
Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Immune-Modulatory Function of Platelets
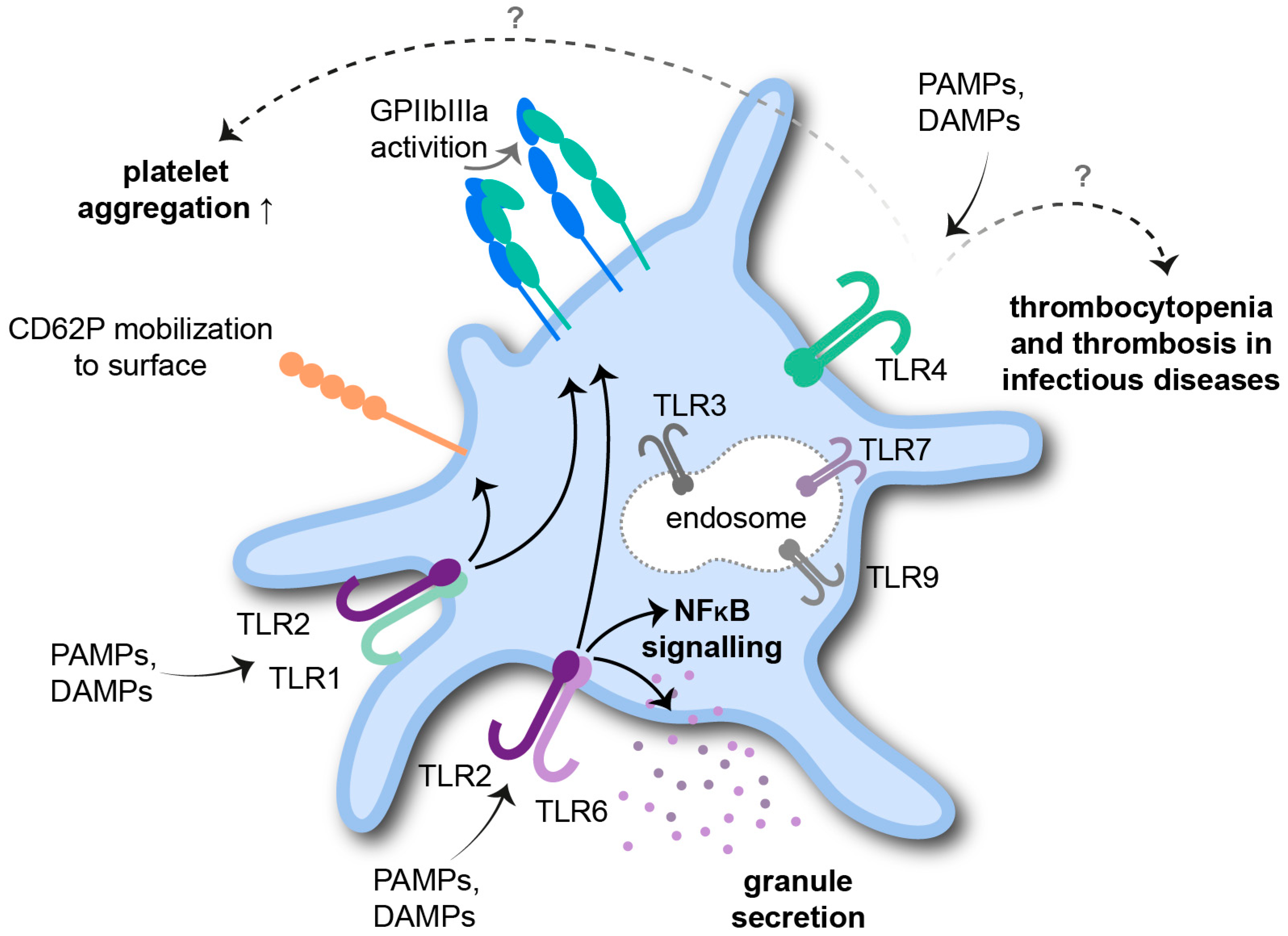
2.1. Platelet Toll-like Receptors
2.2. Platelet Granules
2.3. Platelet-Derived Microvesicles
2.4. Platelet-Leukocyte Interactions
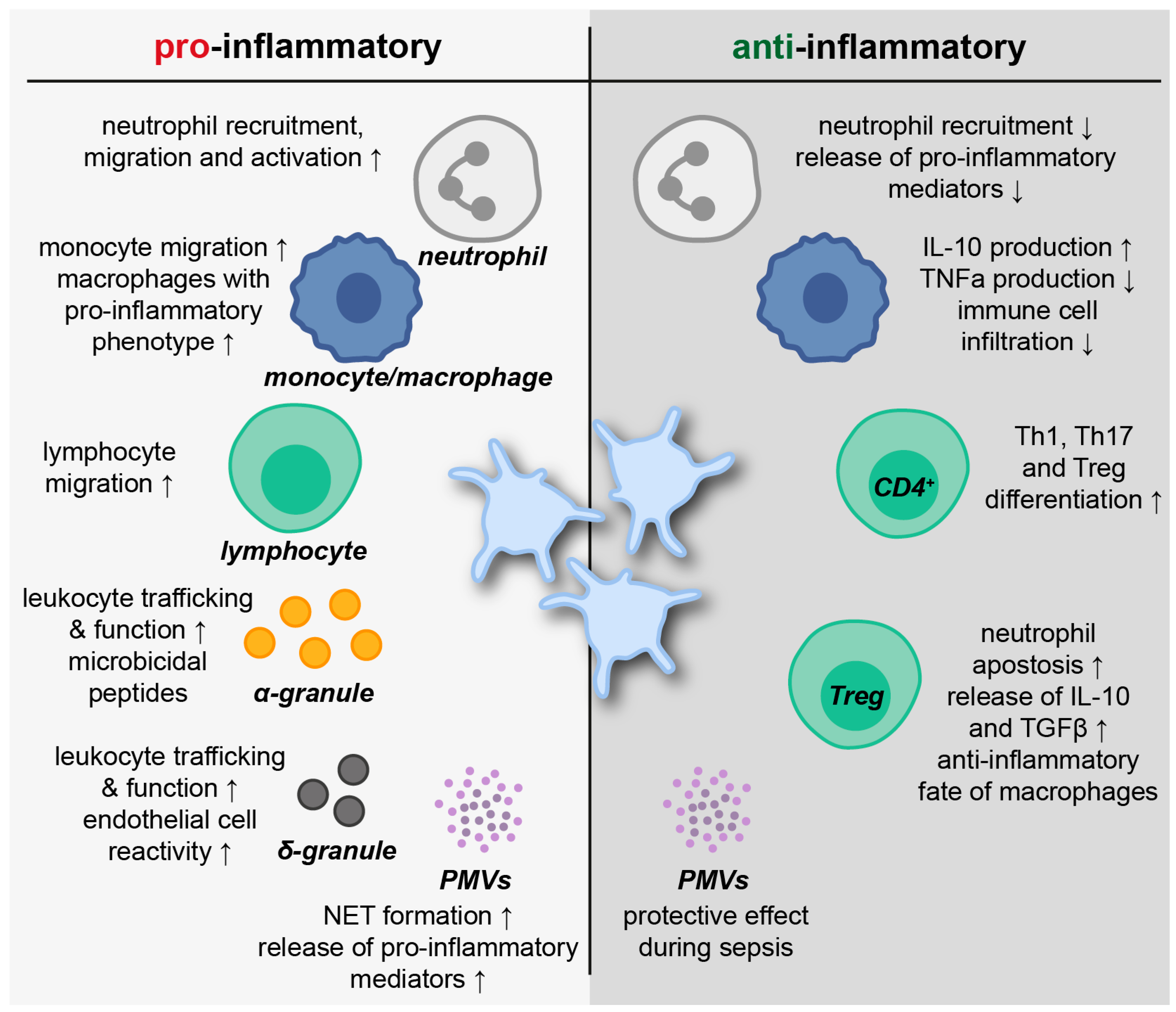
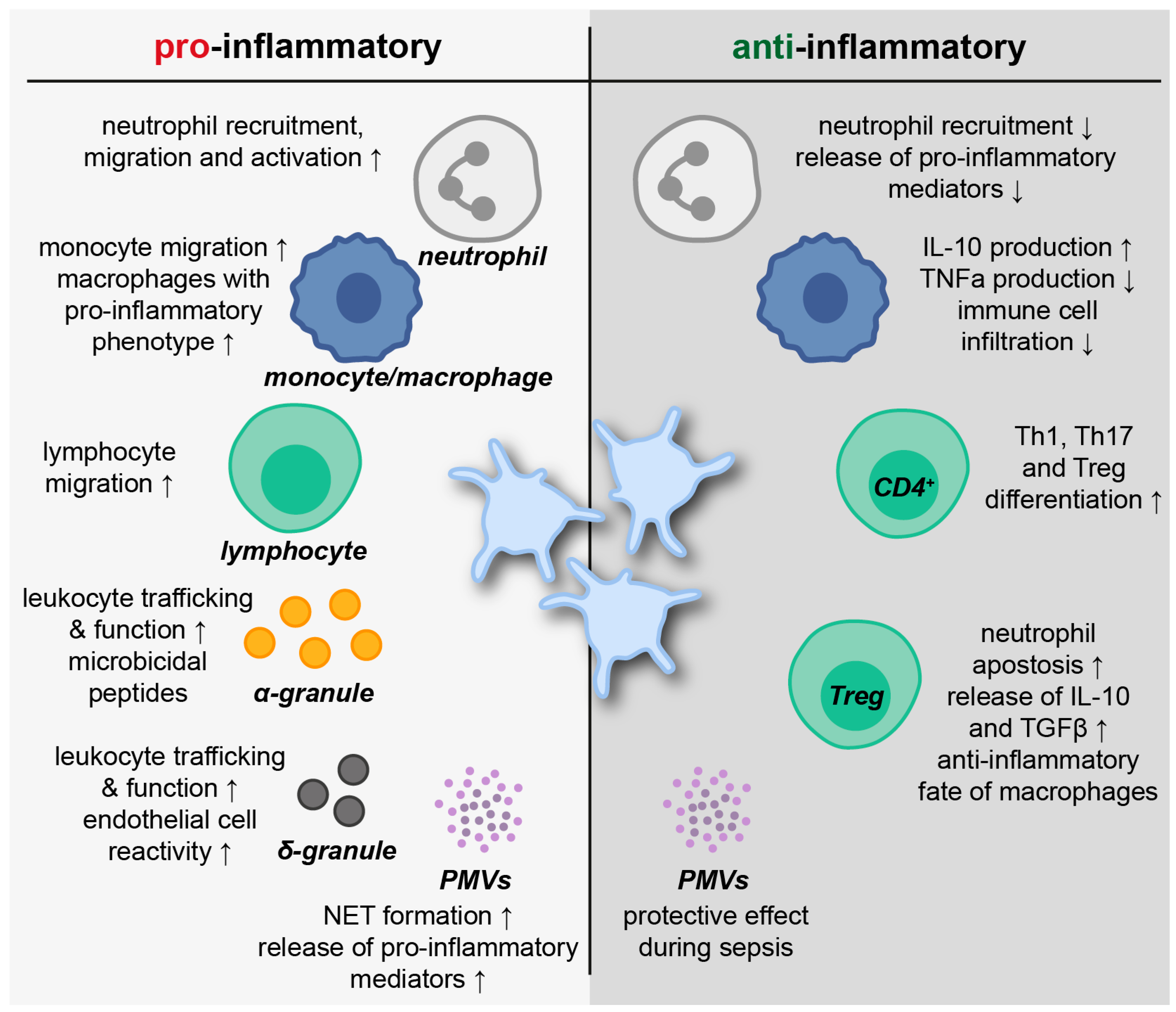
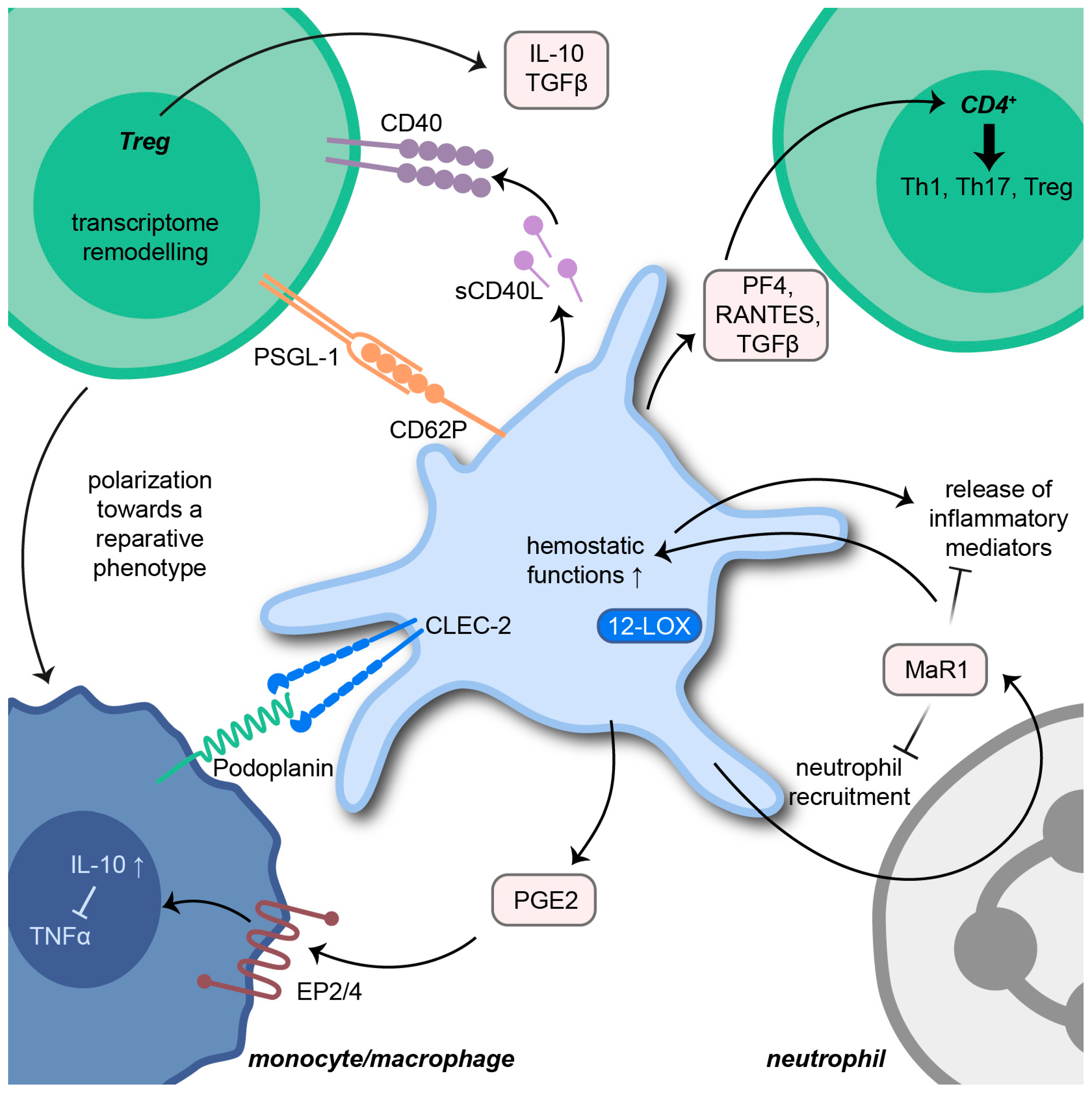
2.5. Platelets in Inflammatory Resolution
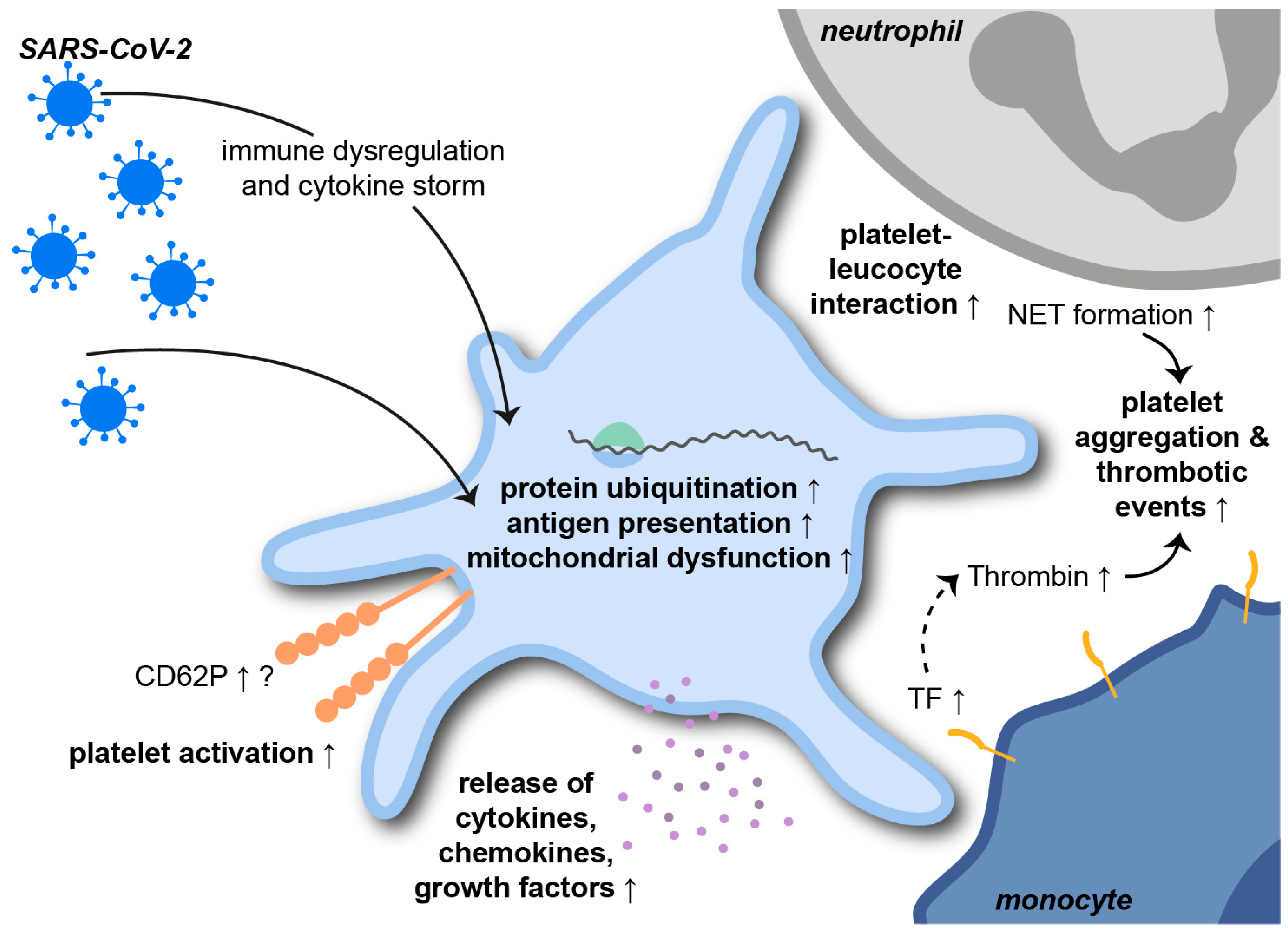
3. The Role of Platelets in Pulmonary Inflammation
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Brewer, D.B. Max Schultze (1865), G. Bizzozero (1882) and the Discovery of the Platelet. Br. J. Haematol. 2006, 133, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Kelemen, E.; Cserhati, I.; Tanos, B. Demonstration and Some Properties of Human Thrombopoietin in Thrombocythaemic Sera. Acta Haematol. 1958, 20, 350–355. [Google Scholar] [CrossRef]
- Lefrançais, E.; Ortiz-Muñoz, G.; Caudrillier, A.; Mallavia, B.; Liu, F.; Sayah, D.M.; Thornton, E.E.; Headley, M.B.; David, T.; Coughlin, S.R.; et al. The Lung Is a Site of Platelet Biogenesis and a Reservoir for Haematopoietic Progenitors. Nature 2017, 544, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.K.; Villacorta-Martin, C.; Hon, S.; Rock, J.R.; Murphy, G.J. Lung Megakaryocytes Display Distinct Transcriptional and Phenotypic Properties. Blood Adv. 2020, 4, 6204–6217. [Google Scholar] [CrossRef] [PubMed]
- Vannucchi, A.M.; Barbui, T. Thrombocytosis and Thrombosis. Hematology 2007, 1, 363–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harker, L.A.; Roskos, L.K.; Marzec, U.M.; Carter, R.A.; Cherry, J.K.; Sundell, B.; Cheung, E.N.; Terry, D.; Sheridan, W. Effects of Megakaryocyte Growth and Development Factor on Platelet Production, Platelet Life Span, and Platelet Function in Healthy Human Volunteers. Blood 2000, 95, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Bender, M.; Palankar, R. Platelet Shape Changes during Thrombus Formation: Role of Actin-Based Protrusions. Hamostaseologie 2021, 41, 14–21. [Google Scholar] [CrossRef]
- Springer, T.A.; Wang, J.H. The Three-Dimensional Structure of Integrins and Their Ligands, and Conformational Regulation of Cell Adhesion. Adv. Protein Chem. 2004, 68, 29–63. [Google Scholar] [CrossRef]
- Rossaint, J.; Margraf, A.; Zarbock, A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Chen, Y.; Yuan, Y.; Li, W. Sorting Machineries: How Platelet-Dense Granules Differ from α-Granules. Biosci. Rep. 2018, 38, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Margraf, A.; Zarbock, A. Platelets in Inflammation and Resolution. J. Immunol. 2019, 203, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Akinosoglou, K.; Alexopoulos, D. Use of Antiplatelet Agents in Sepsis: A Glimpse into the Future. Thromb. Res. 2014, 133, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, J.; Ni, H. Crosstalk Between Platelets and Microbial Pathogens. Front. Immunol. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- O’Brien, L.; Kerrigan, S.W.; Kaw, G.; Hogan, M.; Penadés, J.; Litt, D.; Fitzgerald, D.J.; Foster, T.J.; Cox, D. Multiple Mechanisms for the Activation of Human Platelet Aggregation by Staphylococcus Aureus: Roles for the Clumping Factors ClfA and ClfB, the Serine-Aspartate Repeat Protein SdrE and Protein A. Mol. Microbiol. 2002, 44, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Kerrigan, S.W.; Douglas, I.; Wray, A.; Heath, J.; Byrne, M.F.; Fitzgerald, D.; Cox, D. A Role for Glycoprotein Ib in Streptococcus Sanguis-Induced Platelet Aggregation. Blood 2002, 100, 509–516. [Google Scholar] [CrossRef]
- de Stoppelaar, S.F.; Claushuis, T.A.M.; Jansen, M.P.B.; Hou, B.; Roelofs, J.J.T.H.; van ’t Veer, C.; van der Poll, T. The Role of Platelet MyD88 in Host Response during Gram-Negative Sepsis. J. Thromb. Haemost. 2015, 13, 1709–1720. [Google Scholar] [CrossRef]
- Fitzgerald, J.R.; Loughman, A.; Keane, F.; Brennan, M.; Knobel, M.; Higgins, J.; Visai, L.; Speziale, P.; Cox, D.; Foster, T.J. Fibronectin-Binding Proteins of Staphylococcus Aureus Mediate Activation of Human Platelets via Fibrinogen and Fibronectin Bridges to Integrin GPIIb/IIIa and IgG Binding to the FcγRIIa Receptor. Mol. Microbiol. 2006, 59, 212–230. [Google Scholar] [CrossRef]
- Fitzgerald, J.R.; Foster, T.J.; Cox, D. The Interaction of Bacterial Pathogens with Platelets. Nat. Rev. Microbiol. 2006, 4, 445–457. [Google Scholar] [CrossRef]
- Byrne, M.F.; Kerrigan, S.W.; Corcoran, P.A.; Atherton, J.C.; Murray, F.E.; Fitzgerald, D.J.; Cox, D.M. Helicobacter Pylori Binds von Willebrand Factor and Interacts with GPIb to Induce Platelet Aggregation. Gastroenterology 2003, 124, 1846–1854. [Google Scholar] [CrossRef]
