
The Good and the Bad: Monocytes’ and Macrophages’ Diverse Functions in Inflammation


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Proinflammatory Immune Response
1.1. Inflammatory Monocytes and Macrophages
1.2. S100-Alarmins: Potent Effector Proteins of Inflammatory Macrophages
2. Anti-Inflammatory Immune Response
3. The Yin and Yang of Pro- and Anti-Inflammatory Macrophages under Pathological Conditions
3.1. Macrophages in SIRS: A Prototype of Overwhelming Inflammation
3.2. Pro- and Anti-Inflammatory Macrophages in SARS-CoV-2 Infection
3.3. Macrophages in Chronic Inflammatory Conditions
4. Modulation of the Macrophage Response as Innovative Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hörner, C.; Bouchon, A.; Bierhaus, A.; Nawroth, P.P.; Martin, E.; Bardenheuer, H.J.; Weigand, M.A. Bedeutung der angeborenen Immunantwort in der Sepsis. Anaesthesist 2004, 53, 10–28. [Google Scholar] [CrossRef] [PubMed]
- Jakubzick, C.V.; Randolph, G.J.; Henson, P.M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 2017, 17, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Mildner, A.; Yona, S. Developmental and Functional Heterogeneity of Monocytes. Immunity 2018, 49, 595–613. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion During Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.-F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Cox, N.; Pokrovskii, M.; Vicario, R.; Geissmann, F. Origins, Biology, and Diseases of Tissue Macrophages. Annu. Rev. Immunol. 2021, 39, 313–344. [Google Scholar] [CrossRef]
- Hoeffel, G.; Chen, J.; Lavin, Y.; Low, D.; Almeida, F.F.; See, P.; Beaudin, A.E.; Lum, J.; Low, I.; Forsberg, E.C.; et al. C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 2015, 42, 665–678. [Google Scholar] [CrossRef] [Green Version]
- Bain, C.C.; Bravo-Blas, A.; Scott, C.L.; Perdiguero, E.G.; Geissmann, F.; Henri, S.; Malissen, B.; Osborne, L.C.; Artis, D.; Mowat, A.M. Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat. Immunol. 2014, 15, 929–937. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Rosas, M.; Jenkins, S.J.; Liao, C.-T.; Scurr, M.J.; Brombacher, F.; Fraser, D.J.; Allen, J.E.; Jones, S.A.; Taylor, P.R. Distinct bone marrow-derived and tissue-resident macrophage lineages proliferate at key stages during inflammation. Nat. Commun. 2013, 4, 1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Janeway, C. Innate immune recognition: Mechanisms and pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [Green Version]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Bowie, A.G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like receptor signaling pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Goyal, A.; Wang, Y.; Graham, M.M.; Doseff, A.I.; Bhatt, N.Y.; Marsh, C.B. Monocyte survival factors induce Akt activation and suppress caspase-3. Am. J. Respir. Cell Mol. Biol. 2002, 26, 224–230. [Google Scholar] [CrossRef]
- Takeuchi, O.; Hemmi, H.; Akira, S. Interferon response induced by Toll-like receptor signaling. J. Endotoxin Res. 2004, 10, 252–256. [Google Scholar] [CrossRef]
- Biswas, S.K.; Lopez-Collazo, E. Endotoxin tolerance: New mechanisms, molecules and clinical significance. Trends Immunol. 2009, 30, 475–487. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Hessian, P.A.; Edgeworth, J.; Hogg, N. MRP-8 and MRP-14, two abundant Ca(2+)-binding proteins of neutrophils and monocytes. J. Leukoc. Biol. 1993, 53, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Frosch, M.; Ahlmann, M.; Vogl, T.; Wittkowski, H.; Wulffraat, N.; Foell, D.; Roth, J. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009, 60, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Van Zoelen, M.A.D.; Vogl, T.; Foell, D.; van Veen, S.Q.; van Till, J.W.O.; Florquin, S.; Tanck, M.W.; Wittebole, X.; Laterre, P.-F.; Boermeester, M.A.; et al. Expression and role of myeloid-related protein-14 in clinical and experimental sepsis. Am. J. Respir. Crit. Care Med. 2009, 180, 1098–1106. [Google Scholar] [CrossRef] [Green Version]
- Austermann, J.; Spiekermann, C.; Roth, J. S100 proteins in rheumatic diseases. Nat. Rev. Rheumatol. 2018, 14, 528–541. [Google Scholar] [CrossRef]
- Austermann, J.; Zenker, S.; Roth, J. S100-alarmins: Potential therapeutic targets for arthritis. Expert Opin. Ther. Targets 2017, 21, 739–751. [Google Scholar] [CrossRef]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; van Zoelen, M.A.D.; Nacken, W.; Foell, D.; van der Poll, T.; Sorg, C.; et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef]
