
Contribution of Model Organisms to Investigating the Far-Reaching Consequences of PRPP Metabolism on Human Health and Well-Being



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
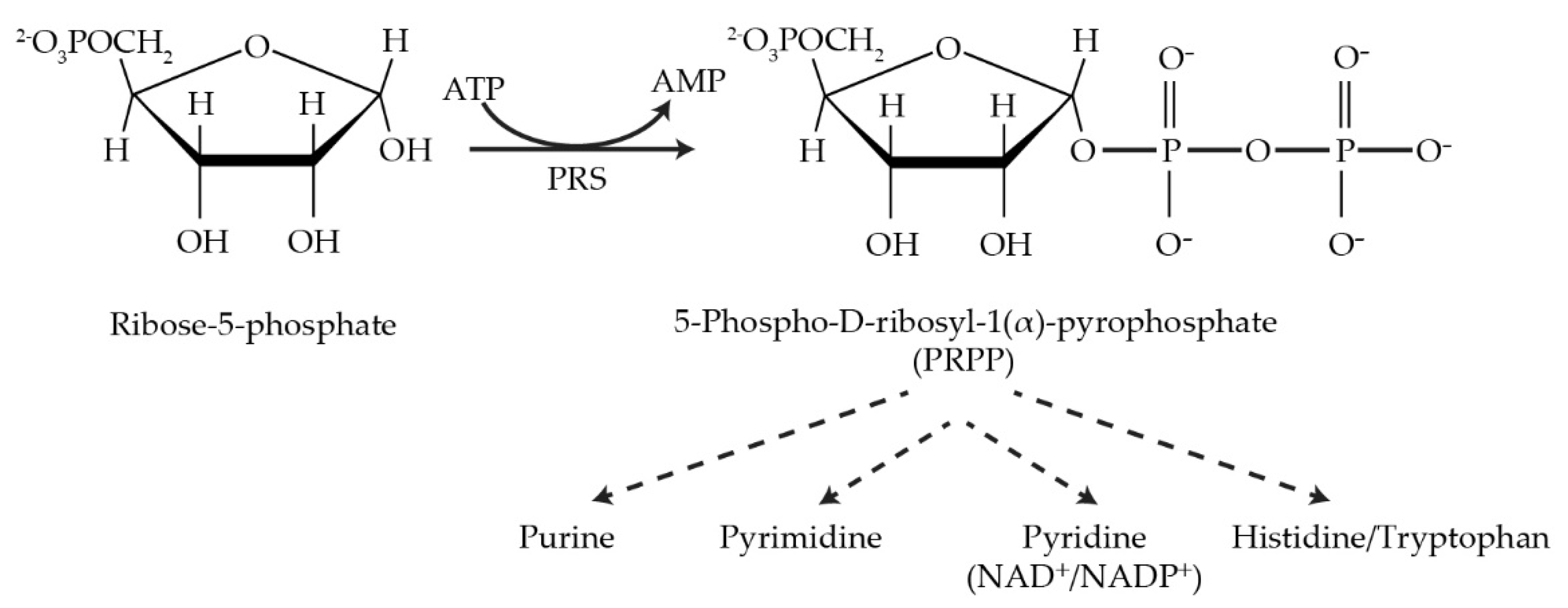
1. Introduction
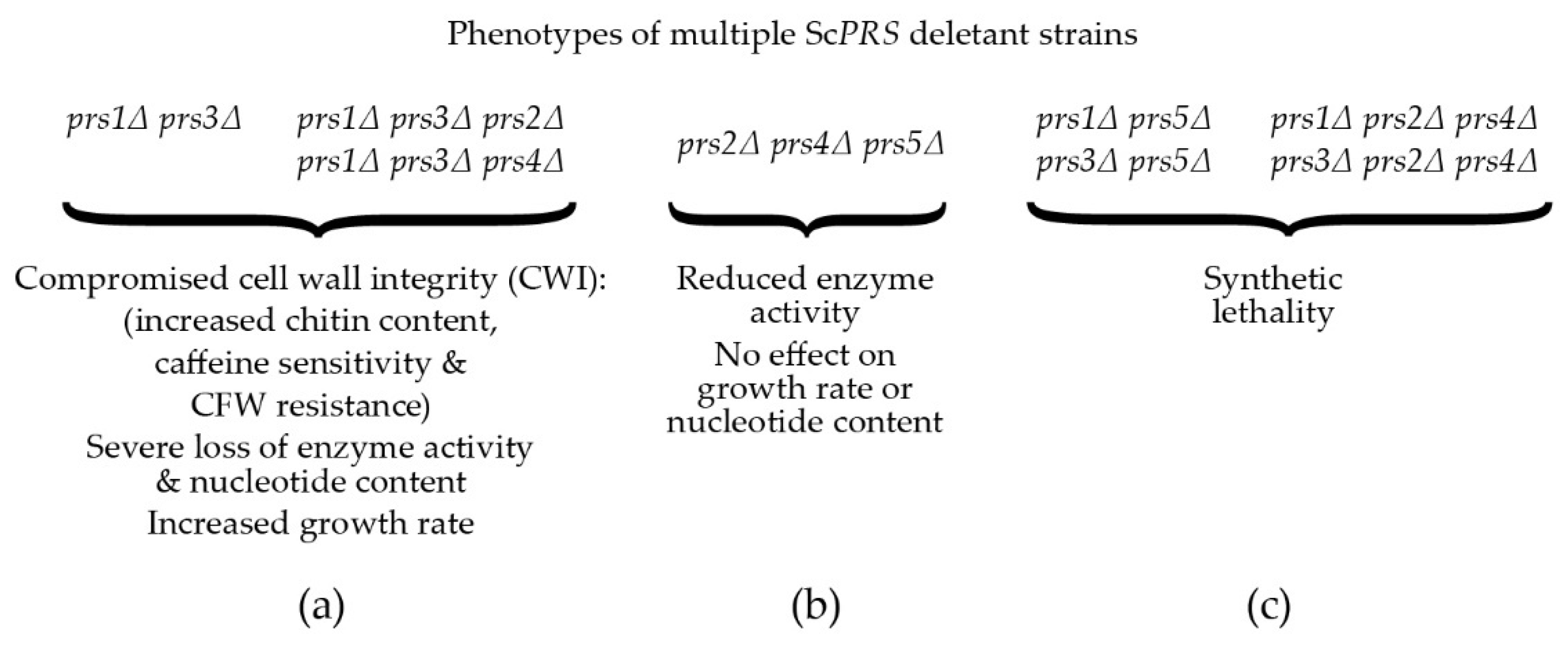
2. PRS-Encoding Genes in Saccharomyces cerevisiae
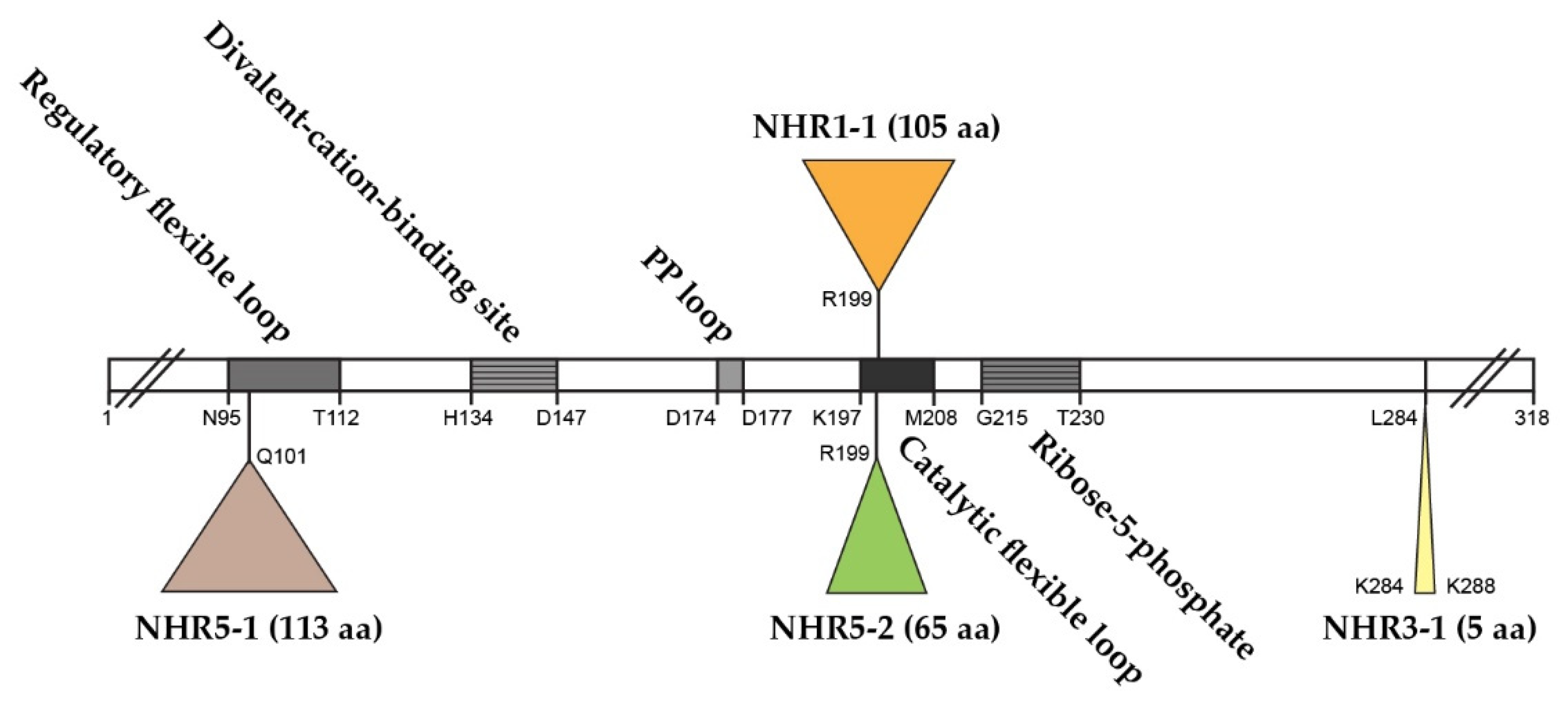
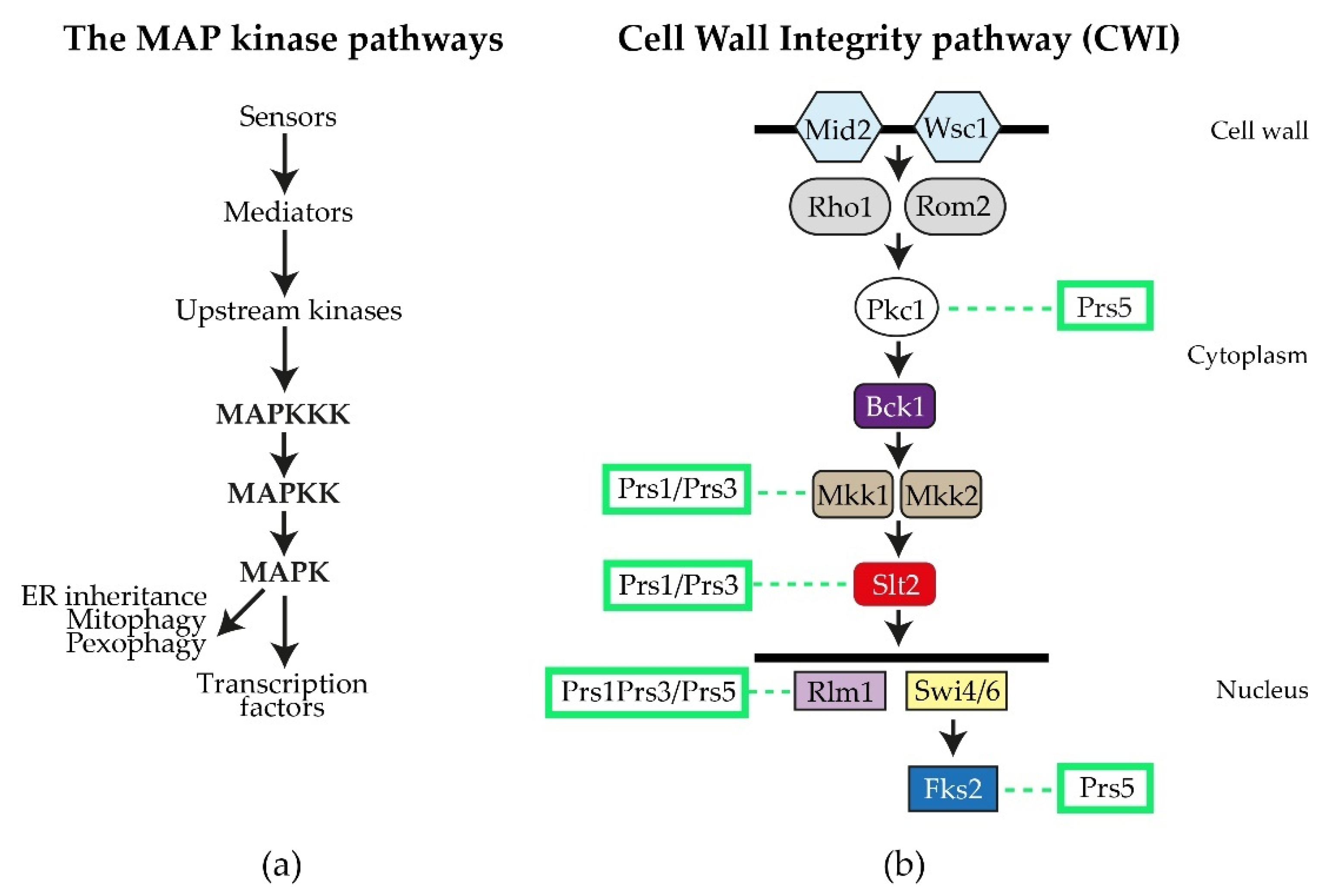
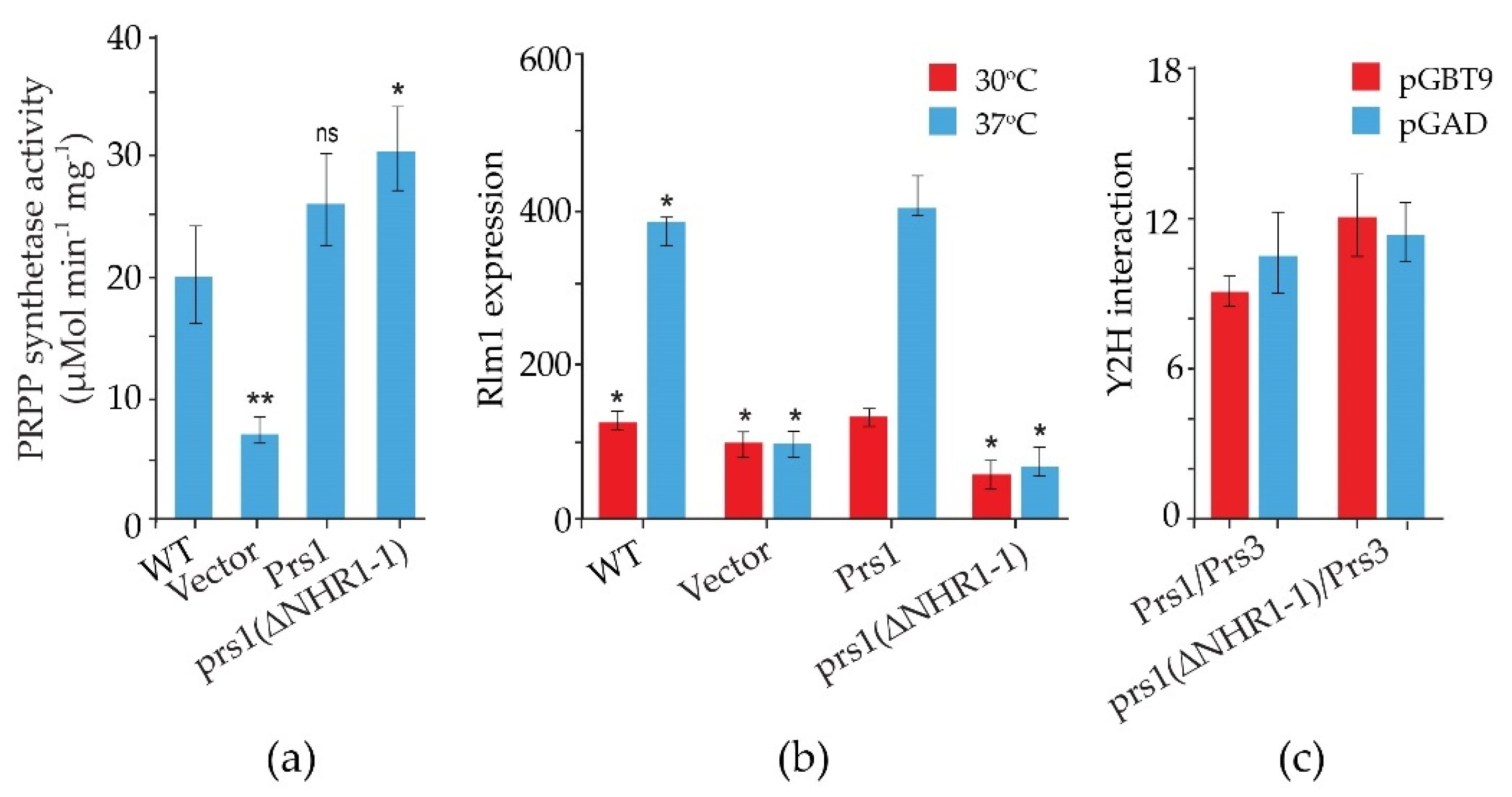
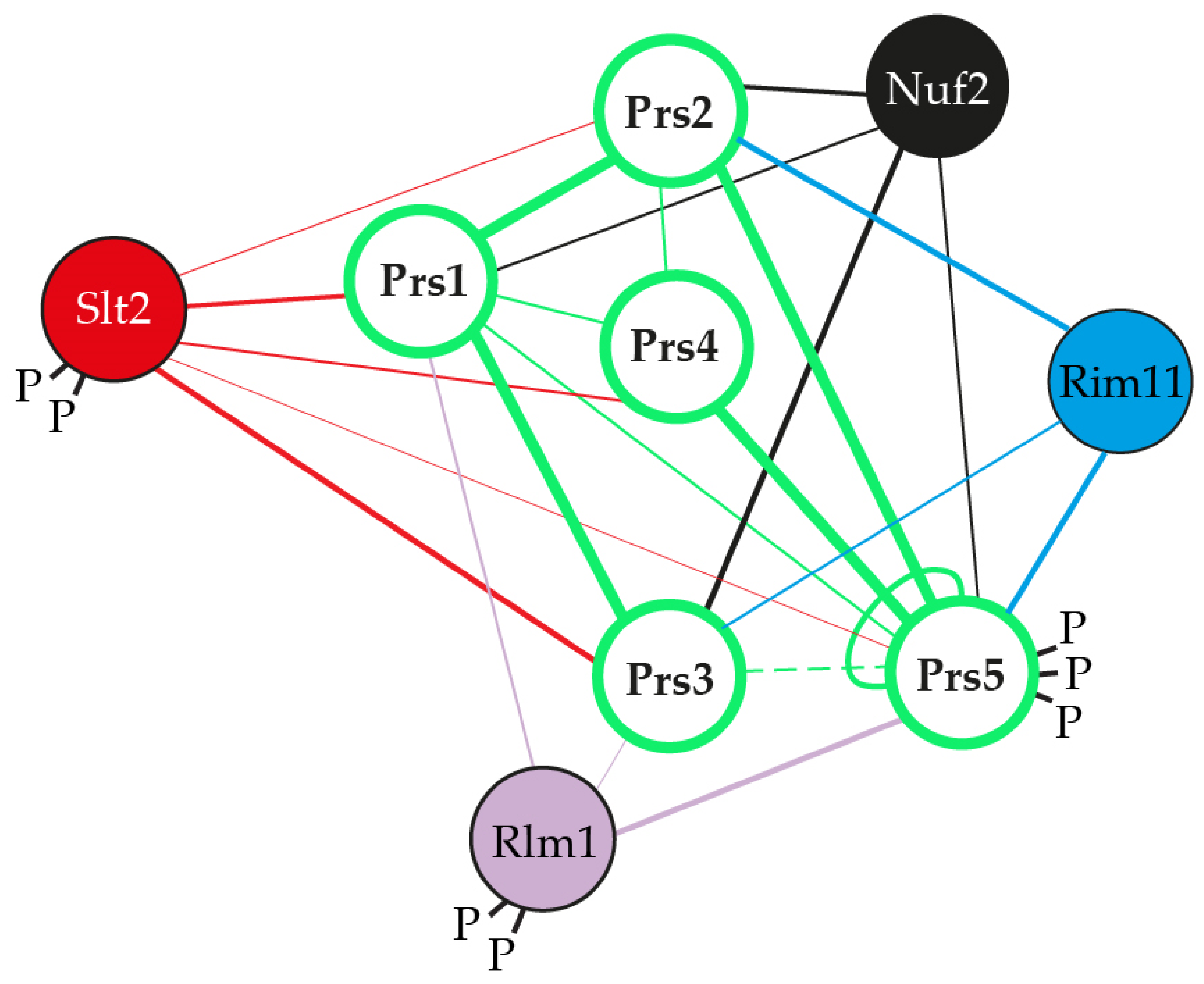
2.1. The Role of NHR1-1 in the Provision of PRPP and Maintenance of CWI
2.2. The Presence of NHR5-2 Influences Rlm1 Expression, the Phosphorylation Status of Slt2 and the Interaction with the Gsk3 Kinase Rim11
2.3. The Role of NHR3-1, the 284KKCPK288 Motif in Prs3
2.4. Prs1 and Prs5 Influence Fks2 Expression
2.5. Yeast Genocopies of PRPS-Associated Human Neuropathies Interfere with CWI
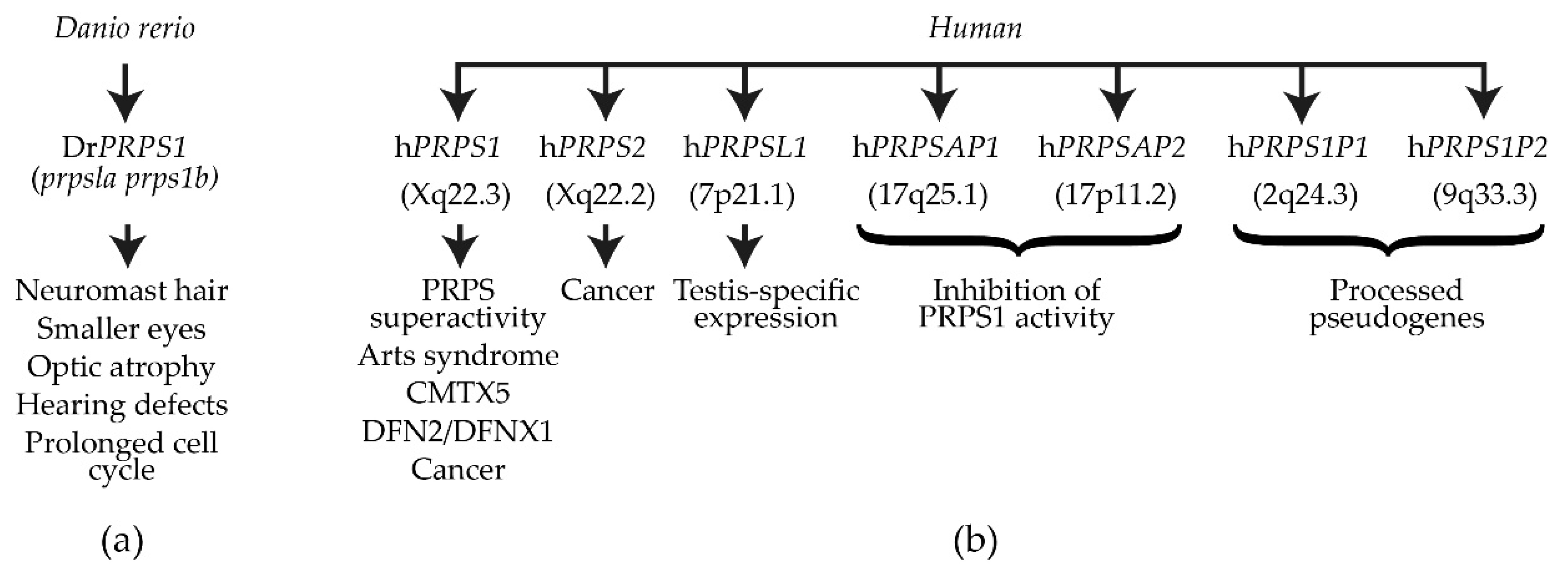
3. DrPRPS1 in the Model Organism Danio rerio
4. Human PRPS and Associated Disorders
4.1. PRPP Synthetase Superactivity: hPRPS1 (OMIM 300661)
4.2. Reduced PRPP Synthetase in Humans
4.2.1. Arts Syndrome
4.2.2. Charcot-Marie-Tooth Inherited Neuropathy (CMTX5)
4.2.3. Sensorineural Deafness 2 (DFN2/DFNX1)
5. Retinal Dystrophy
6. Roles of PRPS in Cancer
7. Role of PRPS1 in Ageing
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
NB
Abbreviations
| aa | Amino acid(s) |
| AMPK Atg32 | AMP-activated protein kinase Autophagy related |
| ATM | Ataxia-telangiectasia-mutated kinase |
| (B-)ALL | (B-cell) acute lymphoblastic leukaemia |
| Bck1 Bcl-2 | Bypass of C kinaseB-cell lymphoma 2 |
| BLASTN | Basic Local Alignment Search Tool for nucleotide sequences |
| BOR | Branchio-Oto-Renal |
| BTICs | Brain tumour initiating cells |
| CAD | Carbamoylphosphate synthetase II |
| CDK1 | Cyclin-dependent kinase 1 |
| CFW circKIF2A | Caclcoflour white circular RNA (circRNA) kinesin superfamily protein 2A |
| CMTX5 | Charcot-Marie-Tooth type 5 inherited neuropathy |
| c-Myc (MYC) | cellular-Myc, a family member of transcription factors |
| CRC | Colorectal cancer cells |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CWI | Cell wall integrity |
| DFN2/DNFX1 | Nonsyndromic hearing loss and deafness |
| DIC | Differential interference contrast |
| DLEU1 | lnc RNA deleted in lymphocytic leukaemia 1 |
| dpf | Days post-fertilization |
| eIF-4E | eukaryotic translation initiation factor-4E |
| ERK5 | Extracellular signal-regulated kinase 5 |
| ERN1 | Endoribonuclease activity of endoplasmic reticulum to nuclei 1 named in humans: serine/threonine-protein kinase/endoribonuclease inositol-requiring enzyme 1α (IRE1α) |
| Esa1 | Catalytic subunit of the histone acetyltransferase complex NuA4 |
| Fks2 Gal4 | Catalytic subunit of 1,3-β-D-glucan synthase DNA-binding transcription factor required for activating GAL genes |
| Gsk3 HAS2-AS1 | Glycogen synthase kinase 3 Hyaluronan synthase 2-antisense RNA 1 |
| Hb HCC | Hevea brasiliensis Müll Hepatocellular carcinoma |
| HGNC hnRNPH1/2 HGPS | HUGO (Human Genome Organization) Gene Nomenclature Heterogeneous Nuclear Ribonucleoprotein H1/2 Hutchinson–Gilford progeria syndrome |
| HOG | High-osmolarity Glycerol |
| hPRPS1/2 or mPRPS1/2 | Human or mouse phosphoribosyl pyrophosphate synthetase 1/2 |
| hPRPS1/2 or mPRPS1/2 | Genes encoding human or mouse phosphoribosyl pyrophosphate synthetase 1/2 |
| HTP | High-throughput score |
| IRD iTRAQ | Inherited Retinal Dystrophy high-resolution quantitative proteomics |
| KHK-A/C | Ketohexokinase-A/C |
| KIX | Kinase-inducible domain |
| KSHV | Kaposi’s sarcoma-associated herpes virus |
| lncRNA | long noncoding RNA |
| lncRNA HAS2-AS1 | lncRNA HAS2 (hyaluronan synthase 2)-antisense RNA 1 |
| LOVD | Leiden Open Variation Database |
| MADS-box | MCM1, Agamous, Deficiens, Serum response factor-box |
| MAPK MAPKK MAPKKK | Mitogen-activated protein kinase MAPK kinase MAPK kinase kinase |
| MCM1 MD MGI | Mini-Chromosome Maintenance 1 Molecular dynamics Mouse Genome Information |
| Mid2 | Mating pheromone-induced death |
| miRNA | microRNA |
| Mkk1/2 Mlp1 | Mitogen-activated protein kinase kinase 1/2 Mpk1-like protein |
| Mpk1 MRI | Mitogen-activated protein kinase 1 Magnetic Resonance Imaging |
| mTOR p-mTOR mTORC1/2 | mechanistic Target Of Rapamycin phosphorylated mTOR mechanistic Target Of Rapamycin Complex |
| NAC | N-acetyl cysteine |
| NGS | Next-generation sequencing |
| NHRs | Non-homologous regions |
| NLS | Nuclear localization signal |
| NMN | Nicotinamide mononucleotide |
| NR Nuf2 | Nicotinamide riboside Component of the kinetochore-associated Ndc80 complex; |
| OMIM | Online Mendelian Inheritance in Man |
| PAP | PRS-associated protein |
| PAT Pbs2 | PRPP amidotransferase Polymyxin B sensitivity |
| PDB | Protein Data Bank |
| Pfa1 | Polymerase associated factor 1 complex |
| pGAT pGBT9 Pkc1 | GAL4 activation domain cloning vector GAL4 DNA-binding domain cloning vector Protein kinase C |
| PRPP | Phosphoribosyl pyrophosphate |
| PRS/PRPS | Phosphoribosyl pyrophosphate synthetase |
| Prs1-5 P-Prs5 PTRE Ras1/2 | 5-phospho-D-ribosyl-1(α)-pyrophosphate synthetase Phosphorylated Prs5 Pyrimidine-rich translational element GTPase involved in G-protein signalling in adenylate cyclase activation |
| RefSeq NM_*, NP_* | NCBI Reference Sequence Gene (RefSeqGene) curated subsets (NM_*, NP_*) |
| Rheb | a Ras homolog enriched in brain GTP binding protein |
| Rho1 Rim11 Rlm1 Rom2 | GTP-binding protein of the rho subfamily of ras-like proteins Regulator of Ime1 Resistance to lethality of MKK1p386 overexpression Guanine nucleotide exchange factor (GEF) for Rho1 |
| SAM | S-adenosylmethionine |
| SBF | Swi4/Swi6 protein complex that binds to the Swi4/6 cell cycle box (SCB) promoter |
| Sc SCOS SGD | Saccharomyces cerevisiae Sertoli cell-only syndrome Saccharomyces Genome Database |
| shRNA | short-hairpin RNA |
| siRNA | small interfering RNA |
| Slt2 P-Slt2 ScPRS1-ScPRS5 | Suppressor of the lytic phenotype Phosphorylated Slt2 Genes encoding 5-phospho-D-ribosyl-1(α)-pyrophosphate synthetase in Saccharomyces cerevisiae |
| SNHL | Sporadic sensorineural hearing loss |
| SNP | Single Nucleotide Polymorphism |
| U87 | Human primary glioblastoma cell line |
| UTR | Untranslated region |
| Varsome Wsc1 | The Human Genomics Community Sensor-transducer of the stress-activated Pkc1-Mpk1 kinase pathway |
| WT | Wild type |
| Y2H | Yeast-two-hybrid |
| ZF ZFIN | Zebrafish Zebrafish Information Network |
References
- Kornberg, A.; Lieberman, I.; Simms, E.S. Enzymatic synthesis and properties of 5-phosphoribosylpyrophosphate. J. Biol. Chem. 1955, 215, 389–402. [Google Scholar] [CrossRef]
- Khorana, H.G.; Fernandes, J.F.; Kornberg, A. Pyrophosphorylation of ribose 5-phosphate in the enzymatic synthesis of 5-phosphorylribose 1-pyrophosphate. J. Biol. Chem. 1958, 230, 941–948. [Google Scholar] [CrossRef]
- Hove-Jensen, B.; Andersen, K.R.; Kilstrup, M.; Martinussen, J.; Switzer, R.L.; Willemoes, M. Phosphoribosyl Diphosphate (PRPP): Biosynthesis, Enzymology, Utilization, and Metabolic Significance. Microbiol. Mol. Biol. Rev. 2017, 81, e00040-16. [Google Scholar] [CrossRef]
- Fox, I.H.; Kelley, W.N. Human phosphoribosylpyrophosphate synthetase. Kinetic mechanism and end product inhibition. J. Biol. Chem. 1972, 247, 2126–2131. [Google Scholar] [CrossRef]
- Becker, M.A.; Ahmed, M. Cell type-specific differential expression of human PRPP synthetase (PRPS) genes. In Purine and Pyrimidine Metabolism in Man X. Advances in Experimental Medicine and Biology; Zoref-Shani, E., Sperling, O., Eds.; Springer: Boston, MA, USA, 2002; Volume 486, pp. 5–10. [Google Scholar]