- O’Seaghdha, M.; Van Schooten, C.J.; Kerrigan, S.W.; Emsley, J.; Silverman, G.J.; Cox, D.; Lenting, P.J.; Foster, T.J. Staphylococcus Aureus Protein A Binding to von Willebrand Factor A1 Domain Is Mediated by Conserved IgG Binding Regions. FEBS J. 2006, 273, 4831–4841. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A Human Homologue of the Drosophila Toll Protein Signals Activation of Adaptive Immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like Receptors and Innate Immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, R.; Inoue, N.; Kawasaki, S.; Takei, A.; Kadotani, M.; Ohnishi, Y.; Ejiri, J.; Kobayashi, S.; Hirata, K.I.; Kawashima, S.; et al. Expression of Toll-like Receptors on Human Platelets. Thromb. Res. 2004, 113, 379–385. [Google Scholar] [CrossRef]
- Cognasse, F.; Hamzeh, H.; Chavarin, P.; Acquart, S.; Genin, C.; Garraud, O. Evidence of Toll-like Receptor Molecules on Human Platelets. Immunol. Cell Biol. 2005, 83, 196–198. [Google Scholar] [CrossRef]
- Anabel, A.S.; Eduardo, P.C.; Pedro Antonio, H.C.; Carlos, S.M.; Juana, N.M.; Honorio, T.A.; Nicolás, V.S.; Sergio Roberto, A.R. Human Platelets Express Toll-like Receptor 3 and Respond to Poly I:C. Hum. Immunol. 2014, 75, 1244–1251. [Google Scholar] [CrossRef]
- Koupenova, M.; Vitseva, O.; MacKay, C.R.; Beaulieu, L.M.; Benjamin, E.J.; Mick, E.; Kurt-Jones, E.A.; Ravid, K.; Freedman, J.E. Platelet-TLR7 Mediates Host Survival and Platelet Count during Viral Infection in the Absence of Platelet-Dependent Thrombosis. Blood 2014, 124, 791–802. [Google Scholar] [CrossRef] [Green Version]
- Andonegui, G.; Kerfoot, S.M.; McNagny, K.; Ebbert, K.V.J.; Patel, K.D.; Kubes, P. Platelets Express Functional Toll-like Receptor-4. Blood 2005, 106, 2417–2423. [Google Scholar] [CrossRef]
- Hally, K.; Fauteux-Daniel, S.; Hamzeh-Cognasse, H.; Larsen, P.; Cognasse, F. Revisiting Platelets and Toll-like Receptors (TLRS): At the Interface of Vascular Immunity and Thrombosis. Int. J. Mol. Sci. 2020, 21, 6150. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.L.; Casey, A.E.; Pollock, S.J.; Gertz, J.M.; McMillan, D.H.; Narasipura, S.D.; Mody, N.A.; King, M.R.; Maggirwar, S.B.; Francis, C.W.; et al. Platelets and Megakaryocytes Contain Functional Nuclear Factor-ΚB. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivadeneyra, L.; Carestia, A.; Etulain, J.; Pozner, R.G.; Fondevila, C.; Negrotto, S.; Schattner, M. Regulation of Platelet Responses Triggered by Toll-like Receptor 2 and 4 Ligands Is Another Non-Genomic Role of Nuclear Factor-KappaB. Thromb. Res. 2014, 133, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Farhat, K.; Riekenberg, S.; Heine, H.; Debarry, J.; Lang, R.; Mages, J.; Buwitt-Beckmann, U.; Röschmann, K.; Jung, G.; Wiesmüller, K.-H.; et al. Heterodimerization of TLR2 with TLR1 or TLR6 Expands the Ligand Spectrum but Does Not Lead to Differential Signaling. J. Leukoc. Biol. 2008, 83, 692–701. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Rex, S.; Vitseva, O.; Beaulieu, L.; Tanriverdi, K.; Chakrabarti, S.; Hayashi, C.; Genco, C.A.; Iafrati, M.; Freedman, J.E. Stimulation of Toll-Like Receptor 2 in Human Platelets Induces a Thromboinflammatory Response Through Activation of Phosphoinositide 3-Kinase. Circ. Res. 2009, 104, 346–354. [Google Scholar] [CrossRef] [Green Version]
- Damien, P.; Cognasse, F.; Payrastre, B.; Spinelli, S.L.; Blumberg, N.; Arthaud, C.A.; Eyraud, M.A.; Phipps, R.P.; McNicol, A.; Pozzetto, B.; et al. NF-ΚB Links TLR2 and PAR1 to Soluble Immunomodulator Factor Secretion in Human Platelets. Front. Immunol. 2017, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Parra-Izquierdo, I.; Lakshmanan, H.H.S.; Melrose, A.R.; Pang, J.; Zheng, T.J.; Jordan, K.R.; Reitsma, S.E.; McCarty, O.J.T.; Aslan, J.E. The Toll-Like Receptor 2 Ligand Pam2CSK4 Activates Platelet Nuclear Factor-ΚB and Bruton’s Tyrosine Kinase Signaling to Promote Platelet-Endothelial Cell Interactions. Front. Immunol. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Zhang, G.; Han, J.; Welch, E.J.; Ye, R.D.; Voyno-Yasenetskaya, T.A.; Malik, A.B.; Du, X.; Li, Z. Lipopolysaccharide Stimulates Platelet Secretion and Potentiates Platelet Aggregation via TLR4/MyD88 and the CGMP-Dependent Protein Kinase Pathway. J. Immunol. 2009, 182, 7997–8004. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, B.; Liang, Y.; Cao, S.; Liu, L.; Xu, X. Role of Platelet TLR4 Expression in Pathogensis of Septic Thrombocytopenia. World J. Emerg. Med. 2011, 2, 13–17. [Google Scholar] [CrossRef]
- Nocella, C.; Carnevale, R.; Bartimoccia, S.; Novo, M.; Cangemi, R.; Pastori, D.; Calvieri, C.; Pignatelli, P.; Violi, F. Lipopolysaccharide as Trigger of Platelet Aggregation via Eicosanoid Over-Production. Thromb. Haemost. 2017, 117, 1558–1570. [Google Scholar] [CrossRef] [Green Version]
