
Generating iPSCs with a High-Efficient, Non-Invasive Method—An Improved Way to Cultivate Keratinocytes from Plucked Hair for Reprogramming

 ,
,  ,
,  and
and
Abstract
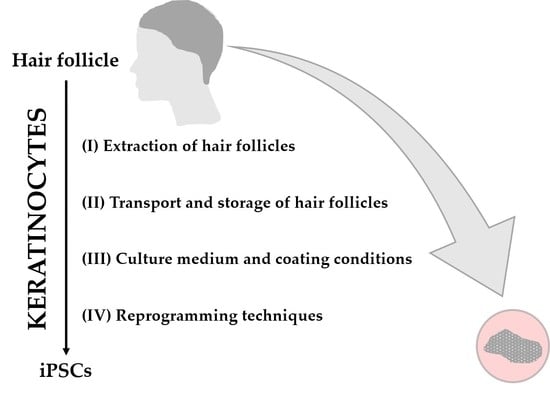
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Plucking and Preparation of Human Hair Follicles (HFs)
2.2. Cultivation of HF-Derived Keratinocytes
2.3. Media Used in This Study
2.4. Coating Solution and Mounting Droplet Used in This Study
2.5. Defining Outgrowing Keratinocytes from HF “Circle Condition”
2.6. Reprogramming of Keratinocytes
2.7. Quality Control of Generated iPSCs
2.8. Histological H&E Staining
2.9. Alkaline Phosphatase Staining
2.10. Immunofluorescence
2.11. Quantitative RT-PCR
2.12. Microscopic Imaging
2.13. Statistics
3. Results
3.1. Extraction of Human Hair Follicles (HFs)
3.1.1. Anatomical Structure of Human HFs Used for This Study
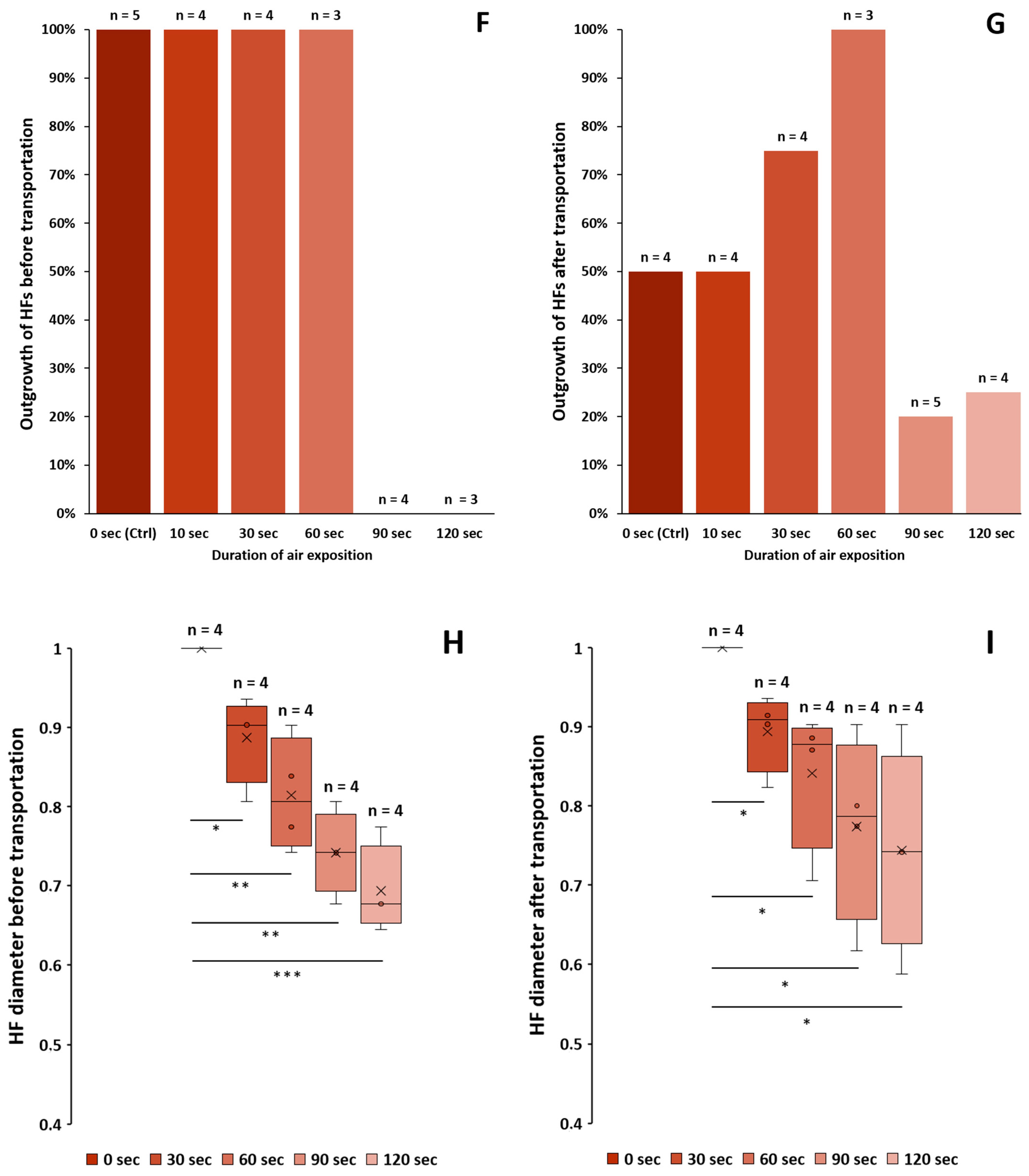
3.1.2. Dehydration of Plucked HFs
3.1.3. Outgrowth from HFs before and after Transportation
3.1.4. Diameter of the HFs before and after Transportation
- ➔
- Dehydration of ORS can be observed with the naked eye.
