gene Therapy. Cancer Gene Ther. 2019, 26, 345–346. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chira, S.; Jackson, C.S.; Oprea, I.; Ozturk, F.; Pepper, M.S.; Diaconu, I.; Braicu, C.; Raduly, L.Z.; Calin, G.A.; Berindan-Neagoe, I. Progresses towards Safe and Efficient Gene Therapy Vectors. Oncotarget 2015, 6, 30675–30703. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richardss, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR Provides Against Viruses in Prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Bano, S.; Kapse, P.; Kundu, G.C. CRISPR Based Therapeutics: A New Paradigm in Cancer Precision Medicine. Mol. Cancer 2022, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, G.; Gohil, N.; Khambhati, K.; Mani, I.; Maurya, R.; Karapurkar, J.K.; Gohil, J.; Chu, D.T.; Vu-Thi, H.; Alzahrani, K.J.; et al. Current Approaches in CRISPR-Cas9 Mediated Gene Editing for Biomedical and Therapeutic Applications. J. Control. Release 2022, 343, 703–723. [Google Scholar] [CrossRef] [PubMed]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakatura, A. Nucleotide Sequence of the Iap Gene, Responsible for Alkaline Phosphatase Isoenzyme Conversion in Escherichia Coli, and Identification of the Gene Product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.; Van Embden, J.D.A.; Gaastra, W.; Schouls, L.M. Identification of Genes That Are Associated with DNA Repeats in Prokaryotes. Mol. Microbiol. 2002, 43, 1565–1575. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Ferrer, C.; Juez, G.; Rodríguez-Valera, F. Long Stretches of Short Tandem Repeats Are Present in the Largest Replicons of the Archaea Haloferax Mediterranei and Haloferax Volcanii and Could Be Involved in Replicon Partitioning. Mol. Microbiol. 1995, 17, 85–93. [Google Scholar] [CrossRef]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Dusko Ehrlich, S. Clustered Regularly Interspaced Short Palindrome Repeats (CRISPRs) Have Spacers of Extrachromosomal Origin. Microbiology 2005, 151, 2551–2561. [Google Scholar] [CrossRef]
- Pourcel, C.; Salvignol, G.; Vergnaud, G. CRISPR Elements in Yersinia Pestis Acquire New Repeats by Preferential Uptake of Bacteriophage DNA, and Provide Additional Tools for Evolutionary Studies. Microbiology 2005, 151, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary Classification of CRISPR–Cas Systems: A Burst of Class 2 and Derived Variants. Nat. Rev. Microbiol. 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H.; Callaway, E. Pioneers of Revolutionary CRISPR Gene Editing Win Chemistry Nobel. Nature 2020, 586, 346–347. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; Dicarlo, J.E.; Norville, J.E.; Church, G.M. RNA-Guided Human Genome Engineering via Cas9. Science 2013, 339, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Mani, M.; Kandavelou, K.; Dy, F.J.; Durai, S.; Chandrasegaran, S. Design, Engineering, and Characterization of Zinc Finger Nucleases. Biochem. Biophys. Res. Commun. 2005, 335, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE Nuclease Architecture for Efficient Genome Editing. Nat. Biotechnol. 2011, 29, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Der, C.J. Ras History: The Saga Continues. Small GTPases 2010, 1, 2–27. [Google Scholar] [CrossRef]
- Nagasaka, M.; Potugari, B.; Nguyen, A.; Sukari, A.; Azmi, A.S.; Ou, S.H.I. KRAS Inhibitors—Yes but What next? Direct Targeting of KRAS—Vaccines, Adoptive T Cell Therapy and Beyond. Cancer Treat. Rev. 2021, 101, 102309. [Google Scholar] [CrossRef]