- Schmid, W.; Novacek, G.; Vogelsang, H.; Papay, P.; Primas, C.; Eser, A.; Panzer, S. Platelets Toll-like Receptor-4 in Crohns Disease. Eur. J. Clin. Investig. 2017, 47, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Stark, R.J.; Aghakasiri, N.; Rumbaut, R.E. Platelet-Derived Toll-like Receptor 4 (Tlr-4) Is Sufficient to Promote Microvascular Thrombosis in Endotoxemia. PLoS ONE 2012, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claushuis, T.A.M.; Van Der Veen, A.I.P.; Horn, J.; Schultz, M.J.; Houtkooper, R.H.; Van ’T Veer, C.; Van Der Poll, T. Platelet Toll-like Receptor Expression and Activation Induced by Lipopolysaccharide and Sepsis. Platelets 2019, 30, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manne, B.K.; Xiang, S.C.; Rondina, M.T. Platelet Secretion in Inflammatory and Infectious Diseases. Platelets 2017, 28, 155–164. [Google Scholar] [CrossRef]
- Lemons, P.P.; Chen, D.; Bernstein, A.M.; Bennett, M.K.; Whiteheart, S.W. Regulated Secretion in Platelets: Identification of Elements of the Platelet Exocytosis Machinery. Blood 1997, 90, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Blair, P.; Flaumenhaft, R. Platelet α-Granules: Basic Biology and Clinical Correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Battinelli, E.M.; Thon, J.N.; Okazaki, R.; Peters, C.G.; Vijey, P.; Wilkie, A.R.; Noetzli, L.J.; Flaumenhaft, R.; Italiano, J.E. Megakaryocytes Package Contents into Separate A-Granules That Are Differentially Distributed in Platelets. Blood Adv. 2019, 3, 3092–3098. [Google Scholar] [CrossRef] [Green Version]
- Aziz, K.A.; Cawley, J.C.; Zdzel, M. Platelets Prime PMN via Released PF4: Mechanism of Priming and Synergy with GM-CSF. Br. J. Haematol. 1995, 91, 846–853. [Google Scholar] [CrossRef]
- Mayadas, T.N.; Johnson, R.C.; Rayburn, H.; Hynes, R.O.; Wagner, D.D. Leukocyte Rolling and Extravasation Are Severely Compromised in P Selectin-Deficient Mice. Cell 1993, 74, 541–545. [Google Scholar] [CrossRef]
- Santarlasci, V.; Maggi, L.; Capone, M.; Frosali, F.; Querci, V.; De Palma, R.; Liotta, F.; Cosmi, L.; Maggi, E.; Romagnani, S.; et al. TGF-β Indirectly Favors the Development of Human Th17 Cells by Inhibiting Th1 Cells. Eur. J. Immunol. 2009, 39, 207–215. [Google Scholar] [CrossRef]
- Morrell, C.N.; Aggrey, A.A.; Chapman, L.M.; Modjeski, K.L. Emerging Roles for Platelets as Immune and Inflammatory Cells. Blood 2014, 123, 2759–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.Q.; Yeaman, M.R.; Selsted, M.E. Antimicrobial Peptides from Human Platelets. Infect. Immun. 2002, 70, 6524–6533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krijgsveld, J.; Zaat, S.A.J.; Meeldijk, J.; Van Veelen, P.A.; Fang, G.; Poolman, B.; Brandt, E.; Ehlert, J.E.; Kuijpers, A.J.; Engbers, G.H.M.; et al. Thrombocidins, Microbicidal Proteins from Human Blood Platelets, Are C- Terminal Deletion Products of CXC Chemokines. J. Biol. Chem. 2000, 275, 20374–20381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, M.K.; Shahinian, H.K.; Michelassi, F. Lymphatic Smooth Muscle Responses to Leukotrienes, Histamine and Platelet Activating Factor. J. Surg. Res. 1988, 44, 172–177. [Google Scholar] [CrossRef]
- León-Ponte, M.; Ahern, G.P.; O’Connell, P.J. Serotonin Provides an Accessory Signal to Enhance T-Cell Activation by Signaling through the 5-HT7 Receptor. Blood 2007, 109, 3139–3146. [Google Scholar] [CrossRef] [Green Version]
- Swaim, A.F.; Field, D.J.; Fox-Talbot, K.; Baldwin, W.M.; Morrell, C.N. Platelets Contribute to Allograft Rejection through Glutamate Receptor Signaling. J. Immunol. 2010, 185, 6999–7006. [Google Scholar] [CrossRef] [Green Version]
- Heijnen, H.; van der Sluijs, P. Platelet Secretory Behaviour: As Diverse as the Granules… or Not? J. Thromb. Haemost. 2015, 13, 2141–2151. [Google Scholar] [CrossRef]
- Thon, J.N.; Peters, C.G.; Machlus, K.R.; Aslam, R.; Rowley, J.; Macleod, H.; Devine, M.T.; Fuchs, T.A.; Weyrich, A.S.; Semple, J.W.; et al. T Granules in Human Platelets Function in TLR9 Organization and Signaling. J. Cell Biol. 2012, 198, 561–574. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular Vesicles: Exosomes, Microvesicles, and Friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Słomka, A.; Urban, S.K.; Lukacs-Kornek, V.; Żekanowska, E.; Kornek, M. Large Extracellular Vesicles: Have We Found the Holy Grail of Inflammation? Front. Immunol. 2018, 9, 1–22. [Google Scholar] [CrossRef]
