
GLIS1-3: Links to Primary Cilium, Reprogramming, Stem Cell Renewal, and Disease



Abstract
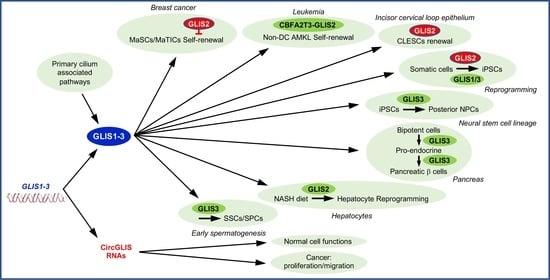
:
{kind=link}
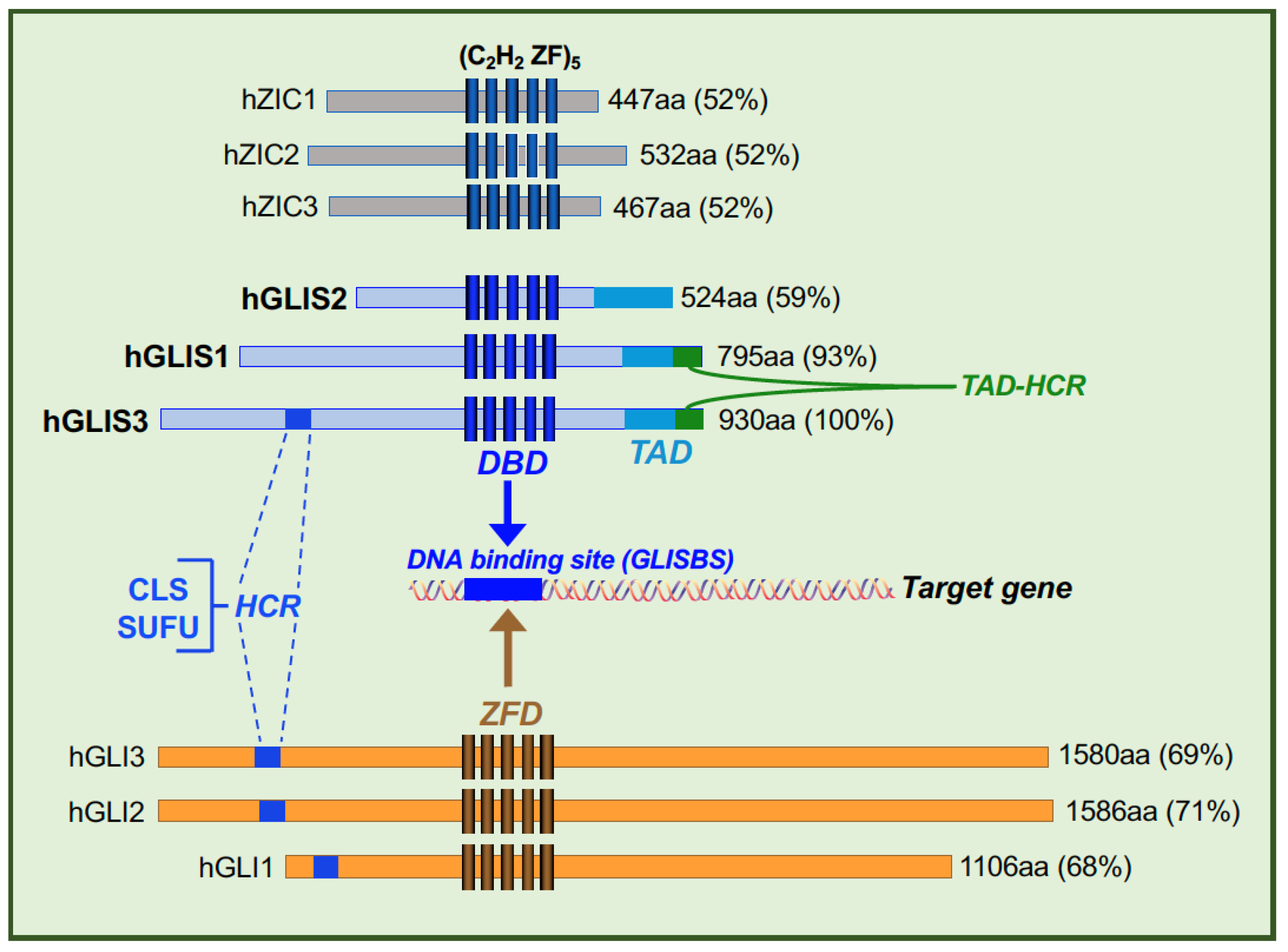
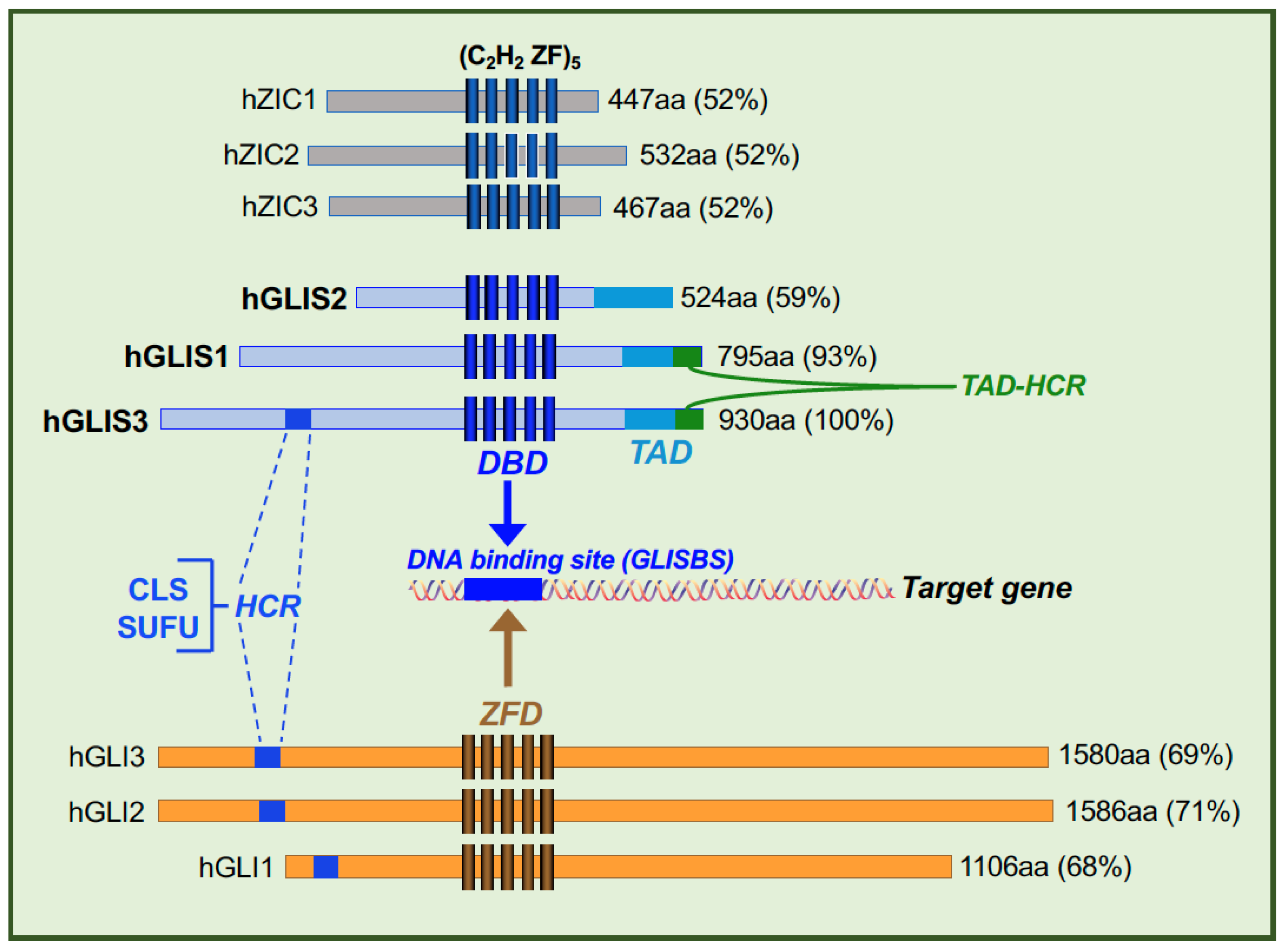{kind=link}
{kind=link}
{kind=link}
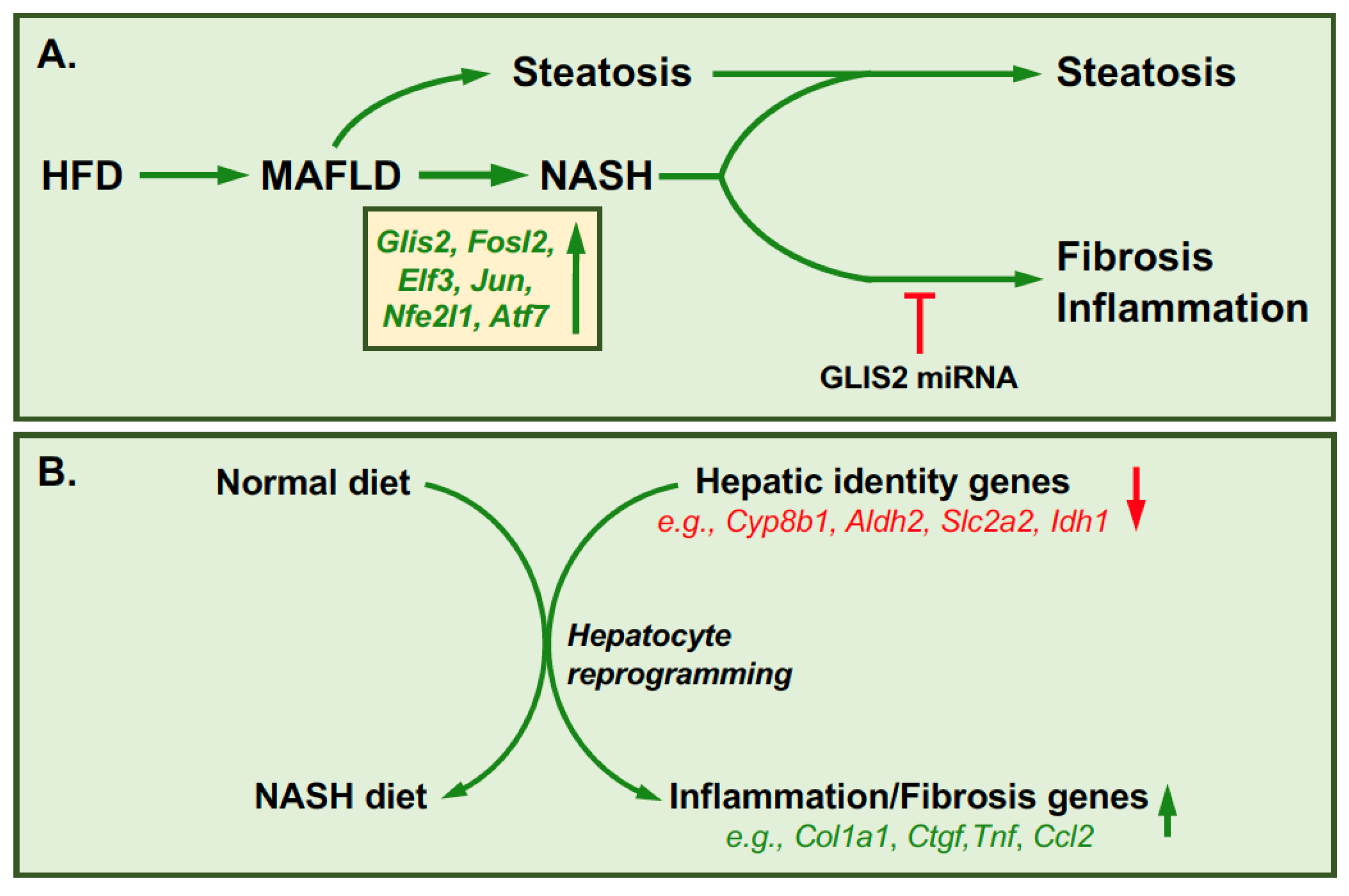{kind=link}
1. Introduction
2. GLIS Proteins and Primary Cilium
3. GLIS1-3: Regulation of Self-Renewal and EMT in Relation to Tumorigenesis
4. GLIS2, Primary Cilium, PROM1 and Cell Renewal
5. Effects of GLIS1-3 on iPSC Reprogramming and Differentiation
6. GLIS2 and Reprogramming in Hepatic Fibrosis
7. Additional Roles for GLIS3 in Stem and Progenitor Cells
8. GLIS Circular RNAs
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, F.; Jetten, A.M. Genomic structure of the gene encoding the human GLI-related, Kruppel-like zinc finger protein GLIS2. Gene 2001, 280, 49–57. [Google Scholar] [CrossRef]
- Kim, Y.S.; Lewandoski, M.; Perantoni, A.O.; Kurebayashi, S.; Nakanishi, G.; Jetten, A.M. Identification of Glis1, a novel Gli-related, Kruppel-like zinc finger protein containing transactivation and repressor functions. J. Biol. Chem. 2002, 277, 30901–30913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, F.; Nakanishi, G.; Kurebayashi, S.; Yoshino, K.; Perantoni, A.; Kim, Y.S.; Jetten, A.M. Characterization of Glis2, a novel gene encoding a Gli-related, Kruppel-like transcription factor with transactivation and repressor functions. Roles in kidney development and neurogenesis. J. Biol. Chem. 2002, 277, 10139–10149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Nakanishi, G.; Lewandoski, M.; Jetten, A.M. GLIS3, a novel member of the GLIS subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. 2003, 31, 5513–5525. [Google Scholar] [CrossRef]
- Kim, S.C.; Kim, Y.S.; Jetten, A.M. Kruppel-like zinc finger protein Gli-similar 2 (Glis2) represses transcription through interaction with C-terminal binding protein 1 (CtBP1). Nucleic Acids Res. 2005, 33, 6805–6815. [Google Scholar] [CrossRef] [Green Version]
- Jetten, A.M. GLIS1-3 transcription factors: Critical roles in the regulation of multiple physiological processes and diseases. Cell Mol. Life Sci. 2018, 75, 3473–3494. [Google Scholar] [CrossRef]
- Scoville, D.W.; Kang, H.S.; Jetten, A.M. Transcription factor GLIS3: Critical roles in thyroid hormone biosynthesis, hypothyroidism, pancreatic beta cells and diabetes. Pharmacol Ther 2020, 215, 107632. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kang, H.S.; Herbert, R.; Beak, J.Y.; Collins, J.B.; Grissom, S.F.; Jetten, A.M. Kruppel-like zinc finger protein Glis2 is essential for the maintenance of normal renal functions. Mol. Cell Biol. 2008, 28, 2358–2367. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Rauhauser, A.A.; Dai, J.; Sakthivel, R.; Igarashi, P.; Jetten, A.M.; Attanasio, M. Increased hedgehog signaling in postnatal kidney results in aberrant activation of nephron developmental programs. Hum. Mol. Genet. 2011, 20, 4155–4166. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Kang, H.S.; Jetten, A.M. The Kruppel-like zinc finger protein Glis2 functions as a negative modulator of the Wnt/beta-catenin signaling pathway. FEBS Lett. 2007, 581, 858–864. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.M.; Callens, C.; Le Gallo, M.; Mironov, S.; Ding, Q.; Salamagnon, A.; Chavarria, T.E.; Viel, R.; Peasah, A.D.; Bhutkar, A.; et al. An EMT-primary cilium-GLIS2 signaling axis regulates mammogenesis and claudin-low breast tumorigenesis. Sci. Adv. 2021, 7, eabf6063. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, M.; Uhlenhaut, N.H.; Sousa, V.H.; O’Toole, J.F.; Otto, E.; Anlag, K.; Klugmann, C.; Treier, A.C.; Helou, J.; Sayer, J.A.; et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat. Genet. 2007, 39, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Thirant, C.; Ignacimouttou, C.; Lopez, C.K.; Diop, M.; Le Mouel, L.; Thiollier, C.; Siret, A.; Dessen, P.; Aid, Z.; Riviere, J.; et al. ETO2-GLIS2 Hijacks Transcriptional Complexes to Drive Cellular Identity and Self-Renewal in Pediatric Acute Megakaryoblastic Leukemia. Cancer Cell 2017, 31, 452–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinnadurai, G. Transcriptional regulation by C-terminal binding proteins. Int. J. Biochem. Cell Biol. 2007, 39, 1593–1607. [Google Scholar] [CrossRef] [PubMed]