- Wan, T.; Chen, Y.; Pan, Q.; Xu, X.; Kang, Y.; Gao, X.; Huang, F.; Wu, C.; Ping, Y. Genome Editing of Mutant KRAS through Supramolecular Polymer-Mediated Delivery of Cas9 Ribonucleoprotein for Colorectal Cancer Therapy. J. Control. Release 2020, 322, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Ouyang, W.; Kang, B.; Han, X.; Xiong, Y.; Ding, R.; Li, Y.; Wang, F.; Huang, L.; Chen, L.; et al. Selective Targeting of the Oncogenic \textit KRAS G12S Mutant Allele by CRISPR/Cas9 Induces Efficient Tumor Regression. Theranostics 2020, 10, 5137–5153. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.H.-K.; Chow, C.; Zhang, J.; Zhou, Y.; Huang, T.; Ng, K.C.-K.; Or, T.C.-T.; Yao, Y.Y.; Dong, Y.; Fung, J.M.-W.; et al. Specific Targeting of Point Mutations in EGFR L858R—Positive Lung Cancer by CRISPR/Cas9. Lab. Investig. 2018, 98, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Koo, T.; Yoon, A.-R.; Cho, H.-Y.; Bae, S.; Yun, C.-O.; Kim, J.-S. Selective Disruption of an Oncogenic Mutant Allele by CRISPR/Cas9 Induces Efficient Tumor Regression. Nucleic Acids Res. 2017, 45, 7897–7908. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Sun, Y. Targeting P53 for Novel Anticancer Therapy. Transl. Oncol. 2010, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.A.J.; Vousden, K.H. Mutant P53 in Cancer: New Functions and Therapeutic Opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Min, L.; Seebacher, N.A.; Li, X.; Zhou, Y.; Hornicek, F.J.; Wei, Y.; Tu, C.; Duan, Z. Targeting Mutant TP53 as a Potential Therapeutic Strategy for the Treatment of Osteosarcoma. J. Orthop. Res. 2019, 37, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Singhal, J.; Chikara, S.; Horne, D.; Awasthi, S.; Salgia, R.; Singhal, S.S. Targeting RLIP with CRISPR/Cas9 Controls Tumor Growth. Carcinogenesis 2021, 42, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.W.; Jang, M.; Kim, S.J.; Kim, S.S.; Rhee, J.E. Degradation of P53 by Natural Variants of the E6 Protein of Human Papillomavirus Type 16. Oncol. Rep. 2013, 29, 1617–1622. [Google Scholar] [CrossRef][Green Version]
- Yoshiba, T.; Saga, Y.; Urabe, M.; Uchibor, R.; Matsubara, S.; Fujiwara, H.; Mizukami, H. CRISPR/Cas9-mediated Cervical Cancer Treatment Targeting Human Papillomavirus E6. Oncol. Lett. 2018, 17, 2197–2206. [Google Scholar] [CrossRef]
- Ehrke-Schulz, E.; Heinemann, S.; Schulte, L.; Schiwon, M.; Ehrhardt, A. Adenoviral Vectors Armed with PAPILLOMAVIRUs Oncogene Specific CRISPR/Cas9 Kill Human—Papillomavirus—Induced Cervical Cancer Cells. Cancers 2020, 12, 1934. [Google Scholar] [CrossRef]
- Noroozi, Z.; Shamsara, M.; Valipour, E.; Esfandyari, S.; Ehghaghi, A.; Monfaredan, A.; Azizi, Z.; Motevaseli, E.; Modarressi, M.H. Antiproliferative Effects of AAV—Delivered CRISPR/Cas9—Based Degradation of the HPV18—E6 Gene in HeLa Cells. Sci. Rep. 2022, 12, 2224. [Google Scholar] [CrossRef]
- Jubair, L.; Fallaha, S.; McMillan, N.A.J. Systemic Delivery of CRISPR/Cas9 Targeting HPV Oncogenes Is Effective at Eliminating Established Tumors. Mol. Ther. 2019, 27, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Lu, J.; Liu, Y.-H.; Chen, W.; Li, X. Synergistic Antitumor Effect on Cervical Cancer by Rational Combination of PD1 Blockade and CRISPR—Cas9—Mediated HPV Knockout. Cancer Gene Ther. 2020, 27, 168–178. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef]
- Yew, C.W.; Lee, P.; Chan, W.K.; Lim, V.K.J.; Tay, S.K.; Tan, T.M.C.; Deng, L.W. A Novel MLL5 Isoform That Is Essential to Activate E6 and E7 Transcription in HPV16/18-Associated Cervical Cancers. Cancer Res. 2011, 71, 6696–6707. [Google Scholar] [CrossRef] [PubMed]
- Pirouzfar, M.; Amiri, F.; Dianatpour, M.; Takhshid, M.A. CRISPR/Cas9—Mediated Knockout of MLL5 Enhances Apoptotic Effect of Cisplatin in HeLa Cells in Vitro. EXCLI J. 2020, 19, 170–182. [Google Scholar] [CrossRef]
- Lin, H.; Li, G.; Peng, X.; Deng, A.; Ye, L.; Shi, L.; Wang, T.; He, J. The Use of CRISPR/Cas9 as a Tool to Study Human Infectious Viruses. Front. Cell. Infect. Microbiol. 2021, 11, 774. [Google Scholar] [CrossRef] [PubMed]
- Binnie, A.; Fernandes, E.; Almeida-Lousada, H.; de Mello, R.A.; Castelo-Branco, P. CRISPR-Based Strategies in Infectious Disease Diagnosis and Therapy. Infection 2021, 49, 377. [Google Scholar] [CrossRef] [PubMed]
- Najafi, S.; Tan, S.C.; Aghamiri, S.; Raee, P.; Ebrahimi, Z.; Jahromi, Z.K.; Rahmati, Y.; Sadri Nahand, J.; Piroozmand, A.; Jajarmi, V.; et al. Therapeutic Potentials of CRISPR-Cas Genome Editing Technology in Human Viral Infections. Biomed. Pharmacother. 2022, 148, 112743. [Google Scholar] [CrossRef] [PubMed]