- Hove-Jensen, B. Mutation in the phosphoribosylpyrophosphate synthetase gene (prs) that results in simultaneous requirements for purine and pyrimidine nucleosides, nicotinamide nucleotide, histidine, and tryptophan in Escherichia coli. J. Bacteriol. 1988, 170, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.; Dandanell, G.; Hove-Jensen, B.; Willemoës, M. Nucleotides, Nucleosides, and Nucleobases. EcoSal Plus 2008, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of purine metabolism: Clinical update and therapies. J. Inherit. Metab. Dis. 2014, 37, 669–686. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Duley, J.A.; Christodoulou, J. Inborn errors of pyrimidine metabolism: Clinical update and therapy. J. Inherit. Metab. Dis. 2014, 37, 687–698. [Google Scholar] [CrossRef]
- Fasullo, M.; Endres, L. Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int. J. Mol. Sci. 2015, 16, 9431–9449. [Google Scholar] [CrossRef]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer cells tune the signaling pathways to empower de novo synthesis of nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef]
- Ullman, B.; Carter, D. Molecular and biochemical studies on the hypoxanthine-guanine phosphoribosyltransferases of the pathogenic haemoflagellates. Int. J. Parasitol. 1997, 27, 203–213. [Google Scholar] [CrossRef]
- Tatibana, M.; Kita, K.; Taira, M.; Ishijima, S.; Sonoda, T.; Ishizuka, T.; Iizasa, T.; Ahmad, I. Mammalian phosphoribosyl-pyrophosphate synthetase. Adv. Enzym. Regul. 1995, 35, 229–249. [Google Scholar] [CrossRef]
- Roessler, B.J.; Nosal, J.M.; Smith, P.R.; Heidler, S.A.; Palella, T.D.; Switzer, R.L.; Becker, M.A. Human X-linked phosphoribosylpyrophosphate synthetase superactivity is associated with distinct point mutations in the PRPS1 gene. J. Biol. Chem. 1993, 268, 26476–26481. [Google Scholar] [CrossRef]
- Taira, M.; Iizasa, T.; Shimada, H.; Kudoh, J.; Shimizu, N.; Tatibana, M. A human testis-specific mRNA for phosphoribosylpyrophosphate synthetase that initiates from a non-AUG codon. J. Biol. Chem. 1990, 265, 16491–16497. [Google Scholar] [CrossRef]
- Kita, K.; Otsuki, T.; Ishizuka, T.; Tatibana, M. Rat liver phosphoribosyl pyrophosphate synthetase: Existence of the purified enzyme as heterogeneous aggregates and identification of the catalytic subunit. J. Biochem. 1989, 105, 736–741. [Google Scholar] [CrossRef]
- Becker, M.A. Phosphoribosylpyrophosphate synthetase and the regulation of phosphoribosylpyrophosphate production in human cells. Prog. Nucleic Acid Res. Mol. Biol. 2001, 69, 115–148. [Google Scholar]
- Krath, B.N.; Hove-Jensen, B. Organellar and cytosolic localization of four phosphoribosyl diphosphate synthase isozymes in spinach. Plant Physiol. 1999, 119, 497–506. [Google Scholar] [CrossRef]
- Krath, B.N.; Eriksen, T.A.; Poulsen, T.S.; Hove-Jensen, B. Cloning and sequencing of cDNAs specifying a novel class of phosphoribosyl diphosphate synthase in Arabidopsis thaliana. Biochim. Biophys. Acta 1999, 1430, 403–408. [Google Scholar] [CrossRef]
- Lin, X.; Kaul, S.; Rounsley, S.; Shea, T.P.; Benito, M.I.; Town, C.D.; Fujii, C.Y.; Mason, T.; Bowman, C.L.; Barnstead, M.; et al. Sequence and analysis of chromosome 2 of the plant Arabidopsis thaliana. Nature 1999, 402, 761–768. [Google Scholar] [CrossRef]
- Eriksen, T.A.; Kadziola, A.; Bentsen, A.K.; Harlow, K.W.; Larsen, S. Structural basis for the function of Bacillus subtilis phosphoribosyl-pyrophosphate synthetase. Nat. Struct. Biol. 2000, 7, 303–308. [Google Scholar]
- Baugh, L.; Gallagher, L.A.; Patrapuvich, R.; Clifton, M.C.; Gardberg, A.S.; Edwards, T.E.; Armour, B.; Begley, D.W.; Dieterich, S.H.; Dranow, D.M.; et al. Combining functional and structural genomics to sample the essential Burkholderia structome. PLoS ONE 2013, 8, e53851. [Google Scholar] [CrossRef]
- Carter, A.T.; Narbad, A.; Pearson, B.M.; Beck, K.F.; Logghe, M.; Contreras, R.; Schweizer, M. Phosphoribosylpyrophosphate synthetase (PRS): A new gene family in Saccharomyces cerevisiae. Yeast 1994, 10, 1031–1044. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.T.; Beiche, F.; Narbad, A.; Hove-Jensen, B.; Schweizer, L.M.; Schweizer, M. Are all four yeast PRS genes essential? Biochem. Soc. Trans. 1995, 23, 621S. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.T.; Beiche, F.; Hove-Jensen, B.; Narbad, A.; Barker, P.J.; Schweizer, L.M.; Schweizer, M. PRS1 is a key member of the gene family encoding phosphoribosylpyrophosphate synthetase in Saccharomyces cerevisiae. Mol. Gen. Genet. 1997, 254, 148–156. [Google Scholar] [CrossRef]
- Hernando, Y.; Parr, A.; Schweizer, M. PRS5, the fifth member of the phosphoribosyl pyrophosphate synthetase gene family in Saccharomyces cerevisiae, is essential for cell viability in the absence of either PRS1 or PRS3. J. Bacteriol. 1998, 180, 6404–6407. [Google Scholar] [CrossRef]
- Wood, V.; Gwilliam, R.; Rajandream, M.A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef]
- DeSmidt, A.A.; Zou, B.; Grati, M.; Yan, D.; Mittal, R.; Yao, Q.; Richmond, M.T.; Denyer, S.; Liu, X.Z.; Lu, Z. Zebrafish Model for Nonsyndromic X-Linked Sensorineural Deafness, DFNX1. Anat. Rec. 2020, 303, 544–555. [Google Scholar] [CrossRef]
- Pei, W.; Xu, L.; Varshney, G.K.; Carrington, B.; Bishop, K.; Jones, M.; Huang, S.C.; Idol, J.; Pretorius, P.R.; Beirl, A.; et al. Additive reductions in zebrafish PRPS1 activity result in a spectrum of deficiencies modeling several human PRPS1-associated diseases. Sci. Rep. 2016, 6, 29946. [Google Scholar] [CrossRef]
- Hove-Jensen, B. Cloning and characterization of the prs gene encoding phosphoribosylpyrophosphate synthetase of Escherichia coli. Mol. Gen. Genet. 1985, 201, 269–276. [Google Scholar] [CrossRef]
- Lau, N.S.; Makita, Y.; Kawashima, M.; Taylor, T.D.; Kondo, S.; Othman, A.S.; Shu-Chien, A.C.; Matsui, M. The rubber tree genome shows expansion of gene family associated with rubber biosynthesis. Sci. Rep. 2016, 6, 28594. [Google Scholar] [CrossRef]
- Tang, C.R.; Yang, M.; Fang, Y.J.; Luo, Y.F.; Gao, S.H.; Xiao, X.H.; An, Z.W.; Zhou, B.H.; Zhang, B.; Tan, X.Y.; et al. The rubber tree genome reveals new insights into rubber production and species adaptation. Nat. Plants 2016, 2, 16073. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, Y.; Zhang, D.; Lu, Y.; He, H.; Zheng, F.; Wang, M. Identification of a Ribose-Phosphate Pyrophosphokinase that Can Interact In Vivo with the Anaphase Promoting Complex/Cyclosome. Int. J. Mol. Sci. 2017, 18, 617. [Google Scholar] [CrossRef]
- Amalou, Z.; Bangratz, J.; Chrestin, H. Ethrel (Ethylene Releaser)-Induced Increases in the Adenylate Pool and Transtonoplast DeltapH within Hevea Latex Cells. Plant Physiol. 1992, 98, 1270–1276. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liu, Z.L.; Weber, S.A.; Li, S. Signature pathway expression of xylose utilization in the genetically engineered industrial yeast Saccharomyces cerevisiae. PLoS ONE 2018, 13, e0195633. [Google Scholar] [CrossRef]
- Cunha, J.T.; Aguiar, T.Q.; Romani, A.; Oliveira, C.; Domingues, L. Contribution of PRS3, RPB4 and ZWF1 to the resistance of industrial Saccharomyces cerevisiae CCUG53310 and PE-2 strains to lignocellulosic hydrolysate-derived inhibitors. Bioresour. Technol. 2015, 191, 7–16. [Google Scholar] [CrossRef]
- Cunha, J.T.; Costa, C.E.; Ferraz, L.; Romani, A.; Johansson, B.; Sa-Correia, I.; Domingues, L. HAA1 and PRS3 overexpression boosts yeast tolerance towards acetic acid improving xylose or glucose consumption: Unravelling the underlying mechanisms. Appl. Microbiol. Biotechnol. 2018, 102, 4589–4600. [Google Scholar] [CrossRef] [PubMed]
- Cunha, J.T.; Soares, P.O.; Baptista, S.L.; Costa, C.E.; Domingues, L. Engineered Saccharomyces cerevisiae for lignocellulosic valorization: A review and perspectives on bioethanol production. Bioengineered 2020, 11, 883–903. [Google Scholar] [CrossRef]
- Lucarelli, A.P.; Buroni, S.; Pasca, M.R.; Rizzi, M.; Cavagnino, A.; Valentini, G.; Riccardi, G.; Chiarelli, L.R. Mycobacterium tuberculosis phosphoribosylpyrophosphate synthetase: Biochemical features of a crucial enzyme for mycobacterial cell wall biosynthesis. PLoS ONE 2010, 5, e15494. [Google Scholar] [CrossRef] [PubMed]
- Alderwick, L.J.; Lloyd, G.S.; Lloyd, A.J.; Lovering, A.L.; Eggeling, L.; Besra, G.S. Biochemical characterization of the Mycobacterium tuberculosis phosphoribosyl-1-pyrophosphate synthetase. Glycobiology 2011, 21, 410–425. [Google Scholar] [CrossRef]