- Scoville, D.W.; Jetten, A.M. GLIS3: A Critical Transcription Factor in Islet beta-Cell Generation. Cells 2021, 10, 3471. [Google Scholar] [CrossRef] [PubMed]
- Jetten, A.M. Emerging Roles of GLI-Similar Kruppel-like Zinc Finger Transcription Factors in Leukemia and Other Cancers. Trends Cancer 2019, 5, 547–557. [Google Scholar] [CrossRef]
- Dimitri, P. The role of GLIS3 in thyroid disease as part of a multisystem disorder. Best Pract. Res. Clin. Endocrinol. Metab. 2017, 31, 175–182. [Google Scholar] [CrossRef]
- Kang, H.S.; Beak, J.Y.; Kim, Y.S.; Herbert, R.; Jetten, A.M. Glis3 is associated with primary cilia and Wwtr1/TAZ and implicated in polycystic kidney disease. Mol. Cell Biol. 2009, 29, 2556–2569. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Chen, L.Y.; Lichti-Kaiser, K.; Liao, G.; Gerrish, K.; Bortner, C.D.; Yao, H.H.; Eddy, E.M.; Jetten, A.M. Transcription Factor GLIS3: A New and Critical Regulator of Postnatal Stages of Mouse Spermatogenesis. Stem Cells 2016, 34, 2772–2783. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Kim, Y.S.; ZeRuth, G.; Beak, J.Y.; Kilic, G.; Jensen, J.; Sosa-Pineda, B.; Jetten, A.M. Transcription factor Glis3: A novel critical player in the regulation of pancreatic β-cell development. Mol. Cell Biol. 2009, 29, 6366–6379. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Kumar, D.; Liao, G.; Lichti-Kaiser, K.; Gerrish, K.; Liao, X.H.; Refetoff, S.; Jothi, R.; Jetten, A.M. GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J. Clin. Investig. 2017, 127, 4326–4337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.S.; Takeda, Y.; Jeon, K.; Jetten, A.M. The Spatiotemporal Pattern of Glis3 Expression Indicates a Regulatory Function in Bipotent and Endocrine Progenitors during Early Pancreatic Development and in Beta, PP and Ductal Cells. PLoS ONE 2016, 11, e0157138. [Google Scholar] [CrossRef]
- Senee, V.; Chelala, C.; Duchatelet, S.; Feng, D.; Blanc, H.; Cossec, J.C.; Charon, C.; Nicolino, M.; Boileau, P.; Cavener, D.R.; et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat. Genet. 2006, 38, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chang, B.H.; Samson, S.L.; Li, M.V.; Chan, L. The Kruppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res. 2009, 37, 2529–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khor, C.C.; Do, T.; Jia, H.; Nakano, M.; George, R.; Abu-Amero, K.; Duvesh, R.; Chen, L.J.; Li, Z.; Nongpiur, M.E.; et al. Genome-wide association study identifies five new susceptibility loci for primary angle closure glaucoma. Nat. Genet. 2016, 48, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Zhang, Y.; Liu, D.; Wang, S.S.; Ding, Q.; Rastogi, P.; Purvis, M.; Wang, A.; Elhadi, S.; Ren, C.; et al. Innate Immune Signaling Contributes to Tubular Cell Senescence in the Glis2 Knockout Mouse Model of Nephronophthisis. Am. J. Pathol. 2020, 190, 176–189. [Google Scholar] [CrossRef]
- Lu, D.; Rauhauser, A.; Li, B.; Ren, C.; McEnery, K.; Zhu, J.; Chaki, M.; Vadnagara, K.; Elhadi, S.; Jetten, A.M.; et al. Loss of Glis2/NPHP7 causes kidney epithelial cell senescence and suppresses cyst growth in the Kif3a mouse model of cystic kidney disease. Kidney Int. 2016, 89, 1307–1323. [Google Scholar] [CrossRef] [Green Version]