- Ju, E.; Li, T.; Ramos da Silva, S.; Markazi, A.; Gao, S.J. Reversible Switching of Primary Cells between Normal and Malignant State by Oncogenic Virus KSHV and CRISPR/Cas9-Mediated Targeting of a Major Viral Latent Protein. J. Med. Virol. 2021, 93, 5065. [Google Scholar] [CrossRef]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. Car T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef]
- Guo, Z.; Tu, S.; Yu, S.; Wu, L.; Pan, W.; Chang, N.; Zhou, X.; Song, C.; Li, Y.; He, Y. Preclinical and Clinical Advances in Dual-Target Chimeric Antigen Receptor Therapy for Hematological Malignancies. Cancer Sci. 2021, 112, 1357–1368. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T Cells in Solid Tumors: Challenges and Opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-Tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, T.; Natsume, A.; Nishimura, F.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Motoyama, Y.; Park, Y.-S.; et al. Effect of CRISPR/Cas9—Mediated PD-1-Disrupted Primary Human Third—Generation CAR—T Cells Targeting EGFRvIII on In Vitro Human Glioblastoma Cell Growth. Cells 2020, 9, 998. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.-F.; Han, W.; Wang, H. TGF-β Inhibition via CRISPR Promotes the Long-Term Efficacy of CAR T Cells against Solid Tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol. Immunother. 2019, 68, 365–377. [Google Scholar] [CrossRef]
- Jung, I.-Y.; Kim, Y.-Y.; Yu, H.-S.; Lee, M.; Kim, S.; Lee, J. CRISPR/Cas9—Mediated Knockout of DGK Improves Antitumor Activities of Human T Cells. Cancer Res. 2018, 78, 4692–4703. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Meng, T.; Zhao, Z.; Han, J.; Zhang, W.; Gao, F.; Cai, J. CRISPR Knock out CTLA-4 Enhances the Anti-Tumor Activity of Cytotoxic T Lymphocytes. Gene 2017, 636, 36–41. [Google Scholar] [CrossRef]
- Wu, X.; Li, Y.; Liu, X.; Chen, C.; Harrington, S.M.; Cao, S.; Xie, T.; Pham, T.; Mansfield, A.S.; Yan, Y.; et al. Targeting B7—H1 (PD—L1) Sensitizes Cancer Cells to Chemotherapy. Heliyon 2018, 4, e01039. [Google Scholar] [CrossRef]
- Ma, S.; Zhang, J.; Guo, Q.; Cao, C.; Bao, K.; Liu, L.; Chen, C.D.; Liu, Z.; Yang, J.; Yang, N.; et al. Disrupting PHF8—TOPBP1 Connection Elicits a Breast Tumor-Specific Vulnerability to Chemotherapeutics. Cancer Lett. 2022, 530, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Fukui, T.; Otani, S.; Hataishi, R.; Jiang, S.X.; Nishii, Y.; Igawa, S.; Mitsufuji, H.; Kubota, M.; Katagiri, M.; Masuda, N. Successful Rechallenge with Erlotinib in a Patient with EGFR-Mutant Lung Adenocarcinoma Who Developed Gefitinib-Related Interstitial Lung Disease. Cancer Chemother. Pharmacol. 2010, 65, 803–806. [Google Scholar] [CrossRef]
- Greaves, W.O.; Verma, S.; Patel, K.P.; Davies, M.A.; Barkoh, B.A.; Galbincea, J.M.; Yao, H.; Lazar, A.J.; Aldape, K.D.; Medeiros, L.J.; et al. Frequency and Spectrum of BRAF Mutations in a Retrospective, Single-Institution Study of 1112 Cases of Melanoma. J. Mol. Diagn. 2013, 15, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Huang, H.; Yu, B.; Zhang, J. A Blue Light—Inducible CRISPR—Cas9 System for Inhibiting Progression of Melanoma Cells. Front. Mol. Biosci. 2020, 7, 606593. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Bonazzina, E.; Amatu, A.; Tosi, F.; Bencardino, K.; Gori, V.; Massihnia, D.; Cipani, T.; Spina, F.; Ghezzi, S.; et al. The Evolutionary Landscape of Treatment for BRAFv600e Mutant Metastatic Colorectal Cancer. Cancers 2021, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Wilding, C.P.; Elms, M.L.; Judson, I.; Tan, A.C.; Jones, R.L.; Huang, P.H. The Landscape of Tyrosine Kinase Inhibitors in Sarcomas: Looking beyond Pazopanib. Expert Rev. Anticancer Ther. 2019, 19, 971. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; De Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing Sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 5. [Google Scholar] [CrossRef]