- Foell, D.; Wittkowski, H.; Roth, J. Mechanisms of disease: A ‘DAMP’ view of inflammatory arthritis. Nat. Clin. Pract. Rheumatol. 2007, 3, 382–390. [Google Scholar] [CrossRef]
- Austermann, J.; Friesenhagen, J.; Fassl, S.K.; Petersen, B.; Ortkras, T.; Burgmann, J.; Barczyk-Kahlert, K.; Faist, E.; Zedler, S.; Pirr, S.; et al. Alarmins MRP8 and MRP14 induce stress tolerance in phagocytes under sterile inflammatory conditions. Cell Rep. 2014, 9, 2112–2123. [Google Scholar] [CrossRef] [Green Version]
- Freise, N.; Burghard, A.; Ortkras, T.; Daber, N.; Imam Chasan, A.; Jauch, S.-L.; Fehler, O.; Hillebrand, J.; Schakaki, M.; Rojas, J.; et al. Signaling mechanisms inducing hyporesponsiveness of phagocytes during systemic inflammation. Blood 2019, 134, 134–146. [Google Scholar] [CrossRef]
- Goerdt, S.; Politz, O.; Schledzewski, K.; Birk, R.; Gratchev, A.; Guillot, P.; Hakiy, N.; Klemke, C.D.; Dippel, E.; Kodelja, V.; et al. Alternative versus classical activation of macrophages. Pathobiology 1999, 67, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Topoll, H.H.; Zwadlo, G.; Lange, D.E.; Sorg, C. Phenotypic dynamics of macrophage subpopulations during human experimental gingivitis. J. Periodontal Res. 1989, 24, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Benoit, M.; Desnues, B.; Mege, J.-L. Macrophage polarization in bacterial infections. J. Immunol. 2008, 181, 3733–3739. [Google Scholar] [CrossRef] [Green Version]
- Shirey, K.A.; Pletneva, L.M.; Puche, A.C.; Keegan, A.D.; Prince, G.A.; Blanco, J.C.G.; Vogel, S.N. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. 2010, 3, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Rimoldi, M.; Raes, G.; Brys, L.; Ghezzi, P.; Di Liberto, D.; Dieli, F.; Ghisletti, S.; Natoli, G.; de Baetselier, P.; et al. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc. Natl. Acad. Sci. USA 2009, 106, 14978–14983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehrchen, J.; Steinmüller, L.; Barczyk, K.; Tenbrock, K.; Nacken, W.; Eisenacher, M.; Nordhues, U.; Sorg, C.; Sunderkötter, C.; Roth, J. Glucocorticoids induce differentiation of a specifically activated, anti-inflammatory subtype of human monocytes. Blood 2007, 109, 1265–1274. [Google Scholar] [CrossRef] [Green Version]
- Doyle, A.G.; Herbein, G.; Montaner, L.J.; Minty, A.J.; Caput, D.; Ferrara, P.; Gordon, S. Interleukin-13 alters the activation state of murine macrophages in vitro: Comparison with interleukin-4 and interferon-gamma. Eur. J. Immunol. 1994, 24, 1441–1445. [Google Scholar] [CrossRef]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef]
- Pesce, J.T.; Ramalingam, T.R.; Mentink-Kane, M.M.; Wilson, M.S.; El Kasmi, K.C.; Smith, A.M.; Thompson, R.W.; Cheever, A.W.; Murray, P.J.; Wynn, T.A. Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 2009, 5, e1000371. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.; Yu, X.; Gao, P.; Wang, Y.; Baek, S.-H.; Chen, X.; Kim, H.L.; Subjeck, J.R.; Wang, X.-Y. Pattern recognition scavenger receptor SRA/CD204 down-regulates Toll-like receptor 4 signaling-dependent CD8 T-cell activation. Blood 2009, 113, 5819–5828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azad, A.K.; Rajaram, M.V.S.; Schlesinger, L.S. Exploitation of the Macrophage Mannose Receptor (CD206) in Infectious Disease Diagnostics and Therapeutics. J. Cytol. Mol. Biol. 2014, 1, 1000003. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Lo, H.-M.; Chen, C.-L.; Yang, C.-M.; Wu, P.-H.; Tsou, C.-J.; Chiang, K.-W.; Wu, W.-B. The carotenoid lutein enhances matrix metalloproteinase-9 production and phagocytosis through intracellular ROS generation and ERK1/2, p38 MAPK, and RARβ activation in murine macrophages. J. Leukoc. Biol. 2013, 93, 723–735. [Google Scholar] [CrossRef]
- Murata, Y.; Shimamura, T.; Hamuro, J. The polarization of T(h)1/T(h)2 balance is dependent on the intracellular thiol redox status of macrophages due to the distinctive cytokine production. Int. Immunol. 2002, 14, 201–212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Choksi, S.; Chen, K.; Pobezinskaya, Y.; Linnoila, I.; Liu, Z.-G. ROS play a critical role in the differentiation of alternatively activated macrophages and the occurrence of tumor-associated macrophages. Cell Res. 2013, 23, 898–914. [Google Scholar] [CrossRef] [Green Version]
- Griess, B.; Mir, S.; Datta, K.; Teoh-Fitzgerald, M. Scavenging reactive oxygen species selectively inhibits M2 macrophage polarization and their pro-tumorigenic function in part, via Stat3 suppression. Free Radic. Biol. Med. 2020, 147, 48–60. [Google Scholar] [CrossRef]