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs that covalently modify the decaprenylphosphoryl-D-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. [Google Scholar] [CrossRef]
- Donini, S.; Garavaglia, S.; Ferraris, D.M.; Miggiano, R.; Mori, S.; Shibayama, K.; Rizzi, M. Biochemical and structural investigations on phosphoribosylpyrophosphate synthetase from Mycobacterium smegmatis. PLoS ONE 2017, 12, e0175815. [Google Scholar] [CrossRef] [PubMed]
- Taira, M.; Kudoh, J.; Minoshima, S.; Iizasa, T.; Shimada, H.; Shimizu, Y.; Tatibana, M.; Shimizu, N. Localization of human phosphoribosylpyrophosphate synthetase subunit I and II genes (PRPS1 and PRPS2) to different regions of the X chromosome and assignment of two PRPS1-related genes to autosomes. Somat. Cell Mol. Genet. 1989, 15, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.A.; Heidler, S.A.; Bell, G.I.; Seino, S.; Le Beau, M.M.; Westbrook, C.A.; Neuman, W.; Shapiro, L.J.; Mohandas, T.K.; Roessler, B.J.; et al. Cloning of cDNAs for human phosphoribosylpyrophosphate synthetases 1 and 2 and X chromosome localization of PRPS1 and PRPS2 genes. Genomics 1990, 8, 555–561. [Google Scholar] [CrossRef]
- Taira, M.; Iizasa, T.; Yamada, K.; Shimada, H.; Tatibana, M. Tissue-differential expression of two distinct genes for phosphoribosyl pyrophosphate synthetase and existence of the testis-specific transcript. Biochim. Biophys. Acta 1989, 1007, 203–208. [Google Scholar] [CrossRef]
- de Brouwer, A.P.M.; Christodoulou, J. Phosphoribosylpyrophosphate snthetase superactivity. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2008; (updated 17 February 2022). [Google Scholar]
- Arts, W.F.; Loonen, M.C.; Sengers, R.C.; Slooff, J.L. X-linked ataxia, weakness, deafness, and loss of vision in early childhood with a fatal course. Ann. Neurol. 1993, 33, 535–539. [Google Scholar] [CrossRef] [PubMed]
- de Brouwer, A.P.; Williams, K.L.; Duley, J.A.; van Kuilenburg, A.B.; Nabuurs, S.B.; Egmont-Petersen, M.; Lugtenberg, D.; Zoetekouw, L.; Banning, M.J.; Roeffen, M.; et al. Arts syndrome is caused by loss-of-function mutations in PRPS1. Am. J. Hum. Genet. 2007, 81, 507–518. [Google Scholar] [CrossRef]
- Liu, X.; Han, D.; Li, J.; Han, B.; Ouyang, X.; Cheng, J.; Li, X.; Jin, Z.; Wang, Y.; Bitner-Glindzicz, M.; et al. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am. J. Hum. Genet. 2010, 86, 65–71. [Google Scholar] [CrossRef]
- Liu, X.Z.; Xie, D.; Yuan, H.J.; de Brouwer, A.P.; Christodoulou, J.; Yan, D. Hearing loss and PRPS1 mutations: Wide spectrum of phenotypes and potential therapy. Int. J. Audiol. 2013, 52, 23–28. [Google Scholar] [CrossRef][Green Version]
- Kim, H.J.; Sohn, K.M.; Shy, M.E.; Krajewski, K.M.; Hwang, M.; Park, J.H.; Jang, S.Y.; Won, H.H.; Choi, B.O.; Hong, S.H.; et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (CMTX5). Am. J. Hum. Genet. 2007, 81, 552–558. [Google Scholar] [CrossRef]
- Cunningham, J.T.; Moreno, M.V.; Lodi, A.; Ronen, S.M.; Ruggero, D. Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 2014, 157, 1088–1103. [Google Scholar] [CrossRef]
- Donini, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.Z.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar]
- Qiu, Z.; Guo, W.; Wang, Q.; Chen, Z.; Huang, S.; Zhao, F.; Yao, M.; Zhao, Y.; He, X. MicroRNA-124 reduces the pentose phosphate pathway and proliferation by targeting PRPS1 and RPIA mRNAs in human colorectal cancer cells. Gastroenterology 2015, 149, 1587–1598.e1511. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Song, L.; Huang, X.; Feng, Y.; Zhang, Y.; Liu, Y.; Li, S.; Zhan, Z.; Zheng, L.; Feng, H.; et al. PRPS1-mediated purine biosynthesis is critical for pluripotent stem cell survival and stemness. Aging 2021, 13, 4063–4078. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.K.; Ellis, T. The second decade of synthetic biology: 2010–2020. Nat. Commun. 2020, 11, 5174. [Google Scholar] [CrossRef]
- Dai, J.B.; Boeke, J.D.; Luo, Z.Q.; Jiang, S.Y.; Cai, Y.Z. Sc3.0: Revamping and minimizing the yeast genome. Genome Biol. 2020, 21, 205. [Google Scholar] [CrossRef]
- Schindler, D. Genetic Engineering and Synthetic Genomics in Yeast to Understand Life and Boost Biotechnology. Bioengineering 2020, 7, 137. [Google Scholar] [CrossRef]
- Wolfe, K.H.; Shields, D.C. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 1997, 387, 708–713. [Google Scholar] [CrossRef]
- Wolfe, K.H. Origin of the Yeast Whole-Genome Duplication. PLoS Biol. 2015, 13, e1002221. [Google Scholar] [CrossRef]
- Botstein, D.; Fink, G.R. Yeast: An experimental organism for 21st Century biology. Genetics 2011, 189, 695–704. [Google Scholar] [CrossRef]
- Burgess, S. Genomics: A matched set of frog sequences. Nature 2016, 538, 320–321. [Google Scholar] [CrossRef][Green Version]
- Ehrenreich, I.M. Evolution after genome duplication. Science 2020, 368, 1424–1425. [Google Scholar] [CrossRef] [PubMed]
- Schneiter, R.; Carter, A.T.; Hernando, Y.; Zellnig, G.; Schweizer, L.M.; Schweizer, M. The importance of the five phosphoribosyl-pyrophosphate synthetase (Prs) gene products of Saccharomyces cerevisiae in the maintenance of cell integrity and the subcellular localization of Prs1p. Microbiology 2000, 146, 3269–3278. [Google Scholar] [CrossRef]
- Bodenmiller, B.; Wanka, S.; Kraft, C.; Urban, J.; Campbell, D.; Pedrioli, P.G.; Gerrits, B.; Picotti, P.; Lam, H.; Vitek, O.; et al. Phosphoproteomic Analysis Reveals Interconnected System-Wide Responses to Perturbations of Kinases and Phosphatases in Yeast. Sci. Signal. 2010, 3, rs4. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Chatr-Aryamontri, A.; Boucher, L.; Oughtred, R.; Livstone, M.S.; Nixon, J.; Van Auken, K.; Wang, X.; Shi, X.; et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011, 39, D698–D704. [Google Scholar] [CrossRef] [PubMed]
- Swaney, D.L.; Beltrao, P.; Starita, L.; Guo, A.; Rush, J.; Fields, S.; Krogan, N.J.; Villen, J. Global analysis of phosphorylation and ubiquitylation cross-talk in protein degradation. Nat. Methods 2013, 10, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Soulard, A.; Cremonesi, A.; Moes, S.; Schütz, F.; Jenö, P.; Hall, M.N. The Rapamycin-sensitive Phosphoproteome Reveals That TOR Controls Protein Kinase A Toward Some But Not All Substrates. Mol. Biol. Cell 2010, 21, 3475–3486. [Google Scholar] [CrossRef] [PubMed]
- Gnad, F.; de Godoy, L.M.F.; Cox, J.; Neuhauser, N.; Ren, S.; Olsen, J.V.; Mann, M. High-accuracy identification and bioinformatic analysis of in vivo protein phosphorylation sites in yeast. Proteomics 2009, 9, 4642–4652. [Google Scholar] [CrossRef] [PubMed]
- Ficarro, S.B.; McCleland, M.L.; Stukenberg, P.T.; Burke, D.J.; Ross, M.M.; Shabanowitz, J.; Hunt, D.F.; White, F.M. Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 2002, 20, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Huber, A.; Bodenmiller, B.; Uotila, A.; Stahl, M.; Wanka, S.; Gerrits, B.; Aebersold, R.; Loewith, R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009, 23, 1929–1943. [Google Scholar] [CrossRef]
- Loewith, R. A brief history of TOR. Biochem. Soc. Trans. 2011, 39, 437–442. [Google Scholar] [CrossRef]
- Jiménez, A.; Santos, M.A.; Revuelta, J.L. Phosphoribosyl pyrophosphate synthetase activity affects growth and riboflavin production in Ashbya gossypii. BMC Biotechnol. 2008, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Ugbogu, E.A.; Wippler, S.; Euston, M.; Kouwenhoven, E.N.; de Brouwer, A.P.; Schweizer, L.M.; Schweizer, M. The contribution of the nonhomologous region of Prs1 to the maintenance of cell wall integrity and cell viability. FEMS Yeast Res. 2013, 13, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Herrero, P.; Martinez-Campa, C.; Moreno, F. The hexokinase 2 protein participates in regulatory DNA-protein complexes necessary for glucose repression of the SUC2 gene in Saccharomyces cerevisiae. FEBS Lett. 1998, 434, 71–76. [Google Scholar] [CrossRef]