- Doyle, L.M.; Wang, Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menck, K.; Bleckmann, A.; Schulz, M.; Ries, L.; Binder, C. Isolation and Characterization of Microvesicles from Peripheral Blood. J. Vis. Exp. 2017, 119, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Berckmans, R.J.; Nieuwland, R.; Böing, A.N.; Romijn, F.P.H.T.M.; Hack, C.E.; Sturk, A. Cell-Derived Microparticles Circulate in Healthy Humans and Support Low Grade Thrombin Generation. Thromb. Haemost. 2001, 85, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijnen, H.F.G.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated Platelets Release Two Types of Membrane Vesicles: Microvesicles by Surface Shedding and Exosomes Derived from Exocytosis of Multivesicular Bodies and α-Granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Gkaliagkousi, E.; Nikolaidou, B.; Gavriilaki, E.; Lazaridis, A.; Yiannaki, E.; Anyfanti, P.; Zografou, I.; Markala, D.; Douma, S. Increased Erythrocyte- and Platelet-Derived Microvesicles in Newly Diagnosed Type 2 Diabetes Mellitus. Diabetes Vasc. Dis. Res. 2019, 16, 458–465. [Google Scholar] [CrossRef]
- Bratseth, V.; Chiva-Blanch, G.; Byrkjeland, R.; Solheim, S.; Arnesen, H.; Seljeflot, I. Elevated Levels of Circulating Microvesicles in Coronary Artery Disease Patients with Type 2 Diabetes and Albuminuria: Effects of Exercise Training. Diabetes Vasc. Dis. Res. 2019, 16, 431–439. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ma, K.L.; Gong, Y.X.; Wang, G.H.; Hu, Z.B.; Liu, L.; Lu, J.; Chen, P.P.; Lu, C.C.; Ruan, X.Z.; et al. Platelet Microparticles Mediate Glomerular Endothelial Injury in Early Diabetic Nephropathy. J. Am. Soc. Nephrol. 2018, 29, 2671–2695. [Google Scholar] [CrossRef]
- Boilard, E.; Nigrovic, P.A.; Larabee, K.; Watts, G.F.M.; Coblyn, J.S.; Weinblatt, M.E.; Massarotti, E.M.; Remold-O’Donnell, E.; Farndale, R.W.; Ware, J.; et al. Platelets Amplify Inflammation in Arthritis via Collagen-Dependent Microparticle Production. Science 2010, 327, 580–583. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V. Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Qi, Q.; Yang, B.; Li, H.; Bao, J.; Li, H.; Wang, B.; Mei, Q. Platelet Microparticles Regulate Neutrophil Extracellular Traps in Acute Pancreatitis. Pancreas 2020, 49, 1099–1103. [Google Scholar] [CrossRef]
- Ogura, H.; Kawasaki, T.; Tanaka, H.; Koh, T.; Tanaka, R.; Ozeki, Y.; Hosotsubo, H.; Kuwagata, Y.; Shimazu, T.; Sugimoto, H. Activated Platelets Enhance Microparticle Formation and Platelet-Leukocyte Interaction in Severe Trauma and Sepsis. J. Trauma-Inj. Infect. Crit. Care 2001, 50, 801–809. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, S.; Luo, L.; Norström, E.; Braun, O.; Mörgelin, M.; Thorlacius, H. Platelet-Derived Microparticles Regulates Thrombin Generation via Phophatidylserine in Abdominal Sepsis. J. Cell. Physiol. 2018, 233, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Janiszewski, M.; Do Carmo, A.O.; Pedro, M.A.; Silva, E.; Knobel, E.; Laurindo, F.R.M. Platelet-Derived Exosomes of Septic Individuals Possess Proapoptotic NAD(P)H Oxidase Activity: A Novel Vascular Redox Pathway. Crit. Care Med. 2004, 32, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Soriano, A.O.; Jy, W.; Chirinos, J.A.; Valdivia, M.A.; Velasquez, H.S.; Jimenez, J.J.; Horstman, L.L.; Kett, D.H.; Schein, R.M.H.; Ahn, Y.S. Levels of Endothelial and Platelet Microparticles and Their Interactions with Leukocytes Negatively Correlate with Organ Dysfunction and Predict Mortality in Severe Sepsis. Crit. Care Med. 2005, 33, 2540–2546. [Google Scholar] [CrossRef]
- Chen, X.; Liang, H.; Zhang, J.; Zen, K.; Zhang, C.Y. Secreted MicroRNAs: A New Form of Intercellular Communication. Trends Cell Biol. 2012, 22, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Szilágyi, B.; Fejes, Z.; Rusznyák, Á.; Fenyvesi, F.; Pócsi, M.; Halmi, S.; Griger, Z.; Kunapuli, S.P.; Kappelmayer, J.; Nagy, B. Platelet Microparticles Enriched in MiR-223 Reduce ICAM-1-Dependent Vascular Inflammation in Septic Conditions. Front. Physiol. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Lazar, S.; Goldfinger, L.E. Platelets and Extracellular Vesicles and Their Cross Talk with Cancer. Blood 2021, 137, 3192–3200. [Google Scholar] [CrossRef]
- Kim, H.K.; Song, K.S.; Park, Y.S.; Kang, Y.H.; Lee, Y.J.; Lee, K.R.; Kim, H.K.; Ryu, K.W.; Bae, J.M.; Kim, S. Elevated Levels of Circulating Platelet Microparticles, VEGF, IL-6 and RANTES in Patients with Gastric Cancer: Possible Role of a Metastasis Predictor. Eur. J. Cancer 2003, 39, 184–191. [Google Scholar] [CrossRef]
- Ren, J.G.; Man, Q.W.; Zhang, W.; Li, C.; Xiong, X.P.; Zhu, J.Y.; Wang, W.M.; Sun, Z.J.; Jia, J.; Zhang, W.F.; et al. Elevated Level of Circulating Platelet-Derived Microparticles in Oral Cancer. J. Dent. Res. 2016, 95, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Janowska-Wieczorek, A.; Marquez-Curtis, L.A.; Wysoczynski, M.; Ratajczak, M.Z. Enhancing Effect of Platelet-Derived Microvesicles on the Invasive Potential of Breast Cancer Cells. Transfusion 2006, 46, 1199–1209. [Google Scholar] [CrossRef]
