
FGF10 Therapeutic Administration Promotes Mobilization of Injury-Activated Alveolar Progenitors in a Mouse Fibrosis Model

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Drug Administration
2.2. Histology and Immunohistochemistry and Hydroxyproline Measurement
2.3. Immunofluorescence
2.4. Western Blot
2.5. Lung Dissociation and Preparation of Single Cells
2.6. Magnetic Cell Sorting (MACS) and Flow Cytometry Analysis
2.7. Quantification and Statistical Analysis
3. Results
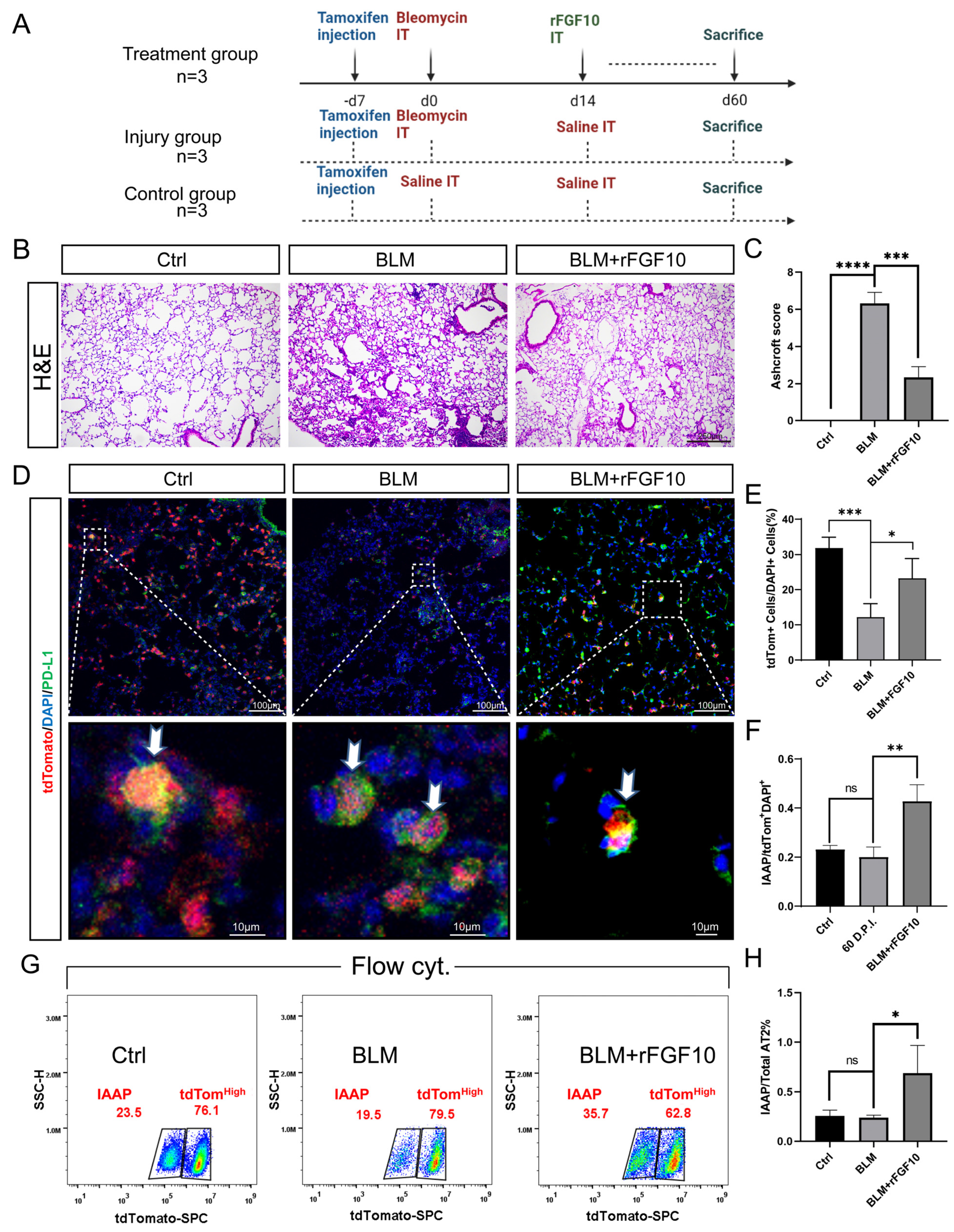
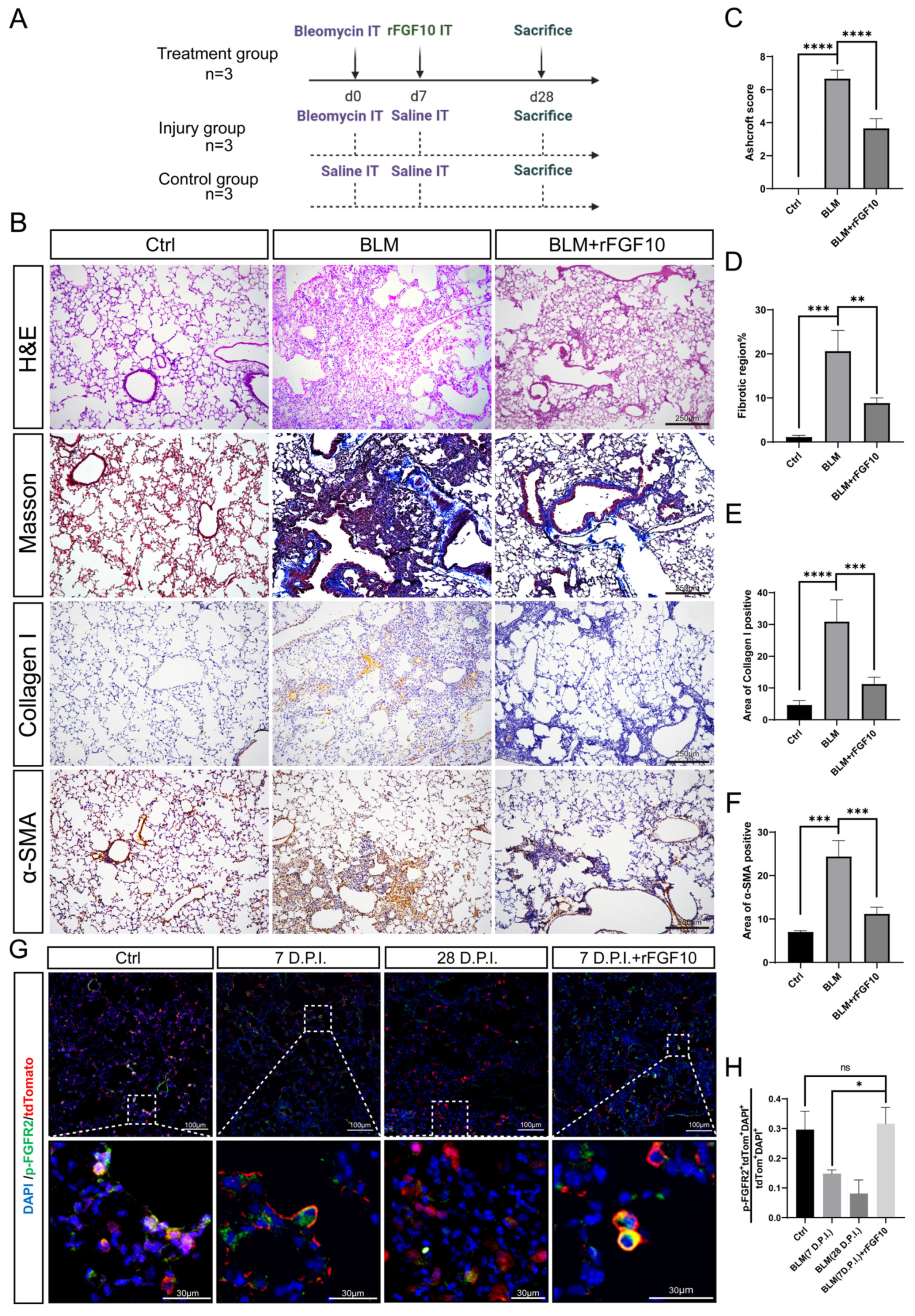
3.1. Preventative rFGF10 Delivery Decreases Fibrosis Formation
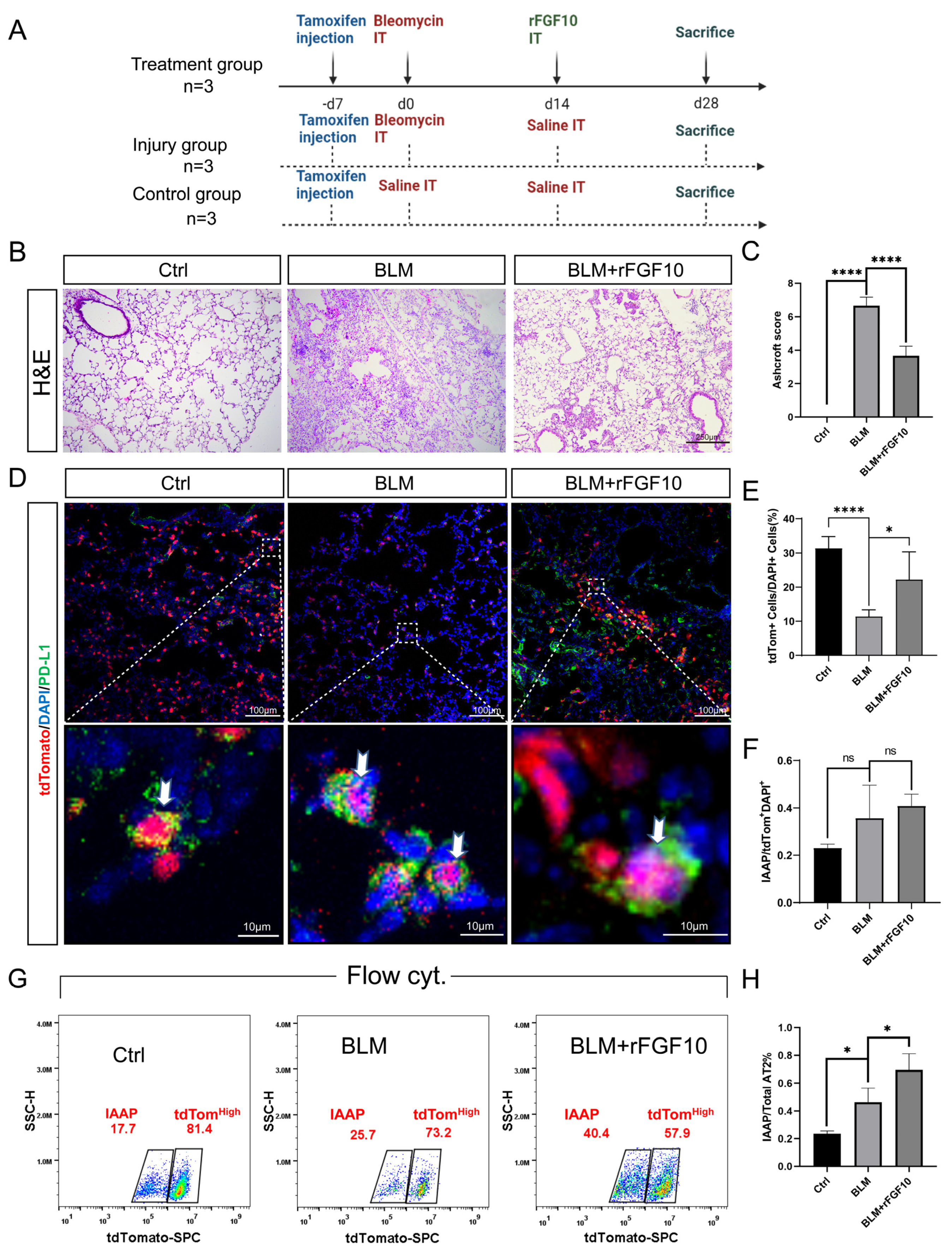
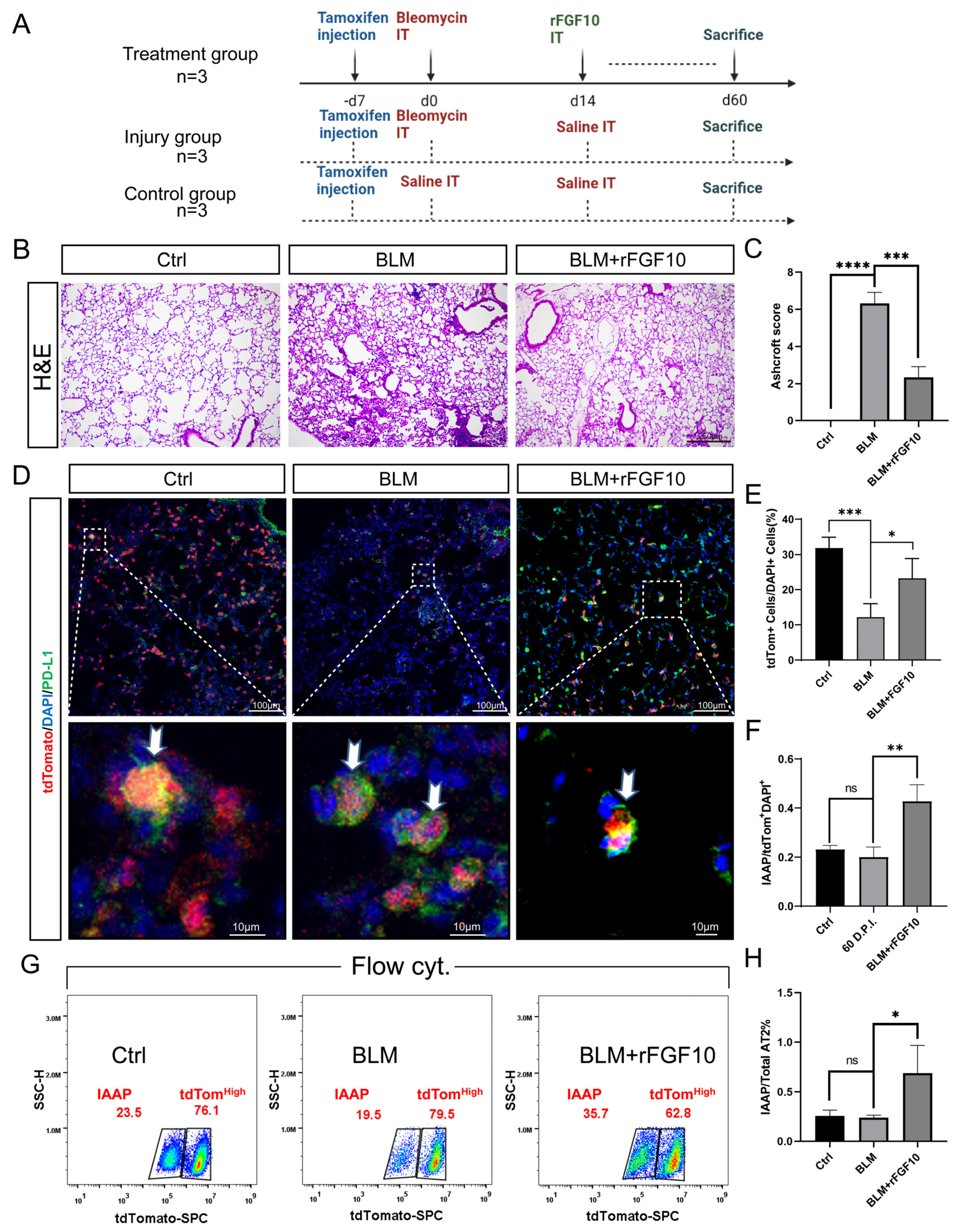
3.2. Therapeutic rFGF10 Delivery at 21 dpi Accelerates Fibrosis Resolution
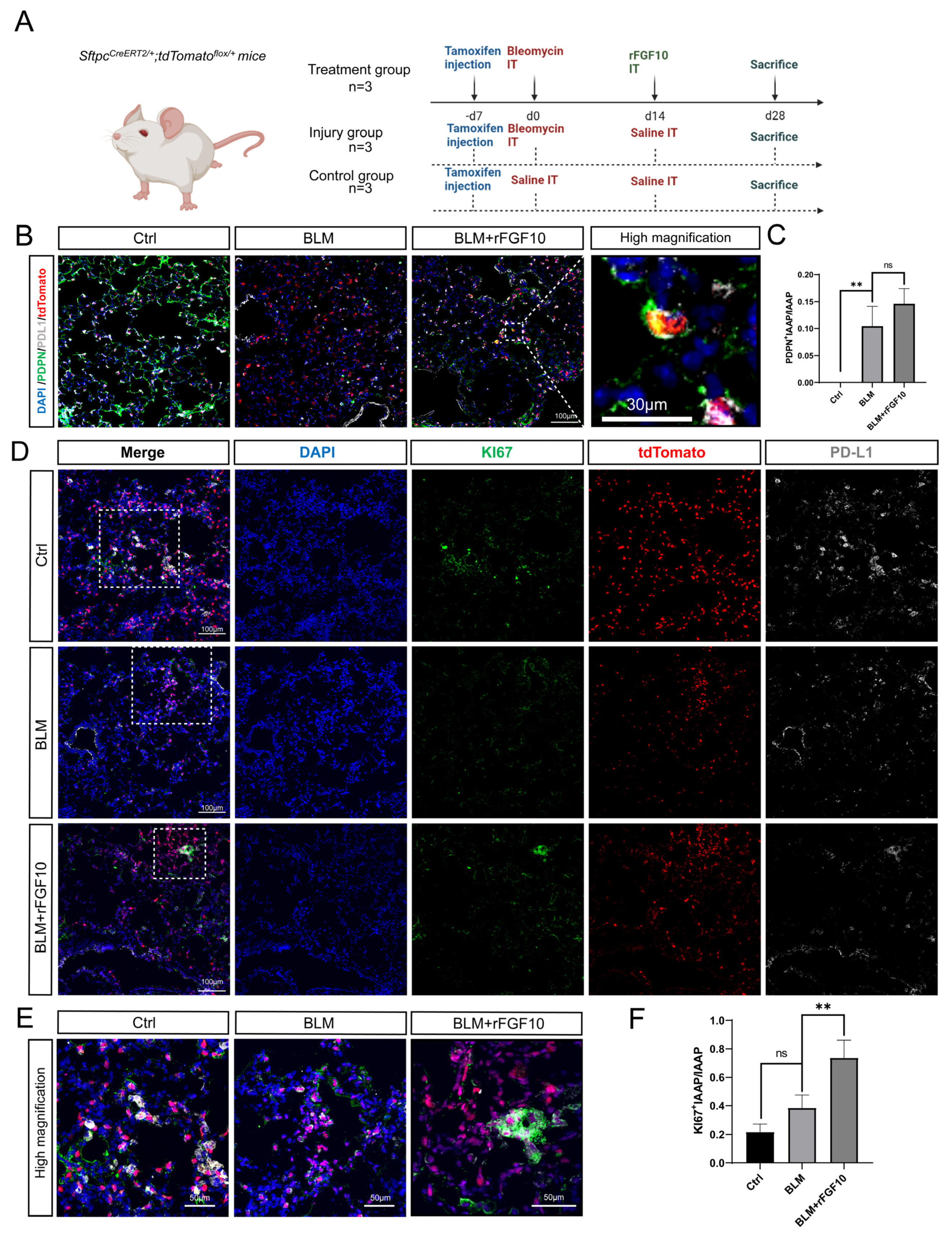
3.3. rFGF10 Promotes Alveolar Epithelial Progenitor Cell Proliferation and Alveolar Repair
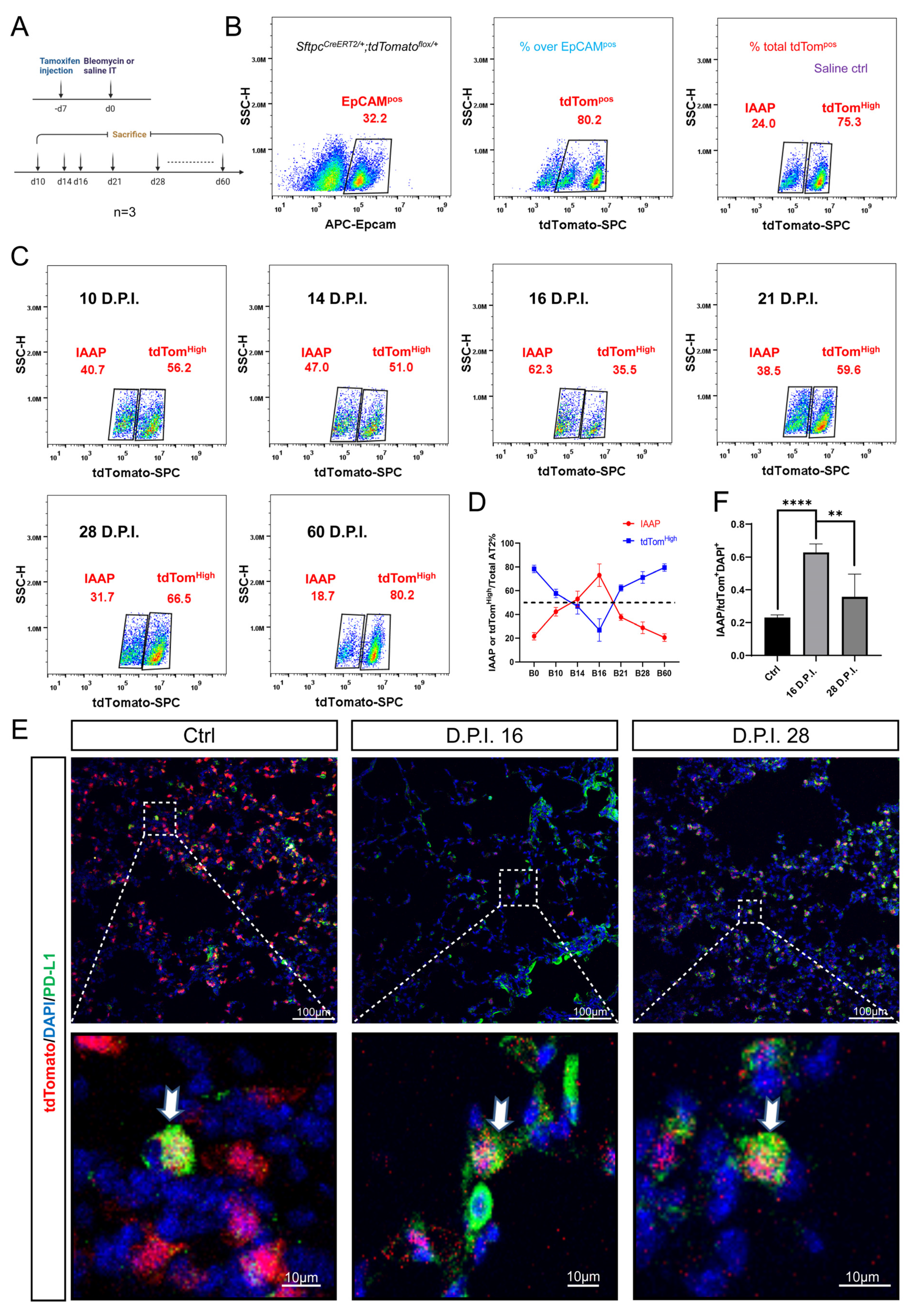
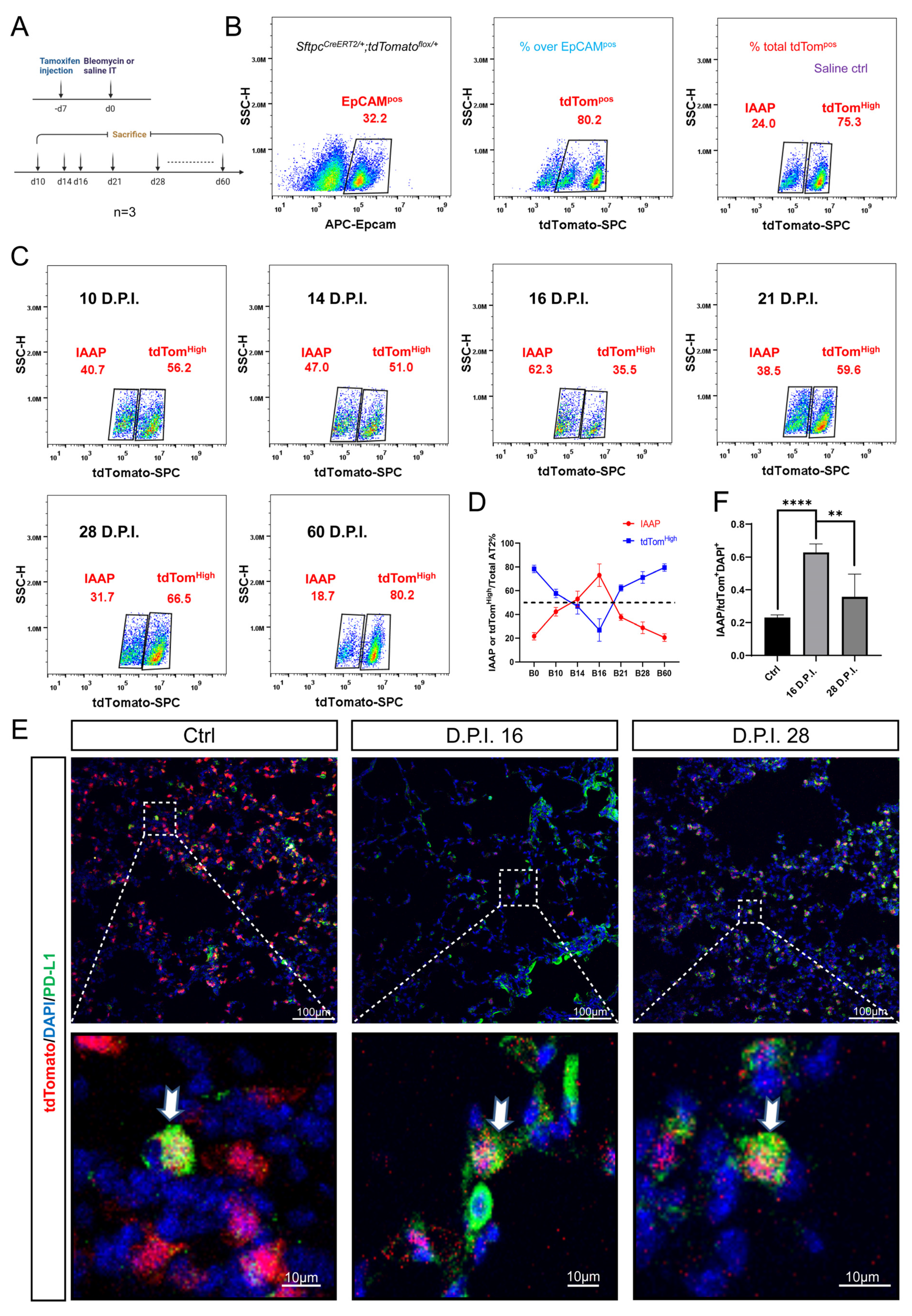
3.4. Dynamic Alteration of IAAP Population during BLM-Induced Lung Fibrosis and Resolution
3.5. rFGF10 Administration Triggers Further IAAPs Expansion
3.6. rFGF10 Increases the Duration of IAAP/AT2 Population Ratio