- Hernando, Y.; Carter, A.T.; Parr, A.; Hove-Jensen, B.; Schweizer, M. Genetic analysis and enzyme activity suggest the existence of more than one minimal functional unit capable of synthesizing phosphoribosyl pyrophosphate in Saccharomyces cerevisiae. J. Biol. Chem. 1999, 274, 12480–12487. [Google Scholar] [CrossRef] [PubMed]
- Hove-Jensen, B. Heterooligomeric phosphoribosyl diphosphate synthase of Saccharomyces cerevisiae: Combinatorial expression of the five PRS genes in Escherichia coli. J. Biol. Chem. 2004, 279, 40345–40350. [Google Scholar] [CrossRef]
- Wang, K.; Vavassori, S.; Schweizer, L.M.; Schweizer, M. Impaired PRPP-synthesizing capacity compromises cell integrity signalling in Saccharomyces cerevisiae. Microbiology 2004, 150, 3327–3339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ugbogu, E.A.; Wang, K.; Schweizer, L.M.; Schweizer, M. Metabolic gene products have evolved to interact with the cell wall integrity pathway in Saccharomyces cerevisiae. FEMS Yeast Res. 2016, 16, fow092. [Google Scholar] [CrossRef]
- Vavassori, S.; Wang, K.; Schweizer, L.M.; Schweizer, M. In Saccharomyces cerevisiae, impaired PRPP synthesis is accompanied by valproate and Li+ sensitivity. Biochem. Soc. Trans. 2005, 33, 1154–1157. [Google Scholar] [CrossRef]
- Vavassori, S. An Investigation of the Influence of Impaired PRPP Production on the Physiology of Saccharomyces cerevisiae. Ph.D. Thesis, Heriot-Watt University, Edinburgh, UK, 2005. [Google Scholar]
- Wang, K. The Involvement of PRPP Synthetase in Cell Integrity Signalling in Saccharomyces cerevisiae. Ph.D. Thesis, Heriot-Watt University, Edinburgh, UK, 2005. [Google Scholar]
- Levin, D.E. Cell Wall Integrity Signaling in Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2005, 69, 262–291. [Google Scholar] [CrossRef]
- Levin, D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175. [Google Scholar] [CrossRef]
- Gonzalez-Rubio, G.; Sellers-Moya, A.; Martin, H.; Molina, M. A walk-through MAPK structure and functionality with the 30-year-old yeast MAPK Slt2. Int. Microbiol. 2021, 24, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, H.P.; Lorberg, A.; Heinisch, J.J. Regulation of yeast protein kinase C activity by interaction with the small GTPase Rho1p through its amino-terminal HR1 domain. Mol. Microbiol. 2002, 44, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, J.J.; Rodicio, R. Protein kinase C in fungi-more than just cell wall integrity. FEMS Microbiol. Rev. 2018, 42, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Sanz, A.B.; García, R.; Pavón-Vergés, M.; Rodriguez-Peña, J.M.; Arroyo, J. Control of Gene Expression via the Yeast CWI Pathway. Int. J. Mol. Sci. 2022, 23, 1791. [Google Scholar] [CrossRef]
- Jiménez-Gutiérrez, E.; Alegria-Carrasco, E.; Sellers-Moya, A.; Molina, M.; Martin, H. Not just the wall: The other ways to turn the yeast CWI pathway on. Int. Microbiol. 2020, 23, 107–119. [Google Scholar] [CrossRef]
- Stark, M.J.R. Protein phosphorylation and dephosphorylation. pp. 284-375; In The Metabolism and Molecuylar Physiology of Saccharomyces cerevisiae, 2nd ed.; Dickinson, J.R., Schweizer, M., Eds.; CRC Press: Boca Raton, FL, USA; London, UK; New York, NY, USA; Washington, DC, USA, 2004; pp. ix–xv, 1–438. [Google Scholar]
- Jiménez-Gutiérrez, E.; Alegria-Carrasco, E.; Alonso-Rodriguez, E.; Fernandez-Acero, T.; Molina, M.; Martin, H. Rewiring the yeast cell wall integrity (CWI) pathway through a synthetic positive feedback circuit unveils a novel role for the MAPKKK Ssk2 in CWI pathway activation. FEBS J. 2020, 287, 4881–4901. [Google Scholar] [CrossRef]
- Sauvaget, M.; Hutton, F.; Coull, R.; Vavassori, S.; Wang, K.; Reznik, A.; Chyker, T.; Newfield, C.G.; Euston, E.; Benary, G.; et al. The NHR1-1 of Prs1 and the pentameric motif 284KKCPK288 of Prs3 permit multi-functionality of the PRPP synthetase in Saccharomyces cerevisiae. FEMS Yeast Res. 2019, 19, foz006. [Google Scholar] [CrossRef]
- Ruta, L.L.; Farcasanu, I.C. Saccharomyces cerevisiae and Caffeine Implications on the Eukaryotic Cell. Nutrients 2020, 12, 2440. [Google Scholar] [CrossRef]
- Kuranda, K.; Leberre, V.; Sokol, S.; Palamarczyk, G.; Francois, J. Investigating the caffeine effects in the yeast Saccharomyces cerevisiae brings new insights into the connection between TOR, PKC and Ras/cAMP signalling pathways. Mol. Microbiol. 2006, 61, 1147–1166. [Google Scholar] [CrossRef]
- Loewith, R.; Hall, M.N. Target of Rapamycin (TOR) in Nutrient Signaling and Growth Control. Genetics 2011, 189, 1177–1201. [Google Scholar] [CrossRef]
- Vavassori, S.; Wang, K.; Schweizer, L.M.; Schweizer, M. Ramifications of impaired PRPP synthesis in Saccharomyces cerevisiae. Biochem. Soc. Trans. 2005, 33, 1418–1420. [Google Scholar] [CrossRef]
- Kim, K.Y.; Levin, D.E. Mpk1 MAPK association with the Paf1 complex blocks Sen1-mediated premature transcription termination. Cell 2011, 144, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; VanderSluis, B.; Koch, E.N.; Baryshnikova, A.; Pons, C.; Tan, G.; Wang, W.; Usaj, M.; Hanchard, J.; Lee, S.D.; et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353, aaf1420. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Sakakibara, K.; Chen, Q.; Okamoto, K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res. 2014, 24, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Nithianandarajah-Jones, G.N.; Wilm, B.; Goldring, C.E.P.; Muller, J.; Cross, M.J. ERK5: Structure, regulation and function. Cell. Signal. 2012, 24, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
- Truman, A.W.; Millson, S.H.; Nuttall, J.M.; King, V.; Mollapour, M.; Prodromou, C.; Pearl, L.H.; Piper, P.W. Expressed in the yeast Saccharomyces cerevisiae, human ERK5 is a client of the Hsp90 chaperone that complements loss of the Slt2D (Mpk1p) cell integrity stress-activated protein kinase. Eukaryot. Cell 2006, 5, 1914–1924. [Google Scholar] [CrossRef]
- Fasolo, J.; Sboner, A.; Sun, M.G.; Yu, H.; Chen, R.; Sharon, D.; Kim, P.M.; Gerstein, M.; Snyder, M. Diverse protein kinase interactions identified by protein microarrays reveal novel connections between cellular processes. Genes Dev. 2011, 25, 767–778. [Google Scholar] [CrossRef]
- Ugbogu, A.E. The PRS Gene Family Links Primary Metabolism and Cell Signalling in Saccharomyces cerevisiae. Ph.D. Thesis, Heriot-Watt University, Edinburgh, UK, 2014. [Google Scholar]
- Uetz, P.; Giot, L.; Cagney, G.; Mansfield, T.A.; Judson, R.S.; Knight, J.R.; Lockshon, D.; Narayan, V.; Srinivasan, M.; Pochart, P.; et al. A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 2000, 403, 623–627. [Google Scholar] [CrossRef]
- Lin, Y.Y.; Lu, J.Y.; Zhang, J.; Walter, W.; Dang, W.; Wan, J.; Tao, S.C.; Qian, J.; Zhao, Y.; Boeke, J.D.; et al. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell 2009, 136, 1073–1084. [Google Scholar] [CrossRef]
- Kleineidam, A.; Vavassori, S.; Wang, K.; Schweizer, L.M.; Griac, P.; Schweizer, M. Valproic acid- and lithium-sensitivity in prs mutants of Saccharomyces cerevisiae. Biochem. Soc. Trans. 2009, 37, 1115–1120. [Google Scholar] [CrossRef]
- Yabuki, Y.; Kodama, Y.; Katayama, M.; Sakamoto, A.; Kanemaru, H.; Wan, K.; Mizuta, K. Glycogen synthase kinase-3 is involved in regulation of ribosome biogenesis in yeast. Biosci. Biotechnol. Biochem. 2014, 78, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen synthase kinase-3 (GSK3): Regulation, actions, and diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef]
- Binley, K.M.; Radcliffe, P.A.; Trevethick, J.; Duffy, K.A.; Sudbery, P.E. The yeast PRS3 gene is required for cell integrity, cell cycle arrest upon nutrient deprivation, ion homeostasis and the proper organization of the actin cytoskeleton. Yeast 1999, 15, 1459–1469. [Google Scholar] [CrossRef]
- Care, A.; Vousden, K.A.; Binley, K.M.; Radcliffe, P.; Trevethick, J.; Mannazzu, I.; Sudbery, P.E. A synthetic lethal screen identifies a role for the cortical actin patch/endocytosis complex in the response to nutrient deprivation in Saccharomyces cerevisiae. Genetics 2004, 166, 707–719. [Google Scholar] [CrossRef]
