
Response to Cysteamine in Osteoclasts Obtained from Patients with Nephropathic Cystinosis: A Genotype/Phenotype Correlation

 , , ,
, , ,
Abstract
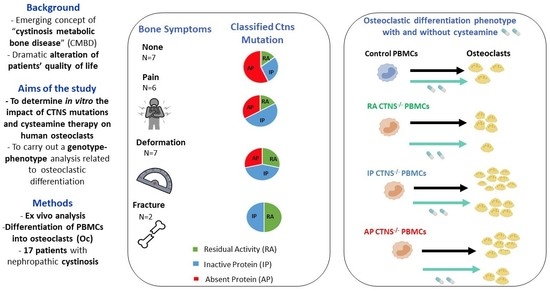
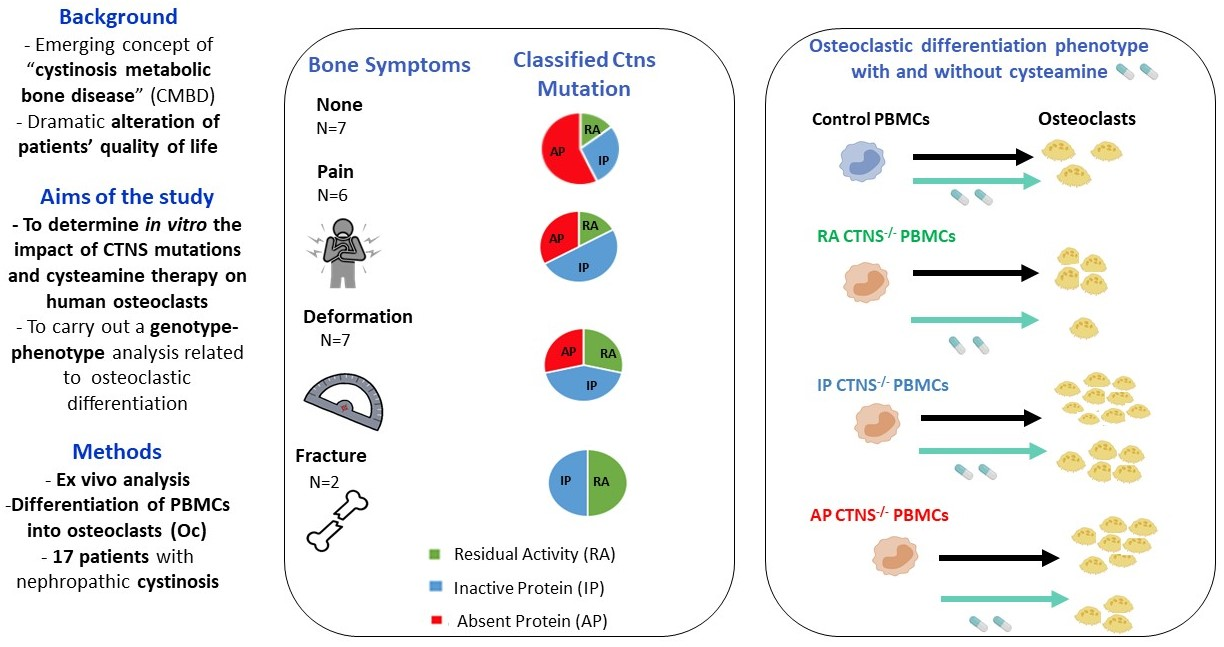
:
1. Introduction
2. Materials and Methods
2.1. Clinical Study
2.2. Primary Cultures of Human Osteoclasts
2.3. Statistical Analyses
2.4. Ethical Considerations
3. Results
3.1. Patients’ Clinical Characteristics
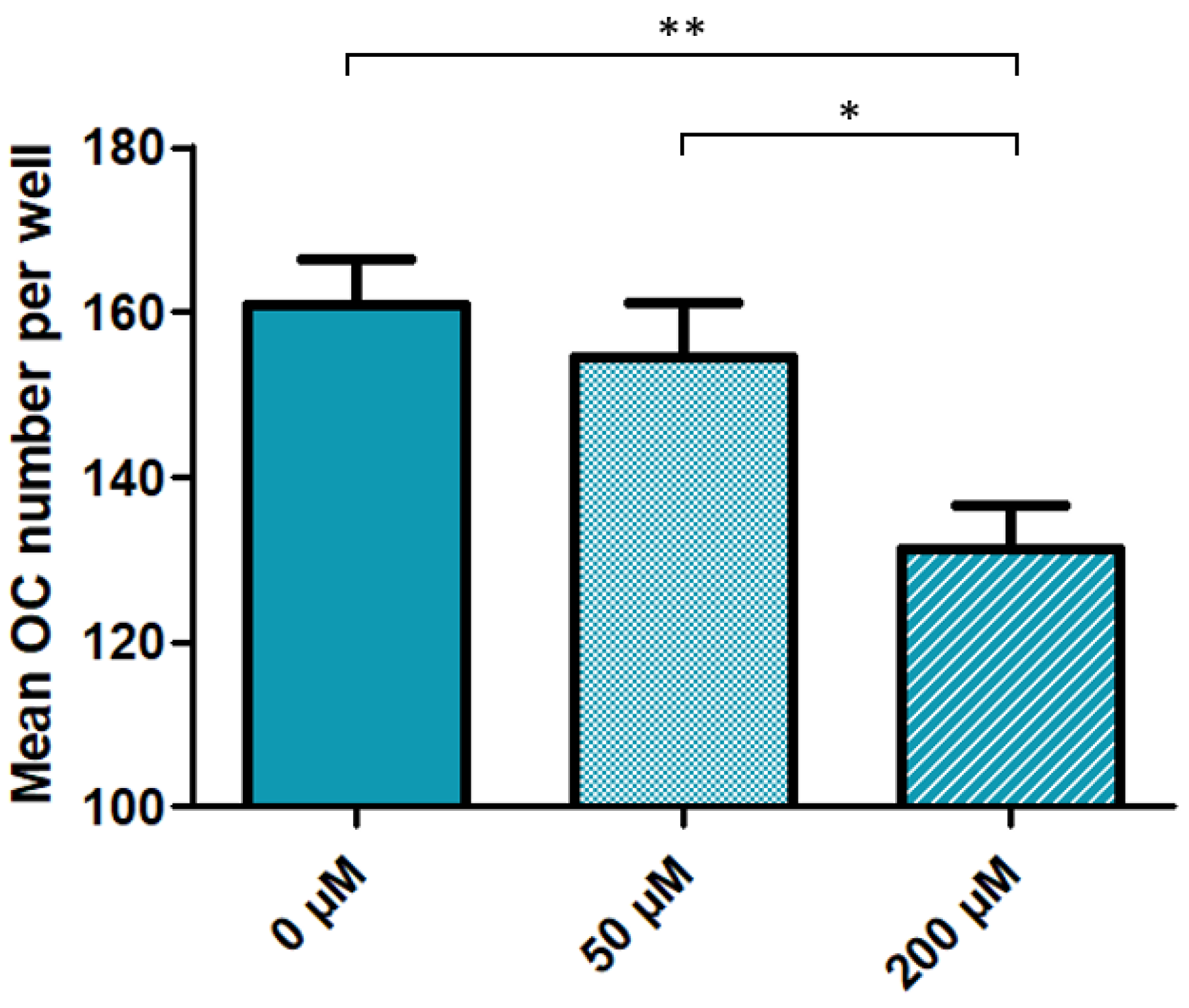
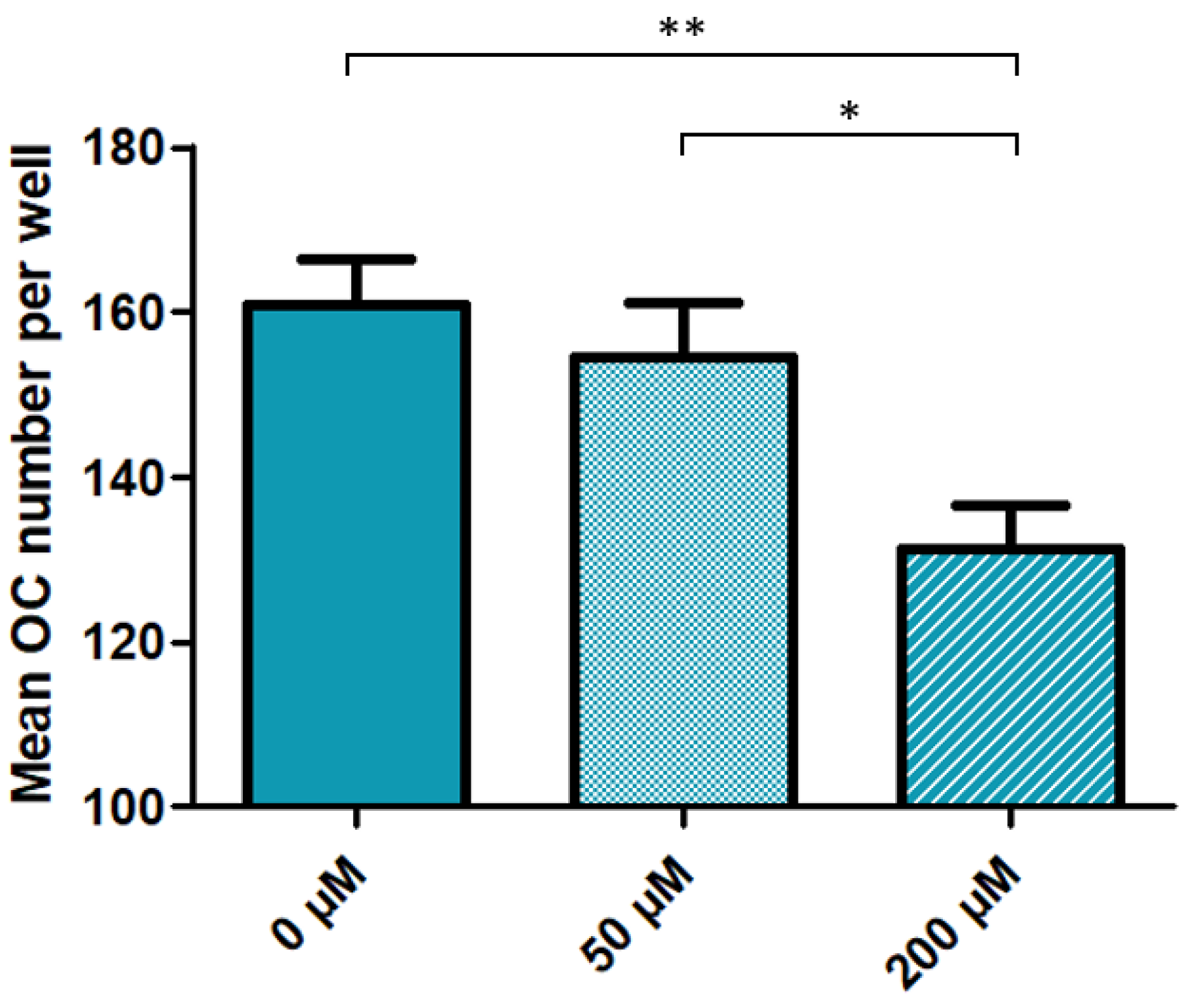
3.2. High Dose Cysteamine Decreases the Propensity of Patients-Derived Mononuclear Progenitors to Generate Osteoclasts
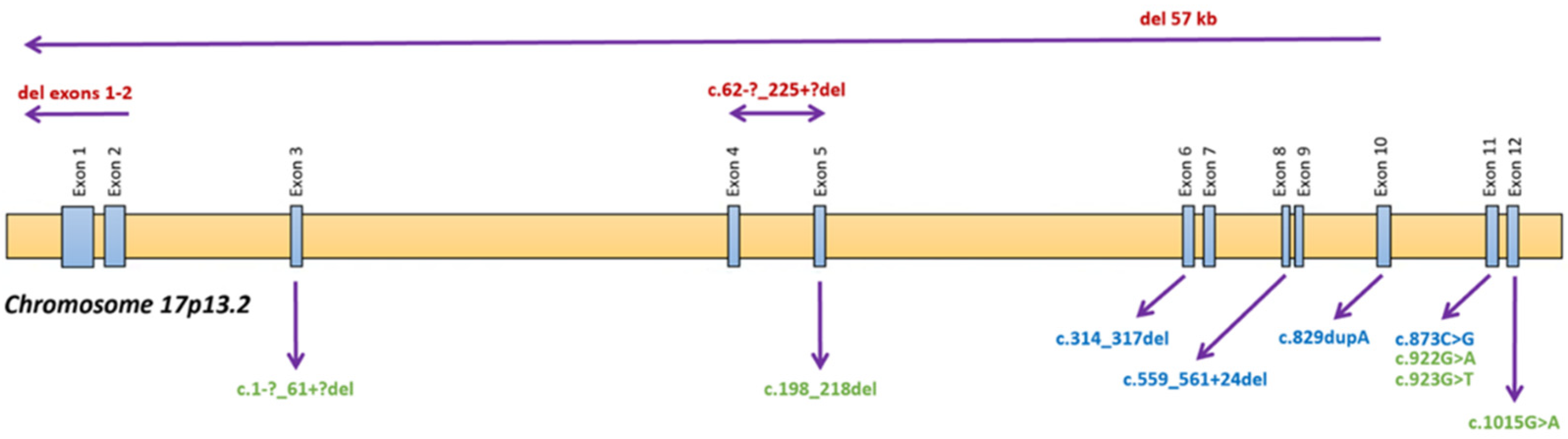
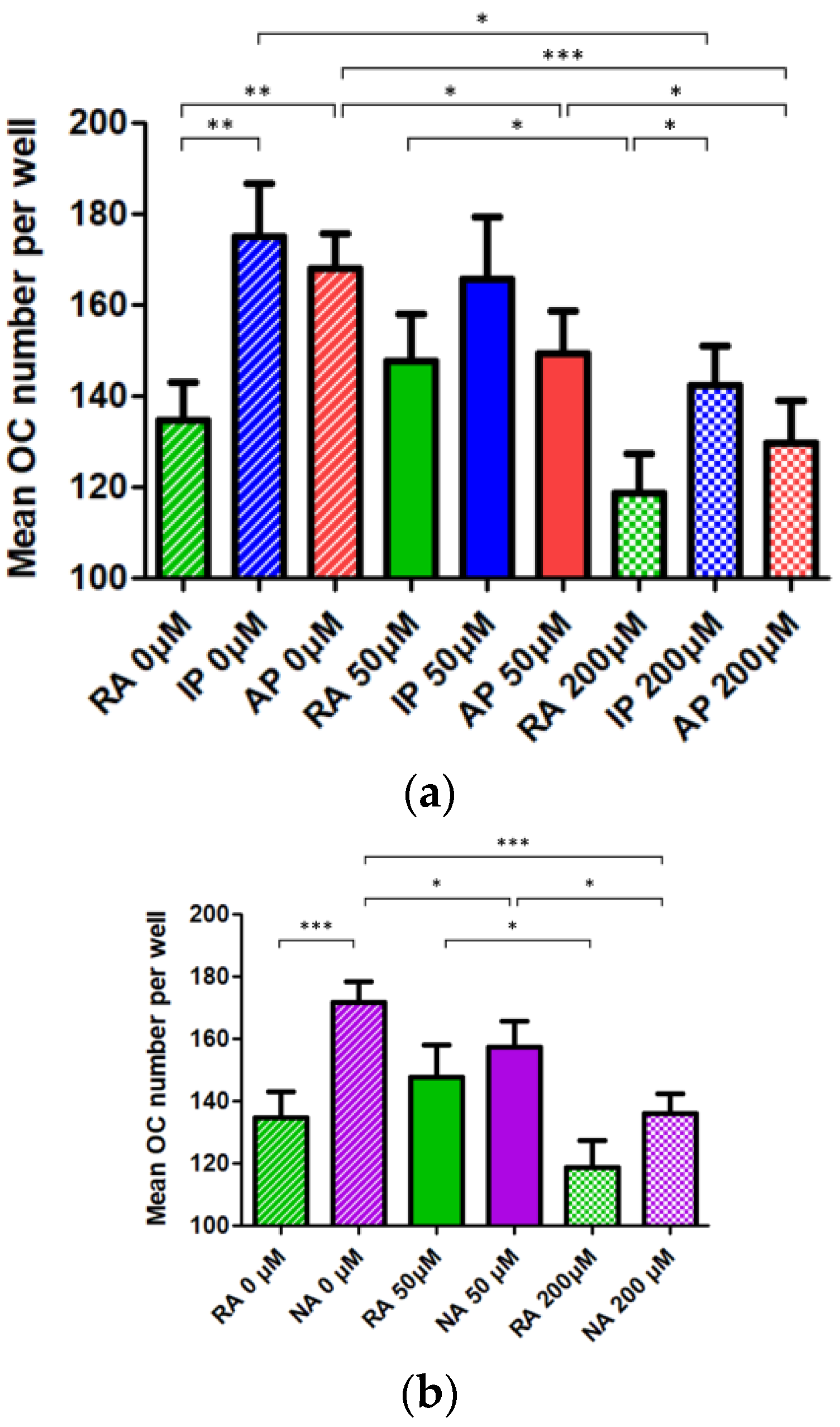
3.3. Inactive or Absent Cystinosin in Monocyte-Macrophage Precursors Favor Osteoclast Formation Whereas Cysteamine Treatment Impairs It Independently of the Genotype
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef]
- David, D.; Princiero Berlingerio, S.; Elmonem, M.A.; Oliveira Arcolino, F.; Soliman, N.; van den Heuvel, B.; Gijsbers, R.; Levtchenko, E. Molecular Basis of Cystinosis: Geographic Distribution, Functional Consequences of Mutations in the CTNS Gene, and Potential for Repair. Nephron 2019, 141, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Thoene, J.G.; Schneider, J.A. Cystinosis. N. Engl. J. Med. 2002, 347, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Emma, F.; Nesterova, G.; Langman, C.; Labbé, A.; Cherqui, S.; Goodyer, P.; Janssen, M.C.; Greco, M.; Topaloglu, R.; Elenberg, E.; et al. Nephropathic cystinosis: An international consensus document. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 4), iv87–iv94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markello, T.C.; Bernardini, I.M.; Gahl, W.A. Improved renal function in children with cystinosis treated with cysteamine. N. Engl. J. Med. 1993, 328, 1157–1162. [Google Scholar] [CrossRef]
- Brodin-Sartorius, A.; Tête, M.-J.; Niaudet, P.; Antignac, C.; Guest, G.; Ottolenghi, C.; Charbit, M.; Moyse, D.; Legendre, C.; Lesavre, P.; et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012, 81, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Gahl, W.A.; Balog, J.Z.; Kleta, R. Nephropathic cystinosis in adults: Natural history and effects of oral cysteamine therapy. Ann. Intern. Med. 2007, 147, 242–250. [Google Scholar] [CrossRef]