- ➔
- Plucked HF samples must be transferred to a transportation medium immediately without extended exposure to air.
3.2. Transport and Safekeeping of Human Hair Follicles
3.2.1. Definition of the “Circle Condition”
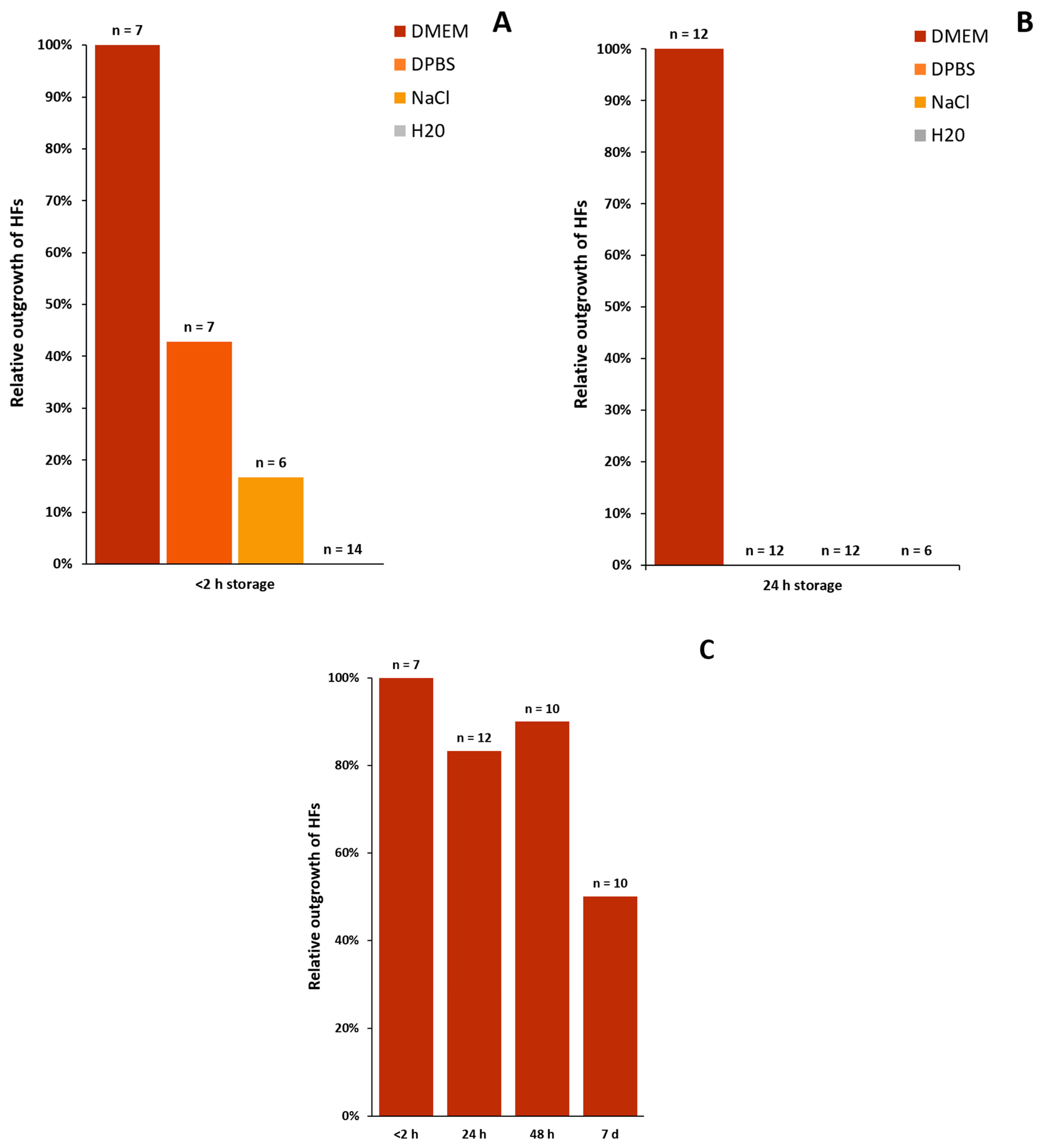
3.2.2. Transport Media
- ➔
- DMEM is the only appropriate media for the transport and storage of plucked HFs when used at room temperature.