- Nair, K.S.; Srivastava, C.; Brown, R.V.; Koli, S.; Choquet, H.; Kang, H.S.; Kuo, Y.M.; Grimm, S.A.; Sutherland, C.; Badea, A.; et al. GLIS1 regulates trabecular meshwork function and intraocular pressure and is associated with glaucoma in humans. Nat. Commun. 2021, 12, 4877. [Google Scholar] [CrossRef]
- Yao, J.; Lei, P.J.; Li, Q.L.; Chen, J.; Tang, S.B.; Xiao, Q.; Lin, X.; Wang, X.; Li, L.Y.; Wu, M. GLIS2 promotes colorectal cancer through repressing enhancer activation. Oncogenesis 2020, 9, 57. [Google Scholar] [CrossRef]
- Rami, F.; Baradaran, A.; Kahnamooi, M.M.; Salehi, M. Alteration of GLIS3 gene expression pattern in patients with breast cancer. Adv. Biomed. Res. 2016, 5, 44. [Google Scholar] [CrossRef]
- Shimamoto, K.; Tanimoto, K.; Fukazawa, T.; Nakamura, H.; Kanai, A.; Bono, H.; Ono, H.; Eguchi, H.; Hirohashi, N. GLIS1, a novel hypoxia-inducible transcription factor, promotes breast cancer cell motility via activation of WNT5A. Carcinogenesis 2020, 41, 1184–1194. [Google Scholar] [CrossRef] [PubMed]
- Masetti, R.; Pigazzi, M.; Togni, M.; Astolfi, A.; Indio, V.; Manara, E.; Casadio, R.; Pession, A.; Basso, G.; Locatelli, F. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood 2013, 121, 3469–3472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruber, T.A.; Larson Gedman, A.; Zhang, J.; Koss, C.S.; Marada, S.; Ta, H.Q.; Chen, S.C.; Su, X.; Ogden, S.K.; Dang, J.; et al. An Inv(16)(p13.3q24.3)-encoded CBFA2T3-GLIS2 fusion protein defines an aggressive subtype of pediatric acute megakaryoblastic leukemia. Cancer Cell 2012, 22, 683–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiollier, C.; Pflumio, F.; Ballerini, P.; Crispino, J.D.; Bernard, O.; Mercher, T. Novel ETO2-GLIS2 fusion and therapeutic strategy in acute megakaryoblastic leukemia. Med. Sci. (Paris) 2012, 28, 1013–1016. [Google Scholar] [CrossRef]
- Thiollier, C.; Lopez, C.K.; Gerby, B.; Ignacimouttou, C.; Poglio, S.; Duffourd, Y.; Guegan, J.; Rivera-Munoz, P.; Bluteau, O.; Mabialah, V.; et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J. Exp. Med. 2012, 209, 2017–2031. [Google Scholar] [CrossRef] [Green Version]
- Masetti, R.; Togni, M.; Astolfi, A.; Pigazzi, M.; Manara, E.; Indio, V.; Rizzari, C.; Rutella, S.; Basso, G.; Pession, A.; et al. DHH-RHEBL1 fusion transcript: A novel recurrent feature in the new landscape of pediatric CBFA2T3-GLIS2-positive acute myeloid leukemia. Oncotarget 2013, 4, 1712–1720. [Google Scholar] [CrossRef] [Green Version]
- Marchio, C.; Da Cruz Paula, A.; Gularte-Merida, R.; Basili, T.; Brandes, A.; da Silva, E.M.; Silveira, C.; Ferrando, L.; Metovic, J.; Maletta, F.; et al. PAX8-GLIS3 gene fusion is a pathognomonic genetic alteration of hyalinizing trabecular tumors of the thyroid. Mod. Pathol. 2019, 32, 1734–1743. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Nikitski, A.V.; Panebianco, F.; Kaya, C.; Yip, L.; Williams, M.; Chiosea, S.I.; Seethala, R.R.; Roy, S.; Condello, V.; et al. GLIS Rearrangement is a Genomic Hallmark of Hyalinizing Trabecular Tumor of the Thyroid Gland. Thyroid 2019, 29, 161–173. [Google Scholar] [CrossRef]
- Pei, D.; Shu, X.; Gassama-Diagne, A.; Thiery, J.P. Mesenchymal-epithelial transition in development and reprogramming. Nat. Cell Biol. 2019, 21, 44–53. [Google Scholar] [CrossRef]
- Jeon, K.; Kumar, D.; Conway, A.E.; Park, K.; Jothi, R.; Jetten, A.M. GLIS3 Transcriptionally Activates WNT Genes to Promote Differentiation of Human Embryonic Stem Cells into Posterior Neural Progenitors. Stem Cells 2019, 37, 202–215. [Google Scholar] [CrossRef] [Green Version]