- Hohenfellner, K.; Rauch, F.; Ariceta, G.; Awan, A.; Bacchetta, J.; Bergmann, C.; Bechtold, S.; Cassidy, N.; Deschenes, G.; Elenberg, E.; et al. Management of bone disease in cystinosis: Statement from an international conference. J. Inherit. Metab. Dis. 2019, 42, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Bertholet-Thomas, A.; Claramunt-Taberner, D.; Gaillard, S.; Deschênes, G.; Sornay-Rendu, E.; Szulc, P.; Cohen-Solal, M.; Pelletier, S.; Carlier, M.-C.; Cochat, P.; et al. Teenagers and young adults with nephropathic cystinosis display significant bone disease and cortical impairment. Pediatr. Nephrol. 2018, 33, 1165–1172. [Google Scholar] [CrossRef]
- Florenzano, P.; Ferreira, C.; Nesterova, G.; Roberts, M.S.; Tella, S.H.; de Castro, L.F.; Brown, S.M.; Whitaker, A.; Pereira, R.C.; Bulas, D.; et al. Skeletal Consequences of Nephropathic Cystinosis. J. Bone Miner. Res. 2018, 33, 1870–1880. [Google Scholar] [CrossRef] [Green Version]
- Machuca-Gayet, I.; Quinaux, T.; Bertholet-Thomas, A.; Gaillard, S.; Claramunt-Taberner, D.; Acquaviva-Bourdain, C.; Bacchetta, J. Bone Disease in Nephropathic Cystinosis: Beyond Renal Osteodystrophy. Int. J. Mol. Sci. 2020, 21, 3109. [Google Scholar] [CrossRef] [PubMed]
- Conforti, A.; Taranta, A.; Biagini, S.; Starc, N.; Pitisci, A.; Bellomo, F.; Cirillo, V.; Locatelli, F.; Bernardo, M.E.; Emma, F. Cysteamine treatment restores the in vitro ability to differentiate along the osteoblastic lineage of mesenchymal stromal cells isolated from bone marrow of a cystinotic patient. J. Transl. Med. 2015, 13, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battafarano, G.; Rossi, M.; Rega, L.R.; Di Giovamberardino, G.; Pastore, A.; D’Agostini, M.; Porzio, O.; Nevo, N.; Emma, F.; Taranta, A.; et al. Intrinsic Bone Defects in Cystinotic Mice. Am. J. Pathol. 2019, 189, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Claramunt-Taberner, D.; Flammier, S.; Gaillard, S.; Cochat, P.; Peyruchaud, O.; Machuca-Gayet, I.; Bacchetta, J. Bone disease in nephropathic cystinosis is related to cystinosin-induced osteoclastic dysfunction. Nephrol. Dial. Transplant. 2017, 33, 1525–1532. [Google Scholar] [CrossRef] [PubMed]
- Ahlenstiel-Grunow, T.; Kanzelmeyer, N.K.; Froede, K.; Kreuzer, M.; Drube, J.; Lerch, C.; Pape, L. Switching from immediate- to extended-release cysteamine in nephropathic cystinosis patients: A retrospective real-life single-center study. Pediatr. Nephrol. 2017, 32, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.J.; Work, D.F. Measurement and estimation of GFR in children and adolescents. Clin. J. Am. Soc. Nephrol. 2009, 4, 1832–1843. [Google Scholar] [CrossRef] [Green Version]