- ➔
- To achieve the best results for outgrowing cells from HFs, the duration between the plucking and processing of the specimen should be a maximum of 2 h.
- ➔
- It is still possible (50%) to cultivate keratinocytes from HFs even after one week in DMEM when stored at room temperature.
3.3. Appropriate Culture Medium
3.3.1. Primary Culture Media in Comparison
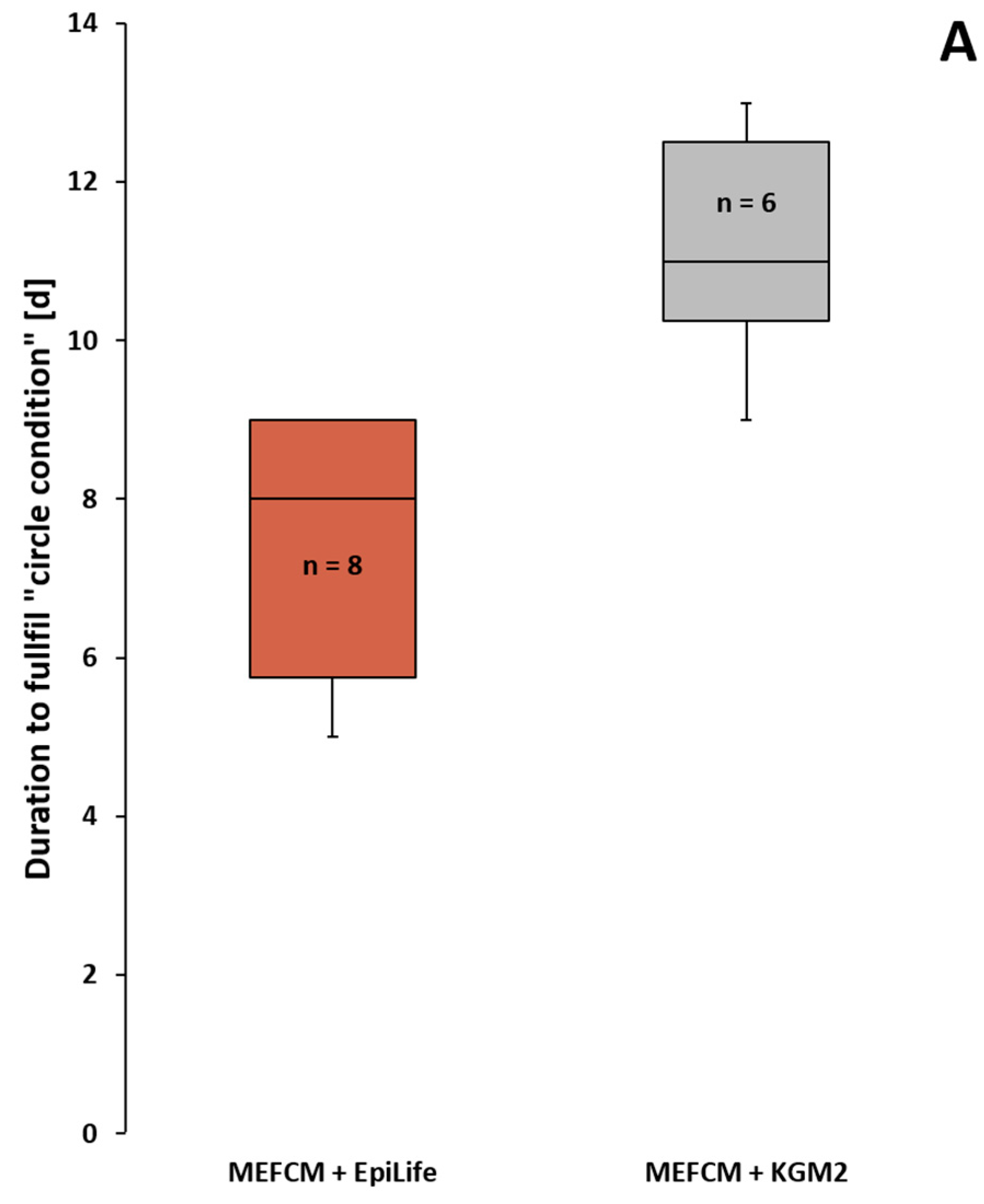
3.3.2. Application of the “Circle Condition” to the Compared Primary Culture Media