- Johann, A.M.; von Knethen, A.; Lindemann, D.; Brüne, B. Recognition of apoptotic cells by macrophages activates the peroxisome proliferator-activated receptor-gamma and attenuates the oxidative burst. Cell Death Differ 2006, 13, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-N.; Surh, Y.-J. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem. Pharmacol. 2013, 86, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Oster, H.; Challet, E.; Ott, V.; Arvat, E.; de Kloet, E.R.; Dijk, D.-J.; Lightman, S.; Vgontzas, A.; van Cauter, E. The Functional and Clinical Significance of the 24-Hour Rhythm of Circulating Glucocorticoids. Endocr. Rev. 2017, 38, 3–45. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.S.; Stewart, P.M. 11Beta-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus-pituitary-adrenal axis, metabolic syndrome, and inflammation. J. Clin. Endocrinol. Metab. 2009, 94, 4645–4654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Nanni, L.; Novakovic, B.; Megchelenbrink, W.; Kuznetsova, T.; Stunnenberg, H.G.; Ceri, S.; Logie, C. Extensive epigenomic integration of the glucocorticoid response in primary human monocytes and in vitro derived macrophages. Sci. Rep. 2019, 9, 2772. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Riepe, L.; Daber, N.; Schulte-Schrepping, J.; Véras De Carvalho, B.C.; Russo, A.; Pohlen, M.; Fischer, J.; Chasan, A.I.; Wolf, M.; Ulas, T.; et al. CD163 expression defines specific, IRF8-dependent, immune-modulatory macrophages in the bone marrow. J. Allergy Clin. Immunol. 2020, 146, 1137–1151. [Google Scholar] [CrossRef] [PubMed]
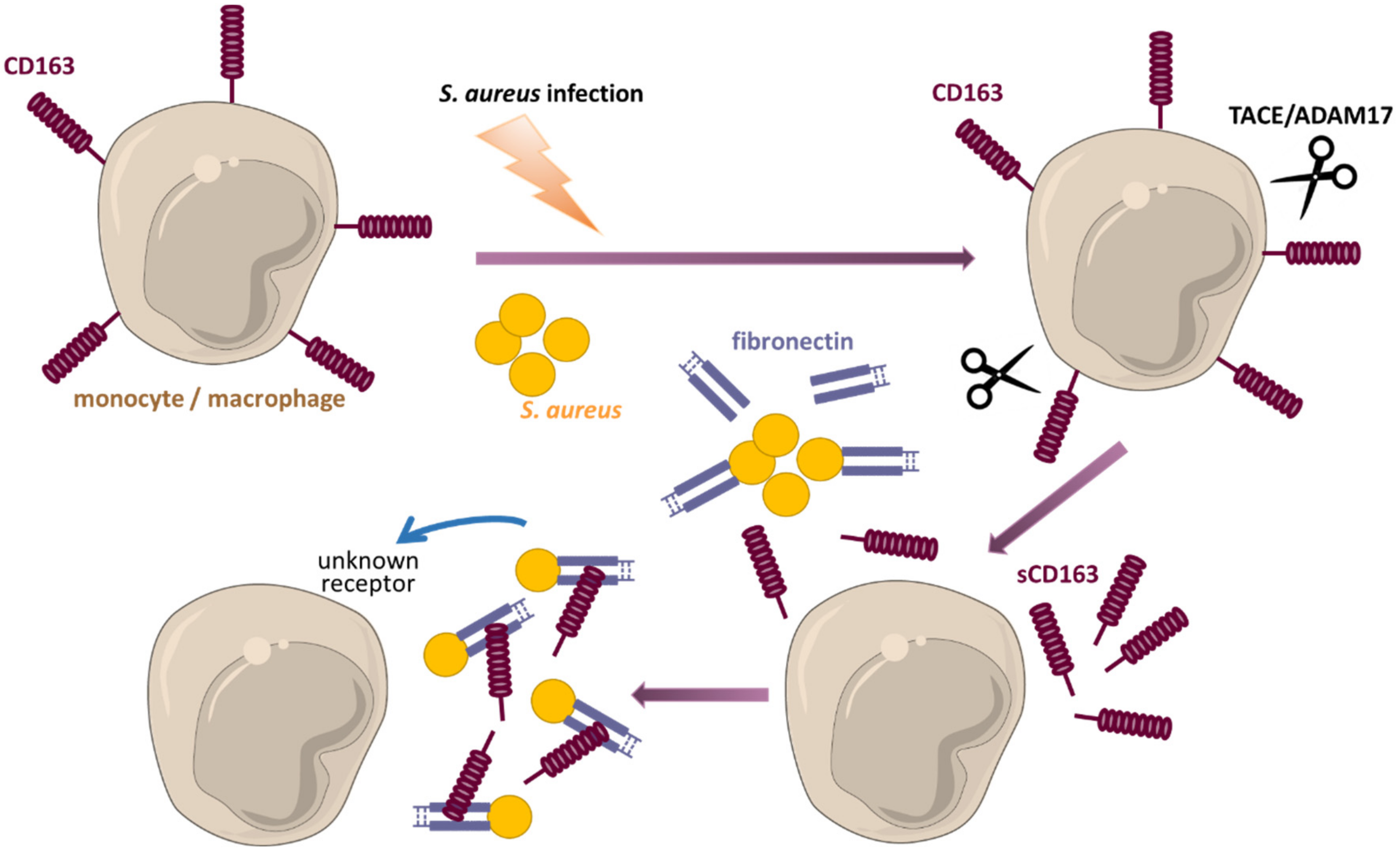
- Kneidl, J.; Löffler, B.; Erat, M.C.; Kalinka, J.; Peters, G.; Roth, J.; Barczyk, K. Soluble CD163 promotes recognition, phagocytosis and killing of Staphylococcus aureus via binding of specific fibronectin peptides. Cell. Microbiol. 2012, 14, 914–936. [Google Scholar] [CrossRef] [PubMed]
- Kneidl, J.; Mysore, V.; Geraci, J.; Tuchscherr, L.; Löffler, B.; Holzinger, D.; Roth, J.; Barczyk-Kahlert, K. Soluble CD163 masks fibronectin-binding protein A-mediated inflammatory activation of Staphylococcus aureus infected monocytes. Cell. Microbiol. 2014, 16, 364–377. [Google Scholar] [CrossRef]
- Etzerodt, A.; Moestrup, S.K. CD163 and inflammation: Biological, diagnostic, and therapeutic aspects. Antioxid. Redox Signal. 2013, 18, 2352–2363. [Google Scholar] [CrossRef] [Green Version]
- Graversen, J.H.; Madsen, M.; Moestrup, S.K. CD163: A signal receptor scavenging haptoglobin–hemoglobin complexes from plasma. Int. J. Biochem. Cell Biol. 2002, 34, 309–314. [Google Scholar] [CrossRef]
- Philippidis, P.; Mason, J.C.; Evans, B.J.; Nadra, I.; Taylor, K.M.; Haskard, D.O.; Landis, R.C. Hemoglobin scavenger receptor CD163 mediates interleukin-10 release and heme oxygenase-1 synthesis: Antiinflammatory monocyte-macrophage responses in vitro, in resolving skin blisters in vivo, and after cardiopulmonary bypass surgery. Circ. Res. 2004, 94, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Svendsen, P.; Etzerodt, A.; Deleuran, B.W.; Moestrup, S.K. Mouse CD163 deficiency strongly enhances experimental collagen-induced arthritis. Sci. Rep. 2020, 10, 12447. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Matsumoto, Y.; Satomi, H.; Tateish, A.; Ohya, M.; Ito, I.; Kanno, H. The ratio of CD163-positive macrophages to Iba1-positive macrophages is low in the intima in the early stage of cutaneous arteritis. Immunol. Res. 2020, 68, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Van der Goes, A.; Hoekstra, K.; van den Berg, T.K.; Dijkstra, C.D. Dexamethasone promotes phagocytosis and bacterial killing by human monocytes/macrophages in vitro. J. Leukoc. Biol. 2000, 67, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Gratchev, A.; Kzhyshkowska, J.; Utikal, J.; Goerdt, S. Interleukin-4 and dexamethasone counterregulate extracellular matrix remodelling and phagocytosis in type-2 macrophages. Scand. J. Immunol. 2005, 61, 10–17. [Google Scholar] [CrossRef]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Weigand, M.A.; Snyder-Ramos, S.A.; Mollers, A.G.; Bauer, J.; Hansen, D.; Kochen, W.; Martin, E.; Motsch, J. Inhaled nitric oxide does not enhance lipid peroxidation in patients with acute respiratory distress syndrome. Crit. Care Med. 2000, 28, 3429–3435. [Google Scholar] [CrossRef]
- Ohlsson, K.; Bjork, P.; Bergenfeldt, M.; Hageman, R.; Thompson, R.C. Interleukin-1 receptor antagonist reduces mortality from endotoxin shock. Nature 1990, 348, 550–552. [Google Scholar] [CrossRef]