- Kamada, Y.; Jung, U.S.; Piotrowski, J.; Levin, D.E. The protein kinase C-activated MAP kinase pathway of Saccharomyces cerevisiae mediates a novel aspect of the heat shock response. Genes Dev. 1995, 9, 1559–1571. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.S.; Thiele, D.J. Regulation of the Saccharomyces cerevisiae Slt2 kinase pathway by the stress-inducible Sdp1 dual specificity phosphatase. J. Biol. Chem. 2002, 277, 21278–21284. [Google Scholar] [CrossRef]
- Shi, X.; Finkelstein, A.; Wolf, A.J.; Wade, P.A.; Burton, Z.F.; Jaehning, J.A. Paf1p, an RNA polymerase II-associated factor in Saccharomyces cerevisiae, may have both positive and negative roles in transcription. Mol. Cell. Biol. 1996, 16, 669–676. [Google Scholar] [CrossRef]
- Chang, M.; French-Cornay, D.; Fan, H.Y.; Klein, H.; Denis, C.L.; Jaehning, J.A. A complex containing RNA polymerase II, Paf1cp, CDc73p, Hpr1p and Ccr4p plays a role in protein kinase C signaling. Mol. Cell. Biol. 1999, 19, 1056–1067. [Google Scholar] [CrossRef]
- Ellison, M.A.; Lederer, A.R.; Warner, M.H.; Mavrich, T.N.; Raupach, E.A.; Heisler, L.E.; Nislow, C.; Lee, M.T.; Arndt, K.M. The Paf1 Complex Broadly Impacts the Transcriptome of Saccharomyces cerevisiae. Genetics 2019, 212, 711–728. [Google Scholar] [CrossRef]
- Porter, S.E.; Washburn, T.M.; Chang, M.P.; Jaehning, J.A. The yeast Paf1-RNA polymerase II complex is required for full expression of a subset of cell cycle-regulated genes. Eukaryot. Cell 2002, 1, 830–842. [Google Scholar] [CrossRef][Green Version]
- Jaehning, J.A. The Paf1 complex: Platform or player in RNA polymerase II transcription? Biochem. Biophys. Res. Commun. 2010, 1799, 379–388. [Google Scholar] [CrossRef] [PubMed]
- de Brouwer, A.P.; van Bokhoven, H.; Nabuurs, S.B.; Arts, W.F.; Christodoulou, J.; Duley, J. PRPS1 mutations: Four distinct syndromes and potential treatment. Am. J. Hum. Genet. 2010, 86, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.A.; Puig, J.G.; Mateos, F.A.; Jimenez, M.L.; Kim, M.; Simmonds, H.A. Inherited superactivity of phosphoribosylpyrophosphate synthetase: Association of uric acid overproduction and sensorineural deafness. Am. J. Med. 1988, 85, 383–390. [Google Scholar] [CrossRef]
- Roessler, B.J.; Golovoy, N.; Palella, T.D.; Heidler, S.; Becker, M.A. Identification of Distinct PRS1 Mutations in Two Patients with X-Linked Phosphoribosylpyrophosphate Synthetase Superactivity. In Purine and Pyrimidine Metabolism in Man VII; Harkness, R.A., Elion, G.B., Zöllner, N., Eds.; Springer: New York, NY, USA, 1991; Volume 309B, pp. 125–128. [Google Scholar] [CrossRef]
- Sperling, O.; Sarapers; Eilam, G.; Devries, A. Accelerated Erythrocyte 5-Phosphoribosyl-1-Pyrophosphate Synthesis. A familial Abnormality Associated with Excessive Uric-Acid Production and Gout. Biochem. Med. 1972, 6, 310–316. [Google Scholar] [CrossRef]
- Chen, P.; Liu, Z.; Wang, X.; Peng, J.; Sun, Q.; Li, J.; Wang, M.; Niu, L.; Zhang, Z.; Cai, G.; et al. Crystal and EM structures of human phosphoribosyl pyrophosphate synthase I (PRS1) provide novel insights into the disease-associated mutations. PLoS ONE 2015, 10, e0120304. [Google Scholar] [CrossRef]
- Varshney, G.K.; Lu, J.; Gildea, D.E.; Huang, H.; Pei, W.H.; Yang, Z.; Huang, S.C.; Schoenfeld, D.; Pho, N.H.; Casero, D.; et al. A large scale zebrafish gene knockout resource for the genome-wide study of gene function. Genome Res. 2013, 23, 727–735. [Google Scholar] [CrossRef]
- Begovich, K.; Yelon, D.; Wilhelm, J.E. PRPS polymerization influences lens fiber organization in zebrafish. Dev. Dyn. 2020, 249, 1018–1031. [Google Scholar] [CrossRef]
- Zoref, E.; Vries, A.; Sperling, O. Mutant feedback-resistant phosphoribosylpyrophosphate synthetase associated with purine overproduction and gout. Phosphoribosylpyrophosphate and purine metabolism in cultured fibroblasts. J. Clin. Investig. 1975, 56, 1093–1099. [Google Scholar] [CrossRef]
- Noree, C.; Begovich, K.; Samilo, D.; Broyer, R.; Monfort, E.; Wilhelm, J.E. A quantitative screen for metabolic enzyme structures reveals patterns of assembly across the yeast metabolic network. Mol. Biol. Cell 2019, 30, 2721–2736. [Google Scholar] [CrossRef]
- Noree, C.; Sato, B.K.; Broyer, R.M.; Wilhelm, J.E. Identification of novel filament-forming proteins in Saccharomyces cerevisiae and Drosophila melanogaster. J. Cell Biol. 2010, 190, 541–551. [Google Scholar] [CrossRef]
- Taira, M.; Ishijima, S.; Kita, K.; Yamada, K.; Iizasa, T.; Tatibana, M. Nucleotide and deduced amino acid sequences of two distinct cDNAs for rat phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1987, 262, 14867–14870. [Google Scholar] [CrossRef]
- Iizasa, T.; Taira, M.; Shimada, H.; Ishijima, S.; Tatibana, M. Molecular cloning and sequencing of human cDNA for phosphoribosyl pyrophosphate synthetase subunit II. FEBS Lett. 1989, 244, 47–50. [Google Scholar] [CrossRef]
- Roessler, B.J.; Bell, G.; Heidler, S.; Seino, S.; Becker, M.; Palella, T.D. Cloning of 2 Distinct Copies of Human Phosphoribosylpyrophosphate Synthetase cDNA. Nucleic Acids Res. 1990, 18, 193. [Google Scholar] [CrossRef] [PubMed]
- Ishizuka, T.; Iizasa, T.; Taira, M.; Ishijima, S.; Sonoda, T.; Shimada, H.; Nagatake, N.; Tatibana, M. Promoter Regions of the Human X-Linked Housekeeping Gene-Prps1 and Gene-Prps2 Encoding Phosphoribosylpyrophosphate Synthetase Subunit-I and Subunit-Ii Isoforms. Biochim. Biophys. Acta 1992, 1130, 139–148. [Google Scholar] [CrossRef]
- Sonoda, T.; Taira, M.; Ishijima, S.; Ishizuka, T.; Iizasa, T.; Tatibana, M. Complete nucleotide sequence of human phosphoribosyl pyrophosphate synthetase subunit I (PRS I) cDNA and a comparison with human and rat PRPS gene families. J. Biochem. 1991, 109, 361–364. [Google Scholar]
- Li, S.; Lu, Y.; Peng, B.; Ding, J. Crystal structure of human phosphoribosylpyrophosphate synthetase 1 reveals a novel allosteric site. Biochem. J. 2007, 401, 39–47. [Google Scholar] [CrossRef]
- Krath, B.N.; Hove-Jensen, B. Class II recombinant phosphoribosyl diphosphate synthase from spinach. Phosphate independence and diphosphoryl donor specificity. J. Biol. Chem. 2001, 276, 17851–17856. [Google Scholar] [CrossRef]
- Krath, B.N.; Hove-Jensen, B. Implications of secondary structure prediction and amino acid sequence comparison of class I and class II phosphoribosyl diphosphate synthases on catalysis, regulation, and quaternary structure. Protein Sci. 2001, 10, 2317–2324. [Google Scholar] [CrossRef]
- Kadziola, A.; Jepsen, C.H.; Johansson, E.; McGuire, J.; Larsen, S.; Hove-Jensen, B. Novel class III phosphoribosyl diphosphate synthase: Structure and properties of the tetrameric, phosphate-activated, non-allosterically inhibited enzyme from Methanocaldococcus jannaschii. J. Mol. Biol. 2005, 354, 815–828. [Google Scholar] [CrossRef]
- Cherney, M.M.; Cherney, L.T.; Garen, C.R.; James, M.N. The structures of Thermoplasma volcanium phosphoribosyl pyrophosphate synthetase bound to ribose-5-phosphate and ATP analogs. J. Mol. Biol. 2011, 413, 844–856. [Google Scholar] [CrossRef]
- Tang, W.; Li, X.; Zhu, Z.; Tong, S.; Zhang, X.; Teng, M.; Niu, L. Expression, purification, crystallization and preliminary X-ray diffraction analysis of human phosphoribosyl pyrophosphate synthetase 1 (PRS1). Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 432–434. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.; Wang, Y. Targeted Quantitative Kinome Analysis Identifies PRPS2 as a Promoter for Colorectal Cancer Metastasis. J. Proteome Res. 2019, 18, 2279–2286. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef] [PubMed]
- Katashima, R.; Iwahana, H.; Fujimura, M.; Yamaoka, T.; Itakura, M. Assignment of the human phosphoribosylpyrophosphate synthetase- associated protein 41 gene (PRPSAP2) to 17p11.2-p12. Genomics 1998, 54, 180–181. [Google Scholar] [CrossRef]
- Katashima, R.; Iwahana, H.; Fujimura, M.; Yamaoka, T.; Ishizuka, T.; Tatibana, M.; Itakura, M. Molecular cloning of a human cDNA for the 41-kDa phosphoribosylpyrophosphate synthetase-associated protein. Biochim. Biophys. Acta 1998, 1396, 245–250. [Google Scholar] [CrossRef]