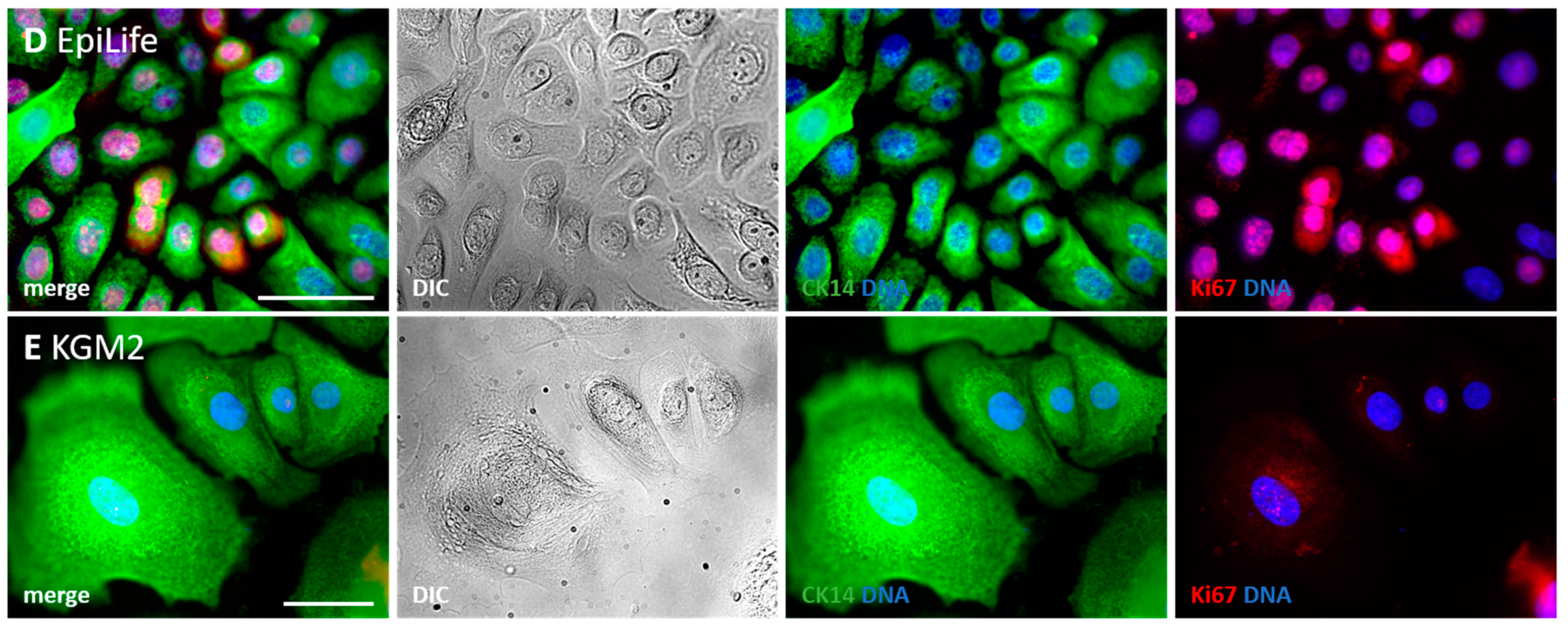
3.3.3. Secondary Culture Media in Comparison
- ➔
- No significant difference in the number of successfully grown keratinocytes from HFs in the tested combinations of MEFCM + EpiLife medium, MEF medium + EpiLife medium or solely EpiLife medium.
- ➔
- DK-SFM and KGM2 are not suitable for the generation and cultivation of keratinocytes from HFs.
- ➔
- Most keratinocytes growing in KGM2 are large, polygonal-shaped and non-dividing cells.