- Ardeshirpour, L.; Cole, D.E.C.; Carpenter, T.O. Evaluation of bone and mineral disorders. Pediatr. Endocrinol Rev. 2007, 5 (Suppl. 1), 584–598. [Google Scholar]
- Shaw, J.L.V.; Cohen, A.; Konforte, D.; Binesh-Marvasti, T.; Colantonio, D.A.; Adeli, K. Validity of establishing pediatric reference intervals based on hospital patient data: A comparison of the modified Hoffmann approach to CALIPER reference intervals obtained in healthy children. Clin. Biochem. 2014, 47, 166–172. [Google Scholar] [CrossRef]
- Bernardor, J.; Flammier, S.; Ranchin, B.; Gaillard, S.; Platel, D.; Peyruchaud, O.; Machuca-Gayet, I.; Bacchetta, J. Inhibition of osteoclast differentiation by 1.25-D and the calcimimetic KP2326 reveals 1.25-D resistance in advanced CKD. J. Bone Miner. Res. 2020, 35, 2265–2274. [Google Scholar] [CrossRef]
- Kalatzis, V.; Nevo, N.; Cherqui, S.; Gasnier, B.; Antignac, C. Molecular pathogenesis of cystinosis: Effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum. Mol. Genet. 2004, 13, 1361–1371. [Google Scholar] [CrossRef]
- Topaloglu, R.; Gulhan, B.; İnözü, M.; Canpolat, N.; Yilmaz, A.; Noyan, A.; Dursun, İ.; Gökçe, İ.; Gürgöze, M.K.; Akinci, N.; et al. The Clinical and Mutational Spectrum of Turkish Patients with Cystinosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 1634–1641. [Google Scholar] [CrossRef] [Green Version]
- Servais, A.; Morinière, V.; Grünfeld, J.-P.; Noël, L.-H.; Goujon, J.-M.; Chadefaux-Vekemans, B.; Antignac, C. Late-onset nephropathic cystinosis: Clinical presentation, outcome, and genotyping. Clin. J. Am. Soc. Nephrol. 2008, 3, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiehntopf, M.; Schickel, J.; von der Gönne, B.; Koch, H.G.; Superti-Furga, A.; Steinmann, B.; Deufel, T.; Harms, E. Analysis of the CTNS gene in patients of German and Swiss origin with nephropathic cystinosis. Hum. Mutat. 2002, 20, 237. [Google Scholar] [CrossRef] [PubMed]
- Kalatzis, V.; Cherqui, S.; Jean, G.; Cordier, B.; Cochat, P.; Broyer, M.; Antignac, C. Characterization of a putative founder mutation that accounts for the high incidence of cystinosis in Brittany. J. Am. Soc. Nephrol. 2001, 12, 2170–2174. [Google Scholar] [CrossRef] [PubMed]