- Totani, L.; Evangelista, V. Platelet-Leukocyte Interactions in Cardiovascular Disease and Beyond. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2357–2361. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Furie, B.C.; Furie, B. The Biology of P-Selectin Glycoprotein Ligand-1: Its Role as a Selectin Counterreceptor in Leukocyte-Endothelial and Leukocyte-Platelet Interaction. Thromb. Haemost. 1999, 81, 1–7. [Google Scholar] [PubMed]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.F.; McIntire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet Glycoprotein Ibα Is a Counterreceptor for the Leukocyte Integrin Mac-1 (CD11b/CD18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Springer, T.A. Neutrophil Accumulation on Activated, Surface-Adherent Platelets in Flow Is Mediated by Interaction of Mac-1 with Fibrinogen Bound to AIIbβ3 and Stimulated by Platelet-Activating Factor. J. Clin. Investig. 1997, 100, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Pluta, K.; Porębska, K.; Urbanowicz, T.; Gąsecka, A.; Olasińska-Wiśniewska, A.; Targoński, R.; Krasińska, A.; Filipiak, K.J.; Jemielity, M.; Krasiński, Z. Platelet–Leucocyte Aggregates as Novel Biomarkers in Cardiovascular Diseases. Biology 2022, 11, 224. [Google Scholar] [CrossRef]
- Finsterbusch, M.; Schrottmaier, W.C.; Kral-Pointner, J.B.; Salzmann, M.; Assinger, A. Measuring and Interpreting Platelet-Leukocyte Aggregates. Platelets 2018, 29, 677–685. [Google Scholar] [CrossRef]
- Kornerup, K.N.; Salmon, G.P.; Pitchford, S.C.; Liu, W.L.; Page, C.P. Circulating Platelet-Neutrophil Complexes Are Important for Subsequent Neutrophil Activation and Migration. J. Appl. Physiol. 2010, 109, 758–767. [Google Scholar] [CrossRef]
- Sreeramkumar, V.; Adrover, J.M.; Ballesteros, I.; Cuartero, M.I.; Rossaint, J.; Bilbao, I.; Nacher, M.; Pitaval, C.; Radovanovic, I.; Fukui, Y.; et al. Neutrophils Scan for Activated Platelets to Initiate Inflammation. Science 2014, 346, 1234–1238. [Google Scholar] [CrossRef] [Green Version]
- Rossaint, J.; Kühne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed Transport of Neutrophil-Derived Extracellular Vesicles Enables Platelet-Mediated Innate Immune Response. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Zuchtriegel, G.; Uhl, B.; Puhr-Westerheide, D.; Pörnbacher, M.; Lauber, K.; Krombach, F.; Reichel, C.A. Platelets Guide Leukocytes to Their Sites of Extravasation. PLoS Biol. 2016, 14, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Kral-Pointner, J.B.; Schrottmaier, W.C.; Salzmann, M.; Mussbacher, M.; Schmidt, G.J.; Moser, B.; Heber, S.; Birnecker, B.; Paar, H.; Zellner, M.; et al. Platelet PI3K Modulates Innate Leukocyte Extravasation during Acid-Induced Acute Lung Inflammation. Thromb. Haemost. 2019, 19, 1642–1654. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Singbartl, K.; Ley, K. Complete Reversal of Acid-Induced Acute Lung Injury by Blocking of Platelet-Neutrophil Aggregation. J. Clin. Investig. 2006, 116, 3211–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passacquale, G.; Vamadevan, P.; Pereira, L.; Hamid, C.; Corrigall, V.; Ferro, A. Monocyte-Platelet Interaction Induces a pro-Inflammatory Phenotype in Circulating Monocytes. PLoS ONE 2011, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diacovo, T.G.; Catalina, M.D.; Siegelman, M.H.; Von Andrian, U.H. Circulating Activated Platelets Reconstitute Lymphocyte Homing and Immunity in L-Selectin-Deficient Mice. J. Exp. Med. 1998, 187, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Ji, Q.; Hjemdahl, P. Platelet-Lymphocyte Conjugation Differs between Lymphocyte Subpopulations. J. Thromb. Haemost. 2006, 4, 874–881. [Google Scholar] [CrossRef]
- Alharbi, A.; Thompson, J.P.; Brindle, N.P.; Stover, C.M. Ex Vivo Modelling of the Formation of Inflammatory Platelet-Leucocyte Aggregates and Their Adhesion on Endothelial Cells, an Early Event in Sepsis. Clin. Exp. Med. 2019, 19, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M.; Fareh-Moghadam, S.; Pilz, G.; Gurland, H.; Werdan, K. Platelet Activation and Interaction with Leucocytes in Patients with Sepsis or Multiple Organ Failure. Eur. J. Clin. Investig. 1995, 25, 843–851. [Google Scholar] [CrossRef]
- Rondina, M.T.; Carlisle, M.; Fraughton, T.; Brown, S.M.; Miller, R.R.; Harris, E.S.; Weyrich, A.S.; Zimmerman, G.A.; Supiano, M.A.; Grissom, C.K. Platelet-Monocyte Aggregate Formation and Mortality Risk in Older Patients with Severe Sepsis and Septic Shock. J. Gerontol.-Ser. A Biol. Sci. Med. Sci. 2015, 70, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Petersen, F.; Bock, L.; Flad, H.-D.; Brandt, E. Platelet Factor 4-Induced Neutrophil-Endothelial Cell Interaction: Involvement of Mechanisms and Functional Consequences Different From Those Elicited by Interleukin-8. Blood 1999, 94, 4020–4028. [Google Scholar] [CrossRef]