3.4. Coating Conditions for the Successful Outgrowth of Keratinocytes
3.4.1. Coating Conditions in Comparison
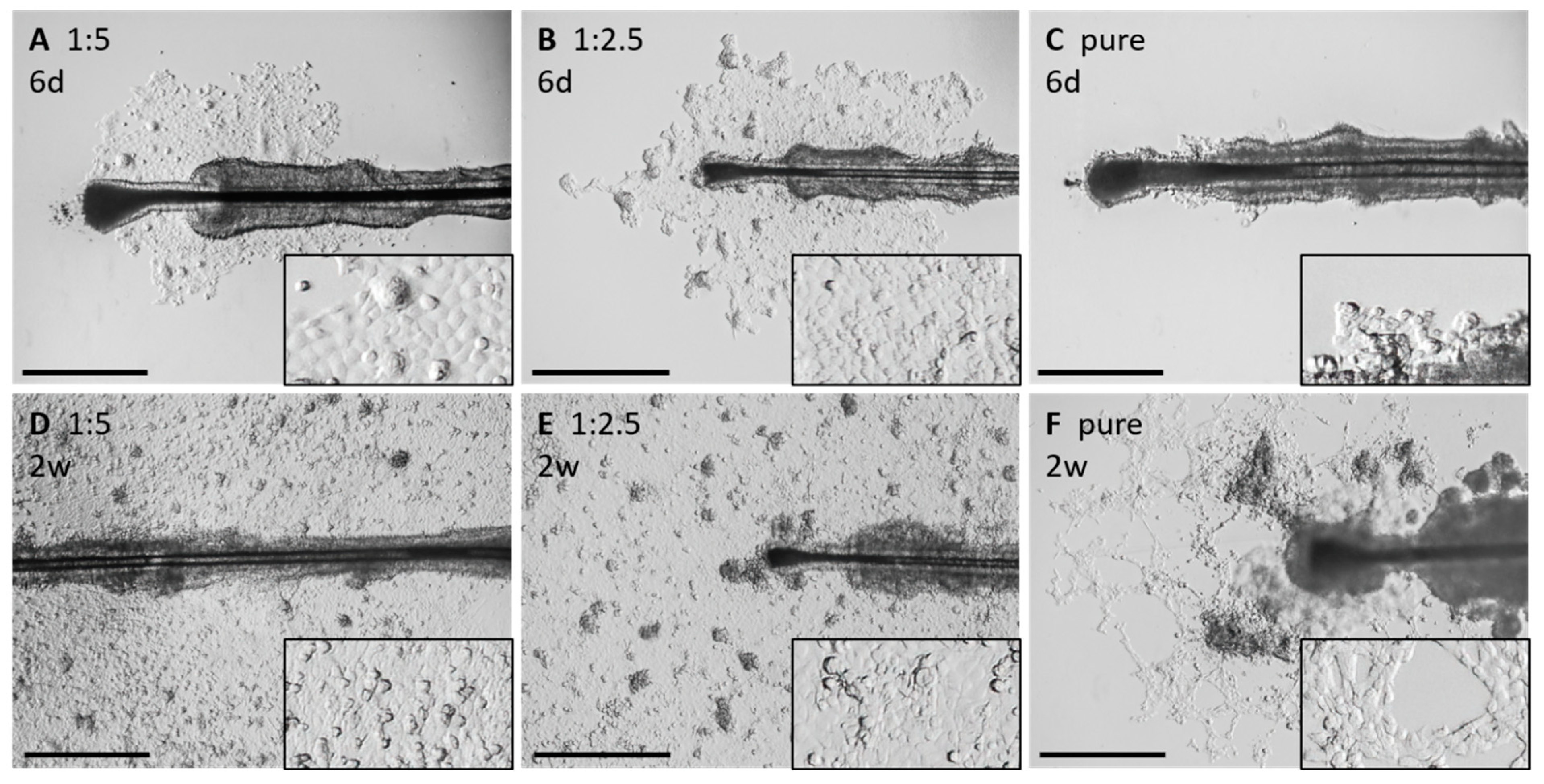
3.4.2. Morphological Alterations under Different Coating Conditions
- ➔
- A 1:5 Matrigel concentration or no Matrigel droplets reduces the attachment of the HFs.
- ➔
- Higher concentrations of Matrigel increase the attachment fidelity of the HFs.
- ➔
- Altered growth behavior and slowed outgrowth of keratinocytes can be observed with higher concentrated Matrigel droplets.