- Soliman, N.A.; Elmonem, M.A.; van den Heuvel, L.; Abdel Hamid, R.H.; Gamal, M.; Bongaers, I.; Marie, S.; Levtchenko, E. Mutational Spectrum of the CTNS Gene in Egyptian Patients with Nephropathic Cystinosis. JIMD Rep. 2014, 14, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shotelersuk, V.; Larson, D.; Anikster, Y.; McDowell, G.; Lemons, R.; Bernardini, I.; Guo, J.; Thoene, J.; Gahl, W.A. CTNS mutations in an American-based population of cystinosis patients. Am. J. Hum. Genet. 1998, 63, 1352–1362. [Google Scholar] [CrossRef] [Green Version]
- Gaillard, S.; Roche, L.; Lemoine, S.; Deschênes, G.; Morin, D.; Vianey-Saban, C.; Acquaviva-Bourdain, C.; Ranchin, B.; Bacchetta, J.; Kassai, B.; et al. Adherence to cysteamine in nephropathic cystinosis: A unique electronic monitoring experience for a better understanding. A prospective cohort study: CrYSTobs. Pediatr. Nephrol. 2021, 36, 581–589. [Google Scholar] [CrossRef]
- Andrzejewska, Z.; Nevo, N.; Thomas, L.; Chhuon, C.; Bailleux, A.; Chauvet, V.; Courtoy, P.J.; Chol, M.; Guerrera, I.C.; Antignac, C. Cystinosin is a Component of the Vacuolar H+-ATPase-Ragulator-Rag Complex Controlling Mammalian Target of Rapamycin Complex 1 Signaling. J. Am. Soc. Nephrol. 2016, 27, 1678–1688. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
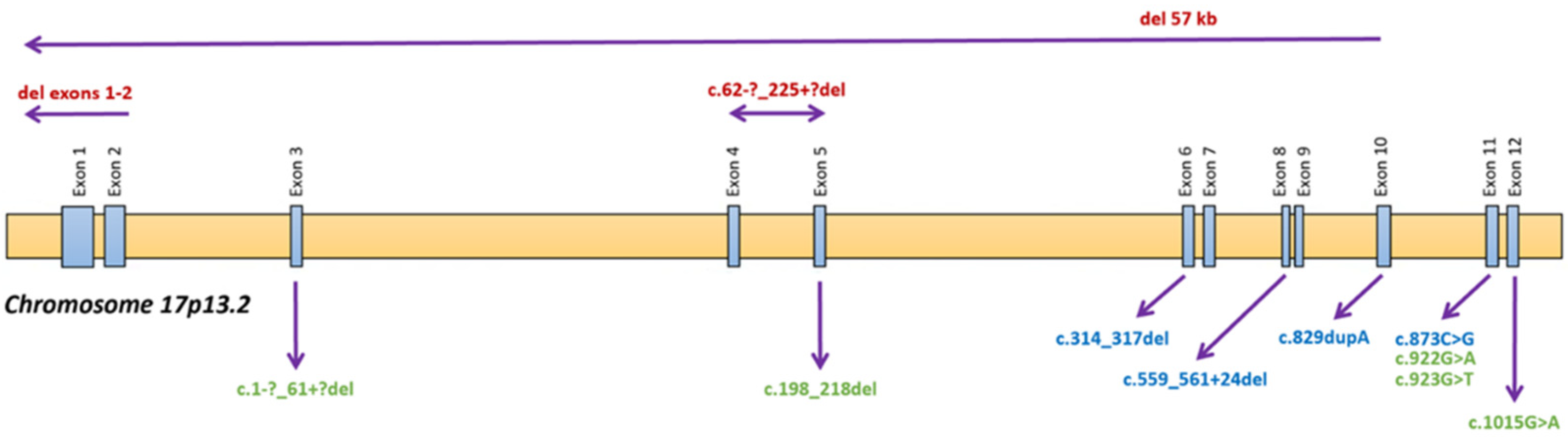
| Pt | Age at Diag (Years) | Renal Status | Age at Eval (Years) | Sex | DNA Mutation | Protein Predicted Effect | Affected Exons | GFR | Cysteamine Daily Dose (mg/m2) | Type of Cysteamine | LHL < 1 | LHL < 2 | rhGH | Past of Fracture | BD | BP | Any Bone Symptoms | Orthopedic Surgery |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 1.3 | C | 4 | M | c.922G > A/c.922G > A | RA | 11/11 | 34 | 564 | DR | 0.7 | No | No | No | No | No | No | |
| 2 | 2.5 | T | 30 | F | c.1015G > A/del 57 kb | RA | 12/1 to 10 | 62 | 1632 | SA | 2.1 | No | No | Yes | No | Yes | No | |
| 3 | 1.7 | C | 15 | F | c.829dupA/c.829dupA | IP | 10/10 | 38 | 1630 | SA | 1.9 | Yes | No | Yes | Yes | Yes | No | |
| 4 | 0.9 | C | 5 | F | del 57 kb/del 57 kb | AP | 1 to 10/1 to 10 | 71 | 954 | DR | 1 | No | No | No | No | No | No | |
| 5 | 1.0 | C | 7 | F | del 57 kb/c.1-?_61 + ?del | RA | 1 to 10/3 | 79 | 580 | DR | 0.2 | No | No | No | No | No | No | |
| 6 | 5.5 | T | 17 | M | c.314_317del/c.314_317del | IP | 6/6 | 112 | 986 | DR | 0.7 | No | Yes | Yes | Yes | Yes | No | |
| 7 | 0.8 | C | 16 | M | del 57 kb/del 57 kb | AP | 1 to 10/1 to 10 | 23 | 1236 | SA | 3.7 | Yes | No | Yes | Yes | Yes | Yes | |
| 8 | 1.2 | C | 3 | F | del 57 kb/c.873C > G | IP | 1 to 10/11 | 149 | 2505 | SA | 1.8 | No | No | No | No | No | No | |