- Ishizuka, T.; Kita, K.; Sonoda, T.; Ishijima, S.; Sawa, K.; Suzuki, N.; Tatibana, M. Cloning and sequencing of human complementary DNA for the phosphoribosylpyrophosphate synthetase-associated protein 39. Biochim. Biophys. Acta 1996, 1306, 27–30. [Google Scholar] [CrossRef]
- Ishizuka, T.; Ahmad, I.; Kita, K.; Sonoda, T.; Ishijima, S.; Sawa, K.; Suzuki, N.; Tatibana, M. The human phosphoribosylpyrophosphate synthetase-associated protein 39 gene (PRPSAP1) is located in the chromosome region 17q24-q25. Genomics 1996, 33, 332–334. [Google Scholar] [CrossRef]
- Sonoda, T.; Ishizuka, T.; Kita, K.; Ishijima, S.; Tatibana, M. Cloning and sequencing of rat cDNA for the 41-kDa phosphoribosylpyrophosphate synthetase-associated protein has a high homology to the catalytic subunits and the 39-kDa associated protein. Biochim. Biophys. Acta 1997, 1350, 6–10. [Google Scholar] [CrossRef]
- Almoguera, B.; He, S.; Corton, M.; Fernandez-San Jose, P.; Blanco-Kelly, F.; Lopez-Molina, M.I.; Garcia-Sandoval, B.; Del Val, J.; Guo, Y.; Tian, L.; et al. Expanding the phenotype of PRPS1 syndromes in females: Neuropathy, hearing loss and retinopathy. Orphanet J. Rare Dis. 2014, 9, 190. [Google Scholar] [CrossRef]
- Synofzik, M.; Muller vom Hagen, J.; Haack, T.B.; Wilhelm, C.; Lindig, T.; Beck-Wodl, S.; Nabuurs, S.B.; van Kuilenburg, A.B.; de Brouwer, A.P.; Schols, L. X-linked Charcot-Marie-Tooth disease, Arts syndrome, and prelingual non-syndromic deafness form a disease continuum: Evidence from a family with a novel PRPS1 mutation. Orphanet J. Rare Dis. 2014, 9, 24. [Google Scholar] [CrossRef]
- Kwiatek, J.M.; Gik-Soo, H.; Carman, G.M. Phosphatidate-mediated regulation of lipid synthesis at the nuclear/endoplasmic reticulum membrane. BBA—Mol. Cell. Biol. Lipids 2020, 1865, 15843. [Google Scholar] [CrossRef]
- Duley, J.A.; Christodoulou, J.; de Brouwer, A.P. The PRPP synthetase spectrum: What does it demonstrate about nucleotide syndromes? Nucleosides Nucleotides Nucleic Acids 2011, 30, 1129–1139. [Google Scholar] [CrossRef]
- García-Pavía, P.; Torres, R.J.; Rivero, M.; Ahmed, M.; García-Puig, J.; Becker, M.A. Phosphoribosylpyrophosphate synthetase overactivity as a cause of uric acid overproduction in a young woman. Arthritis Rheum. 2003, 48, 2036–2041. [Google Scholar] [CrossRef]
- Becker, M.A.; Smith, P.R.; Taylor, W.; Mustafi, R.; Switzer, R.L. The genetic and functional basis of purine nucleotide feedback- resistant phosphoribosylpyrophosphate synthetase superactivity. J. Clin. Investig. 1995, 96, 2133–2141. [Google Scholar] [CrossRef]
- Becker, M.A.; Raivio, K.O.; Bakay, B.; Adams, W.B.; Nyhan, W.L. Superactive phosphoribosylpyrophosphate synthetase with altered regulatory and catalytic properties. Adv. Exp. Med. Biol. 1980, 122A, 387–392. [Google Scholar]
- Becker, M.A.; Losman, M.J.; Wilson, J.; Simmonds, H.A. Superactivity of human phosphoribosyl pyrophosphate synthetase due to altered regulation by nucleotide inhibitors and inorganic phosphate. Biochim. Biophys. Acta 1986, 882, 168–176. [Google Scholar] [CrossRef]
- Becker, M.A.; Taylor, W.; Smith, P.R.; Ahmed, M. Overexpression of the normal phosphoribosylpyrophosphate synthetase 1 isoform underlies catalytic superactivity of human phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1996, 271, 19894–19899. [Google Scholar] [CrossRef]
- Ahmed, M.; Taylor, W.; Smith, P.R.; Becker, M.A. Accelerated transcription of PRPS1 in X-linked overactivity of normal human phosphoribosylpyrophosphate synthetase. J. Biol. Chem. 1999, 274, 7482–7488. [Google Scholar] [CrossRef]
- Gandía, M.; Fernandez-Toral, J.; Solanellas, J.; Dominguez-Ruiz, M.; Gómez-Rosas, E.; Del Castillo, F.J.; Villamar, M.; Moreno-Pelayo, M.A.; Del Castillo, I. Mutations in PRPS1 causing syndromic or nonsyndromic hearing impairment: Intrafamilial phenotypic variation complicates genetic counseling. Pediatr. Res. 2015, 78, 97–102. [Google Scholar] [CrossRef]
- Moran, R.; Kuilenburg, A.B.; Duley, J.; Nabuurs, S.B.; Retno-Fitri, A.; Christodoulou, J.; Roelofsen, J.; Yntema, H.G.; Friedman, N.R.; van Bokhoven, H.; et al. Phosphoribosylpyrophosphate synthetase superactivity and recurrent infections is caused by a p.Val142Leu mutation in PRS-I. Am. J. Med. Genet. A 2012, 158, 455–460. [Google Scholar] [CrossRef]
- Mittal, R.; Patel, K.; Mittal, J.; Chan, B.; Yan, D.; Grati, M.; Liu, X.Z. Association of PRPS1 Mutations with Disease Phenotypes. Dis. Markers 2015, 2015, 127013. [Google Scholar] [CrossRef]
- Porrmann, J.; Betcheva-Krajcir, E.; Di Donato, N.; Kahlert, A.K.; Schallner, J.; Rump, A.; Schrock, E.; Dobritzsch, D.; Roelofsen, J.; van Kuilenburg, A.B.P.; et al. Novel PRPS1 gain-of-function mutation in a patient with congenital hyperuricemia and facial anomalies. Am. J. Med. Genet. A 2017, 173, 2736–2742. [Google Scholar] [CrossRef]
- Disteche, C.M.; Berletch, J.B. X-chromosome inactivation and escape. J. Genet. 2015, 94, 591–599. [Google Scholar] [CrossRef]
- Balaton, B.P.; Dixon-McDougall, T.; Peeters, S.B.; Brown, C.J. The eXceptional nature of the X chromosome. Hum. Mol. Genet. 2018, 27, R242–R249. [Google Scholar] [CrossRef]
- Yang, B.Y.; Yu, H.X.; Min, J.; Song, X.X. A novel mutation in gene of PRPS1 in a young Chinese woman with X-linked gout: A case report and review of the literature. Clin. Rheumatol. 2020, 39, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Zikánová, M.; Wahezi, D.; Hay, A.; Stiburková, B.; Pitts, C.; Mušálková, D.; Škopová, V.; Barešová, V.; Soucková, O.; Hodanová, K.; et al. Clinical manifestations and molecular aspects of phosphoribosylpyrophosphate synthetase superactivity in females. Rheumatology 2018, 57, 1180–1185. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Li, J.; Ma, J.; Teng, M.; Li, X. A small disturbance, but a serious disease: The possible mechanism of D52H-mutant of human PRS1 that causes gout. IUBMB Life 2013, 65, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Ogaya, S.; Kurahashi, N.; Umemura, A.; Yamada, K.; Hashiguchi, A.; Takashima, H.; Torres, R.J.; Aso, K. Arts syndrome with a novel missense mutation in the PRPS1 gene: A case report. Brain Dev. 2016, 38, 954–958. [Google Scholar] [CrossRef] [PubMed]
- de Brouwer, A.P.M.; Christodoulou, J. Arts Syndrome. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Eds.; University of Washington: Seattle, WA, USA, 2008; (updated 22 March 2018). [Google Scholar]
- Puusepp, S.; Reinson, K.; Pajusalu, S.; van Kuilenburg, A.B.P.; Dobritzsch, D.; Roelofsen, J.; Stenzel, W.; Õunap, K. Atypical presentation of Arts syndrome due to a novel hemizygous loss-of-function variant in the PRPS1 gene. Mol. Genet. Metab. Rep. 2020, 25, 100677. [Google Scholar] [CrossRef]
- Emmanuel, N.; Ragunathan, S.; Shan, Q.; Wang, F.; Giannakou, A.; Huser, N.; Jin, G.X.; Myers, J.; Abraham, R.T.; Unsal-Kacmaz, K. Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep. 2017, 19, 2665–2680. [Google Scholar] [CrossRef]
- Lenherr, N.; Christodoulou, J.; Duley, J.; Dobritzsch, D.; Fairbanks, L.; Datta, A.N.; Filges, I.; Gurtler, N.; Roelofsen, J.; van Kuilenburg, A.B.P.; et al. Co-therapy with S-adenosylmethionine and nicotinamide riboside improves t-cell survival and function in Arts Syndrome (PRPS1 deficiency). Mol. Genet. Metab. Rep. 2021, 26, 100709. [Google Scholar] [CrossRef]
- Bottiglieri, T. S-Adenosyl-L-methionine (SAMe): From the bench to the bedside--molecular basis of a pleiotrophic molecule. Am. J. Clin. Nutr. 2002, 76, 1151S–1157S. [Google Scholar] [CrossRef]
- Chi, Y.; Sauve, A.A. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 657–661. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef]
- Nishikura, N.; Yamagata, T.; Morimune, T.; Matsui, J.; Sokoda, T.; Sawai, C.; Sakaue, Y.; Higuchi, Y.; Hashiguchi, A.; Takashima, H.; et al. X-linked Charcot-Marie-Tooth disease type 5 with recurrent weakness after febrile illness. Brain Dev. 2019, 41, 201–204. [Google Scholar] [CrossRef]