- Cervera, S.T.; Rodríguez-Martín, C.; Fernández-Tabanera, E.; de Mera, R.M.; Morin, M.; Fernández-Peñalver, S.; Iranzo-Martínez, M.; Amhih-Cardenas, J.; García-García, L.; González-González, L.; et al. Therapeutic Potential of EWSR1—FLI1 Inactivation by CRISPR/Cas9 in Ewing Sarcoma. Cancers 2021, 13, 3783. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Lage, M.; Torres-Ruiz, R.; Puig-Serra, P.; Moreno-Gaona, P.; Martin, M.C.; Moya, F.J.; Quintana-Bustamante, O.; Garcia-Silva, S.; Carcaboso, A.M.; Petazzi, P.; et al. In Vivo CRISPR/Cas9 Targeting of Fusion Oncogenes for Selective Elimination of Cancer Cells. Nat. Commun. 2020, 11, 5060. [Google Scholar] [CrossRef]
- Shen, N.; Liu, S.; Cui, J.; Li, Q.; You, Y.; Zhong, Z.; Cheng, F.; Guo, A.-Y.; Zou, P.; Yuan, G.; et al. Tumor Necrosis Factor α Knockout Impaired Tumorigenesis in Chronic Myeloid Leukemia Cells Partly by Metabolism Modification and miRNA Regulation. OncoTargets Ther. 2019, 12, 2355–2364. [Google Scholar] [CrossRef]
- Fernández, L.P.; Gómez de Cedrón, M.; Ramírez de Molina, A. Alterations of Lipid Metabolism in Cancer: Implications in Prognosis and Treatment. Front. Oncol. 2020, 10, 577420. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kandori, S.; Sakka, S.; Nitta, S.; Tanuma, K.; Shiga, M.; Nagumo, Y.; Negoro, H.; Kojima, T.; Mathis, B.; et al. ELOVL2 Promotes Cancer Progression by Inhibiting Cell Apoptosis in Renal Cell Carcinoma. Oncol. Rep. 2021, 47, 23. [Google Scholar] [CrossRef]
- Gao, S.; Soares, F.; Wang, S.; Wong, C.C.; Chen, H.; Yang, Z.; Liu, W.; Go, M.Y.Y.; Ahmed, M.; Zeng, Y.; et al. CRISPR Screens Identify Cholesterol Biosynthesis as a Therapeutic Target on Stemness and Drug Resistance of Colon Cancer. Oncogene 2021, 40, 6601–6613. [Google Scholar] [CrossRef]
- Rosenlund, I.A.; Calin, G.A.; Dragomir, M.P.; Knutsen, E. CRISPR/Cas9 to Silence Long Non-Coding RNAs. Methods Mol. Biol. 2021, 2348, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Chai, P.; Yu, J.; Jia, R.; Wen, X.; Ding, T.; Zhang, X.; Ni, H.; Jia, R.; Ge, S.; Zhang, H.; et al. Generation of Onco-Enhancer Enhances Chromosomal Remodeling and Accelerates Tumorigenesis. Nucleic Acids Res. 2020, 48, 12135–12150. [Google Scholar] [CrossRef] [PubMed]
- Głów, D.; Maire, C.L.; Schwarze, L.I.; Lamszus, K.; Fehse, B. CRISPR-to-Kill (C2K)—Employing the Bacterial Immune System to Kill Cancer Cells. Cancers 2021, 13, 6306. [Google Scholar] [CrossRef] [PubMed]
- Deininger, P. Alu Elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-Target Effects in CRISPR/Cas9-Mediated Genome Engineering. Mol. Ther. -Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas Nuclease Specificity Using Truncated Guide RNAs. Nat. Biotechnol. 2014, 32, 279. [Google Scholar] [CrossRef]
- Gopalappa, R.; Suresh, B.; Ramakrishna, S.; Kim, H.H. Paired D10A Cas9 Nickases Are Sometimes More Efficient than Individual Nucleases for Gene Disruption. Nucleic Acids Res. 2018, 46, e71. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by RNA-Guided CRISPR Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380. [Google Scholar] [CrossRef] [PubMed]
- De Santa-Inez, D.C.; Fuziwara, C.S.; Saito, K.C.; Kimura, E.T. Targeting the Highly Expressed microRNA miR-146b with CRISPR/Cas9n Gene Editing System in Thyroid Cancer. Int. J. Mol. Sci. 2021, 22, 7992. [Google Scholar] [CrossRef] [PubMed]
- Biagioni, A.; Chillà, A.; Del Rosso, M.; Fibbi, G.; Scavone, F.; Andreucci, E.; Peppicelli, S.; Bianchini, F.; Calorini, L.; Li Santi, A.; et al. CRISPR/Cas9 uPAR Gene Knockout Results in Tumor Growth Inhibition, EGFR Downregulation and Induction of Stemness Markers in Melanoma and Colon Carcinoma Cell Lines. Front. Oncol. 2021, 11, 663225. [Google Scholar] [CrossRef]
- Cao, Y.; Hu, Q.; Zhang, R.; Li, L.; Guo, M.; Wei, H.; Zhang, L.; Wang, J.; Li, C. Knockdown of Long Non-Coding RNA SNGH3 by CRISPR—dCas9 Inhibits the Progression of Bladder Cancer. Front. Mol. Biosci. 2021, 8, 657145. [Google Scholar] [CrossRef]