- Echtenacher, B.; Falk, W.; Mannel, D.N.; Krammer, P.H. Requirement of endogenous tumor necrosis factor/cachectin for recovery from experimental peritonitis. J. Immunol. 1990, 145, 3762–3766. [Google Scholar]
- Hotchkiss, R.S.; Karl, I.E. The pathophysiology and treatment of sepsis. N. Engl. J. Med. 2003, 348, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Seeley, J.J.; Ghosh, S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 2017, 101, 107–119. [Google Scholar] [CrossRef]
- Foster, S.L.; Medzhitov, R. Gene-specific control of the TLR-induced inflammatory response. Clin. Immunol. 2009, 130, 7–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Fresno, C.; García-Rio, F.; Gómez-Piña, V.; Soares-Schanoski, A.; Fernández-Ruíz, I.; Jurado, T.; Kajiji, T.; Shu, C.; Marín, E.; Del Gutierrez Arroyo, A.; et al. Potent phagocytic activity with impaired antigen presentation identifying lipopolysaccharide-tolerant human monocytes: Demonstration in isolated monocytes from cystic fibrosis patients. J. Immunol. 2009, 182, 6494–6507. [Google Scholar] [CrossRef] [PubMed]
- Mages, J.; Dietrich, H.; Lang, R. A genome-wide analysis of LPS tolerance in macrophages. Immunobiology 2007, 212, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.C.; Gilliam, E.A.; Li, L. Innate immune programing by endotoxin and its pathological consequences. Front. Immunol. 2014, 5, 680. [Google Scholar] [CrossRef] [Green Version]
- Cavaillon, J.M.; Adib-Conquy, M. Bench-to-bedside review: Endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit. Care 2006, 10, 233. [Google Scholar]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef]
- West, M.A.; Heagy, W. Endotoxin tolerance: A review. Crit. Care Med. 2002, 30, S64–S73. [Google Scholar] [CrossRef]
- Biswas, S.K.; Bist, P.; Dhillon, M.K.; Kajiji, T.; Del Fresno, C.; Yamamoto, M.; Lopez-Collazo, E.; Akira, S.; Tergaonkar, V. Role for MyD88-independent, TRIF pathway in lipid A/TLR4-induced endotoxin tolerance. J. Immunol. 2007, 179, 4083–4092. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Tergaonkar, V. Myeloid differentiation factor 88-independent Toll-like receptor pathway: Sustaining inflammation or promoting tolerance? Int. J. Biochem. Cell Biol. 2007, 39, 1582–1592. [Google Scholar] [CrossRef]
- Chang, E.Y.; Guo, B.; Doyle, S.E.; Cheng, G. Cutting edge: Involvement of the type I IFN production and signaling pathway in lipopolysaccharide-induced IL-10 production. J. Immunol. 2007, 178, 6705–6709. [Google Scholar] [CrossRef] [Green Version]
- Ziegler-Heitbrock, L. The p50-homodimer mechanism in tolerance to LPS. J. Endotoxin Res. 2001, 7, 219–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayama, H.; Ramirez-Carrozzi, V.R.; Yamamoto, M.; Mizutani, T.; Kuwata, H.; Iba, H.; Matsumoto, M.; Honda, K.; Smale, S.T.; Takeda, K. Class-specific regulation of pro-inflammatory genes by MyD88 pathways and IκBζ. J. Biol. Chem. 2015, 290, 22446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covic, M.; Hassa, P.O.; Saccani, S.; Buerki, C.; Meier, N.I.; Lombardi, C.; Imhof, R.; Bedford, M.T.; Natoli, G.; Hottiger, M.O. Arginine methyltransferase CARM1 is a promoter-specific regulator of NF-kappaB-dependent gene expression. EMBO J. 2005, 24, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Saccani, S.; Pantano, S.; Natoli, G. p38-Dependent marking of inflammatory genes for increased NF-kappa B recruitment. Nat. Immunol. 2002, 3, 69–75. [Google Scholar] [CrossRef] [PubMed]
- De Santa, F.; Totaro, M.G.; Prosperini, E.; Notarbartolo, S.; Testa, G.; Natoli, G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 2007, 130, 1083–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.-J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, R.M.; Taganov, K.D.; Boldin, M.P.; Cheng, G.; Baltimore, D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. USA 2007, 104, 1604–1609. [Google Scholar] [CrossRef] [Green Version]
- Baltimore, D.; Boldin, M.P.; O’Connell, R.M.; Rao, D.S.; Taganov, K.D. MicroRNAs: New regulators of immune cell development and function. Nat. Immunol. 2008, 9, 839–845. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, R.J.W.; Gresnigt, M.S.; Joosten, L.A.B.; Netea, M.G. Cellular metabolism of myeloid cells in sepsis. J. Leukoc. Biol. 2017, 101, 151–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputa, G.; Castoldi, A.; Pearce, E.J. Metabolic adaptations of tissue-resident immune cells. Nat. Immunol. 2019, 20, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Balk, R.A. Systemic inflammatory response syndrome (SIRS): Where did it come from and is it still relevant today? Virulence 2014, 5, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rani, M.; Nicholson, S.E.; Zhang, Q.; Schwacha, M.G. Damage-associated molecular patterns (DAMPs) released after burn are associated with inflammation and monocyte activation. Burns 2017, 43, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.D.; Lee, Y.L.; Mulekar, S.; Kuck, J.L.; Brevard, S.B.; Gonzalez, R.P.; Gillespie, M.N.; Richards, W.O. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann. Surg. 2013, 258, 591-6; discussion 596-8. [Google Scholar] [CrossRef] [Green Version]