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Steele, M.P.; Schwartz, D.A. Molecular Mechanisms in Progressive Idiopathic Pulmonary Fibrosis. Annu. Rev. Med. 2013, 64, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Noble, P.W. Cellular Mechanisms of Tissue Fibrosis. 7. New insights into the cellular mechanisms of pulmonary fibrosis. Am. J. Physiol. Physiol. 2014, 306, C987–C996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glass, D.S.; Grossfeld, D.; Renna, H.A.; Agarwala, P.; Spiegler, P.; DeLeon, J.; Reiss, A.B. Idiopathic pulmonary fibrosis: Current and future treatment. Clin. Respir. J. 2022, 16, 84–96. [Google Scholar] [CrossRef]
- Karimi-Shah, B.A.; Chowdhury, B.A. Forced Vital Capacity in Idiopathic Pulmonary Fibrosis — FDA Review of Pirfenidone and Nintedanib. New Engl. J. Med. 2015, 372, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- King, T.E., Jr.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A Phase 3 Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Du Bois, R.M.; Fagan, E.A.; Fishman, R.S.; Glaspole, I.; Glassberg, M.K.; Lancaster, L.; et al. Pirfenidone for idiopathic pulmonary fibrosis: Analysis of pooled data from three multinational phase 3 trials. Eur. Respir. J. 2015, 47, 243–253. [Google Scholar] [CrossRef]