- Loft, A.; Alfaro, A.J.; Schmidt, S.F.; Pedersen, F.B.; Terkelsen, M.K.; Puglia, M.; Chow, K.K.; Feuchtinger, A.; Troullinaki, M.; Maida, A.; et al. Liver-fibrosis-activated transcriptional networks govern hepatocyte reprogramming and intra-hepatic communication. Cell Metab. 2021, 33, 1685–1700.e9. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, M.; Yamanaka, S. Glis1, a unique pro-reprogramming factor, may facilitate clinical applications of iPSC technology. Cell Cycle 2011, 10, 3613–3614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoville, D.W.; Kang, H.S.; Jetten, A.M. GLIS1-3: Emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investig. 2017, 4, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, D.; Thamm, K.; Zhuang, H.; Karbanova, J.; Gao, Y.; Walker, J.V.; Jin, H.; Wu, X.; Coveney, C.R.; Marangoni, P.; et al. Prominin-1 controls stem cell activation by orchestrating ciliary dynamics. EMBO J. 2019, 38, e99845. [Google Scholar] [CrossRef]
- Hashimoto, H.; Miyamoto, R.; Watanabe, N.; Shiba, D.; Ozato, K.; Inoue, C.; Kubo, Y.; Koga, A.; Jindo, T.; Narita, T.; et al. Polycystic kidney disease in the medaka (Oryzias latipes) pc mutant caused by a mutation in the Gli-Similar3 (glis3) gene. PLoS ONE 2009, 4, e6299. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.U.; Rodrigues, R.B.; Leib, R.D.; Adams, C.M.; Chien, A.S.; Gygi, S.P.; Nachury, M.V. Proteomics of Primary Cilia by Proximity Labeling. Dev. Cell 2015, 35, 497–512. [Google Scholar] [CrossRef] [Green Version]
- Yasuoka, Y.; Matsumoto, M.; Yagi, K.; Okazaki, Y. Evolutionary History of GLIS Genes Illuminates their Roles in Cell Reprogramming and Ciliogenesis. Mol. Biol. Evol. 2019, 37, 100–109. [Google Scholar] [CrossRef] [Green Version]
- Schou, K.B.; Pedersen, L.B.; Christensen, S.T. Ins and outs of GPCR signaling in primary cilia. EMBO Rep. 2015, 16, 1099–1113. [Google Scholar] [CrossRef] [Green Version]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef]
- Nachury, M.V.; Mick, D.U. Establishing and regulating the composition of cilia for signal transduction. Nat. Rev. Mol. Cell Biol. 2019, 20, 389–405. [Google Scholar] [CrossRef]
- Anvarian, Z.; Mykytyn, K.; Mukhopadhyay, S.; Pedersen, L.B.; Christensen, S.T. Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol. 2019, 15, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Guen, V.J.; Chavarria, T.E.; Kroger, C.; Ye, X.; Weinberg, R.A.; Lees, J.A. EMT programs promote basal mammary stem cell and tumor-initiating cell stemness by inducing primary ciliogenesis and Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E10532–E10539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyu, R.; Zhou, J. The Multifaceted Roles of Primary Cilia in the Regulation of Stem Cell Properties and Functions. J. Cell. Physiol. 2017, 232, 935–938. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.A.; Hildebrandt, F. Ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028191. [Google Scholar] [CrossRef]
- McConnachie, D.J.; Stow, J.L.; Mallett, A.J. Ciliopathies and the Kidney: A Review. Am. J. Kidney Dis. 2021, 77, 410–419. [Google Scholar] [CrossRef]
- Reiter, J.F.; Leroux, M.R. Genes and molecular pathways underpinning ciliopathies. Nat. Rev. Mol. Cell Biol. 2017, 18, 533–547. [Google Scholar] [CrossRef]
- Bangs, F.; Anderson, K.V. Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harb. Perspect. Biol. 2017, 9, a028175. [Google Scholar] [CrossRef]
- Kong, J.H.; Siebold, C.; Rohatgi, R. Biochemical mechanisms of vertebrate hedgehog signaling. Development 2019, 146, dev166892. [Google Scholar] [CrossRef] [Green Version]
- Ho, E.K.; Stearns, T. Hedgehog signaling and the primary cilium: Implications for spatial and temporal constraints on signaling. Development 2021, 148, dev195552. [Google Scholar] [CrossRef]
- Han, Y.; Xiong, Y.; Shi, X.; Wu, J.; Zhao, Y.; Jiang, J. Regulation of Gli ciliary localization and Hedgehog signaling by the PY-NLS/karyopherin-beta2 nuclear import system. PLoS Biol. 2017, 15, e2002063. [Google Scholar] [CrossRef] [Green Version]