- Fox, J.M.; Kausar, F.; Day, A.; Osborne, M.; Hussain, K.; Mueller, A.; Lin, J.; Tsuchiya, T.; Kanegasaki, S.; Pease, J.E. CXCL4/Platelet Factor 4 Is an Agonist of CCR1 and Drives Human Monocyte Migration. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Kameyoshi, Y.; Dörschner, A.; Mallet, A.I.; Christophers, E.; Schröder, J.M. Cytokine RANTES Released by Thrombin-Stimulated Platelets Is a Potent Attractant for Human E Inophils. J. Exp. Med. 1992, 176, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hundelshausen, P.; Weber, K.S.C.; Huo, Y.; Proudfoot, A.E.I.; Nelson, P.J.; Ley, K.; Weber, C. RANTES Deposition by Platelets Triggers Monocyte Arrest on Inflamed and Atherosclerotic Endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.I.; Hackeng, T.M.; Weber, C. Heterophilic Interactions of Platelet Factor 4 and RANTES Promote Monocyte Arrest on Endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, A.J.; Sepuru, K.M.; Sawant, K.V.; Rajarathnam, K. Platelet-Derived Chemokine CXCL7 Dimer Preferentially Exists in the Glycosaminoglycan-Bound Form: Implications for Neutrophil-Platelet Crosstalk. Front. Immunol. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemzadeh, M.; Kaplan, Z.S.; Alwis, I.; Schoenwaelder, S.M.; Ashworth, K.J.; Westein, E.; Hosseini, E.; Salem, H.H.; Slattery, R.; McColl, S.R.; et al. The CXCR1/2 Ligand NAP-2 Promotes Directed Intravascular Leukocyte Migration through Platelet Thrombi. Blood 2013, 121, 4555–4566. [Google Scholar] [CrossRef] [Green Version]
- Schenk, B.I.; Petersen, F.; Flad, H.-D.; Brandt, E. Platelet-Derived Chemokines CXC Chemokine Ligand (CXCL)7, Connective Tissue-Activating Peptide III, and CXCL4 Differentially Affect and Cross-Regulate Neutrophil Adhesion and Transendothelial Migration. J. Immunol. 2002, 169, 2602–2610. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-Derived HMGB1 Is a Critical Mediator of Thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Rath, D.; Borst, O.; Mack, A.; Loughran, P.; Lotze, M.T.; Neal, M.D.; Billiar, T.R.; Gawaz, M. Platelet-Derived High-Mobility Group Box 1 Promotes Recruitment and Suppresses Apoptosis of Monocytes. Biochem. Biophys. Res. Commun. 2016, 478, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 Activates Neutrophil Extracellular Traps to Ensnare Bacteria in Septic Blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets Induce Neutrophil Extracellular Traps in Transfusion-Related Acute Lung Injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Döring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized Integrin Engagement and Chemokine Activation Is Crucial in Neutrophil Extracellular Trap-Mediated Sterile Inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef] [PubMed]
- Vajen, T.; Koenen, R.R.; Werner, I.; Staudt, M.; Projahn, D.; Curaj, A.; Sönmez, T.T.; Simsekyilmaz, S.; Schumacher, D.; Möllmann, J.; et al. Blocking CCL5-CXCL4 Heteromerization Preserves Heart Function after Myocardial Infarction by Attenuating Leukocyte Recruitment and NETosis. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fullerton, J.N.; Gilroy, D.W. Resolution of Inflammation: A New Therapeutic Frontier. Nat. Rev. Drug Discov. 2016, 15, 551–567. [Google Scholar] [CrossRef] [PubMed]
- Headland, S.E.; Norling, L.V. The Resolution of Inflammation: Principles and Challenges. Semin. Immunol. 2015, 27, 149–160. [Google Scholar] [CrossRef]
- Sugimoto, M.A.; Sousa, L.P.; Pinho, V.; Perretti, M.; Teixeira, M.M. Resolution of Inflammation: What Controls Its Onset? Front. Immunol. 2016, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Schrottmaier, W.C.; Kral, J.B.; Badrnya, S.; Assinger, A. Aspirin and P2Y12 Inhibitors in Platelet-Mediated Activation of Neutrophils and Monocytes. Thromb. Haemost. 2015, 114, 478–489. [Google Scholar] [CrossRef]
- Ortiz-Muñoz, G.; Mallavia, B.; Bins, A.; Headley, M.; Krummel, M.F.; Looney, M.R. Aspirin-Triggered 15-Epi-Lipoxin A4 Regulates Neutrophil-Platelet Aggregation and Attenuates Acute Lung Injury in Mice. Blood 2014, 124, 2625–2634. [Google Scholar] [CrossRef] [Green Version]
- Evangelista, V.; Manarini, S.; Dell’Elba, G.; Martelli, N.; Napoleone, E.; Di Santo, A.; Savi, P.; Lorenzet, R. Clopidogrel Inhibits Platelet-Leukocyte Adhesion and Platelet-Dependent Leukocyte Activation. Thromb. Haemost. 2005, 94, 568–577. [Google Scholar] [CrossRef]
- Graff, J.; Harder, S.; Wahl, O.; Scheuermann, E.H.; Gossmann, J. Anti-Inflammatory Effects of Clopidogrel Intake in Renal Transplant Patients: Effects on Platelet-Leukocyte Interactions, Platelet CD40 Ligand Expression, and Proinflammatory Biomarkers. Clin. Pharmacol. Ther. 2005, 78, 468–476. [Google Scholar] [CrossRef]