| 9 | 0.1 | C | 2 | F | del 57 kb/c.873C > G | IP | 1 to 10/11 | 105 | 3607 | SA | 2.2 | No | No | No | No | No | No | |
| 10 | 4.0 | T | 18 | F | del 57 kb/c.62-?_225 + ?del | AP | 1 to 10/4 and 5 | 105 | 1457 | SA | 0.2 | No | No | No | No | No | No | |
| 11 | 1.0 | C | 14 | F | del 57 kb/c.62-?_225 + ?del | AP | 1 to 10/4 and 5 | 64 | 1761 | SA | 1.1 | Yes | No | No | No | No | No | |
| 12 | 2.0 | C | 9 | M | del 57 kb/del 57 kb | AP | 1 to 10/1 to 10 | 84 | 1420 | DR | 0.8 | Yes | No | No | No | No | No | |
| 13 | 5.3 | C | 8 | M | c.198_218del/c.559_561 + 24del | RA | 5/8 | 62 | 1111 | DR | 1.4 | Yes | No | Yes | No | Yes | No | |
| 14 | 1.3 | C | 15 | M | del 57 kb/del 57 kb | AP | 1 to 10/1 to 10 | 127 | 1232 | DR | 1 | Yes | No | No | No | No | No | |
| 15 | 1.1 | T | 61 | F | del 57 kb/c.923G > T | RA | 368 (theory) but 0 in reality | DR | 0.7 | No | Yes | No | No | Yes | No | |||
| 16 | 6.5 | T | 18 | M | Del exons 1–2/del exons 1–2 | AP | 1 and 2 | 59 | 1039 | DR | 3.3 | No | No | Yes | Yes | Yes | Yes | |
| 17 | 2.0 | C | 14 | M | Del exons 1–2/del exons 1–2 | AP | 1 and 2 | 28 | 1902 | DR | 1.2 | No | No | Yes | No | Yes | Yes |
| Nephropathic Cystinosis Patients | Short Acting Cysteamine | Delayed Release Cysteamine |
|---|---|---|
| Number of patients | 7 | 10 |
| Age (y/o) | 15 (2–30) | 12 (4–61) |
| Cysteamine daily dose (mg/m2) * | 1632 (1236–3607) | 1012 (368–1902) |
| Patients in the target for LHL * | 3 (43%) | 9 (90%) |
| GFR (mL/min per 1.73 m2) | 46 (16–149) | 65 (33–84) |
| Calcium (mmol/L) | 2.27 (2.11–2.50) | 2.42 (2.23–2.92) |
| Phosphate (standard deviation for age) | −1.8 (−4.2;1.7) | −1.5 (−3.6;2.4) |
| PTH (ng/L) | 34 (18–127) | 20 (5–90) |
| 25-D (ng/mL) | 28 (10–42) | 26 (21–49) |
| Total ALP (times the upper physiological value for gender and age) | 0.87 (0.41–4.29) | 0.74 (0.28–1.19) |
| Any bone symptoms (%) | 3 (43%) | 5 (50%) |
| Number of osteoclasts obtained at the end of the differentiation process | 168 (97–187) | 162 (111–203) |
| Nephropathic Cystinosis Patients | Conservative Management | Renal Transplantation |
|---|---|---|
| Number of patients | 12 | 5 |
| Patients receiving SA cysteamine | 5 | 2 |
| Age (y/o) * | 9 (2–16) | 18 (17–61) |
| Cysteamine daily dose (mg/m2) | 1328 (564–3607) | 1039 (368–1632) |
| Patients in the target for LHL | 9 (75%) | 3 (60%) |
| GFR (mL/min per 1.73 m2) | 65 (16–149) | 56 (45–76) |
| Calcium (mmol/L) | 2.40 (2.23–2.92) | 2.42 (2.11–2.57) |
| Phosphate (standard deviation for age) | −1.6 (−4.2;2.4) | −1.4 (−2.8;−0.5) |