- Lu, L.; Madugula, V. Mechanisms of ciliary targeting: Entering importins and Rabs. Cell Mol. Life Sci. 2018, 75, 597–606. [Google Scholar] [CrossRef] [PubMed]
- ZeRuth, G.T.; Yang, X.P.; Jetten, A.M. Modulation of the transactivation function and stability of Kruppel-like zinc finger protein Gli-similar 3 (Glis3) by Suppressor of Fused. J. Biol. Chem. 2011, 286, 22077–22089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tukachinsky, H.; Lopez, L.V.; Salic, A. A mechanism for vertebrate Hedgehog signaling: Recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 2010, 191, 415–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosking, C.R.; Ulloa, F.; Hogan, C.; Ferber, E.; Figueroa, A.; Gevaert, K.; Birchmeier, W.; Briscoe, J.; Fujita, Y. The Transcriptional Repressor Glis2 Is a Novel Binding Partner for p120 Catenin. Mol. Biol. Cell 2007, 18, 1918–1927. [Google Scholar] [CrossRef] [Green Version]
- ZeRuth, G.T.; Williams, J.G.; Cole, Y.C.; Jetten, A.M. HECT E3 Ubiquitin Ligase Itch Functions as a Novel Negative Regulator of Gli-Similar 3 (Glis3) Transcriptional Activity. PLoS ONE 2015, 10, e0131303. [Google Scholar] [CrossRef]
- Hoard, T.M.; Yang, X.P.; Jetten, A.M.; ZeRuth, G.T. PIAS-family proteins negatively regulate Glis3 transactivation function through SUMO modification in pancreatic beta cells. Heliyon 2018, 4, e00709. [Google Scholar] [CrossRef] [Green Version]
- Bertuccio, S.N.; Boudia, F.; Cambot, M.; Lopez, C.K.; Lordier, L.; Donada, A.; Robert, E.; Thirant, C.; Aid, Z.; Serravalle, S.; et al. The Pediatric Acute Leukemia Fusion Oncogene ETO2-GLIS2 Increases Self-Renewal and Alters Differentiation in a Human Induced Pluripotent Stem Cells-Derived Model. Hemasphere 2020, 4, e319. [Google Scholar] [CrossRef] [Green Version]
- Shima, H.; Takamatsu-Ichihara, E.; Shino, M.; Yamagata, K.; Katsumoto, T.; Aikawa, Y.; Fujita, S.; Koseki, H.; Kitabayashi, I. Ring1A and Ring1B inhibit expression of Glis2 to maintain murine MOZ-TIF2 AML stem cells. Blood 2018, 131, 1833–1845. [Google Scholar] [CrossRef]
- Vadnais, C.; Shooshtarizadeh, P.; Rajadurai, C.V.; Lesurf, R.; Hulea, L.; Davoudi, S.; Cadieux, C.; Hallett, M.; Park, M.; Nepveu, A. Autocrine Activation of the Wnt/beta-Catenin Pathway by CUX1 and GLIS1 in Breast Cancers. Biol. Open 2014, 3, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Tao, S.; Li, H.; Ma, X.; Ma, Y.; He, J.; Gao, Y.; Li, J. Elevating microRNA-1-3p shuttled by cancer-associated fibroblasts-derived extracellular vesicles suppresses breast cancer progression and metastasis by inhibiting GLIS1. Cancer Gene Ther. 2021, 28, 634–648. [Google Scholar] [CrossRef]
- Fu, N.Y.; Nolan, E.; Lindeman, G.J.; Visvader, J.E. Stem Cells and the Differentiation Hierarchy in Mammary Gland Development. Physiol. Rev. 2020, 100, 489–523. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Tam, W.L.; Shibue, T.; Kaygusuz, Y.; Reinhardt, F.; Ng Eaton, E.; Weinberg, R.A. Distinct EMT programs control normal mammary stem cells and tumour-initiating cells. Nature 2015, 525, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rokkam, P.; Gugalavath, S.; Gift Kumar, D.K.; Vempati, R.K.; Malla, R.R. Prognostic Role of Hedgehog-GLI1 Signaling Pathway in Aggressive and Metastatic Breast Cancers. Curr. Drug Metab. 2020, 21, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Nassour, M.; Idoux-Gillet, Y.; Selmi, A.; Come, C.; Faraldo, M.L.; Deugnier, M.A.; Savagner, P. Slug controls stem/progenitor cell growth dynamics during mammary gland morphogenesis. PLoS ONE 2012, 7, e53498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Rooij, J.D.; Branstetter, C.; Ma, J.; Li, Y.; Walsh, M.P.; Cheng, J.; Obulkasim, A.; Dang, J.; Easton, J.; Verboon, L.J.; et al. Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat. Genet. 2017, 49, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Shiba, N.; Ohki, K.; Tabuchi, K.; Yamato, G.; Park, M.J.; Tomizawa, D.; Kinoshita, A.; Shimada, A.; Arakawa, H.; et al. Prognostic Impact of Specific Molecular Profiles in Pediatric Acute Megakaryoblastic Leukemia in Non-Down Syndrome. Genes Chromosomes Cancer 2017, 56, 394–404. [Google Scholar] [CrossRef]