- Yoshida, M.; Yokota, E.; Sakuma, T.; Yamatsuji, T.; Takigawa, N.; Ushijima, T.; Yamamoto, T.; Fukazawa, T.; Naomoto, Y. Development of an Integrated CRISPRi Targeting ΔNp63 for Treatment of Squamous Cell Carcinoma. Oncotarget 2018, 9, 29220–29232. [Google Scholar] [CrossRef] [PubMed]
- Gallinari, P.; Di Marco, S.; Jones, P.; Pallaoro, M.; Steinkühler, C. HDACs, Histone Deacetylation and Gene Transcription: From Molecular Biology to Cancer Therapeutics. Cell Res. 2007, 17, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, M.; Cho, K.B.; Gao, X.; Guo, B. A CRISPR—Cas9 Repressor for Epigenetic Silencing of KRAS. Pharmacol. Res. 2021, 164, 105304. [Google Scholar] [CrossRef] [PubMed]
- Anson, D.S. The Use of Retroviral Vectors for Gene Therapy-What Are the Risks? A Review of Retroviral Pathogenesis and Its Relevance to Retroviral Vector-Mediated Gene Delivery. Genet. Vaccines Ther. 2004, 2, 9. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical Use of Lentiviral Vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Wilson, M.H.; Coates, C.J.; George, A.L. PiggyBac Transposon-Mediated Gene Transfer in Human Cells. Mol. Ther. J. Am. Soc. Gene Ther. 2007, 15, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Belay, E.; Dastidar, S.; VandenDriessche, T.; Chuah, M.K.L. Transposon-Mediated Gene Transfer into Adult and Induced Pluripotent Stem Cells. Curr. Gene Ther. 2011, 11, 406–413. [Google Scholar] [CrossRef]
- Bokhoven, M.; Stephen, S.L.; Knight, S.; Gevers, E.F.; Robinson, I.C.; Takeuchi, Y.; Collins, M.K. Insertional Gene Activation by Lentiviral and Gammaretroviral Vectors. J. Virol. 2009, 83, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.J.; Ammon, T.; Gosmann, D.; Audehm, S.; Thoene, S.; Winter, C.; Secci, R.; Wolf, A.; Stelzl, A.; Steiger, K.; et al. Gene Editing Enables T-Cell Engineering to Redirect Antigen Specificity for Potent Tumor Rejection. Life Sci. Alliance 2019, 2, e201900367. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Bao, C.; Xie, Y.; Guo, H.; Liu, Y.; Li, J.; Liu, R.; Li, P.; Bai, J.; Yan, Y.; et al. Targeted Core-Shell Nanoparticles for Precise CTCF Gene Insert in Treatment of Metastatic Breast Cancer. Bioact. Mater. 2022, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-H.; Yu, Y.P.; Zuo, Z.-H.; Nelson, J.B.; Michalopoulos, G.K.; Monga, S.; Liu, S.; Tseng, G.; Luo, J.-H. Targeting Genomic Rearrangements in Tumor Cells through Cas9-Mediated Insertion of a Suicide Gene. Nat. Biotechnol. 2017, 35, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and Feasibility of CRISPR-Edited T Cells in Patients with Refractory Non-Small-Cell Lung Cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Chen, Z.H.; Yu, Y.P.; Tao, J.; Liu, S.; Tseng, G.; Nalesnik, M.; Hamilton, R.; Bhargava, R.; Nelson, J.B.; Pennathur, A.; et al. MAN2A1–FER Fusion Gene Is Expressed by Human Liver and Other Tumor Types and Has Oncogenic Activity in Mice. Gastroenterology 2017, 153, 1120–1132.e15. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Tsung, A.; Liu, S.; Nalesnick, M.; Geller, D.; Michalopoulos, G.; Luo, J.H. Detection of Fusion Transcripts in the Serum Samples of Patients with Hepatocellular Carcinoma. Oncotarget 2019, 10, 3352–3360. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Ding, Y.; Chen, Z.; Liu, S.; Michalopoulos, A.; Chen, R.; Gulzar, Z.G.; Yang, B.; Cieply, K.M.; Luvison, A.; et al. Novel Fusion Transcripts Associate with Progressive Prostate Cancer. Am. J. Pathol. 2014, 184, 2840–2849. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Luo, T.; Lin, N.; Zhang, S.; Wang, J. A New Tool for CRISPR—Cas13a—Based Cancer Gene Therapy. Mol. Ther. -Oncolytics 2020, 19, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-L.; Tsai, M.-L.; Lin, C.-Y.; Hsu, K.-W.; Hsieh, W.-S.; Chi, W.-M.; Huang, L.-C.; Lee, C.-H. HDAC1 and HDAC2 Double Knockout Triggers Cell Apoptosis in Advanced Thyroid Cancer. Int. J. Mol. Sci. 2019, 20, 454. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wang, C.; Zheng, Y.; Zhao, Y.; Wang, Y.; Hao, J.; Zhao, X.; Yi, K.; Shi, L.; Kang, C.; et al. Virus-like Nanoparticle as a Co-Delivery System to Enhance Efficacy of CRISPR/Cas9-Based Cancer Immunotherapy. Biomaterials 2020, 258, 120275. [Google Scholar] [CrossRef] [PubMed]