| PTH (ng/L) | 21 (8–90) | 32 (5–127) |
| 25-D (ng/mL) | 28 (10–49) | 26 (22–26) |
| Total ALP (times the upper physiological value for gender and age) | 0.9 (0.3–4.2) | 0.5 (0.4–0.8) |
| Any bone symptoms (%) * | 33 | 80 |
| Number of osteoclasts obtained at the end of the differentiation process | 159 (94–203) | 165 (105–181) |
| Patient Number | Protein Functionality | Cysteamine Concentration | ||
|---|---|---|---|---|
| 0 µM | 50 µM | 200 µM | ||
| 1 | RA | 203 ± 41 | 185 ± 44 | 147 ± 31 |
| 2 | RA | 105 ± 11 | 122 ± 13 | 67 ± 6 |
| 5 | RA | 111 ± 12 | 121 ± 19 | 97 ± 14 |
| 13 | RA | 117 ± 7 | 125 ± 12 | 107 ± 9 |
| 15 | RA | 197 ± 9 | 210 ± 20 | 178 ± 8 |
| 3 | IP | 44 ± 4 | 36 ± 5 | 29 ± 3 |
| 6 | IP | 181 ± 14 | 183 ± 23 | 147 ± 14 |
| 8 | IP | 171 ± 16 | 180 ± 13 | 160 ± 13 |
| 9 | IP | 187 ± 19 | 206 ± 22 | 205 ± 25 |
| 4 | AP | 65 ± 7 | 75 ± 6 | 52 ± 7 |
| 7 | AP | 94 ± 11 | 90 ± 6 | 100 ± 6 |
| 10 | AP | 168 ± 14 | 173 ± 17 | 127 ± 5 |
| 11 | AP | 48 ± 3 | 48 ± 4 | 42 ± 5 |
| 12 | AP | 148 ± 9 | 115 ± 9 | 87 ± 6 |
| 14 | AP | 44 ± 5 | 48 ± 4 | 39 ± 3 |
| 16 | AP | 162 ± 16 | 129 ± 17 | 114 ± 14 |
| 17 | AP | 172 ± 20 | 121 ± 14 | 105 ± 10 |
| DNA Mutation | Protein Mutation | Protein Predicted Effect | Justification Based on Experimental Data and Clinical Phenotype | |
|---|---|---|---|---|
| 1 | c.922G > A/c.922G > A | p.G308R/p.G308R | RA | Clinically quite severe (advanced CKD at 4 years of age) despite early diagnosis and satisfactory compliance, quite low and stable cysteamine doses with LHL within the target, in experimental models prediction of abolished transport [20]. |
| 2 | c.1015G > A/del 57 kb | p.G339R/p.? | RA | Heterozygous form of the large deletion of CTNS + point mutation on the last exon. Transplantation at the age of 11 years in 2000 (median age at that time for transplantation in historical cohorts), standard cysteamine daily dose, and prediction of severe impact (but no functional analysis of transport) [21]. Point mutation in the last transmembrane domain in the C-terminal part may be important for protein–protein interaction. |
| 3 | c.829dupA/c.829dupA | p.T277NfsX19/p.T277NfsX19 | IP | Premature stop [21] |
| 4 | del 57 kb/del 57 kb | p.?/p.? | AP | Homozygous form of the large deletion of CTNS |
| 5 | del 57 kb/c.1-?_61 + ?del | p.?/p.? | RA | Heterozygous form of the large deletion of CTNS + deletion exon 3 (first coding exon). This second mutation was never described. Is there an alternative start? Clinically stable, satisfactory compliance, and quite low and stable cysteamine doses with LHL within the target. |