- Sherwood, E.R.; Koutrouvelis, A. Trauma-Induced Immunosuppression: Pathogenesis and Treatment. In Yearbook of Intensive Care and Emergency Medicine 2000; Vincent, J.-L., Ed.; Springer: Berlin/Heidelberg, Germany, 2000; pp. 51–62. ISBN 978-3-540-66830-5. [Google Scholar]
- Hensler, H.; Hecker, H.; Heeg, K.; Heidecke, C.D.; Bartels, H.; Barthlen, W.; Wagner, H.; Siewert, J.R.; Holzmann, B. Distinct mechanisms of immunosuppression as a consequence of major surgery. Infect. Immun. 1997, 65, 2283–2291. [Google Scholar] [CrossRef] [Green Version]
- Zakkar, M.; Guida, G.; Suleiman, M.S.; Angelini, G.D. Cardiopulmonary bypass and oxidative stress. Oxidative Med. Cell. Longev. 2015, 2015, 189863. [Google Scholar] [CrossRef]
- Loser, K.; Vogl, T.; Voskort, M.; Lueken, A.; Kupas, V.; Nacken, W.; Klenner, L.; Kuhn, A.; Foell, D.; Sorokin, L.; et al. The Toll-like receptor 4 ligands Mrp8 and Mrp14 are crucial in the development of autoreactive CD8+ T cells. Nat. Med. 2010, 16, 713–717. [Google Scholar] [CrossRef]
- Melo, E.S.; Barbeiro, H.V.; Ariga, S.; Goloubkova, T.; Curi, T.; Velasco, I.T.; Vasconcelos, D.; Soriano, F.G. Immune cells and oxidative stress in the endotoxin tolerance mouse model. Braz. J. Med. Biol. Res. 2010, 43, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Etzerodt, A.; Maniecki, M.B.; Møller, K.; Møller, H.J.; Moestrup, S.K. Tumor necrosis factor α-converting enzyme (TACE/ADAM17) mediates ectodomain shedding of the scavenger receptor CD163. J. Leukoc. Biol. 2010, 88, 1201–1205. [Google Scholar] [CrossRef] [PubMed]
- Etzerodt, A.; Berg, R.M.G.; Plovsing, R.R.; Andersen, M.N.; Bebien, M.; Habbeddine, M.; Lawrence, T.; Møller, H.J.; Moestrup, S.K. Soluble ectodomain CD163 and extracellular vesicle-associated CD163 are two differently regulated forms of ‘soluble CD163’ in plasma. Sci. Rep. 2017, 7, 40286. [Google Scholar] [CrossRef] [PubMed]
- Kjærgaard, A.G.; Rødgaard-Hansen, S.; Dige, A.; Krog, J.; Møller, H.J.; Tønnesen, E. Monocyte expression and soluble levels of the haemoglobin receptor (CD163/sCD163) and the mannose receptor (MR/sMR) in septic and critically ill non-septic ICU patients. PLoS ONE 2014, 9, e92331. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Zhou, X.; Su, L.-X.; Feng, D.; Jia, Y.-H.; Xie, L.-X. Clinical significance of soluble hemoglobin scavenger receptor CD163 (sCD163) in sepsis, a prospective study. PLoS ONE 2012, 7, e38400. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Xiong, X.; Ren, Y.; Wang, F.; Wang, C.; Zhang, Y. CD163 as a valuable diagnostic and prognostic biomarker of sepsis-associated hemophagocytic lymphohistiocytosis in critically ill children. Pediatr. Blood Cancer 2019, 66, e27909. [Google Scholar] [CrossRef]
- Knoll, R.; Schultze, J.L.; Schulte-Schrepping, J. Monocytes and Macrophages in COVID-19. Front. Immunol. 2021, 12, 720109. [Google Scholar] [CrossRef]
- Silvin, A.; Chapuis, N.; Dunsmore, G.; Goubet, A.-G.; Dubuisson, A.; Derosa, L.; Almire, C.; Hénon, C.; Kosmider, O.; Droin, N.; et al. Elevated Calprotectin and Abnormal Myeloid Cell Subsets Discriminate Severe from Mild COVID-19. Cell 2020, 182, 1401–1418.e18. [Google Scholar] [CrossRef]
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L.; et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell. Mol. Immunol. 2020, 17, 992–994. [Google Scholar] [CrossRef]
- Mellett, L.; Khader, S.A. S100A8/A9 in COVID-19 pathogenesis: Impact on clinical outcomes. Cytokine Growth Factor Rev. 2022, 63, 90–97. [Google Scholar] [CrossRef]
- LGuo, Q.; Zhao, Y.; Li, J.; Liu, J.; Yang, X.; Guo, X.; Kuang, M.; Xia, H.; Zhang, Z.; Cao, L.; et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Microbe 2021, 29, 222–235.e4. [Google Scholar]
- Trombetta, A.C.; Farias, G.B.; Gomes, A.M.C.; Godinho-Santos, A.; Rosmaninho, P.; Conceição, C.M.; Laia, J.; Santos, D.F.; Almeida, A.R.M.; Mota, C.; et al. Severe COVID-19 Recovery Is Associated with Timely Acquisition of a Myeloid Cell Immune-Regulatory Phenotype. Front. Immunol. 2021, 12, 691725. [Google Scholar] [CrossRef] [PubMed]