- Ishibashi, M.; Yokosuka, T.; Yanagimachi, M.D.; Iwasaki, F.; Tsujimoto, S.I.; Sasaki, K.; Takeuchi, M.; Tanoshima, R.; Kato, H.; Kajiwara, R.; et al. Clinical Courses of Two Pediatric Patients with Acute Megakaryoblastic Leukemia Harboring the CBFA2T3-GLIS2 Fusion Gene. Turk. J. Haematol. 2016, 33, 331–334. [Google Scholar] [CrossRef]
- Thirant, C.; Lopez, C.; Malinge, S.; Mercher, T. Molecular pathways driven by ETO2-GLIS2 in aggressive pediatric leukemia. Mol. Cell Oncol. 2017, 4, e1345351. [Google Scholar] [CrossRef]
- Gruber, T.A.; Downing, J.R. The biology of pediatric acute megakaryoblastic leukemia. Blood 2015, 126, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.L.; Ries, R.E.; Hylkema, T.; Alonzo, T.A.; Gerbing, R.B.; Santaguida, M.T.; Eidenschink Brodersen, L.; Pardo, L.; Cummings, C.L.; Loeb, K.R.; et al. Comprehensive Transcriptome Profiling of Cryptic CBFA2T3-GLIS2 Fusion-Positive AML Defines Novel Therapeutic Options: A COG and TARGET Pediatric AML Study. Clin. Cancer Res. 2020, 26, 726–737. [Google Scholar] [CrossRef]
- Benbarche, S.; Lopez, C.K.; Salataj, E.; Aid, Z.; Thirant, C.; Laiguillon, M.C.; Lecourt, S.; Belloucif, Y.; Vaganay, C.; Antonini, M.; et al. Screening of ETO2-GLIS2-induced Super Enhancers identifies targetable cooperative dependencies in acute megakaryoblastic leukemia. Sci. Adv. 2022, 8, eabg9455. [Google Scholar] [CrossRef] [PubMed]
- Corbeil, D.; Marzesco, A.M.; Wilsch-Brauninger, M.; Huttner, W.B. The intriguing links between prominin-1 (CD133), cholesterol-based membrane microdomains, remodeling of apical plasma membrane protrusions, extracellular membrane particles, and (neuro)epithelial cell differentiation. FEBS Lett. 2010, 584, 1659–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, G.Y. CD133 as a regulator of cancer metastasis through the cancer stem cells. Int. J. Biochem. Cell Biol. 2019, 106, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jaszai, J.; Thamm, K.; Karbanova, J.; Janich, P.; Fargeas, C.A.; Huttner, W.B.; Corbeil, D. Prominins control ciliary length throughout the animal kingdom: New lessons from human prominin-1 and zebrafish prominin-3. J. Biol. Chem. 2020, 295, 6007–6022. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.T.; Li, X.F.; Wang, L.; Yang, J.L. CD133 expressionand clinicopathologic significance in benign and malignant breast lesions. Cancer Biomark. 2020, 28, 293–299. [Google Scholar] [CrossRef]
- Wang, L.; Su, Y.; Huang, C.; Yin, Y.; Chu, A.; Knupp, A.; Tang, Y. NANOG and LIN28 dramatically improve human cell reprogramming by modulating LIN41 and canonical WNT activities. Biol Open 2019, 8, bio047225. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.Y.; Noh, H.B.; Kim, H.T.; Lee, K.I.; Hwang, D.Y. Glis family proteins are differentially implicated in the cellular reprogramming of human somatic cells. Oncotarget 2017, 8, 77041–77049. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, M.; Yamaguchi, K.; Nakamura, T.; Shibukawa, R.; Kodanaka, I.; Ichisaka, T.; Kawamura, Y.; Mochizuki, H.; Goshima, N.; Yamanaka, S. Direct reprogramming of somatic cells is promoted by maternal transcription factor Glis1. Nature 2011, 474, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, N.; Dowdy, S.F. Enhanced generation of iPSCs from older adult human cells by a synthetic five-factor self-replicative RNA. PLoS ONE 2017, 12, e0182018. [Google Scholar] [CrossRef]