- Liverani, E.; Rico, M.C.; Tsygankov, A.Y.; Kilpatrick, L.E.; Kunapuli, S.P. P2Y12 Receptor Modulates Sepsis-Induced Inflammation. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 961–971. [Google Scholar] [CrossRef] [Green Version]
- Borges-Rodriguez, M.; Shields, C.A.; Travis, O.K.; Tramel, R.W.; Baik, C.H.; Giachelli, C.A.; Tardo, G.A.; Williams, J.M.; Cornelius, D.C. Platelet Inhibition Prevents Nlrp3 Inflammasome Activation and Sepsis-Induced Kidney Injury. Int. J. Mol. Sci. 2021, 22, 10330. [Google Scholar] [CrossRef] [PubMed]
- Abdulnour, R.E.E.; Dalli, J.; Colby, J.K.; Krishnamoorthy, N.; Timmons, J.Y.; Tan, S.H.; Colas, R.A.; Petasis, N.A.; Serhan, C.N.; Levy, B.D. Maresin 1 Biosynthesis during Platelet-Neutrophil Interactions Is Organ-Protective. Proc. Natl. Acad. Sci. USA 2014, 111, 16526–16531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannan, K.L.; Spinelli, S.L.; Blumberg, N.; Phipps, R.P. Maresin 1 Induces a Novel Pro-Resolving Phenotype in Human Platelets. J. Thromb. Haemost. 2017, 15, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Van Dyke, T.E.; Serhan, C.N. Resolvin E1 Regulates Adenosine Diphosphate Activation of Human Platelets. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2005–2013. [Google Scholar] [CrossRef] [Green Version]
- Linke, B.; Schreiber, Y.; Picard-Willems, B.; Slattery, P.; Nüsing, R.M.; Harder, S.; Geisslinger, G.; Scholich, K. Activated Platelets Induce an Anti-Inflammatory Response of Monocytes/Macrophages through Cross-Regulation of PGE2 and Cytokines. Mediators Inflamm. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Rayes, J.; Lax, S.; Wichaiyo, S.; Watson, S.K.; Di, Y.; Lombard, S.; Grygielska, B.; Smith, S.W.; Skordilis, K.; Watson, S.P. The Podoplanin-CLEC-2 Axis Inhibits Inflammation in Sepsis. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Corken, A.; Russell, S.; Dent, J.; Post, S.R.; Ware, J. Platelet Glycoprotein Ib-IX as a Regulator of Systemic Inflammation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 996–1001. [Google Scholar] [CrossRef] [Green Version]
- Gerdes, N.; Zhu, L.; Ersoy, M.; Hermansson, A.; Hjemdahl, P.; Hu, H.; Hansson, G.K.; Nailin, L. Platelets Regulate CD4+ T-Cell Differentiation via Multiple Chemokines in Humans. Thromb. Haemost. 2011, 106, 353–362. [Google Scholar] [CrossRef]
- Rossaint, J.; Thomas, K.; Mersmann, S.; Skupski, J.; Margraf, A.; Tekath, T.; Jouvene, C.C.; Dalli, J.; Hidalgo, A.; Meuth, S.G.; et al. Platelets Orchestrate the Resolution of Pulmonary Inflammation in Mice by T Reg Cell Repositioning and Macrophage Education. J. Exp. Med. 2021, 218, 1–21. [Google Scholar] [CrossRef]
- Matthay, M.A.; Zemans, R.L.; Zimmerman, G.A.; Arabi, Y.M.; Beitler, J.R.; Mercat, A.; Herridge, M.; Randolph, A.G.; Calfee, C.S. Acute Respiratory Distress Syndrome. Nat. Rev. Dis. Prim. 2018, 5, 1–22. [Google Scholar] [CrossRef]
- Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute Respiratory Distress Syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [PubMed]
- Lee, K.Y. Pneumonia, Acute Respiratory Distress Syndrome, and Early Immune-Modulator Therapy. Int. J. Mol. Sci. 2017, 18, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.J.M.; Leong, J.Y.; Lee, J.H.; Albani, S.; Yeo, J.G. Insights into the Immuno-Pathogenesis of Acute Respiratory Distress Syndrome. Ann. Transl. Med. 2019, 7, 504. [Google Scholar] [CrossRef] [PubMed]
- Cleary, S.J.; Hobbs, C.; Amison, R.T.; Arnold, S.; O’Shaughnessy, B.G.; Lefrançais, E.; Mallavia, B.; Looney, M.R.; Page, C.P.; Pitchford, S.C. LPS-Induced Lung Platelet Recruitment Occurs Independently from Neutrophils, PSGL-1, and P-Selectin. Am. J. Respir. Cell Mol. Biol. 2019, 61, 232–243. [Google Scholar] [CrossRef] [PubMed]
- de Stoppelaar, S.F.; van’t Veer, C.; Roelofs, J.J.T.H.; Claushuis, T.A.M.; de Boer, O.J.; Tanck, M.W.T.; Hoogendijk, A.J.; van der Poll, T. Platelet and Endothelial Cell P-Selectin Are Required for Host Defense against Klebsiella Pneumoniae-Induced Pneumosepsis. J. Thromb. Haemost. 2015, 13, 1128–1138. [Google Scholar] [CrossRef]
- De Stoppelaar, S.F.; Claushuis, T.A.M.; Schaap, M.C.L.; Hou, B.; Van Der Poll, T.; Nieuwland, R.; Van ’T Veer, C. Toll-like Receptor Signalling Is Not Involved in Platelet Response to Streptococcus Pneumoniae in Vitro or in Vivo. PLoS ONE 2016, 11, 1–17. [Google Scholar] [CrossRef]
- Letsiou, E.; Alves, L.G.T.; Felten, M.; Mitchell, T.J.; Müller-Redetzky, H.C.; Dudek, S.M.; Witzenrath, M. Neutrophil-Derived Extracellular Vesicles Activate Platelets after Pneumolysin Exposure. Cells 2021, 10, 3581. [Google Scholar] [CrossRef]