| 6 | c.314_317del/c.314_317del | p.H105PfsX12/p.H105PfsX12 | IP | Stop in exon 6, this second mutation was not described. |
| 7 | del 57 kb/del 57 kb | p.?/p.? | AP | Homozygous form of the large deletion of CTNS |
| 8 | del 57 kb/c.873C > G | p.?/p.Tyr291X | IP | Heterozygous form of the large deletion of CTNS + early stop in exon 11. Severe clinical phenotype. |
| 9 | del 57 kb/c.873C > G | p.?/p.Tyr291X | IP | Heterozygous form of the large deletion of CTNS + early stop in exon 11. Severe clinical phenotype. |
| 10 | del 57 kb/c.62-?_225 + ?del | p.?/p.? | AP | Heterozygous form of the large deletion of CTNS + exons 4 and 5 missing at the beginning of the protein. This second mutation was never described. |
| 11 | del 57 kb/c.62-?_225 + ?del | p.?/p.? | AP | Heterozygous form of the large deletion of CTNS + exons 4 and 5 missing at the beginning of the protein. This second mutation was never described. |
| 12 | del 57 kb/del 57 kb | p.?/p.? | AP | Homozygous form of the large deletion of CTNS. |
| 13 | c.198_218del/c.559_561 + 24del | p.Ile67_Pro73del/splicing | RA | Clinically stable, satisfactory compliance, and quite low and stable cysteamine doses with LHL within the target. The first mutation is described with residual activity [22]. The second mutation induces a splicing and leads to a truncated protein [24]. |
| 14 | del 57 kb/del 57 kb | p.?/p.? | AP | Homozygous form of the large deletion of CTNS. |
| 15 | del 57 kb/c.923G > T | p.?/p.G308V | RA | Diagnosis at 13 months, ESRD 14 years, transplantation 18 years, still on the first graft, bad compliance, two pregnancies. Initiation of CYSTAGON at 37 years of age, switch to PROCYSBI at the age of 60 years. Very atypical clinical course with mild phenotype. Moreover, the described functional impact of the second mutation favors the existence of residual activity [23]. |
| 16 | Del exons 1–2/del exons 1–2 | p.?/p.? | AP | Severe clinical phenotype with muscular impairment. Likely corresponds to the homozygous form of the large CTNS deletion. Could correspond to a contiguous gene syndrome. |
| 17 | Del exons 1–2/del exons 1–2 | p.?/p.? | AP | Severe clinical phenotype. Likely corresponds to the homozygous form of the large CTNS deletion. Could correspond to a contiguous gene syndrome. |
| Nephropathic Cystinosis Patients | RA | IP | AP |