- Chira, S.; Nutu, A.; Bica, C.; Pop, L.; Gherman, M.; Angheluta, M.; Berindan-Neagoe, I. Turning tables for CRISPR/Cas9 editing system: From scratch to advanced delivery platforms. Handbook of Cancer and Immunology, Springer: Cham, Switzerland, 2022; accepted for publication. [Google Scholar]
- Peng, L.; Pan, P.; Chen, J.; Yu, X.; Wu, J.; Chen, Y. A Tetracycline-inducible CRISPR/Cas9 System, Targeting Two Long Non-coding RNAs, Suppresses the Malignant Behavior of Bladder Cancer Cells. Oncol. Lett. 2018. [Google Scholar] [CrossRef] [PubMed]
- Che, W.; Ye, S.; Cai, A.; Cui, X.; Sun, Y. CRISPR—Cas13a Targeting the Enhancer RNA—SMAD7e Inhibits Bladder Cancer Development Both in Vitro and in Vivo. Front. Mol. Biosci. 2020, 7, 607740. [Google Scholar] [CrossRef]
- Zhuang, C.; Zhuang, C.; Zhou, Q.; Huang, X.; Gui, Y.; Lai, Y.; Yang, S. Engineered CRISPR/Cas13d Sensing hTERT Selectively Inhibits the Progression of Bladder Cancer In Vitro. Front. Mol. Biosci. 2021, 8, 646412. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Hao, L.; Han, X.; Wu, Z.-X.; Pang, K.; Dong, Y.; Qin, J.; Wang, G.; Zhang, X.; Xia, T.; et al. Targeting HNRNPU to Overcome Cisplatin Resistance in Bladder Cancer. Mol. Cancer 2022, 21, 37. [Google Scholar] [CrossRef]
- Wen, L.; Zhao, C.; Song, J.; Ma, L.; Ruan, J.; Xia, X.; Chen, Y.E.; Zhang, J.; Ma, P.X.; Xu, J. CRISPR/Cas9—Mediated TERT Disruption in Cancer Cells. Int. J. Mol. Sci. 2020, 21, 653. [Google Scholar] [CrossRef] [PubMed]
- Tian, R.; Liu, J.; Fan, W.; Li, R.; Cui, Z.; Jin, Z.; Huang, Z.; Xie, H.; Li, L.; Huang, Z.; et al. Gene Knock-out Chain Reaction Enables High Disruption Efficiency of HPV18 E6/E7 Genes in Cervical Cancer Cells. Mol. Ther.-Oncolytics 2022, 24, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Stöckl, S.; Lindner, G.; Li, S.; Schuster, P.; Haferkamp, S.; Wagner, F.; Prodinger, P.M.; Multhoff, G.; Boxberg, M.; Hillmann, A.; et al. SOX9 Knockout Induces Polyploidy and Changes Sensitivity to Tumor Treatment Strategies in a Chondrosarcoma Cell Line. Int. J. Mol. Sci. 2020, 21, 7627. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, X.; Li, Z.; Huang, Q.; Li, F.; Li, C.-Y. Caspase-3 Regulates the Migration, Invasion and Metastasis of Colon Cancer Cells: Metastasis of Colon Cancer Cells. Int. J. Cancer 2018, 143, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Ramasamy, K.; Pillai, S.M.A.; Santhamma, B.; Konda, S.; Pitta Venkata, P.; Blankenship, L.; Liu, J.; Liu, Z.; Altwegg, K.A.; et al. LIF/LIFR Oncogenic Signaling Is a Novel Therapeutic Target in Endometrial Cancer. Cell Death Discov. 2021, 7, 216. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Fdez, A.; Re-Louhau, M.F.; Rodríguez-Núñez, P.; Ludeña, D.; Matilla-Almazán, S.; Pandiella, A.; Esparís-Ogando, A. Clinical, Genetic and Pharmacological Data Support Targeting the MEK5/ERK5 Module in Lung Cancer. npj Precis. Oncol. 2021, 5, 78. [Google Scholar] [CrossRef] [PubMed]
- Yoon, A.-R.; Jung, B.-K.; Choi, E.; Chung, E.; Hong, J.; Kim, J.-S.; Koo, T.; Yun, C.-O. CRISPR—Cas12a with an oAd Induces Precise and Cancer—Specific Genomic Reprogramming of EGFR and Efficient Tumor Regression. Mol. Ther. 2020, 28, 2286–2296. [Google Scholar] [CrossRef] [PubMed]
- Saifullah; Sakari, M.; Suzuki, T.; Yano, S.; Tsukahara, T. Effective RNA Knockdown Using CRISPR—Cas13a and Molecular Targeting of the EML4—ALK Transcript in H3122 Lung Cancer Cells. Int. J. Mol. Sci. 2020, 21, 8904. [Google Scholar] [CrossRef] [PubMed]
- Haase-Kohn, C.; Laube, M.; Donat, C.K.; Belter, B.; Pietzsch, J. CRISPR/Cas9 Mediated Knockout of Cyclooxygenase -2 Gene Inhibits Invasiveness in A2058 Melanoma Cells. Cells 2022, 11, 749. [Google Scholar] [CrossRef] [PubMed]