- Yoshioka, N.; Gros, E.; Li, H.R.; Kumar, S.; Deacon, D.C.; Maron, C.; Muotri, A.R.; Chi, N.C.; Fu, X.D.; Yu, B.D.; et al. Efficient generation of human iPSCs by a synthetic self-replicative RNA. Cell Stem Cell 2013, 13, 246–254. [Google Scholar] [CrossRef] [Green Version]
- Borisova, E.; Nishimura, K.; An, Y.; Takami, M.; Li, J.; Song, D.; Matsuo-Takasaki, M.; Luijkx, D.; Aizawa, S.; Kuno, E.; et al. Structurally-discovered KLF4 variants accelerate and stabilize reprogramming to pluripotency. iScience 2021, 25, 103525. [Google Scholar] [CrossRef]
- Kondrateva, E.; Demchenko, A.; Slesarenko, Y.; Pozhitnova, V.; Yasinovsky, M.; Amelina, E.; Tabakov, V.; Voronina, E.; Lavrov, A.; Smirnikhina, S. Generation of two induced pluripotent stem cell lines (RCMGi004-A and -B) from human skin fibroblasts of a cystic fibrosis patient with compound heterozygous F508del/W1282X mutations in CFTR gene. Stem Cell Res. 2021, 52, 102232. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Y.; Guan, C.; Zhao, Z.; Li, Q.; Yang, J.; Mo, J.; Wang, B.; Wu, W.; Yang, X.; et al. Using low-risk factors to generate non-integrated human induced pluripotent stem cells from urine-derived cells. Stem Cell. Res. Ther. 2017, 8, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Liang, J.; Ni, S.; Zhou, T.; Qing, X.; Li, H.; He, W.; Chen, J.; Li, F.; Zhuang, Q.; et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell 2010, 7, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Chen, K.; Wang, T.; Wu, Y.; Xing, G.; Chen, M.; Hao, Z.; Zhang, C.; Zhang, J.; Ma, B.; et al. Glis1 facilitates induction of pluripotency via an epigenome-metabolome-epigenome signalling cascade. Nat. Metab. 2020, 2, 882–892. [Google Scholar] [CrossRef]
- Liu, J.; Han, Q.; Peng, T.; Peng, M.; Wei, B.; Li, D.; Wang, X.; Yu, S.; Yang, J.; Cao, S.; et al. The oncogene c-Jun impedes somatic cell reprogramming. Nat. Cell Biol. 2015, 17, 856–867. [Google Scholar] [CrossRef]
- Pells, S.; Koutsouraki, E.; Morfopoulou, S.; Valencia-Cadavid, S.; Tomlinson, S.R.; Kalathur, R.; Futschik, M.E.; De Sousa, P.A. Novel Human Embryonic Stem Cell Regulators Identified by Conserved and Distinct CpG Island Methylation State. PLoS ONE 2015, 10, e0131102. [Google Scholar] [CrossRef] [Green Version]
- Dimitri, P.; Habeb, A.M.; Gurbuz, F.; Millward, A.; Wallis, S.; Moussa, K.; Akcay, T.; Taha, D.; Hogue, J.; Slavotinek, A.; et al. Expanding the Clinical Spectrum Associated With GLIS3 Mutations. J. Clin. Endocrinol. Metab. 2015, 100, E1362–E1369. [Google Scholar] [CrossRef] [Green Version]
- Scoville, D.W.; Lichti-Kaiser, K.; Grimm, S.; Jetten, A. GLIS3 binds pancreatic beta cell regulatory regions alongside other islet transcription factors. J. Endocrinol. 2019, 243, 1–14. [Google Scholar] [CrossRef]
- Yang, Y.; Bush, S.P.; Wen, X.; Cao, W.; Chan, L. Differential Gene Dosage Effects of Diabetes-Associated Gene GLIS3 in Pancreatic beta Cell Differentiation and Function. Endocrinology 2017, 158, 9–20. [Google Scholar] [CrossRef]
- Kim, Y.S.; Kang, H.S.; Takeda, Y.; Hom, L.; Song, H.Y.; Jensen, J.; Jetten, A.M. Glis3 regulates neurogenin 3 expression in pancreatic beta-cells and interacts with its activator, Hnf6. Mol. Cells 2012, 34, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Gao, D.; Feng, T.; Tremblay, J.R.; Ghazalli, N.; Luo, A.; Rawson, J.; Quijano, J.C.; Chai, J.; Wedeken, L.; et al. Cells with surface expression of CD133highCD71low are enriched for tripotent colony-forming progenitor cells in the adult murine pancreas. Stem Cell Res. 2016, 16, 40–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tremblay, J.R.; Lopez, K.; Ku, H.T. A GLIS3-CD133-WNT-signaling axis regulates the self-renewal of adult murine pancreatic progenitor-like cells in colonies and organoids. J. Biol. Chem. 2019, 294, 16634–16649. [Google Scholar] [CrossRef]