3.4.3. Application of the “Circle Condition” to the Compared Matrigel Droplet Dilution
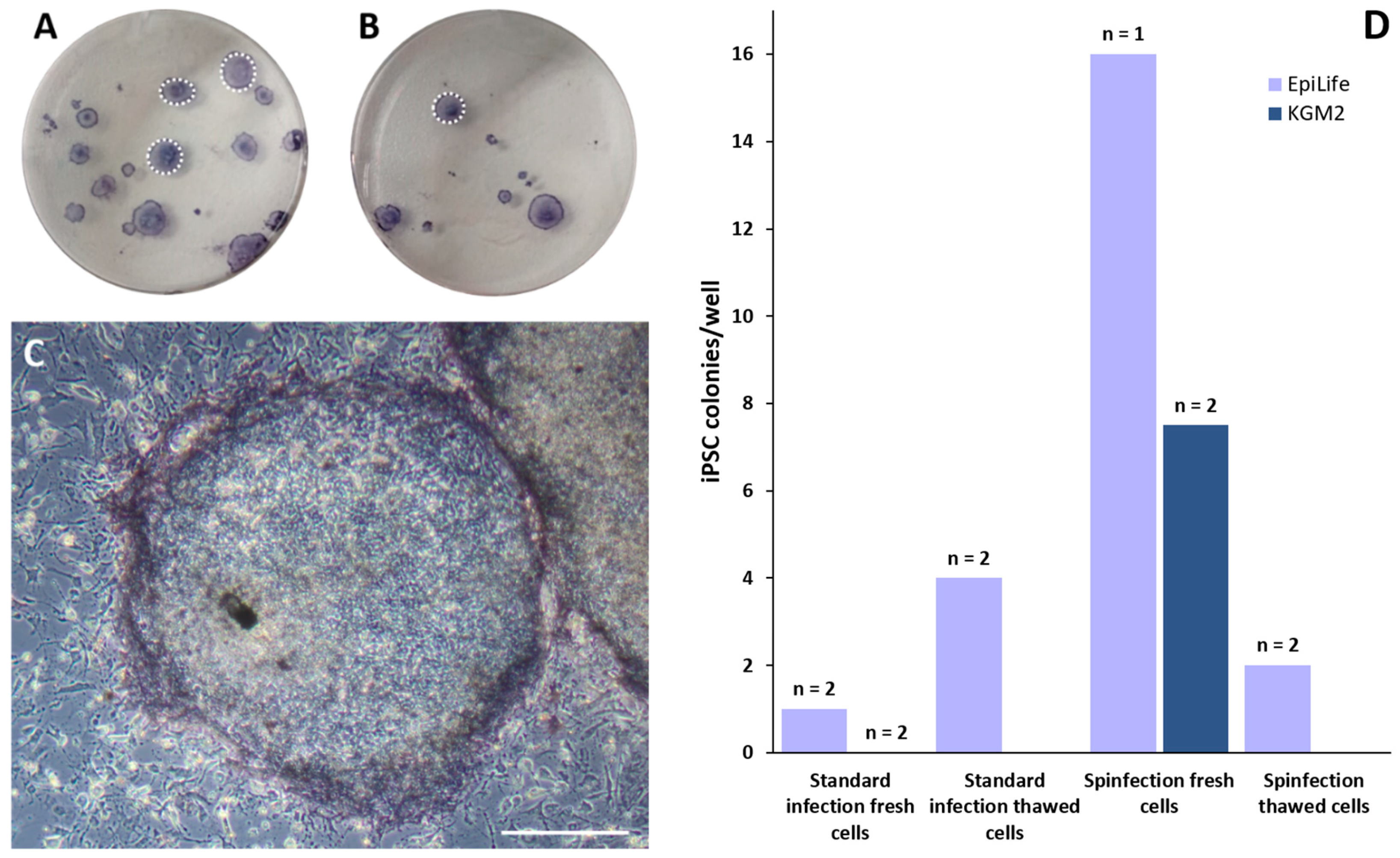
3.5. Reprogramming Keratinocytes to iPSCs
- ➔
- The most efficient way to generate keratinocyte-derived iPSCs with lentivirus induction is via fresh, non-thawed cells using the EpiLife medium and the spinfection protocol.
- ➔
- All generated iPSC colonies are pluripotent and able to differentiate into all three germ layers.