- Shekoohi, S.; Rajasekaran, S.; Patel, D.; Yang, S.; Liu, W.; Huang, S.; Yu, X.; Witt, S.N. Knocking out Alpha-Synuclein in Melanoma Cells Dysregulates Cellular Iron Metabolism and Suppresses Tumor Growth. Sci. Rep. 2021, 11, 5267. [Google Scholar] [CrossRef]
- Garcia-Peterson, L.M.; Ndiaye, M.A.; Chhabra, G.; Singh, C.K.; Guzmán-Pérez, G.; Iczkowski, K.A.; Ahmad, N. CRISPR/Cas9-Mediated Knockout of SIRT6 Imparts Remarkable Anti-Proliferative Response in Human Melanoma Cells in Vitro and in Vivo. Photochem. Photobiol. 2020, 96, 1314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, L.; Leng, Y.; Dai, Y.; Orlowski, R.Z.; Grant, S. Positive Transcription Elongation Factor b (P—TEFb) Is a Therapeutic Target in Human Multiple Myeloma. Oncotarget 2017, 8, 59476–59491. [Google Scholar] [CrossRef][Green Version]
- Pan, S.; Su, Y.; Sun, B.; Hao, R.; Gao, X.; Han, B. Knockout of CD147 Inhibits the Proliferation, Invasion, and Drug Resistance of Human Oral Cancer CAL27 Cells in Vitro and in Vivo. Int. J. Biol. Macromol. 2021, 181, 378–389. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR—Cas9 Genome Editing Using Targeted Lipid Nanoparticles for Cancer Therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Zhao, G.; Zhang, P.; Huo, W.; Dong, P.; Watari, H.; Jia, L.; Pfeffer, L.M.; Yue, J.; Zheng, J. Knockout of MTF1 Inhibits the Epithelial to Mesenchymal Transition in Ovarian Cancer Cells. J. Cancer 2018, 9, 4578–4585. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Xie, H.; Liu, Y.; Xia, C.; Cun, X.; Long, Y.; Chen, X.; Deng, M.; Guo, R.; Zhang, Z.; et al. Knockdown of Hypoxia-Inducible Factor-1 Alpha by Tumor Targeted Delivery of CRISPR/Cas9 System Suppressed the Metastasis of Pancreatic Cancer. J. Control. Release 2019, 304, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Li, H.; Liu, X.; Zhang, J.; Zhang, W.; Li, T.; Liu, L.; Yu, X. Precise and Efficient Silencing of Mutant Kras $^ \textrm G12D $ by CRISPR—CasRx Controls Pancreatic Cancer Progression. Theranostics 2020, 10, 11507–11519. [Google Scholar] [CrossRef] [PubMed]
- Albayrak, G.; Konac, E.; Ugras Dikmen, A.; Bilen, C.Y. FOXA1 Knock-out via CRISPR/Cas9 Altered Casp -9, Bax, CCND1, CDK4, and Fibronectin Expressions in LNCaP Cells. Exp. Biol. Med. 2018, 243, 990–994. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Pathological Condition | Phase | Status | CRISPR-Engineered Therapeutic Agent | Other Therapies | Identifier |
|---|---|---|---|---|---|
| B-cell acute lymphoblastic leukemia | Phase 1 | Recruiting | Allogenic transplantation of engineered T cells—PBLTT52CAR19 | - | NCT04557436 |
| CD19+ leukemia and lymphoma | Phase 1 | Withdrawn | Allogenic transplantation of engineered T cells—PACE CART19 | - | NCT05037669 |
| Gastrointestinal cancers | Ph½ 1/2 | Recruiting | Autologous transplantation of CISH CRISPR TILs | Chemotherapy—cyclophosphamide and fludarabine Immunotherapy—aldesleukin | NCT04426669 |
| HIV-infected subjects with hematological malignances | N/A | Unknown | Allogenic transplantation of CRISPR/Cas9 CCR5 gene modified CD34+ hematopoietic stem/progenitor cells | - | NCT03164135 |
| Human papillomavirus-related malignant neoplasm | Phase 1 | Unknown | Local direct application of HPV16 E6/E7T1 or CRISPR/Cas9-HPV18 E6/E7T2 | - | NCT03057912 |
| Epstein-Barr virus (EBV)-associated malignancies | Ph½ 1/2 | Recruiting | Autologous transplantation of PD-1 knockout EBV-CTL cells | Chemotherapy—cyclophosphamide and fludarabine Immunotherapy—interleukin-2 | NCT03044743 |
| Non-small cell lung cancer | Phase 1 | Completed | Autologous transplantation of PD-1 knockout T cells | Chemotherapy—cyclophosphamide | NCT02793856 |
| Renal cell carcinoma | Phase 1 | Withdrawn (no funding) | Autologous transplantation of PD-1 knockout T cells | Chemotherapy—cyclophosphamide Immunotherapy—interleukin-2 | NCT02867332 |