- Sugano, M.; Hirayama, K.; Saito, T.; Tsukamoto, T.; Yamamoto, T. Optic atrophy, sensorineural hearing loss and polyneuropathy—A case of sporadic Rosenberg-Chutorian syndrome. Fukushima J. Med. Sci. 1992, 38, 57–65. [Google Scholar]
- Agrahari, A.K.; Sneha, P.; George Priya Doss, C.; Siva, R.; Zayed, H. A profound computational study to prioritize the disease-causing mutations in PRPS1 gene. Metab. Brain Dis. 2018, 33, 589–600. [Google Scholar] [CrossRef]
- Robusto, M.; Fang, M.; Asselta, R.; Castorina, P.; Previtali, S.C.; Caccia, S.; Benzoni, E.; De Cristofaro, R.; Yu, C.; Cesarani, A.; et al. The expanding spectrum of PRPS1-associated phenotypes: Three novel mutations segregating with X-linked hearing loss and mild peripheral neuropathy. Eur. J. Hum. Genet. 2015, 23, 766–773. [Google Scholar] [CrossRef]
- Park, J.; Hyun, Y.S.; Kim, Y.J.; Nam, S.H.; Kim, S.H.; Hong, Y.B.; Park, J.M.; Chung, K.W.; Choi, B.O. Exome Sequencing Reveals a Novel PRPS1 Mutation in a Family with CMTX5 without Optic Atrophy. J. Clin. Neurol. 2013, 9, 283–288. [Google Scholar] [CrossRef]
- Lerat, J.; Magdelaine, C.; Derouault, P.; Beauvais-Dzugan, H.; Bieth, E.; Acket, B.; Arne-Bes, M.C.; Sturtz, F.; Lia, A.S. New PRPS1 variant p.(Met68Leu) located in the dimerization area identified in a French CMTX5 patient. Mol. Genet. Genom. Med. 2019, 7, e875. [Google Scholar] [CrossRef]
- Meng, L.; Wang, K.; Lv, H.; Wang, Z.; Zhang, W.; Yuan, Y. A novel mutation in PRPS1 causes X-linked Charcot-Marie-Tooth disease-5. Neuropathology 2019, 39, 342–347. [Google Scholar] [CrossRef]
- Shirakawa, S.; Murakami, T.; Hashiguchi, A.; Takashima, H.; Hasegawa, H.; Ichida, K.; Sunada, Y. A Novel PRPS1 Mutation in a Japanese Patient with CMTX5. Intern. Med. 2021, 8029-21. [Google Scholar] [CrossRef]
- Corvino, V.; Apisa, P.; Malesci, R.; Laria, C.; Auletta, G.; Franze, A. X-Linked Sensorineural Hearing Loss: A Literature Review. Curr. Genom. 2018, 19, 327–338. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, A.R.; Kim, N.K.; Lee, C.; Han, J.H.; Kim, M.Y.; Jeon, E.H.; Park, W.Y.; Mittal, R.; Yan, D.; et al. Functional characterization of a novel loss-of-function mutation of PRPS1 related to early-onset progressive nonsyndromic hearing loss in Koreans (DFNX1): Potential implications on future therapeutic intervention. J. Gene Med. 2016, 18, 353–358. [Google Scholar] [CrossRef]
- Song, M.H.; Lee, K.Y.; Choi, J.Y.; Bok, J.; Kim, U.K. Nonsyndromic X-linked hearing loss. Front. Biosci. (Elite Ed.) 2012, 4, 924–933. [Google Scholar] [CrossRef]
- Mercati, O.; Abi Warde, M.T.; Lina-Granade, G.; Rio, M.; Heide, S.; de Lonlay, P.; Ceballos-Picot, I.; Robert, M.P.; Couloigner, V.; Beltrand, J.; et al. PRPS1 loss-of-function variants, from isolated hearing loss to severe congenital encephalopathy: New cases and literature review. Eur. J. Med. Genet. 2020, 63, 104033. [Google Scholar] [CrossRef]
- Fiorentino, A.; Fujinami, K.; Arno, G.; Robson, A.G.; Pontikos, N.; Arasanz Armengol, M.; Plagnol, V.; Hayashi, T.; Iwata, T.; Parker, M.; et al. Missense variants in the X-linked gene PRPS1 cause retinal degeneration in females. Hum. Mutat. 2018, 39, 80–91. [Google Scholar] [CrossRef]
- Al-Maawali, A.; Dupuis, L.; Blaser, S.; Heon, E.; Tarnopolsky, M.; Al-Murshedi, F.; Marshall, C.R.; Paton, T.; Scherer, S.W.; Roelofsen, J.; et al. Prenatal growth restriction, retinal dystrophy, diabetes insipidus and white matter disease: Expanding the spectrum of PRPS1-related disorders. Eur. J. Hum. Genet. 2015, 23, 310–316. [Google Scholar] [CrossRef]
- Georgiou, M.; Grewal, P.S.; Narayan, A.; Alser, M.; Ali, N.; Fiujinami, K.; Webster, A.R.; Michaelides, M. Sector Retinitis Pigmentosa: Extending the Molecular Genetics Basis and Elucidating the Natural History. Am. J. Ophthal. 2021, 221, 299. [Google Scholar] [CrossRef]
- Diñeiro, M.; Capín, R.; Cifuentes, G.; Fernández-Vega, B.; Villota, E.; Otero, A.; Santiago, A.; Pruneda, P.C.; Castillo, D.; Viejo-Díaz, M.; et al. Comprehensive genomic diagnosis of inherited retinal and optical nerve disorders reveals hidden syndromes and personalized therapeutic options. Acta Ophthalmol. 2020, 98, e1034–e1048. [Google Scholar] [CrossRef]
- Liu, R.; Li, J.Y.; Shao, J.C.; Lee, J.H.; Qiu, X.M.; Xiao, Y.X.; Zhang, B.W.; Hao, Y.L.; Li, M.; Chen, Q.M. Innate immune response orchestrates phosphoribosyl pyrophosphate synthetases to support DNA repair. Cell Metab. 2021, 33, 2076–2089.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Brady, S.W.; Ma, X.; Shen, S.; Zhang, Y.; Li, Y.; Szlachta, K.; Dong, L.; Liu, Y.; Yang, F.; et al. Therapy-induced mutations drive the genomic landscape of relapsed acute lymphoblastic leukemia. Blood 2020, 135, 41–55. [Google Scholar] [CrossRef]
- Mullighan, C.G. Mutant PRPS1: A new therapeutic target in relapsed acute lymphoblastic leukemia. Nat. Med. 2015, 21, 553–554. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Song, L.; Zhang, Y.; Han, Y.; Zhan, Z.; Xv, Z.; Li, Y.; Tang, Y.; Yang, Y.; Wang, S.; et al. Molecular mechanism of c-Myc and PRPS1/2 against thiopurine resistance in Burkitt’s lymphoma. J. Cell. Mol. Med. 2020, 24, 6704–6715. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Li, H.; Bai, Y.; Kirschner-Schwabe, R.; Yang, J.J.; Chen, Y.; Lu, G.; Tzoneva, G.; Ma, X.; Wu, T.; et al. Negative feedback-defective PRPS1 mutants drive thiopurine resistance in relapsed childhood ALL. Nat. Med. 2015, 21, 563–571. [Google Scholar] [CrossRef]
- Xue, C.; Yu, D.M.; Gherardi, S.; Koach, J.; Milazzo, G.; Gamble, L.; Liu, B.; Valli, E.; Russell, A.J.; London, W.B.; et al. MYCN promotes neuroblastoma malignancy by establishing a regulatory circuit with transcription factor AP4. Oncotarget 2016, 7, 54937–54951. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hsieh, A.C.; Ruggero, D. Targeting Eukaryotic Translation Initiation Factor 4E (eIF4E) in Cancer. Clin. Cancer Res. 2010, 16, 4914–4920. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61. [Google Scholar] [CrossRef]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef]
- Lei, B.; Xie, L.X.; Zhang, S.B.; Wan, B.; Zhong, L.R.; Zhou, X.M.; Mao, X.M.; Shu, F.P. Phosphoribosyl-pyrophosphate synthetase 2 (PRPS2) depletion regulates spermatogenic cell apoptosis and is correlated with hypospermatogenesis. Asian J. Androl. 2020, 22, 493–499. [Google Scholar] [CrossRef]
- Luo, Y.; Yuan, J.; Huang, J.; Yang, T.; Liu, M.; Chen, J.; Huang, W.; Zhang, H. Role of PRPS2 as a prognostic and therapeutic target in osteosarcoma. J. Clin. Pathol. 2021, 74, E12. [Google Scholar] [CrossRef]
- Ma, Y.; An, X.; Guan, X.; Kong, Q.; Wang, Y.; Li, P.; Meng, Y.; Cui, Y.; Wen, X.; Guo, Y.; et al. High expression of PRPS1 induces an anti-apoptotic effect in B-ALL cell lines and predicts an adverse prognosis in Chinese children with B-ALL. Oncol. Lett. 2018, 15, 4314–4322. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Chao, L.; You, Y.P. PRPS1 silencing reverses cisplatin resistance in human breast cancer cells. Biochem. Cell Biol. 2017, 95, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Minchenko, O.H.; Garmash, I.A.; Kovalevska, O.V.; Tsymbal, D.O.; Minchenko, D.O. Expression of phosphoribosyl pyrophosphate synthetase genes in U87 glioma cells with ERN1 knockdown: Effect of hypoxia and endoplasmic reticulum stress. Ukr. Biochem. J. 2014, 86, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, T.; Ramos da Silva, S.; Lee, J.J.; Lu, C.; Eoh, H.; Jung, J.U.; Gao, S.J. A Critical Role of Glutamine and Asparagine γ-Nitrogen in Nucleotide Biosynthesis in Cancer Cells Hijacked by an Oncogenic Virus. mBio 2017, 8, e01179-17. [Google Scholar] [CrossRef]
- Angius, A.; Uva, P.; Pira, G.; Muroni, M.R.; Sotgiu, G.; Saderi, L.; Uleri, E.; Caocci, M.; Ibba, G.; Cesaraccio, M.R.; et al. Integrated Analysis of miRNA and mRNA Endorses a Twenty miRNAs Signature for Colorectal Carcinoma. Int. J. Mol. Sci. 2019, 20, 4067. [Google Scholar] [CrossRef]
- Preethi, B.; Ramanathan, K. In silico analysis of functional nsSNPs of the human PRPS1 gene. Res. J. Pharm. Biol. Chem. Sci. 2015, 6, 846–851. [Google Scholar]