- Wendisch, D.; Dietrich, O.; Mari, T.; von Stillfried, S.; Ibarra, I.L.; Mittermaier, M.; Mache, C.; Chua, R.L.; Knoll, R.; Timm, S.; et al. SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell 2021, 184, 6243–6261.e27. [Google Scholar] [CrossRef] [PubMed]
- Laria, A.; Lurati, A.; Marrazza, M.; Mazzocchi, D.; Re, K.A.; Scarpellini, M. The macrophages in rheumatic diseases. J. Inflamm. Res. 2016, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Youssef, P.; Roth, J.; Frosch, M.; Costello, P.; Fitzgerald, O.; Sorg, C.; Bresnihan, B. Expression of myeloid related proteins (MRP) 8 and 14 and the MRP8/14 heterodimer in rheumatoid arthritis synovial membrane. J. Rheumatol. 1999, 26, 2523–2528. [Google Scholar] [PubMed]
- Van Lent, P.L.; Grevers, L.C.; Schelbergen, R.; Blom, A.; Geurts, J.; Sloetjes, A.; Vogl, T.; Roth, J.; van den Berg, W.B. S100A8 causes a shift toward expression of activatory Fcγ receptors on macrophages via toll-like receptor 4 and regulates Fcγ receptor expression in synovium during chronic experimental arthritis. Arthritis Rheum. 2010, 62, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Odink, K.; Cerletti, N.; Brüggen, J.; Clerc, R.G.; Tarcsay, L.; Zwadlo, G.; Gerhards, G.; Schlegel, R.; Sorg, C. Two calcium-binding proteins in infiltrate macrophages of rheumatoid arthritis. Nature 1987, 330, 80–82. [Google Scholar] [CrossRef]
- Sunahori, K.; Yamamura, M.; Yamana, J.; Takasugi, K.; Kawashima, M.; Yamamoto, H.; Chazin, W.J.; Nakatani, Y.; Yui, S.; Makino, H. The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, R69. [Google Scholar] [CrossRef] [Green Version]
- Grevers, L.C.; de Vries, T.J.; Vogl, T.; Abdollahi-Roodsaz, S.; Sloetjes, A.W.; Leenen, P.J.M.; Roth, J.; Everts, V.; van den Berg, W.B.; van Lent, P.L.E.M. S100A8 enhances osteoclastic bone resorption in vitro through activation of Toll-like receptor 4: Implications for bone destruction in murine antigen-induced arthritis. Arthritis Rheum. 2011, 63, 1365–1375. [Google Scholar] [CrossRef]
- Van Lent, P.L.E.M.; Grevers, L.; Blom, A.B.; Sloetjes, A.; Mort, J.S.; Vogl, T.; Nacken, W.; van den Berg, W.B.; Roth, J. Myeloid-related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann. Rheum. Dis. 2008, 67, 1750–1758. [Google Scholar] [CrossRef]
- Vogl, T.; Eisenblätter, M.; Völler, T.; Zenker, S.; Hermann, S.; van Lent, P.; Faust, A.; Geyer, C.; Petersen, B.; Roebrock, K.; et al. Alarmin S100A8/S100A9 as a biomarker for molecular imaging of local inflammatory activity. Nat. Commun. 2014, 5, 4593. [Google Scholar] [CrossRef] [Green Version]
- Völler, T.; Faust, A.; Roth, J.; Schäfers, M.; Vogl, T.; Hermann, S. A Non-Peptidic S100A9 Specific Ligand for Optical Imaging of Phagocyte Activity In Vivo. Mol. Imaging Biol. 2018, 20, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Faust, A.; Völler, T.; Busch, F.; Schäfers, M.; Roth, J.; Hermann, S.; Vogl, T. Development and evaluation of a non-peptidic ligand for the molecular imaging of inflammatory processes using S100A9 (MRP14) as a novel target. Chem. Commun. 2015, 51, 15637–15640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helfen, A.; Rieß, J.; Fehler, O.; Stölting, M.; An, Z.; Kocman, V.; Schnepel, A.; Geyer, C.; Gerwing, M.; Masthoff, M.; et al. In vivo imaging of microenvironmental and anti-PD-L1-mediated dynamics in cancer using S100A8/S100A9 as an imaging biomarker. Neoplasia 2022, 28, 100792. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Große Hokamp, N.; Zenker, S.; Flores-Borja, F.; Barzcyk, K.; Varga, G.; Roth, J.; Geyer, C.; Heindel, W.; Bremer, C.; et al. Optical in vivo imaging of the alarmin S100A9 in tumor lesions allows for estimation of the individual malignant potential by evaluation of tumor-host cell interaction. J. Nucl. Med. 2015, 56, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenblaetter, M.; Flores-Borja, F.; Lee, J.J.; Wefers, C.; Smith, H.; Hueting, R.; Cooper, M.S.; Blower, P.J.; Patel, D.; Rodriguez-Justo, M.; et al. Visualization of Tumor-Immune Interaction - Target-Specific Imaging of S100A8/A9 Reveals Pre-Metastatic Niche Establishment. Theranostics 2017, 7, 2392–2401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Møller, H.J. Soluble CD163. Scand. J. Clin. Lab. Invest. 2012, 72, 1–13. [Google Scholar] [CrossRef]