- Nishat, S.; Wuescher, L.M.; Worth, R.G. Platelets Enhance Dendritic Cell Responses against Staphylococcus Aureus through CD40-CD40L. Infect. Immun. 2018, 86, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Bain, W.; Olonisakin, T.; Yu, M.; Qu, Y.; Hulver, M.; Xiong, Z.; Li, H.; Pilewski, J.; Mallampalli, R.K.; Nouraie, M.; et al. Platelets Inhibit Apoptotic Lung Epithelial Cell Death and Protect Mice against Infection-Induced Lung Injury. Blood Adv. 2019, 3, 432–445. [Google Scholar] [CrossRef]
- Wang, T.; Liu, Z.; Wang, Z.; Duan, M.; Li, G.; Wang, S.; Li, W.; Zhu, Z.; Wei, Y.; Christiani, D.C.; et al. Thrombocytopenia Is Associated with Acute Respiratory Distress Syndrome Mortality: An International Study. PLoS ONE 2014, 9, 1–10. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middelton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet Gene Expression and Function in Patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Abdulnour, R.E.E.; Gunderson, T.; Barkas, I.; Timmons, J.Y.; Barnig, C.; Gong, M.; Kor, D.J.; Gajic, O.; Talmor, D.; Carter, R.E.; et al. Early Intravascular Events Are Associated with Development of Acute Respiratory Distress Syndrome a Substudy of the Lips-A Clinical Trial. Am. J. Respir. Crit. Care Med. 2018, 197, 1575–1585. [Google Scholar] [CrossRef] [PubMed]
- Gries, A.; Herr, A.; Kirsch, S.; Günther, C.; Weber, S.; Szabo, G.; Holzmann, A.; Böttiger, B.W.; Martin, E. Inhaled Nitric Oxide Inhibits Platelet-Leukocyte Interactions in Patients with Acute Respiratory Distress Syndrome. Crit. Care Med. 2003, 31, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Burley, D.S.; Ferdinandy, P.; Baxter, G.F. Cyclic GMP and Protein Kinase-G in Myocardial Ischaemia-Reperfusion: Opportunities and Obstacles for Survival Signaling. Br. J. Pharmacol. 2007, 152, 855–869. [Google Scholar] [CrossRef] [Green Version]
- Pulavendran, S.; Rudd, J.M.; Maram, P.; Thomas, P.G.; Akhilesh, R.; Malayer, J.R.; Chow, V.T.K.; Teluguakula, N. Combination Therapy Targeting Platelet Activation and Virus Replication Protects Mice against Lethal Influenza Pneumonia. Am. J. Respir. Cell Mol. Biol. 2019, 61, 689–701. [Google Scholar] [CrossRef]
- Sugiyama, M.G.; Gamage, A.; Zyla, R.; Armstrong, S.M.; Advani, S.; Advani, A.; Wang, C.; Lee, W.L. Influenza Virus Infection Induces Platelet-Endothelial Adhesion Which Contributes to Lung Injury. J. Virol. 2016, 90, 1812–1823. [Google Scholar] [CrossRef] [Green Version]
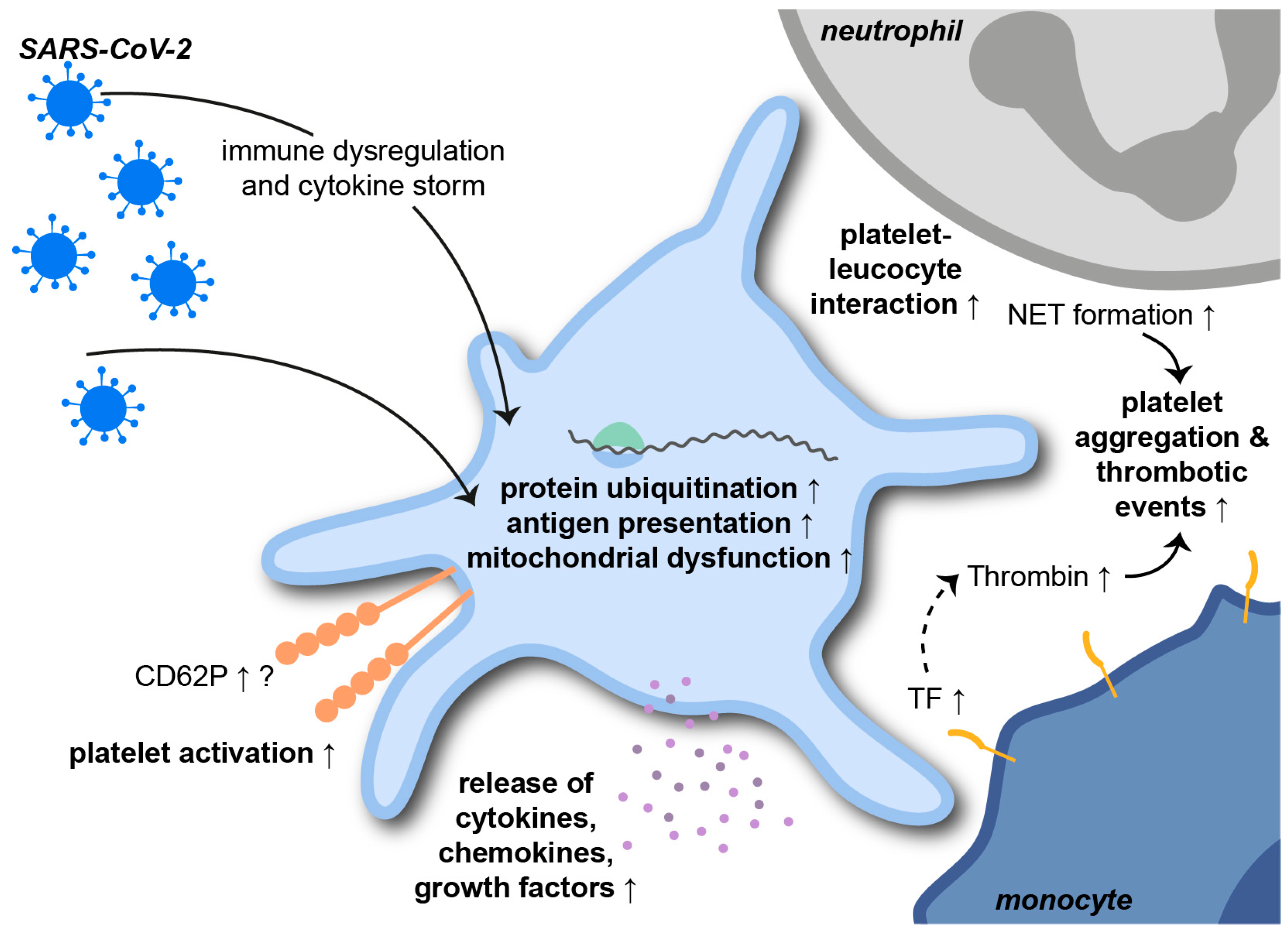
- Rohlfing, A.-K.; Rath, D.; Geisler, T.; Gawaz, M. Platelets and COVID-19. Hamostaseologie 2021, 41, 379–385. [Google Scholar] [CrossRef]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Tanriverdi, K.; Somasundaran, M.; Liu, P.; Soofi, S.; Bhandari, R.; Godwin, M.; Parsi, K.M.; et al. SARS-CoV-2 Initiates Programmed Cell Death in Platelets. Circ. Res. 2021, 129, 631–646. [Google Scholar] [CrossRef]
- Taus, F.; Salvagno, G.; Canè, S.; Fava, C.; Mazzaferri, F.; Carrara, E.; Petrova, V.; Barouni, R.M.; Dima, F.; Dalbeni, A.; et al. Platelets Promote Thromboinflammation in SARS-CoV-2 Pneumonia. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2975–2989. [Google Scholar] [CrossRef]
- Middleton, A.E.; He, Y.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil Extracellular Traps Contribute to Immunothrombosis in COVID-19 Acute Respiratory Distress Syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and Tissue Factor-Enriched Neutrophil Extracellular Traps Are Key Drivers in COVID-19 Immunothrombosis. J. Clin. Investig. 2020, 130, 6151. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet Activation and Platelet-Monocyte Aggregate Formation Trigger Tissue Factor Expression in Patients with Severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Caillon, A.; Trimaille, A.; Favre, J.; Jesel, L.; Morel, O.; Kauffenstein, G. Role of Neutrophils, Platelets, and Extracellular Vesicles and Their Interactions in COVID-19-Associated Thrombopathy. J. Thromb. Haemost. 2022, 20, 17–31. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ludwig, N.; Hilger, A.; Zarbock, A.; Rossaint, J. Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation. Cells 2022, 11, 1957. https://doi.org/10.3390/cells11121957
Ludwig N, Hilger A, Zarbock A, Rossaint J. Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation. Cells. 2022; 11(12):1957. https://doi.org/10.3390/cells11121957
Chicago/Turabian StyleLudwig, Nadine, Annika Hilger, Alexander Zarbock, and Jan Rossaint. 2022. "Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation" Cells 11, no. 12: 1957. https://doi.org/10.3390/cells11121957
APA StyleLudwig, N., Hilger, A., Zarbock, A., & Rossaint, J. (2022). Platelets at the Crossroads of Pro-Inflammatory and Resolution Pathways during Inflammation. Cells, 11(12), 1957. https://doi.org/10.3390/cells11121957