| Prostate cancer | Phase 1 | Withdrawn (no funding or financial support | Autologous transplantation of PD-1 knockout T cells | Chemotherapy—cyclophosphamide Immunotherapy—interleukin-2 | NCT02867345 |
| Bladder cancer | Phase 1 | Withdrawn (no funding) | Autologous transplantation of PD-1 knockout T cells | Chemotherapy—cyclophosphamide Immunotherapy—interleukin-2 | NCT02863913 |
| Hepatocellular carcinoma | Phase 1 | Recruiting | Autologous transplantation of PD-1 knockout T cell | TACE | NCT04417764 |
| Esophageal cancer | Phase 1 | Completed | Autologous transplantation of PD-1 knockout T cells | - | NCT03081715 |
| CD19+ leukemia and lymphoma | Ph½ 1/2 | Recruiting | Allogenic transplantation of UCART019 | - | NCT03166878 |
| Leukemia and lymphoma | Ph½ 1/2 | Recruiting | Allogenic transplantation of CAR-T Cells Targeting CD19 and CD20 or CD22 | - | NCT03398967 |
| T or B-cell malignancies | Phase 1 | Recruiting | Allogenic transplantation of CTX130 | Prior lymphodepleting chemotherapy | NCT04502446 |
| B-cell malignancies | Phase 1 | Recruiting | Allogenic transplantation of CTX110 | Prior lymphodepleting chemotherapy | NCT04035434 |
| Solid tumors | Phase 1 | Unknown | Mesothelin-directed CAR-T cells | Prior conditioning regimen of paclitaxel and cyclophosphamide | NCT03747965 |
| Renal cell carcinoma | Phase 1 | Recruiting | Allogenic transplantation of CTX130 | Prior lymphodepleting chemotherapy | NCT04438083 |
| Multiple myeloma | Phase 1 | Recruiting | Allogenic transplantation of CTX120 | Prior lymphodepleting chemotherapy | NCT04244656 |
| Solid tumors | Phase 1 | Recruiting | Anti-mesothelin CAR-T cells | - | NCT03545815 |
| Multiple myeloma Melanoma Synovial sarcoma Myxoid/round cell liposarcoma | Phase 1 | Terminated | Autologous T cells were transduced with a lentiviral vector to express NY-ESO-1 and electroporated with CRISPR guide RNA to disrupt the expression of endogenous TCRα, TCRβ, and PD-1 (NYCE T Cells) | Cyclophosphamide Fludarabine | NCT03399448 |
| Relapsed or refractory CD19+ leukemia or lymphoma | Phase 1 | Recruiting | Autologous T cells engineered to specify CD19 transduced with a lentiviral vector and electroporated with CRISPR guide RNA to disrupt the expression of endogenous HPK1 administered by IV injection | Cyclophosphamide Fludarabine | NCT04037566 |
| Non-Hodgkin lymphoma | Phase 1 | Recruiting | CRISPR-edited allogeneic CAR-T cell therapy targeting CD19 | Cyclophosphamide Fludarabine | NCT04637763 |
| Acute myeloid leukemia | Ph½ 1/2 | Recruiting | Autologous WT1-directed TCR T cells engineered ex vivo using CRISPR/Cas9 | Pre-conditioning chemotherapy: cyclophosphamide Fludarabine | NCT05066165 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chira, S.; Nutu, A.; Isacescu, E.; Bica, C.; Pop, L.; Ciocan, C.; Berindan-Neagoe, I. Genome Editing Approaches with CRISPR/Cas9 for Cancer Treatment: Critical Appraisal of Preclinical and Clinical Utility, Challenges, and Future Research. Cells 2022, 11, 2781. https://doi.org/10.3390/cells11182781
Chira S, Nutu A, Isacescu E, Bica C, Pop L, Ciocan C, Berindan-Neagoe I. Genome Editing Approaches with CRISPR/Cas9 for Cancer Treatment: Critical Appraisal of Preclinical and Clinical Utility, Challenges, and Future Research. Cells. 2022; 11(18):2781. https://doi.org/10.3390/cells11182781
Chicago/Turabian StyleChira, Sergiu, Andreea Nutu, Ecaterina Isacescu, Cecilia Bica, Laura Pop, Cristina Ciocan, and Ioana Berindan-Neagoe. 2022. "Genome Editing Approaches with CRISPR/Cas9 for Cancer Treatment: Critical Appraisal of Preclinical and Clinical Utility, Challenges, and Future Research" Cells 11, no. 18: 2781. https://doi.org/10.3390/cells11182781
APA StyleChira, S., Nutu, A., Isacescu, E., Bica, C., Pop, L., Ciocan, C., & Berindan-Neagoe, I. (2022). Genome Editing Approaches with CRISPR/Cas9 for Cancer Treatment: Critical Appraisal of Preclinical and Clinical Utility, Challenges, and Future Research. Cells, 11(18), 2781. https://doi.org/10.3390/cells11182781