- Van Baarsen, L.G.M.; Wijbrandts, C.A.; Timmer, T.C.G.; van der Pouw Kraan, T.C.T.M.; Tak, P.P.; Verweij, C.L. Synovial tissue heterogeneity in rheumatoid arthritis in relation to disease activity and biomarkers in peripheral blood. Arthritis Rheum. 2010, 62, 1602–1607. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- King, R.J.; Shukla, S.K.; He, C.; Vernucci, E.; Thakur, R.; Attri, K.S.; Dasgupta, A.; Chaika, N.V.; Mulder, S.E.; Abrego, J.; et al. CD73 induces GM-CSF/MDSC-mediated suppression of T cells to accelerate pancreatic cancer pathogenesis. Oncogene 2022, 41, 971–982. [Google Scholar] [CrossRef]
- Roth, F.; de La Fuente, A.C.; Vella, J.L.; Zoso, A.; Inverardi, L.; Serafini, P. Aptamer-mediated blockade of IL4Rα triggers apoptosis of MDSCs and limits tumor progression. Cancer Res. 2012, 72, 1373–1383. [Google Scholar] [CrossRef] [Green Version]
- Beury, D.W.; Parker, K.H.; Nyandjo, M.; Sinha, P.; Carter, K.A.; Ostrand-Rosenberg, S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J. Leukoc. Biol. 2014, 96, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W.; et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020, 33, 108571. [Google Scholar] [CrossRef]
- Ortiz, M.L.; Kumar, V.; Martner, A.; Mony, S.; Donthireddy, L.; Condamine, T.; Seykora, J.; Knight, S.C.; Malietzis, G.; Lee, G.H.; et al. Immature myeloid cells directly contribute to skin tumor development by recruiting IL-17-producing CD4+ T cells. J. Exp. Med. 2015, 212, 351–367. [Google Scholar] [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef]
- Fujii, W.; Ashihara, E.; Hirai, H.; Nagahara, H.; Kajitani, N.; Fujioka, K.; Murakami, K.; Seno, T.; Yamamoto, A.; Ishino, H.; et al. Myeloid-derived suppressor cells play crucial roles in the regulation of mouse collagen-induced arthritis. J. Immunol. 2013, 191, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Brudecki, L.; Ferguson, D.A.; McCall, C.E.; El Gazzar, M. Myeloid-derived suppressor cells evolve during sepsis and can enhance or attenuate the systemic inflammatory response. Infect. Immun. 2012, 80, 2026–2034. [Google Scholar] [CrossRef] [Green Version]
- Dorhoi, A.; Du Plessis, N. Monocytic Myeloid-Derived Suppressor Cells in Chronic Infections. Front. Immunol. 2017, 8, 1895. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.-I.; Cheng, P.; Cho, H.-I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Mondini, M.; Loyher, P.-L.; Hamon, P.; Gerbé de Thoré, M.; Laviron, M.; Berthelot, K.; Clémenson, C.; Salomon, B.L.; Combadière, C.; Deutsch, E.; et al. CCR2-Dependent Recruitment of Tregs and Monocytes Following Radiotherapy Is Associated with TNFα-Mediated Resistance. Cancer Immunol. Res. 2019, 7, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Twum, D.Y.; Colligan, S.H.; Hoffend, N.C.; Katsuta, E.; Cortes Gomez, E.; Hensen, M.L.; Seshadri, M.; Nemeth, M.J.; Abrams, S.I. IFN regulatory factor-8 expression in macrophages governs an antimetastatic program. JCI Insight 2019, 4, e124267. [Google Scholar] [CrossRef]
- Sidibe, A.; Ropraz, P.; Jemelin, S.; Emre, Y.; Poittevin, M.; Pocard, M.; Bradfield, P.F.; Imhof, B.A. Angiogenic factor-driven inflammation promotes extravasation of human proangiogenic monocytes to tumours. Nat. Commun. 2018, 9, 355. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Li, X.; Tan, S.; Zhou, H.J.; Ji, W.; Bellone, S.; Xu, X.; Zhang, H.; Santin, A.D.; Lou, G.; et al. Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Invest. 2016, 126, 4157–4173. [Google Scholar] [CrossRef] [Green Version]
- Annamalai, R.T.; Turner, P.A.; Carson, W.F.; Levi, B.; Kunkel, S.; Stegemann, J.P. Harnessing macrophage-mediated degradation of gelatin microspheres for spatiotemporal control of BMP2 release. Biomaterials 2018, 161, 216–227. [Google Scholar] [CrossRef]
- Wei, Y.; Zhao, Q.; Gao, Z.; Lao, X.-M.; Lin, W.-M.; Chen, D.-P.; Mu, M.; Huang, C.-X.; Liu, Z.-Y.; Li, B.; et al. The local immune landscape determines tumor PD-L1 heterogeneity and sensitivity to therapy. J. Clin. Invest. 2019, 129, 3347–3360. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; Meng, J.; Zhang, Y.; Liu, J.; Nie, X.; Wu, F.; Yang, Y.; Wang, C.; Gu, N.; Xu, H. Macrophage phenotypic mechanomodulation of enhancing bone regeneration by superparamagnetic scaffold upon magnetization. Biomaterials 2017, 140, 16–25. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, N.; Li, Q.; Zhang, W.; Ke, F.; Leng, Q.; Wang, H.; Chen, J.; Wang, H. Tumor-associated macrophages recruit CCR6+ regulatory T cells and promote the development of colorectal cancer via enhancing CCL20 production in mice. PLoS ONE 2011, 6, e19495. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Perego, M.; Tyurin, V.A.; Tyurina, Y.Y.; Yellets, J.; Nacarelli, T.; Lin, C.; Nefedova, Y.; Kossenkov, A.; Liu, Q.; Sreedhar, S.; et al. Reactivation of dormant tumor cells by modified lipids derived from stress-activated neutrophils. Sci. Transl. Med. 2020, 12, eabb5817. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Kohanbash, G.; Liu, S.J.; Alvarado, B.; Carrera, D.; Bhaduri, A.; Watchmaker, P.B.; Yagnik, G.; Di Lullo, E.; Malatesta, M.; et al. Single-cell profiling of human gliomas reveals macrophage ontogeny as a basis for regional differences in macrophage activation in the tumor microenvironment. Genome Biol. 2017, 18, 234. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogl, T.; Stratis, A.; Wixler, V.; Völler, T.; Thurainayagam, S.; Jorch, S.K.; Zenker, S.; Dreiling, A.; Chakraborty, D.; Fröhling, M.; et al. Autoinhibitory regulation of S100A8/S100A9 alarmin activity locally restricts sterile inflammation. J. Clin. Invest. 2018, 128, 1852–1866. [Google Scholar] [CrossRef]
- Wei, S.; Chen, X.; Zhou, J.; Zhang, L.; Fortenbery, N.R.; Rasche, S.S.; McGraw, K.; Eksioglu, E.A.; Epling-Burnette, P.K.; Djeu, J.Y.; et al. Microenvironment Induced Myelodysplastic Syndrome (MDS) in S100A9 Transgenic Mice Caused by Myeloid-Derived Suppressor Cells (MDSC). Blood 2011, 118, 788. [Google Scholar] [CrossRef]
- Björk, P.; Björk, A.; Vogl, T.; Stenström, M.; Liberg, D.; Olsson, A.; Roth, J.; Ivars, F.; Leanderson, T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3-carboxamides. PLoS Biol. 2009, 7, e97. [Google Scholar] [CrossRef] [Green Version]
- Pruenster, M.; Kurz, A.R.M.; Chung, K.-J.; Cao-Ehlker, X.; Bieber, S.; Nussbaum, C.F.; Bierschenk, S.; Eggersmann, T.K.; Rohwedder, I.; Heinig, K.; et al. Extracellular MRP8/14 is a regulator of β2 integrin-dependent neutrophil slow rolling and adhesion. Nat. Commun. 2015, 6, 6915. [Google Scholar] [CrossRef] [Green Version]
- Comi, G.; Jeffery, D.; Kappos, L.; Montalban, X.; Boyko, A.; Rocca, M.A.; Filippi, M. Placebo-controlled trial of oral laquinimod for multiple sclerosis. N. Engl. J. Med. 2012, 366, 1000–1009. [Google Scholar] [CrossRef] [Green Version]
- D’Haens, G.; Sandborn, W.J.; Colombel, J.F.; Rutgeerts, P.; Brown, K.; Barkay, H.; Sakov, A.; Haviv, A.; Feagan, B.G. A phase II study of laquinimod in Crohn’s disease. Gut 2015, 64, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rammes, A.; Roth, J.; Goebeler, M.; Klempt, M.; Hartmann, M.; Sorg, C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J. Biol. Chem. 1997, 272, 9496–9502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzhndoian, Z.T. Serum levels of myeloid-related protein MRP 8/14 (calprotectin) in Armenian patients with familial mediterranean fever. Ter. Arkh. 2012, 84, 45–48. [Google Scholar] [PubMed]
- Hatterer, E.; Shang, L.; Simonet, P.; Herren, S.; Daubeuf, B.; Teixeira, S.; Reilly, J.; Elson, G.; Nelson, R.; Gabay, C.; et al. A specific anti-citrullinated protein antibody profile identifies a group of rheumatoid arthritis patients with a toll-like receptor 4-mediated disease. Arthritis Res. Ther. 2016, 18, 224. [Google Scholar] [CrossRef] [Green Version]
- Monnet, E.; Shang, L.; Lapeyre, G.; deGraaf, K.; Hatterer, E.; Buatois, V.; Elson, G.; Ferlin, W.; Gabay, C.; Sokolove, J.; et al. AB0451 NI-0101, a Monoclonal Antibody Targeting Toll Like Receptor 4 (TLR4) Being Developed for Rheumatoid Arthritis (RA) Treatment with a Potential for Personalized Medicine. Ann. Rheum. Dis. 2015, 74, 1046. [Google Scholar] [CrossRef]
- Hudson, B.I.; Lippman, M.E. Targeting RAGE Signaling in Inflammatory Disease. Annu. Rev. Med. 2018, 69, 349–364. [Google Scholar] [CrossRef]
- Sabbagh:, M.N.; Agro, A.; Bell, J.; Aisen, P.S.; Schweizer, E.; Galasko, D. PF-04494700, an oral inhibitor of receptor for advanced glycation end products (RAGE), in Alzheimer disease. Alzheimer Dis. Assoc. Disord. 2011, 25, 206–212. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Austermann, J.; Roth, J.; Barczyk-Kahlert, K. The Good and the Bad: Monocytes’ and Macrophages’ Diverse Functions in Inflammation. Cells 2022, 11, 1979. https://doi.org/10.3390/cells11121979
Austermann J, Roth J, Barczyk-Kahlert K. The Good and the Bad: Monocytes’ and Macrophages’ Diverse Functions in Inflammation. Cells. 2022; 11(12):1979. https://doi.org/10.3390/cells11121979
Chicago/Turabian StyleAustermann, Judith, Johannes Roth, and Katarzyna Barczyk-Kahlert. 2022. "The Good and the Bad: Monocytes’ and Macrophages’ Diverse Functions in Inflammation" Cells 11, no. 12: 1979. https://doi.org/10.3390/cells11121979
APA StyleAustermann, J., Roth, J., & Barczyk-Kahlert, K. (2022). The Good and the Bad: Monocytes’ and Macrophages’ Diverse Functions in Inflammation. Cells, 11(12), 1979. https://doi.org/10.3390/cells11121979