- Richeldi, L.; Costabel, U.; Selman, M.; Kim, D.S.; Hansell, D.M.; Nicholson, A.G.; Brown, K.K.; Flaherty, K.R.; Noble, P.W.; Raghu, G.; et al. Efficacy of a Tyrosine Kinase Inhibitor in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2011, 365, 1079–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and Safety of Nintedanib in Idiopathic Pulmonary Fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [Green Version]
- Azuma, A.; Nukiwa, T.; Tsuboi, E.; Suga, M.; Abe, S.; Nakata, K.; Taguchi, Y.; Nagai, S.; Itoh, H.; Ohi, M.; et al. Double-blind, Placebo-controlled Trial of Pirfenidone in Patients with Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2005, 171, 1040–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corte, T.J.; Bonella, F.; Crestani, B.; Demedts, M.G.; Richeldi, L.; Coeck, C.; Pelling, K.; Quaresma, M.; Lasky, J.A. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir. Res. 2015, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costabel, U.; Inoue, Y.; Richeldi, L.; Collard, H.R.; Tschoepe, I.; Stowasser, S.; Azuma, A. Efficacy of Nintedanib in Idiopathic Pulmonary Fibrosis across Prespecified Subgroups in INPULSIS. Am. J. Respir. Crit. Care Med. 2016, 193, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Salton, F.; Braga, L.; Wade, B.; Confalonieri, P.; Volpe, M.C.; Baratella, E.; Maiocchi, S.; Confalonieri, M. The History and Mystery of Alveolar Epithelial Type II Cells: Focus on Their Physiologic and Pathologic Role in Lung. Int. J. Mol. Sci. 2021, 22, 2566. [Google Scholar] [CrossRef] [PubMed]