4. Discussion
4.1. Why Use Keratinocytes as the Primary Cell Source?
4.2. Evaluating the Best Generation and Culture Conditions for Keratinocytes
4.3. Which Coating Conditions Are the Best?
4.4. How to Achieve a Successful Lentiviral-Based Reprogramming of Keratinocytes?
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix A.1. Culture and Pluripotency Analysis of iPSCs
Appendix A.2. Quantitative RT-PCR
Appendix B
References
- Feng, C.; Jia, Y.D.; Zhao, X.Y. Pluripotency of induced pluripotent stem cells. Genom. Proteom. Bioinform. 2013, 11, 299–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Dawud, R.; Graffmann, N.; Ferber, S.; Wruck, W.; Adjaye, J. Pluripotent stem cells: Induction and self-renewal. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobhani, A.; Khanlarkhani, N.; Baazm, M.; Mohammadzadeh, F.; Najafi, A.; Mehdinejadiani, S.; Aval, F.S. Multipotent Stem Cell and Current Application. Acta Med. Iran. 2017, 55, 6–23. [Google Scholar] [PubMed]
- Shafa, M.; Yang, F.; Fellner, T.; Rao, M.S.; Baghbaderani, B.A. Human-Induced Pluripotent Stem Cells Manufactured Using a Current Good Manufacturing Practice-Compliant Process Differentiate Into Clinically Relevant Cells From Three Germ Layers. Front. Med. 2018, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Re, S.; Dogan, A.A.; Ben-Shachar, D.; Berger, G.; Werling, A.M.; Walitza, S.; Grunblatt, E. Improved Generation of Induced Pluripotent Stem Cells From Hair Derived Keratinocytes—A Tool to Study Neurodevelopmental Disorders as ADHD. Front. Cell. Neurosci. 2018, 12, 321. [Google Scholar] [CrossRef] [Green Version]
- Seitz, I.P.; Achberger, K.; Liebau, S.; Fischer, M.D. Stem Cells for Retina Replacement. Klin. Monbl. Augenheilkd. 2016, 233, 1350–1356. [Google Scholar] [CrossRef]
- Raab, S.; Klingenstein, M.; Liebau, S.; Linta, L. A Comparative View on Human Somatic Cell Sources for iPSC Generation. Stem Cells Int. 2014, 2014, 768391. [Google Scholar] [CrossRef] [Green Version]
- Aasen, T.; Raya, A.; Barrero, M.J.; Garreta, E.; Consiglio, A.; Gonzalez, F.; Vassena, R.; Bilic, J.; Pekarik, V.; Tiscornia, G.; et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat. Biotechnol. 2008, 26, 1276–1284. [Google Scholar] [CrossRef]
- Normand, J.; Karasek, M.A. A method for the isolation and serial propagation of keratinocytes, endothelial cells, and fibroblasts from a single punch biopsy of human skin. In Vitro Cell Dev. Biol. Anim. 1995, 31, 447–455. [Google Scholar] [CrossRef]
- Hybbinette, S.; Bostrom, M.; Lindberg, K. Enzymatic dissociation of keratinocytes from human skin biopsies for in vitro cell propagation. Exp. Dermatol. 1999, 8, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Sciezynska, A.; Nogowska, A.; Sikorska, M.; Konys, J.; Karpinska, A.; Komorowski, M.; Oldak, M.; Malejczyk, J. Isolation and culture of human primary keratinocytes-a methods review. Exp. Dermatol. 2019, 28, 107–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingenstein, S.; Klingenstein, M.; Kleger, A.; Liebau, S. From Hair to iPSCs-A Guide on How to Reprogram Keratinocytes and Why. Curr. Protoc. Stem Cell Biol. 2020, 55, e121. [Google Scholar] [CrossRef] [PubMed]
- Warlich, E.; Kuehle, J.; Cantz, T.; Brugman, M.H.; Maetzig, T.; Galla, M.; Filipczyk, A.A.; Halle, S.; Klump, H.; Scholer, H.R.; et al. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol. Ther. 2011, 19, 782–789. [Google Scholar] [CrossRef]
- Linta, L.; Stockmann, M.; Kleinhans, K.N.; Bockers, A.; Storch, A.; Zaehres, H.; Lin, Q.; Barbi, G.; Bockers, T.M.; Kleger, A.; et al. Rat embryonic fibroblasts improve reprogramming of human keratinocytes into induced pluripotent stem cells. Stem Cells Dev. 2012, 21, 965–976. [Google Scholar] [CrossRef]
- Houschyar, K.S.; Borrelli, M.R.; Tapking, C.; Popp, D.; Puladi, B.; Ooms, M.; Chelliah, M.P.; Rein, S.; Pforringer, D.; Thor, D.; et al. Molecular Mechanisms of Hair Growth and Regeneration: Current Understanding and Novel Paradigms. Dermatology 2020, 236, 271–280. [Google Scholar] [CrossRef]