- Makela, J.A.; Hobbs, R.M. Molecular regulation of spermatogonial stem cell renewal and differentiation. Reproduction 2019, 158, R169–R187. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.; Song, H.W.; Wilkinson, M.F. RHOX10 drives mouse spermatogonial stem cell establishment through a transcription factor signaling cascade. Cell Rep. 2021, 36, 109423. [Google Scholar] [CrossRef] [PubMed]
- Ungewitter, E.K.; Rotgers, E.; Kang, H.S.; Lichti-Kaiser, K.; Li, L.; Grimm, S.A.; Jetten, A.M.; Yao, H.H. Loss of Glis3 causes dysregulation of retrotransposon silencing and germ cell demise in fetal mouse testis. Sci. Rep. 2018, 8, 9662. [Google Scholar] [CrossRef]
- Dunty, W.C., Jr.; Biris, K.K.; Chalamalasetty, R.B.; Taketo, M.M.; Lewandoski, M.; Yamaguchi, T.P. Wnt3a/beta-catenin signaling controls posterior body development by coordinating mesoderm formation and segmentation. Development 2008, 135, 85–94. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, M.; Van, X.H.; Leyns, L. Anterior-posterior patterning of neural differentiated embryonic stem cells by canonical Wnts, Fgfs, Bmp4 and their respective antagonists. Dev. Growth Differ. 2009, 51, 687–698. [Google Scholar] [CrossRef]
- Das, A.; Sinha, T.; Shyamal, S.; Panda, A.C. Emerging Role of Circular RNA-Protein Interactions. Non-Coding RNA 2021, 7, 48. [Google Scholar] [CrossRef]
- Fontemaggi, G.; Turco, C.; Esposito, G.; Di Agostino, S. New Molecular Mechanisms and Clinical Impact of circRNAs in Human Cancer. Cancers 2021, 13, 3154. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, X.; Liu, R.; Wen, C.; Wang, H.; Huang, L.; Li, W.; Zhu, Z.; Zhu, Y.; Liu, H. Circular RNA GLIS2 promotes colorectal cancer cell motility via activation of the NF-kappaB pathway. Cell Death Dis. 2020, 11, 788. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, J.; Chen, Z.; Xu, X.; Weng, J.; Zhang, Y.; Mo, Y.; Liu, Y.; Wang, J.; Ke, Y. CircGLIS3 Promotes High-Grade Glioma Invasion via Modulating Ezrin Phosphorylation. Front. Cell Dev. Biol. 2021, 9, 663207. [Google Scholar] [CrossRef]
- Wu, S.; Yang, J.; Xu, H.; Wang, X.; Zhang, R.; Lu, W.; Yang, J.; Li, X.; Chen, S.; Zou, Y.; et al. Circular RNA circGLIS3 promotes bladder cancer proliferation via the miR-1273f/SKP1/Cyclin D1 axis. Cell Biol. Toxicol. 2022, 38, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Jiang, H.; Fu, H.; Zhang, Y. A circGLIS3/miR-644a/PTBP1 positive feedback loop promotes the malignant biological progressions of non-small cell lung cancer. Am. J. Cancer Res. 2021, 11, 108–122. [Google Scholar]
- Liu, Z.; Liu, L.; Qi, Y.; Li, H.; Pan, S. GLIS family zinc finger 3 promoting cell malignant behaviors and NF-kappaB signaling in glioma. Brain Res. 2021, 1770, 147623. [Google Scholar] [CrossRef] [PubMed]
- Xiong, L.; Chen, L.; Wu, L.; He, W.; Chen, D.; Peng, Z.; Li, J.; Zhu, X.; Su, L.; Li, Y.; et al. Lipotoxicity-induced circGlis3 impairs beta cell function and is transmitted by exosomes to promote islet endothelial cell dysfunction. Diabetologia 2022, 65, 188–205. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jetten, A.M.; Scoville, D.W.; Kang, H.S. GLIS1-3: Links to Primary Cilium, Reprogramming, Stem Cell Renewal, and Disease. Cells 2022, 11, 1833. https://doi.org/10.3390/cells11111833
Jetten AM, Scoville DW, Kang HS. GLIS1-3: Links to Primary Cilium, Reprogramming, Stem Cell Renewal, and Disease. Cells. 2022; 11(11):1833. https://doi.org/10.3390/cells11111833
Chicago/Turabian StyleJetten, Anton M., David W. Scoville, and Hong Soon Kang. 2022. "GLIS1-3: Links to Primary Cilium, Reprogramming, Stem Cell Renewal, and Disease" Cells 11, no. 11: 1833. https://doi.org/10.3390/cells11111833
APA StyleJetten, A. M., Scoville, D. W., & Kang, H. S. (2022). GLIS1-3: Links to Primary Cilium, Reprogramming, Stem Cell Renewal, and Disease. Cells, 11(11), 1833. https://doi.org/10.3390/cells11111833