- Calkovska, A.; Kolomaznik, M.; Calkovsky, V. Alveolar Type II Cells and Pulmonary Surfactant in COVID-19 Era. Physiol. Res. 2021, S195–S208. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar Epithelial Type II Cells as Drivers of Lung Fibrosis in Idiopathic Pulmonary Fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [Green Version]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef]
- Desai, T.J.; Brownfield, D.; Krasnow, M.A. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014, 507, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.J.; Cabral, L.J.; Stephens, R.J.; Freeman, G. Transformation of alveolar Type 2 cells to Type 1 cells following exposure to NO2. Exp. Mol. Pathol. 1975, 22, 142–150. [Google Scholar] [CrossRef]
- Chen, Q.; Liu, Y. Heterogeneous groups of alveolar type II cells in lung homeostasis and repair. Am. J. Physiol. Physiol. 2020, 319, C991–C996. [Google Scholar] [CrossRef] [PubMed]
- Ahmadvand, N.; Khosravi, F.; Lingampally, A.; Wasnick, R.; Vazquez-Armendariz, A.I.; Carraro, G.; Heiner, M.; Rivetti, S.; Lv, Y.; Wilhelm, J.; et al. Identification of a novel subset of alveolar type 2 cells enriched in PD-L1 and expanded following pneumonectomy. Eur. Respir. J. 2021, 58, 2004168. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N.; Ohta, H. Fgf10: A paracrine-signaling molecule in development, disease, and regenerative medicine. Curr. Mol. Med. 2014, 14, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Itoh, N. FGF10: A multifunctional mesenchymal-epithelial signaling growth factor in development, health, and disease. Cytokine Growth Factor Rev. 2016, 28, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Volckaert, T.; Chanda, D.; Thannickal, V.J.; De Langhe, S.P. Fgf10 Signaling in Lung Development, Homeostasis, Disease, and Repair After Injury. Front. Genet. 2018, 9. [Google Scholar] [CrossRef]
- Gupte, V.V.; Ramasamy, S.K.; Reddy, R.; Lee, J.; Weinreb, P.H.; Violette, S.M.; Guenther, A.; Warburton, D.; Driscoll, B.; Minoo, P.; et al. Overexpression of Fibroblast Growth Factor-10 during Both Inflammatory and Fibrotic Phases Attenuates Bleomycin-induced Pulmonary Fibrosis in Mice. Am. J. Respir. Crit. Care Med. 2009, 180, 424–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadvand, N.; Carraro, G.; Jones, M.R.; Shalashova, I.; Wilhelm, J.; Baal, N.; Kosravi, F.; Chen, C.; Zhang, J.; Ruppert, C.; et al. Cell-surface PD-L1 expression identifies a sub-population of distal epithelial cells enriched in idiopathic pulmonary fibrosis. Biorxiv 2022. [Google Scholar] [CrossRef]