- Bikle, D.D.; Xie, Z.; Tu, C.L. Calcium regulation of keratinocyte differentiation. Expert Rev. Endocrinol. Metab. 2012, 7, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.Z.; Li, Z.J.; Du, L.X.; Li, J.; Wang, L.Y. Using bovine pituitary extract to increase proliferation of keratocytes and maintain their phenotype in vitro. Int. J. Ophthalmol. 2013, 6, 758–765. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Aminuddin, B.S.; Ruszymah, B.H. Effect of supplementation of dermal fibroblasts conditioned medium on expansion of keratinocytes through enhancing attachment. Indian J. Exp. Biol. 2012, 50, 332–339. [Google Scholar]
- Hughes, C.S.; Postovit, L.M.; Lajoie, G.A. Matrigel: A complex protein mixture required for optimal growth of cell culture. Proteomics 2010, 10, 1886–1890. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, H.; Duan, E. Epidermal development in mammals: Key regulators, signals from beneath, and stem cells. Int. J. Mol. Sci. 2013, 14, 10869–10895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awe, J.P.; Lee, P.C.; Ramathal, C.; Vega-Crespo, A.; Durruthy-Durruthy, J.; Cooper, A.; Karumbayaram, S.; Lowry, W.E.; Clark, A.T.; Zack, J.A.; et al. Generation and characterization of transgene-free human induced pluripotent stem cells and conversion to putative clinical-grade status. Stem Cell Res. Ther. 2013, 4, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aasen, T.; Izpisua Belmonte, J.C. Isolation and cultivation of human keratinocytes from skin or plucked hair for the generation of induced pluripotent stem cells. Nat. Protoc. 2010, 5, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Lei, Q.; Yin, C.; Wang, H.Y.; Jin, K.; Xiang, M. Generation of Urine Cell-Derived Non-integrative Human iPSCs and iNSCs: A Step-by-Step Optimized Protocol. Front. Mol. Neurosci. 2017, 10, 348. [Google Scholar] [CrossRef]
- Nygaard, U.H.; Niehues, H.; Rikken, G.; Rodijk-Olthuis, D.; Schalkwijk, J.; van den Bogaard, E.H. Antibiotics in cell culture: Friend or foe? Suppression of keratinocyte growth and differentiation in monolayer cultures and 3D skin models. Exp. Dermatol. 2015, 24, 964–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Valk, J.; Bieback, K.; Buta, C.; Cochrane, B.; Dirks, W.G.; Fu, J.; Hickman, J.J.; Hohensee, C.; Kolar, R.; Liebsch, M.; et al. Fetal Bovine Serum (FBS): Past–Present–Future. Altex 2018, 35, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Frank, S.; Zhang, M.; Schöler, H.R.; Greber, B. Small molecule-assisted, line-independent maintenance of human pluripotent stem cells in defined conditions. PLoS ONE 2012, 7, e41958. [Google Scholar] [CrossRef]
- Klingenstein, M.; Raab, S.; Achberger, K.; Kleger, A.; Liebau, S.; Linta, L. TBX3 Knockdown Decreases Reprogramming Efficiency of Human Cells. Stem Cells Int. 2016, 2016, 6759343. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wüstner, L.S.; Klingenstein, M.; Frey, K.G.; Nikbin, M.R.; Milazzo, A.; Kleger, A.; Liebau, S.; Klingenstein, S. Generating iPSCs with a High-Efficient, Non-Invasive Method—An Improved Way to Cultivate Keratinocytes from Plucked Hair for Reprogramming. Cells 2022, 11, 1955. https://doi.org/10.3390/cells11121955
Wüstner LS, Klingenstein M, Frey KG, Nikbin MR, Milazzo A, Kleger A, Liebau S, Klingenstein S. Generating iPSCs with a High-Efficient, Non-Invasive Method—An Improved Way to Cultivate Keratinocytes from Plucked Hair for Reprogramming. Cells. 2022; 11(12):1955. https://doi.org/10.3390/cells11121955
Chicago/Turabian StyleWüstner, Lisa S., Moritz Klingenstein, Karl G. Frey, Mohammad R. Nikbin, Alfio Milazzo, Alexander Kleger, Stefan Liebau, and Stefanie Klingenstein. 2022. "Generating iPSCs with a High-Efficient, Non-Invasive Method—An Improved Way to Cultivate Keratinocytes from Plucked Hair for Reprogramming" Cells 11, no. 12: 1955. https://doi.org/10.3390/cells11121955
APA StyleWüstner, L. S., Klingenstein, M., Frey, K. G., Nikbin, M. R., Milazzo, A., Kleger, A., Liebau, S., & Klingenstein, S. (2022). Generating iPSCs with a High-Efficient, Non-Invasive Method—An Improved Way to Cultivate Keratinocytes from Plucked Hair for Reprogramming. Cells, 11(12), 1955. https://doi.org/10.3390/cells11121955