|---|---|---|---|
| Number of patients | 5 | 4 | 8 |
| Age (y/o) | 22 (4;61) | 9 (2;17) | 14 (5;18) |
| CKM/ Tx (N/N) | 3/2 | 3/1 | 6/2 |
| Past of rhGH therapy (N) | 1 | 1 | 4 |
| Cysteamine daily dose (mg/m2) * | 777 (0;1632) | 1932 (986;3607) | 1375 (954;1902) |
| Number of patients receiving SA cysteamine | 1 (20%) | 3 (75%) | 3 (38%) |
| Proportion of patients in the target for LHL | 4 (80%) | 3 (75%) | 5 (62%) |
| Number of patients with past of rhGH | 1 | 1 | 4 |
| GFR (ml/min per 1.73 m2) | 57 (34;79) | 101 (38;149) | 70 (23;127) |
| Calcium (mmol/L) | 2.5 (2.1;2.9) | 2.5 (2.4;2.5) | 2.3 (2.2;2.6) |
| Phosphate (standard deviation for age) | −1.4 (−3.6;2.4) | −1.9 (−2.9;−1.2) | −1.3 (−4.2;1.7) |
| PTH (ng/L) | 35 (8;127) | 22 (18;37) | 36 (5;90) |
| 25-D (ng/mL) | 28 (22;35) | 30 (26;39) | 28 (10;49) |
| Total ALP (times the upper physiological value for gender and age) | 0.7 (0.3;1.2) | 0.7 (0.4;0.9) | 1.5 (0.4;4.3) |
| Any bone symptoms | 60% | 50% | 38% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quinaux, T.; Bertholet-Thomas, A.; Servais, A.; Boyer, O.; Vrillon, I.; Hogan, J.; Lemoine, S.; Gaillard, S.; Alioli, C.; Vasseur, S.; et al. Response to Cysteamine in Osteoclasts Obtained from Patients with Nephropathic Cystinosis: A Genotype/Phenotype Correlation. Cells 2021, 10, 2498. https://doi.org/10.3390/cells10092498
Quinaux T, Bertholet-Thomas A, Servais A, Boyer O, Vrillon I, Hogan J, Lemoine S, Gaillard S, Alioli C, Vasseur S, et al. Response to Cysteamine in Osteoclasts Obtained from Patients with Nephropathic Cystinosis: A Genotype/Phenotype Correlation. Cells. 2021; 10(9):2498. https://doi.org/10.3390/cells10092498
Chicago/Turabian StyleQuinaux, Thomas, Aurélia Bertholet-Thomas, Aude Servais, Olivia Boyer, Isabelle Vrillon, Julien Hogan, Sandrine Lemoine, Ségolène Gaillard, Candide Alioli, Sophie Vasseur, and et al. 2021. "Response to Cysteamine in Osteoclasts Obtained from Patients with Nephropathic Cystinosis: A Genotype/Phenotype Correlation" Cells 10, no. 9: 2498. https://doi.org/10.3390/cells10092498
APA StyleQuinaux, T., Bertholet-Thomas, A., Servais, A., Boyer, O., Vrillon, I., Hogan, J., Lemoine, S., Gaillard, S., Alioli, C., Vasseur, S., Acquaviva, C., Peyruchaud, O., Machuca-Gayet, I., & Bacchetta, J. (2021). Response to Cysteamine in Osteoclasts Obtained from Patients with Nephropathic Cystinosis: A Genotype/Phenotype Correlation. Cells, 10(9), 2498. https://doi.org/10.3390/cells10092498