- Ahmadvand, N.; Lingampally, A.; Khosravi, F.; Vazquez-Armendariz, I.; Rivetti, S.; Wilhelm, J.; Herold, S.; Barreto, G.; Koepke, J.; Samakovlis, C.; et al. Fgfr2b signaling is essential for the maintenance of the alveolar epithelial type 2 lineage during lung homeostasis in mice. Cell. Mol. Life Sci. 2022, 79, 302. [Google Scholar] [CrossRef] [PubMed]
- Carrington, R.; Jordan, S.; Pitchford, S.C.; Page, C.P. Use of animal models in IPF research. Pulm. Pharmacol. Ther. 2018, 51, 73–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; De Los Santos, F.G.; Phan, S.H. The Bleomycin Model of Pulmonary Fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar] [CrossRef]
- Ashcroft, T.; Simpson, J.M.; Timbrell, V. Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 1988, 41, 467–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Y.-Q.; Dhlamini, Q.; Chen, C.; Li, X.; Bellusci, S.; Zhang, J.-S. FGF10 and Lipofibroblasts in Lung Homeostasis and Disease: Insights Gained From the Adipocytes. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Park, J.-E.; Tsagkogeorga, G.; Yanagita, M.; Koo, B.-K.; Han, N.; Lee, J.-H. Inflammatory Signals Induce AT2 Cell-Derived Damage-Associated Transient Progenitors that Mediate Alveolar Regeneration. Cell Stem Cell 2020, 27, 366–382. [Google Scholar] [CrossRef] [PubMed]