- Li, X.; Qian, X.; Peng, L.X.; Jiang, Y.; Hawke, D.H.; Zheng, Y.; Xia, Y.; Lee, J.H.; Cote, G.; Wang, H.; et al. A splicing switch from ketohexokinase-C to ketohexokinase-A drives hepatocellular carcinoma formation. Nat. Cell Biol. 2016, 18, 561–571. [Google Scholar] [CrossRef]
- Yang, J.; Yang, S.; Wang, Q.; Pang, J.; Wang, Y.; Wang, H.; Fu, X. KHK-A promotes the proliferation of oesophageal squamous cell carcinoma through the up-regulation of PRPS1. Arab J. Gastroenterol. 2021, 22, 40–46. [Google Scholar] [CrossRef]
- Li, C.; Yan, Z.; Cao, X.; Zhang, X.; Yang, L. Phosphoribosylpyrophosphate Synthetase 1 Knockdown Suppresses Tumor Formation of Glioma CD133+ Cells Through Upregulating Cell Apoptosis. J. Mol. Neurosci. 2016, 60, 145–156. [Google Scholar] [CrossRef]
- Yang, L.; Yan, Z.; Wang, Y.; Ma, W.; Li, C. Down-expression of miR-154 suppresses tumourigenesis in CD133(+) glioblastoma stem cells. Cell Biochem. Funct. 2016, 34, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ye, J.; Zhu, S.; Cui, H. Down-Regulation of Phosphoribosyl Pyrophosphate Synthetase 1 Inhibits Neuroblastoma Cell Proliferation. Cells 2019, 8, 955. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Wang, X.J.; Zhang, T.; Zhu, W.; Fang, Y.; Wu, H.; Liu, X.; Ma, D.; Ji, X.; Jiang, Y.; et al. Cell-Cycle-Dependent Phosphorylation of PRPS1 Fuels Nucleotide Synthesis and Promotes Tumorigenesis. Cancer Res. 2019, 79, 4650–4664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Patel, D.M.; Colavita, K.; Rodionova, I.; Buckley, B.; Scott, D.; Kumar, A.; Shabalina, S.A.; Saha, S.; Chernov, M.; et al. Arginylation regulates purine biosynthesis by enhancing the biological activity of phosphoribosyl pyrophospate synthase. Nat. Comm. 2015, 6, 7517. [Google Scholar] [CrossRef]
- Kaida, A.; Ariumi, Y.; Baba, K.; Matsubae, M.; Takao, T.; Shimotohno, K. Identification of a novel p300-specific-associating protein, PRS1 (phosphoribosylpyrophosphate synthetase subunit 1). Biochem. J. 2005, 391, 239–247. [Google Scholar] [CrossRef][Green Version]
- Qiao, H.; Tan, X.; Lv, D.J.; Xing, R.W.; Shu, F.P.; Zhong, C.F.; Li, C.; Zou, Y.G.; Mao, X.M. Phosphoribosyl pyrophosphate synthetases 2 knockdown inhibits prostate cancer progression by suppressing cell cycle and inducing cell apoptosis. J. Cancer 2020, 11, 1027–1037. [Google Scholar] [CrossRef]
- Qian, X.; Li, X.; Tan, L.; Lee, J.H.; Xia, Y.; Cai, Q.; Zheng, Y.; Wang, H.; Lorenzi, P.L.; Lu, Z. Conversion of PRPS Hexamer to Monomer by AMPK-Mediated Phosphorylation Inhibits Nucleotide Synthesis in Response to Energy Stress. Cancer Discov. 2018, 8, 94–107. [Google Scholar] [CrossRef]
- Lei, B.; Wan, B.; Peng, J.; Yang, Y.; Lv, D.; Zhou, X.; Shu, F.; Li, F.; Zhong, L.; Wu, H.; et al. PRPS2 Expression Correlates with Sertoli-Cell Only Syndrome and Inhibits the Apoptosis of TM4 Sertoli Cells. J. Urol. 2015, 194, 1491–1497. [Google Scholar] [CrossRef]
- Wang, X.; Yang, K.; Xie, Q.; Wu, Q.; Mack, S.C.; Shi, Y.; Kim, L.J.Y.; Prager, B.C.; Flavahan, W.A.; Liu, X.; et al. Purine synthesis promotes maintenance of brain tumor initiating cells in glioma. Nat. Neurosci. 2017, 20, 661–673. [Google Scholar] [CrossRef]
- Wang, D.; Chen, Y.; Fang, H.; Zheng, L.; Li, Y.; Yang, F.; Xu, Y.; Du, L.; Zhou, B.S.; Li, H. Increase of PRPP enhances chemosensitivity of PRPS1 mutant acute lymphoblastic leukemia cells to 5-Fluorouracil. J. Cell. Mol. Med. 2018, 22, 6202–6212. [Google Scholar] [CrossRef]
- Merideth, M.A.; Collins, F.S.; Gahl, W.A. Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008, 358, 2410–2411. [Google Scholar] [CrossRef] [PubMed]
- Vermeij, W.P.; Hoeijmakers, J.H.J. Base editor repairs gene of premature-ageing disease. Nature 2021, 589, 522–524. [Google Scholar] [CrossRef] [PubMed]
- Mateos, J.; Fafian-Labora, J.; Morente-López, M.; Lesende-Rodriguez, I.; Monserrat, L.; Ódena, M.; de Oliveira, E.; de Toro, J.; Arufe., M.C. Next-Generation Sequencing and Quantitative Proteomics of Hutchinson-Gilford progeria syndrome-derived cells point to a role of nucleotide metabolism in premature aging. PLoS One 2018, 13, 10e0205878. [Google Scholar] [CrossRef] [PubMed]
- Nosal, J.M.; Switzer, R.L.; Becker, M.A. Overexpression, purification, and characterization of recombinant human 5-phosphoribosyl-1-pyrophosphate synthetase isozymes I and II. J. Biol. Chem. 1993, 268, 10168–10175. [Google Scholar] [CrossRef]
- Chen, J.; Yang, S.; Li, Y.; Zwen, X.; Zhang, P.; Song, Q.; Yao, Y.; Pei, H. De novo nucleotide biosynthetic pathway and cancer. Genes Dis. 2022. [Google Scholar] [CrossRef]
- Jiménez-Guitiérrez, E.; Alegría-Carrasco, E.; Alonso-Rodríguez, E.; Fernández-Acero, T.; Molina, M.; Martín, H. Rewiring the yeast cell wall integrity (CWI) pathway through a synthetic positive feedback circuit unveils a novel role for the MAPKKK Ssk2 in CWI pathway activation. FEBS. J. 2020, 287, 4881–4901. [Google Scholar] [CrossRef] [PubMed]
- Heinisch, J.J. How to study intertwined and autoregulated eukaryotic signal transduction pathways. FEBS J. 2020, 287, 4844–4847. [Google Scholar] [CrossRef]
- Brommage, R.; Liu, J.; Hansen, G.M.; Kirkpatrick, L.L.; Potter, D.G.; Sands, A.T.; Zambrowicz, B.; Powell, D.R.; Vogel, P. High-throughput screening of mouse gene knockouts identifies established and novel skeletal phenotypes. Bone Res. 2014, 2, 14034. [Google Scholar] [CrossRef]
- Yan, D.N.S.; Xing, Y.Z.; Ouyang, X.M.; Zhu, J.H.; Chen, Z.Y.; Lang, H.N.; Liu, X.Z. Analysis of miR-376 RNA cluster members in the mouse inner ear. Int. J. Exp. Pathol. 2012, 93, 450–457. [Google Scholar] [CrossRef]
- Kawahara, Y.; Zinshteyn, B.; Sethupathy, P.; Iizasa, H.; Hatzigeorgiou, A.G.; Nishikura, K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science 2007, 315, 1137–1140. [Google Scholar] [CrossRef]
- Jin, Q.; Li, J.M.; Yang, F.; Feng, L.L.; Du, X. Circular RNA circKIF2A Contributes to the Progression of Neuroblastoma Through Regulating PRPS1 Expression by Sponging miR-377-3p. Biochem. Genet. 2022. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Yang, F.; Fan, Y.C.; Jing, W.L.; Wen, J.F.; Miao, W.; Ding, X.Y.; Yang, H.B. LncRNA DLEU1 Contributes to the Growth and Invasion of Colorectal Cancer via Targeting miR-320b/PRPS1. Front. Oncol. 2021, 11, 1930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Q.; Wang, H.; Xu, M.J.; Chen, F.Y.; Li, W.K.; Hu, H.T.; Yuan, Q.; Su, Y.; Liu, X.X.; Wuri, J.; et al. Long noncoding RNA HAS2-AS1 promotes tumor progression in glioblastoma via functioning as a competing endogenous RNA. J. Cell. Biochem. 2020, 121, 661–671. [Google Scholar] [CrossRef] [PubMed]
- Tarassov, K.; Messier, V.; Landry, C.R.; Radinovic, S.; Molina, M.M.S.; Shames, I.; Malitskaya, Y.; Vogel, J.; Bussey, H.; Michnick, S.W. An in vivo map of the yeast protein interactome. Science 2008, 320, 1465–1470. [Google Scholar] [CrossRef]
- Schlecht, U.; Miranda, M.; Sureh, S.; Davis, R.W.; St Onge, R.P. Multiplex assay for condition-dependent changes in protein-protein interactions. Proc. Natl. Acad. Sci. USA 2012, 109, 9213–9218. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ugbogu, E.A.; Schweizer, L.M.; Schweizer, M. Contribution of Model Organisms to Investigating the Far-Reaching Consequences of PRPP Metabolism on Human Health and Well-Being. Cells 2022, 11, 1909. https://doi.org/10.3390/cells11121909
Ugbogu EA, Schweizer LM, Schweizer M. Contribution of Model Organisms to Investigating the Far-Reaching Consequences of PRPP Metabolism on Human Health and Well-Being. Cells. 2022; 11(12):1909. https://doi.org/10.3390/cells11121909
Chicago/Turabian StyleUgbogu, Eziuche A., Lilian M. Schweizer, and Michael Schweizer. 2022. "Contribution of Model Organisms to Investigating the Far-Reaching Consequences of PRPP Metabolism on Human Health and Well-Being" Cells 11, no. 12: 1909. https://doi.org/10.3390/cells11121909
APA StyleUgbogu, E. A., Schweizer, L. M., & Schweizer, M. (2022). Contribution of Model Organisms to Investigating the Far-Reaching Consequences of PRPP Metabolism on Human Health and Well-Being. Cells, 11(12), 1909. https://doi.org/10.3390/cells11121909