- Kathiriya, J.J.; Wang, C.; Zhou, M.; Brumwell, A.; Cassandras, M.; Le Saux, C.J.; Cohen, M.; Alysandratos, K.-D.; Wang, B.; Wolters, P.; et al. Human alveolar type 2 epithelium transdifferentiates into metaplastic KRT5+ basal cells. Nature 2021, 24, 10–23. [Google Scholar] [CrossRef] [PubMed]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Szibor, M.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 571. [Google Scholar] [CrossRef] [Green Version]
- Nouri-Keshtkar, M.; Taghizadeh, S.; Farhadi, A.; Ezaddoustdar, A.; Vesali, S.; Hosseini, R.; Totonchi, M.; Kouhkan, A.; Chen, C.; Zhang, J.-S.; et al. Potential Impact of Diabetes and Obesity on Alveolar Type 2 (AT2)-Lipofibroblast (LIF) Interactions After COVID-19 Infection. Front. Cell Dev. Biol. 2021, 9, 676150. [Google Scholar] [CrossRef] [PubMed]
- Kheirollahi, V.; Wasnick, R.M.; Biasin, V.; Vazquez-Armendariz, A.I.; Chu, X.; Moiseenko, A.; Weiss, A.; Wilhelm, J.; Zhang, J.-S.; Kwapiszewska, G.; et al. Metformin induces lipogenic differentiation in myofibroblasts to reverse lung fibrosis. Nat. Commun. 2019, 10, 2987. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, Y.-Q.; Cai, G.-F.; Zeng, P.-P.; Dhlamini, Q.; Chen, L.-F.; Chen, J.-J.; Lyu, H.-D.; Mossahebi-Mohammadi, M.; Ahmadvand, N.; Bellusci, S.; et al. FGF10 Therapeutic Administration Promotes Mobilization of Injury-Activated Alveolar Progenitors in a Mouse Fibrosis Model. Cells 2022, 11, 2396. https://doi.org/10.3390/cells11152396
Lv Y-Q, Cai G-F, Zeng P-P, Dhlamini Q, Chen L-F, Chen J-J, Lyu H-D, Mossahebi-Mohammadi M, Ahmadvand N, Bellusci S, et al. FGF10 Therapeutic Administration Promotes Mobilization of Injury-Activated Alveolar Progenitors in a Mouse Fibrosis Model. Cells. 2022; 11(15):2396. https://doi.org/10.3390/cells11152396
Chicago/Turabian StyleLv, Yu-Qing, Ge-Fu Cai, Ping-Ping Zeng, Qhaweni Dhlamini, Le-Fu Chen, Jun-Jie Chen, Han-Deng Lyu, Majid Mossahebi-Mohammadi, Negah Ahmadvand, Saverio Bellusci, and et al. 2022. "FGF10 Therapeutic Administration Promotes Mobilization of Injury-Activated Alveolar Progenitors in a Mouse Fibrosis Model" Cells 11, no. 15: 2396. https://doi.org/10.3390/cells11152396
APA StyleLv, Y.-Q., Cai, G.-F., Zeng, P.-P., Dhlamini, Q., Chen, L.-F., Chen, J.-J., Lyu, H.-D., Mossahebi-Mohammadi, M., Ahmadvand, N., Bellusci, S., Li, X., Chen, C., & Zhang, J.-S. (2022). FGF10 Therapeutic Administration Promotes Mobilization of Injury-Activated Alveolar Progenitors in a Mouse Fibrosis Model. Cells, 11(15), 2396. https://doi.org/10.3390/cells11152396